

Abstract
Traumatic brain injury (TBI) and can lead to persistent hypogonadotropic hypogonadism (PHH) and poor outcomes. We hypothesized that autoimmune and inflammatory mechanisms contribute to PHH pathogenesis. Men with moderate-to-severe TBI (n = 143) were compared with healthy men (n = 39). The TBI group provided blood samples 1–12 months post-injury (n = 1225). TBI and healthy control (n = 39) samples were assayed for testosterone (T) and luteinizing hormone (LH) to adjudicate PHH status. TBI samples 1–6 months post-injury and control samples were assayed for immunoglobulin M (IgM)/immunoglobulin G (IgG) anti-pituitary autoantibodies (APA) and anti-hypothalamus autoantibodies (AHA). Tissue antigen specificity for APA and AHA was confirmed via immunohistochemistry (IHC). IgM and IgG autoantibodies for glial fibrillary acid protein (GFAP) (AGA) were evaluated to gauge APA and AHA production as a generalized autoimmune response to TBI and to evaluate the specificity of APA and AHA to PHH status. An inflammatory marker panel was used to assess relationships to autoantibody profiles and PHH status. Fifty-one men with TBI (36%) had PHH. An age-related decline in T levels by both TBI and PHH status were observed. Injured men had higher APA IgM, APA IgG, AHA IgM, AHA IgG, AGA IgM, and AGA IgG than controls (p < 0.0001 all comparisons). However, only APA IgM (p = 0.03) and AHA IgM (p = 0.03) levels were lower in the PHH than in the non-PHH group in multivariate analysis. There were no differences in IgG levels by PHH status. Multiple inflammatory markers were positively correlated with IgM autoantibody production. PHH was associated with higher soluble tumor-necrosis-factor receptors I/II, (sTNFRI, sTNFRII), regulated on activation, normal T-cell expressed and secreted (RANTES) and soluble interleukin-2-receptor-alpha (sIL-2Rα) levels. Higher IgM APA, and AHA, but not AGA, in the absence of PHH may suggest a beneficial or reparative role for neuroendocrine tissue-specific IgM autoantibody production against PHH development post-TBI.
Keywords: autoantibodies, autoimmunity, hypogonadism, hypopituitarism, IgG autoantibody, IgM autoantibody, inflammation, TBI
Introduction
Traumatic brain injury (TBI) results in at least 2,500,000 visits to hospitals and emergency departments annually in the United States, and the incidence of TBI is increasing.1,2 The long-term effects of TBI are both prevalent and debilitating; it is estimated that 1.1% of people in the United States live with TBI-related disabilities.3 One likely significant contributor to post-injury disability is post-traumatic hypopituitarism, which is a well-documented chronic complication of TBI.4–7 In the largest prospective cohort screening study to date, 340 individuals with TBI receiving inpatient rehabilitation were screened for pituitary hormone deficiencies.4 Thirty-seven percent had laboratory values consistent with hypopituitarism, and the most common deficiency was hypogonadism, where 40% of men were deficient in testosterone (T) (estradiol was not reported in women). Other work suggests that growth hormone may be the most common post-traumatic pituitary deficiency, with gonadotropin, adrenocorticotropic, and thyroid stimulating hormones having lower incidence.5 Despite these differences with incidence, recent work suggests that symptoms specific to hypogonadism are often predictive of other co-occurring neuroendocrinopathies; therefore, hypogonadism symptoms may be a useful screening tool for identifying individuals after TBI who need comprehensive testing for hypopituitarism.8 In our recent work, we reported that 44% of men with severe TBI had persistent hypogonadotropic hypogonadism (PHH).9 We further observed that PHH was associated with worse global outcome scores, more disability, greater fatigue, and reduced functional cognition at 6 and 12 months post-TBI. These results corroborate prior data showing post-traumatic hypogonadism to be a complication associated with worse outcomes.10–13
The mechanism for post-traumatic hypopituitarism, including hypogonadism, remains undefined. Traditionally, it has been held that hypopituitarism may be caused by traumatic lesions and vascular injury of the mechanically vulnerable pituitary or hypothalamus, as early post-mortem studies recorded these findings in the majority of autopsies of patients with fatal TBI.14,15 Among TBI survivors, associations have been reported between post-traumatic hypopituitarism and abnormalities on magnetic resonance imaging (MRI) of the pituitary gland.6 However, gross structural changes do not entirely explain all cases of post-traumatic hypopituitarism. Among individuals with mild TBI, where injury does not cause visible structural abnormalities on imaging studies, rates of post-traumatic hypopituitarism up to 45% have been reported.16,17 These findings suggest that hypopituitarism is not strictly related to direct trauma of the pituitary or hypothalamus or even injury severity. Other mechanisms for post-traumatic hypopituitarism have been explored, including alterations in the chronic inflammatory and autoimmune responses.18–20
Increasing collective evidence, including some of our own work, suggests that blood samples collected after TBI contain autoantibodies to multiple brain proteins including glial fibrillary acidic protein (GFAP), S100 calcium-binding protein B (S100B), myelin basic protein (MBP), and glutamate receptors.21,22 Some evidence also suggests that autoimmunity development against the pituitary gland and hypothalamus could be involved in post-traumatic hypopituitarism.23 Blood–brain barrier dysfunction occurs rapidly after TBI,24,25 which may allow for systemic exposure to otherwise privileged central nervous system (CNS) antigens and subsequent development of autoantibodies.22 One group has reported that higher serum titers of anti-pituitary and anti-hypothalamus immunoglobulin G (IgG) antibodies detected by indirect immunofluorescence are associated with pituitary deficits in 25 individuals with TBI up to 5 years after injury.20,26 Their studies have not yet been replicated in independent populations. The data specifically include patient assessments at 3 or 5 years post-TBI and measure IgG class antibodies. These data support autoantibody production as pathogenic in post-traumatic hypopituitarism, which aligns with classic autoimmune disease pathophysiology.
Immunoglobulin M (IgM) antibodies, however, are the earliest isotype expressed during immune development and can promote B and T cell secretion to reduce tissue inflammation and damage.27 In some cases, IgM antibodies have been linked to directly neutralizing pathogens, while initiating adaptive immune responses from follicular B cells.28,29 Further, some have hypothesized that under certain conditions, autoimmune activity involving IgM class immunoglobulins may be beneficial to repair and recovery after CNS injury.30 Characterizing autoantibody production after CNS injury may help improve our understanding of TBI pathobiology and tissue repair.
In addition to autoantibody production, there are other inflammatory factors that contribute to the adaptive immune response. Prior reports hypothesize that autoantibody production after brain injury may promote tissue repair by facilitating well-regulated neuroinflammation.31,32 There is also an influx of peripheral inflammatory cells into the brain following BBB breach.33 The time frame in which cellular immunity occurs suggests a potentially prolonged period for therapeutic intervention that moderates both pro- and anti-inflammatory responses. The harmful or beneficial impacts of systemic immune responses depend largely on timing post-injury.24 We have done seminal work in a clinical population with moderate to severe TBI in characterizing chronic elevations of serum inflammatory markers and their associations with long-term global outcome.34 However, additional characterization of chronic inflammatory biomarker patterns, including autoantibody production, could inform the recovery mechanisms needed to restore homeostatic balance and mitigate the occurrence and secondary conditions such as PHH.
We hypothesized that IgM anti-pituitary autoantibody (APA) and anti-hypothalamus autoantibody (AHA) could be detected in serum via immunological assays up to 6 months after TBI, and that these levels would be specifically associated with PHH status and T levels over time. We also hypothesized that PHH relationships would be specific to neuroendocrine tissue autoantibody production. We further hypothesized that there may be key inflammatory markers associated with adaptive immunity (including autoantibody production) and PHH status that might begin to inform inflammatory repair and recovery mechanisms after neurological injury.
In a prospective cohort of men with moderate and severe TBI, we characterized T levels over time, PHH status, longitudinal serum APA and AHA levels, and how these levels relate to PHH as markers of hypopituitarism. We also measured longitudinal serum autoantibody levels to the ubiquitous astrocytic glial fibrillary acid protein (GFAP) as a control assay in the same samples as APA and AHA measurements in order to demonstrate a generalized autoantibody response (in addition to APA and AHA) after TBI. Anti-GFAP autoantibodies (AGA) were also measured in this cohort to concurrently demonstrate the specificity of APA and AHA levels to PHH status. We also provide immunohistochemistry (IHC) evidence of the specificity of our APA and AHA autoantibody assays to neuroendocrine tissue type, and we characterized multiple inflammatory markers over this same time period to evaluate associations with PHH and autoantibody production.
Methods
Study design and population
The University of Pittsburgh Institutional Review Board approved this research. Informed consent was provided by next-of-kin for participants with TBI. TBI participants whose cognitive status improved sufficiently during the study period were later given the opportunity to self-consent. Healthy volunteers self-consented to provide blood samples.
The study cohorts presented here build on previous cohorts reported9,13 and represent a prospective, longitudinal, observational study. We prospectively recruited individuals who were not on testosterone therapy from two cohorts. The first cohort presented to our university hospital level 1 trauma center with severe TBI, defined by Glasgow Coma Scale (GCS) score ≤8 at presentation and confirmed computed tomographic findings, survived to acute care discharge, and agreed to be followed for 1 year (∼ 12–15 months) post-injury. The second cohort presented with moderate or severe TBI (GCS <13), were admitted to acute inpatient rehabilitation after acute hospital discharge, and agreed to be followed for 1 year (∼ 12–15 months) post-injury. This analysis included men 17–78 years of age, in whom we were able to collect at least two blood samples beginning at least 2 weeks after injury. Individuals were excluded if they had a history of hypothalamic or pituitary tumors, orchiectomy, luteinizing hormone (LH) therapy, untreated thyroid disease prior to injury, or hormone levels consistent with primary hypogonadism. Individuals were excluded in cases of primary hypogonadism, wherein hormone values for T were lower than our medical center pathology laboratory minimum normal T level while LH values were higher than the maximum cutoff across multiple sample points per subject.
Demographic and injury information were obtained from patient records, including age, body mass index (BMI), education level, race, GCS score (best in 24 h).35 injury severity score,36 length of hospital stay, mechanism of injury, and neuroradiology results from acute admission. Neuroimaging reports for computed tomography (CT) and MRI during acute hospital stay were available and analyzed for 128 men. Initial CT imaging reports were not available for those not presenting to our medical center for acute care.
As psychotropic medication use can impact neuroendocrine function,37–41 we assessed psychotropic medication use in our cohort based on guidelines and medication groups outlined by the National Alliance on Mental Illness.42 Antipsychotics, mood stabilizers, antidepressants, anti-panic/anti-obsessive/anti-anxiety agents, stimulants, and opioid analgesics were reviewed (see Supplementary Table S1). The reported cohort (n = 143) spanned two different data collection periods, which recorded medication data differently. For 64 individuals, medication use data were obtained during monthly collections over the 1 year study period. Medication data for 60 individuals were obtained at 6 and 12 months post-TBI only. Across both collection periods, there were 19 individuals for whom medication data collection was attempted but was unobtainable.
For the entire cohort with medication data available (n = 124), individual research records were reviewed for medication usage in the categories presented in Table S1 at any time in the 1st year and by PHH status. Thus, in the total group (n = 124), we explored associations between frequency of psychotropic medication use (ever reported at any time during the 1 year study period) and neuroendocrine dysfunction post-TBI. When possible, individuals (n = 64) were assessed for monthly use (yes/no) for each medication category based on Supplementary Table S1. Also, in the subgroup with monthly data, a cumulative psychotropic medication burden was assigned to individuals to account for number of medications used in addition to the duration of use (in months) for each medication. This psychotropic medication burden score was tabulated by taking medications reported multiplied by the number of months used for each medication (i.e., medication months). For example, if one medication was reported for 3 months, then a burden score of 3 was assigned. Similarly, if three medications were reported for 3 months each, then a medication burden score of 9 was assigned.
Derivation of the cohorts used for analysis
Figure 1 shows how our cohort was derived based on the data available for analysis. 143 men (n = 1225 samples) had T and LH data generated that were used to clinically adjudicate PHH status over the course of 1 year post-injury. Of these men, 137 had autoantibody data (n = 592 samples), and 138 (n = 930 samples) had inflammation data measured up to 6 months post-TBI. Furthermore, 132 men out of this cohort had both autoantibody and inflammatory data for analysis. Of the 137 men with autoantibody data, 124 had at least two samples at time points over the course of the first 6 months post-TBI, meeting our criteria for group-based trajectory analysis (TRAJ).
FIG. 1.
Consolidated Standards of Reporting Trials (CONSORT) flow chart. Out of the 143 men adjudicated for persistent hypogonadotropic hypogonadism (PHH), 137 had autoantibody data and 138 had inflammation data, and 132 participants had both autoantibody and inflammation data. Out of the 137 with autoantibody data, 124 individuals received trajectory group assignments. AAb, autoantibody; TRAJ, group-based trajectory analysis.
Upon collection, samples were centrifuged, aliquoted in polypropylene cryovials, and stored at −80°C until analysis. Healthy male volunteers provided blood samples to serve as a reference/control. Controls were 18–68 years of age and had no history of brain injury, neurological disorder, or endocrine disorder. Autoantibody levels were measured in 39 male volunteers (median age, 31 years; range 18–68 years). T and LH levels were measured in a subset of 11 male volunteers (median age, 21 years; range, 19–57 years).
T and LH assays and PHH adjudication
Of the 1225 samples utilized for this analysis, a portion of these serum samples (n = 786 samples) were assayed for T and LH levels as described previously9 using a radioimmunoassay with the Coat-A-Count® In-vitro Diagnostic Test Kit (Siemens Healthcare Diagnostics). Kits included a solid-phase 125I radioimmunoassay (RIA) designed for direct, quantitative measurements. T and LH inter-assay and intra-assays percent coefficients of variation (%CV) were all <10%. Samples with undetectable levels were assigned the minimum detection limit of the assay. The remaining samples (n = 439 samples) were assayed for T (Monobind Inc.) and LH (BioVendor) levels using enzyme-linked immunosorbent assays (ELISAs). The inter-assay and intra-assay %CV for both ELISAs were <10%, and any samples that fell below the detection limit were assigned the value of the detection limit for the respective assay. We measured T and LH for a subset of samples (n = 47 for T and n = 44 for LH) using both RIA and ELISA in order to determine a correlation between the two assay methodologies. Linear regression equations were generated for both T and LH data to fit the correlation between hormone measurements by RIA and ELISA. [(RIA T = 1.0023 x (ELISA T) +0.2451)] and [(RIA LH = 1.3248 x (ELISA LH) – 0.2183)]. ELISA sample values were converted using these linear regression equations, and the measurements for the two assay types were pooled into one data set.
PHH status was determined as reported in our prior work.9 Briefly, individuals with at least two blood samples collected between 1 and 12 months post-injury were dichotomized into PHH or non-PHH groups. The average number of T and LH samples used per participant to adjudicate our cohort for PHH status was 8.57 samples. As per our previous reports,9 those with at least 50% of samples meeting criteria for hypogonadotropic hypogonadism (T < 10 nmol/L [minimum normal range level] with LH <5.6 IU/L [maximum normal range level]) were categorized as having PHH. Individuals with <50% of samples meeting these criteria were categorized in the non-PHH group.
Pituitary, hypothalamus, and GFAP autoantibody ELISA protocols
Autoantibody levels were measured in serum (n = 592 samples; n = 137 subjects) using a customized direct ELISA approach. Briefly, custom 96-well ELISA plates were coated with antigens from bovine hypothalamic lysate (Science Cell Research, cat#0613) or bovine pituitary extract lysate (Sigma, cat#P1476) (2 μg/well) or 50 ng/well purified human full length GFAP protein (#DXAG-001, Dx-Sys, CA). After plate preparation, 1 μL of human serum sample was mixed with 99 μL of Start-Block buffer and transferred to each well (1:100 dilution) with incubation at 4°C overnight with shaking. Plates were washed again 4 times with Tris-Buffered Saline and Tween 20 (TBST) wash buffer. Anti-Human IgG/IgM HRP-conjugate (Jackson ImmunoResearch, as 1:10,000 in TBST Start-block blocking buffer) was added as a 100 μL aliquot to each well. These secondary antibodies are specific to either human IgM or human IgG that distinguish subjects' titer for AGA, APA, or AHA IgG and IgM, respectively. Plates were incubated at 25°C, with shaking for 45 min. After plate washing with 4x TBST, 100 μL tetramethylbenzidine (TMB) substrate was added to develop color for 15 min. Stop Solution (100 μL) was then added, and plates were read at 450 nm for yellow color of final product.
As a part of assay development, two standard curves were constructed: one for human IgG, and one for human IgM using their respective antibody isotype (see Fig. 2). It is important to note that these standard curves were not APA, AHA, or AGA specific, but rather, IgM or IgG specific. They were used on the same plate when assaying APA, AHA or AGA levels for our serum samples. Standard curves were introduced by adding 0, 17, 26, 39, 58.5, 88, 131.5, 198, 296, 444, 666, and 1000 ng/mL (50 μL) of either purified human IgG (Sigma Co. cat# I4506) or human IgM (Sigma Co. cat#I8260) to the first rows of the ELISA plate (see Fig. 2). Upon blocking and washing as described, anti-human IgG or IgM HRP conjugate (1:10,000 in TBST Startblock blocking buffer) was added to these wells followed by TMB substrate addition. Thus, optical density readings reflect the presence of human APA, AHA, or AGA, IgG or IgM. These readings were converted to IgG or IgM concentration in μg/mL, and 50 μL of standards were used, while 1 μL of human serum per sample was loaded per assay; thus, a dilution factor of 50 was applied to determine the actual human subject APA, AHA, and AGA serum concentrations. Intra-assay %CV was 5–10%, and inter-assay %CV was 15–20%.
FIG. 2.

Immunoglobulin (Ig)M and IgG autoantibody standard curves. IgM and IgG class autoantibodies enzyme-linked immunosorbent assays (ELISA) standard curves used to calculate the anti-pituitary autoantibodies (APA), anti-hypothalamus autoantibodies (AHA), and anti- glial fibrillary acid protein (GFAP) autoantibody concentrations (titers) in human subject serum. (A) IgM standard curves, (B) IgG standard curves. Calculated IgG or IgM concentrations ± standard deviation (STD) from four independent runs were shown. Linear regression fitting results are shown.
Pituitary and hypothalamic tissue immunohistochemical staining with APA and AHA in human TBI serum
Immunocytochemistry (IHC) analysis was performed on human pituitary and human hypothalamus paraffin sections per vendor instructions (Zyagen, CA). Slides were first de-paraffinized through Trilogy solution (Cell Marque, CA) by incubating for 10 min at 95°C and then blocked for endogenous peroxides with 3% hydrogen peroxide. Then routine staining was performed after a 1-h blocking step in 10% goat serum. TBI and control participants' serum, at a dilution of 1:200, was used and incubated over night at 4°C. Alexa 555-conjugated goat–anti-human IgG or IgM secondary antibody (Invitrogen, CA) was added at a dilution of 1:1000 and incubated for 1 h at room temperature. The tissues were counterstained with 4,6-diamidine-2-phenylindole (DAPI) for 5 min (Vector Laboratories, Burlingame, CA). Fluorescent images were captured with an x40 objective on the OLYMPUS DP71 fluorescent microscope (Olympus America Inc., Center Valley, PA).
Inflammatory marker Luminex™ bead assay
Cytokine levels were measured in serum (n = 930 samples) using a Luminex bead array assay (Millipore, Billerica, MA). These multiplex assays used microsphere technology in which assay beads were tagged with various fluorescent-labeled markers. The binding for each protein onto the multiplex bead was analyzed with a fluorescence detection laser optic system. The human high sensitivity T cell magnetic bead panel included interleukin (IL)-10, IL-12(p70), IL-13, IL-1β, IL-2, IL-21, IL-4, IL-23, IL-5, IL-6, IL-7, IL-8, macrophage inflammatory protein (MIP)-1α, MIP-1β, tumor necrosis factor (TNF)-α, fractalkine (CX3CL1), granulocyte macrophage colony stimulating factor (GM-CSF), interferon (IFN)-inducible T-cell alpha chemoattractant (ITAC), and IFN-γ. The intra-assay %CV was <5%. The inter-assay %CV was <20%. The human neurodegenerative disease magnetic bead included soluble intracellular adhesion molecule (sICAM)-1, regulated upon activation, normal T-cell expressed and secreted (RANTES), neural cell adhesion molecule (NCAM), and soluble vascular adhesion molecule (sVCAM)-1. Intra-assay %CV was <6%, and inter-assay %CV was <13%. The human soluble cytokine receptor magnetic bead panel included soluble (s) sCD30, soluble glycoprotein (sgp)130, soluble IL-1 receptor (sIL-1R)-I, sIL-1RII, sIL-2α, sIL-4R, sIL-6R, sTNFRI, and sTNFRII. The intra-assay %CV was <10% while inter-assay %CV was <15%.
Group-based trajectory analysis
To assess temporal serum APA and AHA profiles over the first 6 months post-injury, we applied TRAJ43 as previously reported.44–49 TRAJ analysis is a data-driven technique that leverages longitudinal patterns of a time-varying variable in order to identify distinct subgroups within the population expressing similar temporal levels over time. Data from samples collected 1–6 months post-injury were binned to create monthly values. TRAJ analysis was conducted after rank transformation of the monthly autoantibody data and yielded two TRAJ groups (high and low) for IgM APA and AHA autoantibodies. The two-group model output for each autoantibody marker had an optimal Bayesian Information Criterion (BIC), and excellent posterior probabilities (> 90%) were achieved for all TRAJ group assignments. We used TRAJ group membership to classify individuals and identify individuals with discordant APA versus AHA autoantibody profiles over time for which to conduct the fluorescent immune-histochemical staining analysis to assess the specificity of APA and AHA for pituitary and hypothalamic tissues respectively. APA and AHA TRAJ group membership were also tested for concordance and for associations with T levels over time.
Statistical analysis
Statistical analyses were performed with SAS (Statistical Analysis Software) version 9.4 (Cary, NC). Reported variables were assessed for normality using Shapiro–Wilk tests. Age, autoantibody levels (APA, AHA, AGA), hormone levels, BMI, GCS score, length of hospital stay, and medications (use duration and cumulative medication burden) were reported as a median with interquartile range (IQR). Mann–Whitney U tests were used to assess differences among PHH and non-PHH groups. Group differences for categorical data, including education level, race, mechanism of injury, radiographic injury type, and psychotropic medication use (yes/no) were assessed using χ2 or Fisher's exact tests as appropriate.
Autoantibody and inflammatory marker levels from multiple serum samples over the 1–6-month period were averaged for each individual. Between-group differences with autoantibodies and inflammatory markers and PHH were examined using Mann–Whitney U tests or Kruskal–Wallis tests. Because repeated measurements were taken on participants over a longitudinal time course, we also used a mixed effects regression model with an unstructured covariance pattern to assess APA and AHA TRAJ group differences in T levels over time. Spearman correlations were used to assess associations between age and 1–6–month mean T, LH, autoantibody, and inflammatory marker levels.
Binary logistical regression models were generated to assess associations between independent variables and a dichotomous dependent variable such as PHH status or autoantibody TRAJ group membership. Multivariable regression was used to test associations between APA and AHA IgM and PHH while adjusting for injury severity (GCS). Additionally, the association between age and PHH was investigated for potential associations with AHA IgM levels as biomarker mediating this relationship. A four-step process was used to test mediation effects outlined by Baron and Kenny.50 Covariate (GCS score) adjusted models were run to test associations between age and PHH directly and after adjustment for AHA IgM, to assess a potential role for this AAb as a mediator. The percent mediation was calculated using Y-standardization method appropriate for logistical regressions with binary outcome as suggested in a recent article by Rijnhart and coworkers.51 P values <0.05 were considered significant.
Results
Demographics and clinical characteristics
We recruited 143 men having had at least two post-acute blood samples drawn in the first 12 months available for hormone analysis. Fifty-one individuals (36%) had persistently low T with low or normal LH levels and were categorized as having PHH. Demographic information for this cohort is reported in Table 1. There were no significant differences in age, BMI, education level, race, GCS score, injury severity score, length of hospitalization, or mechanism of injury by PHH status. Acute care neuroradiology reports from CT and/or MRI were available for 128 of the 143 individuals. Subarachnoid hemorrhage (SAH) on CT imaging was less common in the PHH group than in the non-PHH group (27% vs. 39%, p = 0.05). Diffuse axonal injury (DAI) on CT imaging tended to be less common in the PHH group than in the non-PHH group (28% vs. 42%, p = 0.07). One individual had a subacute hemorrhage in the thalamus and hypothalamus on MRI 9 days post-injury. The other men in the cohort had no evidence of pituitary or hypothalamic injury based on clinically available neuroimaging studies.
Table 1.
TBI Patient Demographics and Characteristics
| All | Non-PHH | PHH | p value | |
|---|---|---|---|---|
| n (%) | 143 | 92 (64.34) | 51 (35.66) | - |
| Age, median (IQR), y | 31 (26) | 28.5 (22.5) | 35 (27) | 0.08 |
| BMI, median (IQR), kg/m2 | 25.87 (5.26) | 25.84 (4.26) | 26.5 (6.51) | 0.34 |
| Education, n (%) | 0.16 | |||
| < HS | 42 (29.37) | 22 (15.38) | 20 (13.99) | |
| HS | 56 (39.16) | 39 (27.27) | 17 (11.89) | |
| > HS | 45 (31.47) | 31 (21.68) | 14 (9.79) | |
| Race, n (%) | 0.75 | |||
| Caucasian | 122 (92.42) | 78 (59.09) | 44 (33.33) | |
| African American | 8 (6.06) | 6 (4.55) | 2 (1.52) | |
| Other | 2 (1.52) | 1 (0.76) | 1 (0.76) | |
| GCS score (best in 24 h), median (IQR) | 8 (4) | 8 (4) | 7.5 (4) | 0.62 |
| Non-brain injury severity score, median (IQR) | 26 (15) | 26 (17) | 29 (16) | 0.37 |
| Length of hospital stay, median (IQR), d | 12 (9) | 19 (14) | 20.5 (18) | 0.1 |
| Mechanism of injury, n (%) | 0.31 | |||
| Motor vehicle accident | 48 (36.09) | 35 (26.32) | 13 (9.77) | |
| Motorcycle accident | 30 (22.56) | 16 (12.03) | 14 (10.53) | |
| Fall/jump | 35 (26.32) | 24 (18.05) | 11 (8.27) | |
| Off-road vehicle | 10 (7.52) | 5 (3.76) | 5 (3.76) | |
| Bicycle | 4 (3.01) | 3 (2.26) | 1 (0.75) | |
| Other | 6 (4.51) | 2 (1.50) | 4 (3.01) | |
| Radiographic injury type, n (%) | ||||
| Subdural hematoma | 88 (69.29) | 53 (41.73) | 35 (27.56) | 0.07 |
| Subarachnoid hemorrhage | 83 (65.87) | 49 (38.89) | 34 (26.98) | 0.05 |
| Diffuse axonal injury | 13 (10.32) | 11 (8.73) | 2 (1.59) | 0.14 |
| Epidural hemorrhage | 27 (21.43) | 19 (15.08) | 8 (6.35) | 0.65 |
| Contusion | 73 (57.94) | 47 (37.30) | 26 (20.63) | 1.0 |
| Intraventricular hemorrhage | 40 (31.75) | 27 (21.43) | 13 (10.32) | 0.84 |
| Intraparenchymal hemorrhage | 63 (50.00) | 43 (34.13) | 20 (15.87) | 0.58 |
| Midline shift | 42 (36.84) | 27 (23.68) | 15 (13.16) | 0.68 |
Italicized values reflect trends between groups at the p < 0.1 level.
TBI, traumatic brain injury; PHH, persistent hypogonadotropic hypogonadism; BMI, body mass index; IQR, interquartile range.
For those in whom any psychotropic medication use (see Table S1) was reported at any point during the 1st year post-TBI (n = 124), medication use was not associated PHH status (χ2 = 0.0895, p = 0.3511) (Table 2).
Table 2.
Any Psychotropic Medication Use during First Year after TBI
| Non-PHH (n = 80) | PHH (n = 44) | |
|---|---|---|
| No medications | 36 (29.03%) | 16 (12.90%) |
| 45%* | 36.36%* | |
| Medications | 44 (35.48%) | 28 (22.58%) |
| 55%* | 63.64%* |
Indicates column percentages (PHH category%).
Parentheses indicate group percentages. (overall cohort).
TBI, traumatic brain injury; PHH, persistent hypogonadotropic hypogonadism.
In the subset of study individuals for whom monthly psychotropic medication use data were specifically collected (n = 64), we graphed and assessed duration of use and cumulative medication burden over the study period (Fig. 3A and B). Neither duration of use reported nor cumulative medication burden were significantly associated with PHH status (Table 3).
FIG. 3.

Frequency distribution for (A) duration of psychotropic medication use and (B) Cumulative psychotropic medication burden (aggregate measure of medication use and duration of each medication) during the first year post-injury for individuals categorized by persistent hypogonadotropic hypogonadism (PHH) status.
Table 3.
Psychotropic Medication Use Duration and Burden
| 12 months post-injury | Non-PHH (n = 44) | PHH (n = 20) | p value |
|---|---|---|---|
| Duration of use reported (months), mean (SE) | 1.7045 (0.33) | 2.0500 (0.55) | 0.6848 |
| Cumulative medication burden, mean (SE) | 2.8409 (0.63) | 3.100 (0.78) | 0.5833 |
PHH, persistent hypogonadotropic hypogonadism; SE, standard error.
T and LH levels in TBI subject serum and non-TBI control serum
A total of 1225 individual T and LH levels for 143 men were used to adjudicate PHH status. We wanted to observe any age-related changes in T levels in the healthy control group versus the TBI group. Figure 4A shows a scatter plot of T level means over the first 6 months post-TBI by age in the entire TBI cohort of 143 men. The linear regression equation (y = -0.1007x + 15.686) has a negative slope (significantly non-zero, F = 15.20, p = 0.0001) which indicates that older participants in our cohort had overall lower T levels than the younger men. Mean T levels for healthy controls (n = 11, 16.64 nmol/L) was also graphed, along with our clinical hospital system's low-normal reference laboratory level (10 nmol/L).52 Only a marginally age-related incline was observed in individuals' month 1–6 mean LH levels post-TBI (non-significant slope deviation from zero, F = 3.84, p = 0.0521) (Fig. 4B). Mean LH levels in healthy controls (n = 10, 4.82 IU/L) and our clinical hospital system's high-normal reference laboratory level (5.6 IU/L)52 accompany this figure.
FIG. 4.

Scatter plot of serum testosterone (T) and luteinizing hormone (LH) by age in the traumatic brain injury (TBI) cohort. Linear regression for (A) 1–6 month average T levels. Also graphed are mean T levels for our healthy controls (16.64 nmol/L) and our clinical laboratory low-normal reference level (10 nmol/L). (B) 1–6 month average LH levels compared with age for men with TBI. There was a significant reduction in serum T with older age (p = 0.0001) but only a trend for increases in LH with older age (p = 0.0521). Also graphed are mean LH (4.82 IU/L) for healthy controls and high normal reference level for our clinical laboratory (5.6 IU/L).
Because of biological associations among older age, sex hormone production, and adaptive immune responses,53–56 we assessed age-related relationships with sex hormones and autoantibodies using Spearman correlations among age, T, LH, and autoantibodies (month 1–6 means). As expected, there were significant inverse associations were observed between age and T (r = -0.27, p = 0.0011), AHA IgM (r = -0.26, p = 0.0025), and AGA IgM (r = -0.18, p = 0.0313). As shown, age had a marginally significant and positive association with LH (p = 0.0521). Also, T was positively associated with LH (r = 0.24, p = 0.0027) and AHA IgM (r = 0.16, p = 0.0567) (Table 4).
Table 4.
Age Correlations to Month 1–6 Mean T, LH, and Autoantibodies Levels
| Age | T | LH | |
|---|---|---|---|
| T | r = -0.27110 | - | - |
| p = 0.0011** | |||
| n = 143 | |||
| LH | r = 0.16279 | r = 0.24921 | - |
| p = 0.0521# | p = 0.0027** | ||
| n = 143 | n = 143 | ||
| APA IgM | r = -0.09551 | r = 0.13385 | r = -0.11587 |
| p = 0.2669 | p = 0.1189 | p = 0.1775 | |
| n = 137 | n = 137 | n = 137 | |
| APA IgG | r = -0.121177 | r = 0.04957 | r = -0.07583 |
| p = 0.1563 | p = 0.5651 | p = 0.3785 | |
| n = 137 | n = 137 | n = 137 | |
| AHA IgM | r = -0.25660 | r = 0.16321 | r = -0.03229 |
| p = 0.0025** | p = 0.0567# | p = 0.7080 | |
| n = 137 | n = 137 | n = 137 | |
| AHA IgG | r = -0.06316 | r = -0.03341 | r = -0.14968 |
| p = 0.4634 | p = 0.6983 | p = 0.0809# | |
| n = 137 | n = 137 | n = 137 | |
| AGA IgM | r = -0.18410 | r = 0.06357 | r = 0.07216 |
| p = 0.0313* | p = 0.4605 | p = 0.4021 | |
| n = 137 | n = 137 | n = 137 | |
| AGA IgG | r = 0.05847 | r = -0.10252 | r = -0.00362 |
| p = 0.4974 | p = 0.2332 | p = 0.9655 | |
| n = 137 | n = 137 | n = 137 |
Statistical significance denoted by: #p < 0.1 (italicized); *p < 0.05, **p < 0.01, ***p < 0.001 (bolded).
T, testosterone; LH, luteinizing hormone; APA, anti-pituitary autoantibodies; AHA, anti-hypothalamus autoantibodies; Ig, immunoglobulin.
T and LH Levels by TBI and PHH status
Mean 1–6 month post-injury T levels were assessed and graphed by PHH status. Figure 5A indicates that the PHH group had significantly lower T levels than the non-PHH group (8.24 vs. 14.61 nmol/L, p < 0.0001). Healthy control men had significantly higher T levels than all men with TBI (16.64 vs. 12.34 nmol/L. p = 0.0307) and the PHH group (16.64 nmol/L vs. 8.24 nmol/L p < 0.0001). Control levels were higher than the non-PHH levels, but were not significantly different (16.64 vs. 14.61 nmol/L, p = 0.3386). All men with TBI, and men without PHH and control groups, had levels that were above the clinical laboratory low-normal reference laboratory value (10 nmol/L),52 while mean T levels for the PHH group fell below this reference value.
FIG. 5.

Mean traumatic brain injury (TBI) (n = 143) and control (n = 11) and testosterone (T) and luteinizing hormone (LH) levels stratified by persistent hypogonadotropic hypogonadism (PHH) status. Individuals in the TBI group were stratified by PHH status. (A)T levels are higher for controls versus all TBI and versus the TBI PHH group. Those without PHH (n = 92) had higher T versus those with PHH (n = 51). (B) Serum LH levels averaged 1–6 months post-injury for TBI (n = 143) versus controls (n = 10). Controls and non-PHH TBI groups (n = 92) had higher mean LH levels than men with PHH (n = 51). #Our clinical laboratory low-normal T reference level (10 nmol/L) is also graphed as is our clinical laboratory high normal for LH (5.6IU/L). (A, B) #p < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001. Data are represented as bar graphs + standard error of mean.
Mean LH levels were also assessed over months 1–6 post-injury and were graphed by PHH status (Fig. 5B). Mean LH levels did not differ significantly between healthy control men and the all TBI group (4.82 vs. 4.98 IU/L, p = 0.9838). Men with PHH had lower LH levels than men without PHH (3.71 vs. 5.67 IU/L, p < 0.0001), and there was a trend for differences compared with the control group (3.71 vs. 4.82 IU/L, p = 0.0883). However, non-PHH and control group LH means did not differ significantly, (5.67 vs. 4.82 IU/L, p = 0.3893). The all TBI group, non-PHH group, and controls had mean LH levels near the high-normal reference laboratory level (5.6 IU/L),52 while mean LH levels for the PHH group were considerably lower than the reference level.
Standard curves and serum IgM/IgG APA, AHA, and AGA autoantibody levels in TBI and non-TBI controls
To quantitatively assess serum IgM and IgG APA, AHA, and AGA autoantibody levels in TBI participants and non-TBI control serum samples, we developed direct ELISAs for APA, AHA, and AGA specific to two immunoglobulin isotypes (IgM and IgG). We also generated IgM and IgG class autoantibody standard curves with each assay run to calculate the actual APA, AHA, and AGA IgM and IgG autoantibody concentrations (titers) in human subject serum. Figure 2 shows the standard curves for IgM (A) and IgG (B) by plotting the added IgG, IgM concentrations (x-axis) versus calculated IgG or IgM concentrations (recovery) based on absorbance (optical density) values (mean ± standard deviation [SD] from four independent runs). Linear regression was performed with a regression coefficient of determination R2 > 0.99 (Fig. 2). From these standard curve plots, we found reliable predictability of IgM and IgG concentrations (based on IgM, IgG recovery) as well as optimal assay robustness (based on the low SD values across the full range of the standard curve).
APA and AHA Relationships to TBI and PHH
A total of 592 individual samples from 137 participants with TBI collected over the study period were used for autoantibody measurement. Mean control and all TBI autoantibody levels (averaged over 1–6 months), as well as individuals with TBI dichotomized by PHH status, are shown in Figure 6. Concentrations of all Ig isotypes measured were significantly higher in TBI participants than in healthy controls for IgM/IgG APA and AHA autoantibodies. Healthy controls (n = 39) had lower levels of all autoantibodies than men with TBI: APA IgM (mean 0.24 vs. 0.94 μg/mL, p < 0.0001), APA IgG (mean 0.56 vs. 1.34 μg/mL, p < 0.0001), AHA IgM (mean 4.32 vs 7.04 μg/mL, p < 0.0001), and AHA IgG (mean 3.13 vs. 7.93 μg/mL, p < 0.0001) (Fig. 6A–D). Compared with the non-PHH group, the PHH group tended to have lower serum concentrations of APA IgM (mean 0.62 vs. 1.11 μg/mL, p = 0.06) and AHA IgM (mean 5.38 vs. 7.94 μg/mL, p = 0.02) (Fig. 6A and B). There were no significant differences in APA or AHA IgG levels between the non-PHH and PHH groups (Fig. 6C and D).
FIG. 6.

Mean control (n = 39) and traumatic brain injury (TBI) (n = 137) anti-pituitary antibody (APA) and anti-hypothalamic antibody (AHA) immunoglobulin (Ig)M and IgG levels stratified by persistent hypogonadotropic hypogonadism (PHH) Status. APA IgM concentrations tended to be lower in the PHH group than in the non-PHH group (A). There is also significantly lower AHA IgM expression in PHH than in non-PHH (B). No statistically significant differences existed between PHH and non-PHH groups in APA IgG, or AHA IgG (C and D). There were significant elevations in mean 1–6-month Ig concentrations in both IgM and IgG APA and AHA with TBI versus controls. The non-PHH group had higher APA and AHA IgM and IgG levels than controls. (A–D) #p < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001. Data are represented as bar graphs + standard error of mean.
Anti-GFAP autoantibody associations with TBI and PHH status
In order to demonstrate a generalized autoantibody response to TBI, while concurrently demonstrating the specificity of APA and AHA levels to PHH status, we assessed AGA levels in the same subjects (n = 137) and sample set described. Figure 7 shows that both 1–6 mo. IgM AGA autoantibody levels (mean 0.97 vs. 0.69 μg/ml; p = 0.0014) and IgG AGA autoantibody levels (mean 3.98 vs. 2.53 μg/ml; p < 0.0001) were elevated in the TBI group compared with controls. When stratifying the TBI cohort by PHH status, however, there were no differences in either AGA IgM or IgG autoantibody levels.
FIG. 7.

Mean immunoglobulin (Ig)M and IgG autoantibody levels for glial fibrillary acid protein (GFAP) for traumatic brain injury (TBI) (n = 137) and control (n = 39) groups as well as for TBI group stratified by persistent hypogonadotropic hypogonadism (PHH) status. (A) Anti-GFAP autoantibodies are increased for TBI group versus control, but levels did not differ by PHH status. (B) IgG GFAP autoantibodies are increased for TBI group versus control, but levels did not differ by PHH status. *p < 0.05, **p < 0.01, ***p < 0.001. Data are represented as bar graphs + standard error of mean.
Autoantibody TRAJ
TRAJ analysis of autoantibody levels over time in this cohort revealed two distinct subgroups of individuals regarding temporal IgM profiles for both APA and AHA. We denote the groups as high and low, as graphed in Figure 8. For APA IgM TRAJ analysis, there were 72 (58%) individuals in the high group, and 52 (42%) in the low group. For AHA IgM TRAJ analysis, 59 (48%) individuals were in the high group, and 65 (52%) were in the low group. APA and AHA IgM TRAJ group membership was significantly concordant (χ2 = 4.3780, p = 0.0364) as presented in Table 5, meaning that a significant number in the high APA TRAJ group are also in the high AHA TRAJ group and vice versa.
FIG. 8.

Immunoglobulin (Ig) levels by group-based trajectory analysis (TRAJ) group membership. (A) IgM anti-pituitary antibody (APA) and (B) anti-hypothalamic antibody (AHA) levels were graphed by the generated TRAJ groups. There were significant differences in autoantibody levels by TRAJ groups for all autoantibodies (A and B, p < 0.05 all comparisons). Dashed lines represent mean control levels (APA IgM, 0.24 μg/mL and AHA IgM, 4.32 μg/mL).
Table 5.
Auto-antibody Group-Based Trajectory Analysis Concordance Frequencies
| Low AHA IgM TRAJ | High AHA IgM TRAJ | |
|---|---|---|
| Low APA IgM TRAJ | 33 (26.61%) | 19 (15.32%) |
| 63.46%# | 36.54%# | |
| 50.77%* | 32.20%* | |
| High APA IgM TRAJ | 32 (25.81%) | 40 (32.26%) |
| 44.44%# | 55.56%# | |
| 49.23%* | 67.80%* |
Indicates row percentages (APA TRAJ%).
Indicates column percentages (AHA TRAJ%).
Parentheses indicate group percentages. (overall cohort).
AHA, anti-hypothalamus autoantibodies; Ig, immunoglobulin; TRAJ, group-based trajectory analysis APA, anti-pituitary autoantibodies.
Mean IgM levels by APA and AHA TRAJ group membership generally were stable over the 6-month time frame (Fig. 8A and B), consistent with a zero-order polynomial fit to the data generated in TRAJ analysis. Both the APA and AHA IgM low TRAJ groups had a modestly higher average age than the high TRAJ groups, but these comparisons were not significant (APA IgM: 38.18 years vs. 34.04 years, p = 0.3987; and AHA IgM: 38.15 years vs. 33.12 years, p = 0.3960). There were significant differences in APA and AHA IgM autoantibody levels by APA and AHA IgM TRAJ group membership, respectively, although at all monthly time points (p < 0.05 for all comparisons). For both APA and AHA IgM analyses, low TRAJ group membership autoantibody levels were similar to controls, while high TRAJ group membership levels were substantially higher than the reference control group (p < 0.05).
T levels by autoantibody trajectory
Mean T levels over the same 6-month time period post-injury were graphed by APA IgM and AHA IgM TRAJ groups. Mixed effects regression analyses, specifically covariance pattern models, were used to test whether T levels differed over time, by TRAJ group membership, and whether the pattern of T levels over a 6-month time period differed by TRAJ group membership. Figure 9a depicts T levels over time by APA IgM TRAJ group membership. The results indicate that T levels differ significantly over time (p < 0.0001) and by APA IgM TRAJ group (p = 0.0285). However, T levels follow the same overall pattern longitudinally in both high and low TRAJ groups (p = 0.866). When assessing AHA IgM TRAJ groups, T levels also differed over time (p < 0.0001) and by TRAJ group membership (p = 0.0142) (Fig. 9b). Similar to the APA IgM results, however, T levels followed the same overall pattern across both high and low AHA IgM TRAJ groups. T levels in both high and low APA/AHA IgM TRAJ groups were lower than control levels (16.64 nmol/L). However, T levels in both low TRAJ groups were similar to the clinical laboratory low-normal reference level (10 nmol/L).
FIG. 9.

Mean testosterone (T) Levels by immunoglobulin (Ig)M anti-pituitary antibody (APA) (A) and IgM anti-hypothalamic antibody (AHA) (B) group-based trajectory analysis (TRAJ) group membership. T levels were graphed by IgM TRAJ group membership. T levels are significantly different over time and T levels differ by high versus low IgM TRAJ groups (A and B). T levels follow the same overall pattern longitudinally across both high and low APA/AHA IgM TRAJ groups. Dashed lines represent mean control levels (T = 16.64 nmol/L). The clinical laboratory low-normal reference level (10 nmol/L) is also graphed.
Individual TBI serum IgM and IgG autoantibody reactivity to human pituitary and hypothalamic tissue sections
To confirm that serum from participants with high ELISA signals for APA and AHA in fact label their respective brain tissue targets (pituitary and hypothalamus, respectively), we obtained pituitary and hypothalamic paraffin sections from human cadaveric controls for fluorescent immune-histochemical staining analysis. For this assessment, we present eight representative IHC slide preparations using cadaveric specimens of hypothalamic and pituitary tissue to measure IgM APA and AHA staining from four participants classified as being within specifically low and/or high APA and AHA IgM TRAJ groups. Figure 10 shows representative results of immunofluorescent autoantibody staining using serum samples from TBI participants. Here we note that serum samples with high APA IgM show robust immunoreactivity with hormone-releasing granule-bearing cells from pituitary tissue, while serum samples with high AHA IgM show robust neuron staining in hypothalamic tissue sections (Fig. 10). Cell labeling was confirmed by counterstaining with DAPI to show nuclei.
FIG. 10.

Anti-pituitary antibody (APA) and anti-hypothalamic antibody (AHA) immunoglobulin (Ig)M fluorescence immunohistochemistry (IHC) staining with selected traumatic brain injury (TBI) participants. Human cavaderic pituitary and hypothalamic tissue was stained with subacute-chronic (1 month) serum samples from four men (subjects 1–4) with moderate to severe TBI. Each serum sample was exposed to human pituitary and hypothalamic tissue sections and then developed with fluorophore Alexa 555-conjugated anti-human IgM (images shown from top to bottom in each column). Yellow arrows point to strongly staining cells. White bar in lower right corner represents 50 μm. Individual subject's membership in APA and AHA IgM TRAJ group (high, low) are shown on top and bottom, respectively, of its IHC images.
Participants 1 and 2 were in the high APA IgM TRAJ group and low AHA IgM TRAJ group, and they showed strong APA IgM pituitary tissue staining (see yellow arrows in Fig. 10) and weaker AHA IgM pituitary staining. Participants 3 and 4 were in the high AHA IgM TRAJ group, but the low APA IgM TRAJ group, and showed strong hypothalamic tissue staining for IgM (see yellow arrows) but weak pituitary APA IgM staining (Fig. 10). These results support the quantification and tissue specificity of the APA and AHA autoantibodies measured with ELISA.
Logistical regression APA and AHA IgM in PHH categorization
Because IgG autoantibodies were not significantly different by PHH status, we performed logistical regression to model the effect of IgM autoantibodies on PHH categorization (Table 6). A binomial logistical regression model was run to test the relationship between APA IgM mean levels and PHH, while adjusting for age and GCS score (best in 24 h). Table 6 shows that for every unit increase in APA IgM there was 51.4% lower odds of PHH (p = 0.03). A logistical regression model was also run to observe the predictive capabilities of AHA IgM levels for PHH. AHA IgM was significantly associated with PHH (p = 0.03), wherein for every unit increase in AHA IgM there was 11% lower odds of PHH.
Table 6.
Multivariable Logistic Regression for APA and AHA IgM Predicting PHH
| Logistical Regression for Outcome of PHH (n = 126) | ||||
|---|---|---|---|---|
| |
APA IgM Model |
AHA IgM Model |
||
| Variable | Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value |
| Age | 1.025 | p = 0.05 | 1.017 | p = 0.17 |
| (1.000, 1.049) | (0.993, 1.043) | |||
| GCS | 0.881 | p = 0.07 | 0.903 | p = 0.14 |
| (0.768, 1.010) | (0.791, 1.033) | |||
| APA IgM levels | 0.486 | p = 0.03 | - | - |
| (0.253, 0.933) | ||||
| AHA IgM levels | - | - | 0.890 | p = 0.03 |
| (0.800, 0.991) | ||||
Statistical significance denoted by italicized value at p < 0.1 and bolded value at p < 0.05 level.
APA, anti-pituitary autoantibodies; AHA, anti-hypothalamus autoantibodies; Ig, immunoglobulin, PHH, persistent hypogonadotropic hypogonadism; CI, confidence interval.
An age-focused mediation analysis adjusting for 24 h best GCS score is presented in Figure 11. Older age was associated with decreased AHA IgM (month 1–6 mean) (p = 0.002) and increased PHH rates (p = 0.0396). However, for every unit increase of AHA IgM there was 11% lower odds of PHH (p = 0.0336). Both age and AHA IgM were independently associated with PHH when controlling for 24 h best GCS score; however, the effects of age on PHH are attenuated by 31.91% after AHA IgM adjustment. In other words, the indirect effect explained 31.91% of the total effect of age on PHH.
FIG. 11.

Anti-hypothalamic antibody (AHA) immunoglobulin (Ig)M mediation model. This brain injury severity adjusted model demonstrates a mediation effect (by 31.91%) on the relationship between age and persistent hypogonadotropic hypogonadism (PHH) by AHA IgM levels. Increasing age was associated with lower levels of AHA IgM (month 1–6 means) and PHH. Greater month 1–6 mean AHA IgM levels were also associated with reduced odds of PHH. Each model leg adjusted for best in 24 h. Glasgow Coma Scale (GCS) score. Statistical significance denoted by: *p < 0.05, **p < 0.01. CI: confidence interval OR: odds ratio; SE: standard error.
Inflammatory cytokine associations with APA/AHA IgM
Motivated by IgM-specific autoantibody differences by PHH status, we investigated potential inflammatory associations related to the differential expression of APA, AHA, and GFAP IgM. Table 7 indicates markers that were both significantly correlated and had at least a moderate relationship (r > 0.25). Fractalkine was significantly correlated with APA, AHA, and GFAP IgM, while IL-2 and IL-7 were significantly correlated with only APA and anti-GFAP antibody. Further, APA IgM was also positively correlated with IL-10 and IL-2, while GFAP IgM was correlated with IL-4, IL-12p70, and sIL-4R.
Table 7.
Correlations between Inflammatory Markers and IgM Autoantibodies
| Variable | APA IgM | AHA IgM | GFAP IgM |
|---|---|---|---|
| IL-2 | r = 0.26 | - | r = 0.36 |
| p = 0.0023 | p<0.0001 | ||
| n = 132 | n = 132 | ||
| IL-4 | - | - | r = 0.33 |
| p = 0.0001 | |||
| n = 131 | |||
| IL-7 | r = 0.35 | - | r = 0.25 |
| p<0.0001 | p = 0.0038 | ||
| n = 132 | n = 132 | ||
| IL-10 | r = 0.27 | - | - |
| p = 0.0018 | |||
| n = 129 | |||
| IL-12p70 | - | - | r = 0.32 |
| p = 0.0002 | |||
| n = 131 | |||
| Fractalkine | r = 0.29 | r = 0.30 | r = 0.26 |
| p = 0.0007 | p = 0.0005 | p = 0.0029 | |
| n = 131 | n = 131 | n = 131 | |
| IL-21 | r = 0.28 | - | - |
| p = 0.0016 | |||
| n = 127 | |||
| sIL-4R | - | - | r = 0.27 |
| p = 0.0015 | |||
| n = 132 |
Statistical significance denoted by bolded value at p < 0.05 level.
Ig, immunoglobulin; APA, anti-pituitary autoantibodies; AHA, anti-hypothalamus autoantibodies; GFAP, glial fibrillary acid protein; IL, interleukin; sIL, soluble interleukin.
Inflammatory cytokine associations to PHH
We then tested our entire inflammatory 33 biomarker panel for associations to PHH. Figure 12 depicts 1–6 month mean levels (pg/mL) by PHH status for the significant associations (p < 0.05) in the panel. Mean levels of each marker were scaled by a multiple of 10x accordingly to fit a 0–20 pg/mL range. sTNFRI, sTNFRII, RANTES, and sIL-2Rα all were higher in the PHH group than in the non-PHH group. Conversely, GM-CSF levels were higher in the non-PHH group than in the PHH group.
FIG. 12.
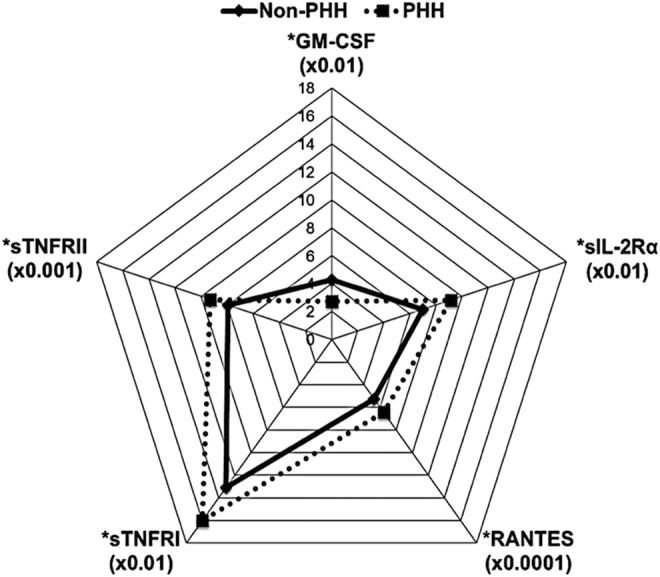
Cytokine biomarkers associated with persistent hypogonadotropic hypogonadism (PHH) status. Of the 33 inflammatory markers assayed, the five graphed above were significantly associated with PHH status (p < 0.05). Each marker was scaled by a multiple of 10x (indicated in the graph) to fit a 0–20 pg/mL range. Only granulocyte macrophage colony stimulating factor (GM-CSF) levels were higher in non-PHH individuals, while soluble tumor-necrosis-factor receptors (sTNFR)I, sTNFRII, regulated on activation, normal T-cell expresse (RANTES), and soluble interleukin receptor (sIL-2R)α levels were significantly higher in PHH.
Discussion
Given growing evidence that post-traumatic hypopituitarism, specifically hypogonadism, is deleterious to long-term outcomes, neuroendocrine dysfunction after TBI is an important topic of study.9–13 Guidelines on screening for post-traumatic hypopituitarism5,57 are based on limited evidence on the time frame in which hypopituitarism develops, and there is a paucity of evidence on the safety and efficacy of hormone replacement therapy after TBI. Despite its clinical relevance, the pathophysiological mechanisms underlying post-traumatic hypogonadism are unclear, and the role of age in contributing to PHH risk and immunity profiles after TBI is not well documented.
Here we provide the first longitudinal quantitative data on APA and AHA levels after TBI, and we report on relationships between these autoantibodies and PHH in men after moderate to severe injury. The scatter plot of T levels by age in our TBI cohort demonstrate that older individuals had lower T levels than younger individuals and that LH levels modestly increased with age (Fig. 4), findings consistent with sex hormone changes observed with normal aging.58,59 T and LH levels were significantly correlated. Furthermore, we show in Figure 5 that compared with both healthy control men and individuals without PHH, those with PHH have significantly lower T and LH levels. The results of this combined cohort study further verify our early work that men with PHH present with an impaired hypothalamic–pituitary–gonadal (HPG) function.9,13
Our analyses of psychotropic medication used within our cohort demonstrate that although these medications can impact neuroendocrine function, their use did not differ by PHH status. One reason for this finding might be evident, particularly in the subgroup with monthly data. That is, there are relatively few individuals on these drugs long-term, and most are not on more than one of these medications. This point is important given that PHH status was clinically adjudicated over a 1-year time period post-injury, lowering the impact of brief medication on a longitudinal measure of year 1 neuroendocrine function. Given the variety of medications used across each of the drug classes shown in Table S1, as well as monthly data only being available for a portion of the population, more granular or medication-specific effects on PHH status are underpowered. However, future studies with larger sample sizes might reveal potential medication effects on concurrent hormone levels and/or PHH status overall.
Men with TBI also have elevated APA and AHA levels compared with healthy volunteers, and individuals without PHH had elevated IgM autoantibody levels compared with both healthy controls and PHH individuals (Fig. 6). Our previous study suggests that men with PHH are older than those without PHH, yet age and PHH are independent risk factors for TBI recovery.9
We show significant concordance between high and low APA and AHA groups (Table 5), suggesting that for many in the cohort, relative APA and AHA autoantibody production was similar. We also show reduced PHH prevalence among those with increased APA and AHA IgM levels, even while adjusting for age and neurological injury severity (Table 6). Further, several markers representing adaptive immunity, chemokine signaling, and microglial activation are associated with autoantibody production and PHH (Table 7 and Fig. 12). These descriptive results begin to provide important biological insight into the immune system in the early chronic phase after TBI and its contribution to PHH development.
APA and AHA associations with post-traumatic hypopituitarism have been studied previously in mixed groups of men and women 3–5 years after mild to severe TBI.20,26 They each showed higher IgG autoantibody titers associated with hypopituitarism. Elevated autoantibody titers and hypopituitarism have also been shown in boxers.60 This same work also has reported many individuals with negative titers using a semiquantitative assay;60 however, we used quantitative ELISA to measure APA and AHA levels in all individuals. With this assay, we were able to quantitatively compare autoantibody levels between individuals with TBI (with and without PHH) and healthy controls, and distinguish associations with IgM and IgG autoantibodies.
In this study, we found that mean APA and AHA IgM levels were higher after TBI compared with control and also higher among those without PHH, compared with those with PHH (Fig. 6). IgG levels were modestly lower in those with PHH, which is in apparent contrast to previous literature showing high levels related to disease. However, these levels are measured much sooner than previous studies, which may contribute to the differences observed. Ig class specificity may be a temporal function of autoantibody production in response to brain-specific antigens associated with blood–brain barrier breach post-injury. The temporal onset of autoimmunity development against pituitary and hypothalamic tissue may differ (years vs. months) post-injury and among those with repeated CNS injuries. Early IgM profiles may be associated with neuroinflammatory processes supporting CNS cell rescue and repair. Also, our data focus on men with moderate-to-severe TBI (GCS <13). Although differences in TBI pathophysiology between men and women are less well understood, sex differences in secondary injury processes do exist and some evidence suggests that women have hormonal derangements and menstrual dysfunction after TBI61,62 which warrants further study examining if/how autoantibodies impact hormone dysfunction in women with TBI.
We also confirmed that serum from TBI participants with high APA or AHA IgM levels can be used as a specific immunofluorescent label for pituitary and hypothalamus tissue sections (Fig. 10). Although we do not know the exact antigens in pituitary and hypothalamus tissues that triggered these autoantibody responses, it is likely there are multiple autoantigens for APA and AHA. Therefore, some post-TBI predominant autoantigens might exist for these tissues, similar to our previous work with GFAP (and its breakdown product) having dominant autoantigens detected in post-TBI serum samples.32 Future work will aim to identify dominant antigens for APA and AHA, which will require detailed proteomic study similar to previous work.32
Previously published work has characterized multiple pituitary axis dysfunction, including growth hormone, adrenocorticotropic hormone, gonadotropin, and thyroid stimulating hormone deficiency.5,63 Because hypogonadism, which is characterized by T deficiency, is arguably the most common type of pituitary disorder after TBI,4 and symptomatic hypogonadism is indicative of other co-occurring neuroendocrinopathies,64 we focused on evaluating PHH as a secondary condition in the context of autoimmunity and inflammation after moderate to severe TBI. Given our results, further study on how autoimmunity affects other pituitary axes after TBI is warranted.
Our results indicate that T levels are significantly different over time and that these levels differ by high versus low APA/AHA IgM TRAJ groups (Fig. 9). However, T levels, regardless of the TRAJ group (high vs. low), follow the same overall longitudinal pattern. In using the mixed effects regression model approach, we were able to show that individuals in the low APA/AHA IgM TRAJ group have lower T levels over time, indicating a potential link between ongoing IgM specific autoimmune processes post-TBI and persistent differences in T that contribute to PHH.
Other studies have shown measurable IgM autoantibodies to CNS antigens in healthy individuals.65–67 Interestingly, significant autoantibody associations to PHH were IgM specific and not IgG specific. Naturally arising IgM autoantibodies to apoptotic cell membranes68,69 and leukocytes70 are generated from B1-cells and are a physiological (evolutionarily conserved) part of the innate immune system68,69; although constitutively expressed,71 these autoantibodies are amplified in environments with high concentrations of apoptotic cells72,73 and in pro-inflammatory states,70 to increase apoptotic cell phagocytosis74 and to activate complement mediated anti-inflammatory pathways75,76 in an attempt to support immune homeostasis and tissue health. Specifically, IgM antibodies bound to antigens on pathogens or dying cells facilitate lectin pathway activation of the complement system, which in turn, can facilitate phagocytosis and inflammatory cell recruitment, as well as modulate the adaptive immune response.77
Adaptive immune molecules such as IL-7 support T-cell lymphoproliferation in producing IgM autoantibodies sensitive to circulating self-antigens.78,79 These processes suggest that these types of reparative autoimmune responses are beneficial. With blood–brain barrier disruption and both systemic and lymphatic exposure to CNS antigens after TBI,80,81 autoantibody production to circulating brain antigens, including pituitary and hypothalamic tissue antigens, may help clear debris and facilitate repair. Further, work on CNS lymphatic drainage and immune surveillance82 suggests brain-periphery crosstalk and a role for systemic immunity with modulating injury and repair responses. Studies such as these are shifting dogma on the physiological role of autoantibodies and CNS immune privilege, demonstrating that even healthy individuals produce autoimmune responses to CNS antigens via immune surveillance systems.80,83
Our study is observational in nature, and it does not prove the mechanisms by which IgM autoantibodies might have beneficial qualities in facilitating cleanup and repair after TBI. However, the fact that AGA are increased after TBI, yet do not discriminate PHH status (Fig. 7), suggests some degree of specificity for APA and AHA to have beneficial effects on repair as an injury-induced immune response that does affect neuroendocrine function after TBI. Therefore, our working hypothesis is that the inverse relationship between both APA and AHA IgM levels and PHH represent an adaptive immune process that supports CNS repair. This hypothesis is based on a growing literature on possible beneficial effects of autoimmunity after CNS injury, in which self-antigen recognizing immune cells contribute to repair.30 Further work, outside the scope of this study, is needed to discern if/how IgM or IgG AGA production might map to other various TBI recovery metrics.
Evidence of protective autoimmunity has been reported in other CNS conditions such as stroke and spinal cord injury.84–87 High IgM autoantibody levels are thought to protect donor organs,70,88 reduce risk for stroke and Alzheimer's disease,89,90 and reduce disease burden in autoimmune disorders,68 whereas low/absent IgM levels are observed with pathogenic IgG production associated with autoimmune and other diseases.69 The idea of protective autoimmunity in TBI is also supported by other work showing autoantibodies binding to breakdown products of injured neural cells.91 We propose that beneficial autoimmune effects may be present after TBI via CNS antigen-specific IgM production to facilitate repair. Together, these data provide an exemplar for future work focused on identifying other key autoantibodies facilitating CNS cleanup and repair and mapping them to secondary conditions sensitive to reparative effects that brain-specific IgM production might provide.
We explored chronic inflammation associated with APA and AHA, as well as PHH status, to begin to understand how systemic inflammation may support IgM associated autoimmunity after TBI. After all, immune responses against brain-specific antigens and cellular debris must be well regulated to promote healing over damage. Also, those with post-injury immunosuppression are more vulnerable to infections and impaired healing, which negatively impacts neurological recovery, which potentially contributes to maladaptive autoantibody responses.92,93 Given the plurality of roles that cytokines and chemokines can play in different contexts, especially following an injury, our goal with these results is to simply describe associations within our data that might inform roles for immunity in recovery and secondary conditions following TBI.
T-lymphocytes are critical to post-injury adaptive immune cytokine production driving autoimmunity.94 Table 7 indicates that fractalkine has a strong positive correlation with APA, AHA, and GFAP IgM. Further, IL-2 and IL-7 are positively correlated with APA and GFAP IgM. After CNS injury, IL-7 production promotes lymphoproliferation of Th-cells sensitive to circulating self-antigens that produce CNS antibodies.78,95 IL-2 supports proliferation of effector T-cells, while also regulating immune system function via regulatory T-cells.96 Fractalkine is a chemokine that promotes monocyte survival and induces cellular chemotaxis, and is a product of proteolytic cleavage of the CX3C chemokine receptor 1 (CX3CR1) membrane-bound receptor.97,98 Cellular immunity is amplified with fractalkine expression, and increased circulating fractalkine is linked to T-cell signaling by sources of local inflammation and infection.99,100 Autoantibody associations with serum IL-2, IL-7, and fractalkine in our study implicate cellular and adaptive immunity (including autoimmunity) as relevant to neuroendocrine dysfunction post-TBI. Other notable associations include APA IgM to IL-10 and IL-21 and GFAP IgM to IL-4, IL-12p70, and sIL-4R, demonstrating other potential and diverse roles of inflammation in the context of autoimmunity. IL-21 can regulate both innate and adaptive immune responses.101 Also, IL-12 regulates T-cell responses and differentiation of T-helper 1 cells.102 IL-10 is expressed on many immune cells,103 and IL-4 supports T-helper cells to control cell-mediated immunity.104 Validating these post-TBI relationships may provide insight into how inflammation facilitates autoimmunity and in relation to neuroendocrine dysfunction.
We also explored if, in addition to autoantibody levels, chronic systemic inflammation markers are associated with PHH in men with moderate-to-severe TBI. Figure 12 demonstrated higher levels of GM CSF in the non-PHH group compared with the PHH group. GM CSF is a pro-inflammatory cytokine produced by T-cells and implicated in several inflammatory diseases105 because of its role in T-cells and macrophage recruitment.106,107 Also, sTNFRI, sTNFRII, RANTES, and sIL-2Rα levels were all higher in the PHH group than in the non-PHH group. sIL-2Rα is an important contributor to autoimmune diseases and also is a potent T-cell growth factor.108,109 RANTES is a chemokine that supports chemotaxis and T-cell expression.110 sTNFRI is ubiquitously expressed on all cells and has a death domain,111,112 whereas sTNFRII is only present on T-cells. However, both receptors are upregulated during chronic inflammation.113 Soluble TNFα receptors are formed via proteolytic cleavage on surface receptors upon TNFα activation.111,114 Together, the literature demonstrates that along with other markers associated with PHH, soluble TNF receptor signaling may be a key process that perpetuates the post-injury innate immune response and impairs adaptive immunity,115 both of which can impact tissue damage and repair after TBI. Therefore, future work is needed to better understand how these inflammatory marker observations might relate to susceptibility to chronic secondary conditions after TBI, such as PHH.
Similar to our previous work,9 men with PHH tended to be older than men without PHH. The aging immune system has decreased capacity to respond to infection and other insults, with limited reactive potential involving both T cells and B cells.116–118 Therefore, we hypothesized and also found that higher autoantibody IgM levels were associated with younger age, a finding that may help protect against PHH incidence when considering the potentially deleterious effects of age and injury severity on pituitary tissue repair. Interestingly, older age was an independent predictor of PHH when modeled with APA IgM, but not with AHA IgM. Age associations with AHA IgM levels (adaptive immunity) as well as T and LH (PHH status) suggested shared variance with regard to age relationships with PHH that were born out in mediation analyses (Fig. 11). Although age and APA correlations were in the expected direction, the relationship was not significant. However, older age was also associated with AGA IgM production, lending further credence to the idea that older age negatively affects the adaptive immune responses underlying IgM production. Together, the findings presented provide a foundation for further development of risk stratification that considers age and autoantibody levels over time, as early chronic biomarkers predicting neuroendocrine dysfunction with TBI. This type of personalized, biomarker-based approach to susceptibility to secondary conditions after TBI fits well within the Rehabilomics framework for rehabilitation research aimed at personalizing treatments that improve health and function for those with disabilities.119–121 The findings also suggest that more mechanistic studies are warranted that explore possible protective or reparative roles for autoimmunity after TBI, while also exploring for temporal inflammatory scenarios that might support traditional IgG autoantibody associations with damage, poor outcome, or increased risk for secondary conditions after TBI.
Limitation of current study
This study should be interpreted within the context of its limitations. Foremost, this is not an experimental study, and we cannot definitively establish causality between autoantibody levels and pituitary dysfunction. However, our observational findings do suggest that APA and AHA IgM are associated with PHH. Although specific antigens in the pituitary and hypothalamus involved in APA and AHA autoantibody production remain unknown, our immunohistochemistry findings support the specificity of these autoantibodies to their target tissues. Further, we did not analyze autoantibody relationships to functional or neuropsychological outcomes. However, we previously demonstrated relationships between PHH status and worse multidimensional outcomes.9 We also did not analyze women in this study. As moderate-to-severe TBI rates for women are less than men, accruing women for similar analyses remains an ongoing effort. Also, the time course of our autoantibody data extends only to 6 months post-injury, and future study should determine the dynamics of these antibodies over longer time periods.
Conclusion
In conclusion, we report novel, longitudinal data on APA and AHA levels in the setting of post-traumatic hypopituitarism, specifically PHH. The higher APA IgM levels observed in those without PHH suggest a protective role for IgM class autoantibodies against pituitary dysfunction during the first 6 months after TBI. It remains unclear how autoantibody production fluctuates and evolves after 6 months post-injury. Biomarker associations with IgM autoantibodies and PHH implicate the importance of studying post-acute inflammation in the context of secondary conditions and autoimmunity after TBI. Additional work investigating the underlying mechanisms supporting autoimmunity and development/prevention of hypopituitarism after TBI is warranted.
Supplementary Material
Funding Information
This work was supported by the Centers for Disease Control and Prevention (CDC) grant number R49 CCR 323155-03 (A.K.W.), The United States Department of Defense (DOD) grant number W81XWH-071-0701 (A.K.W.), the University of Pittsburgh Medical Center Rehabilitation Institute (A.K.W.), and the University of Pittsburgh Physicians Foundation (A.K.W.), and National Institute of Health (NIH) grant number NIH-NINDS R21 NS085455-01 (K.K.W.). The sponsors had no role in the study design; the collection, analysis, and interpretation of data; the writing of the report; or the decision to submit the manuscript for publication.
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control (2016). TBI: Get the facts. http://www.cdc.gov/traumaticbraininjury/get_the_facts.html (last accessed June7, 2016)
- 2. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control (2016). Rates of TBI-related emergency department visits, hospitalizations, and deaths by sex — United States, 2001–2010. http://www.cdc.gov/traumaticbraininjury/data/rates_bysex.html (last accessed June7, 2016)
- 3. Zaloshnja E., Miller T., Langlois J.A., and Selassie A.W. (2008). Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J. Head Trauma Rehabil. 23, 394–400 [DOI] [PubMed] [Google Scholar]
- 4. Kopczak A., Kilimann I., von Rosen F., Krewer C., Schneider H.J., Stalla G.K., and Schneider M. (2014). Screening for Hypopituitarism in 509 Patients with Traumatic Brain Injury or Subarachnoid Hemorrhage. J. Neurotrauma 31, 99–107 [DOI] [PubMed] [Google Scholar]
- 5. Tanriverdi F., Schneider H.J., Aimaretti G., Masel B.E., Casanueva F.F., and Kelestimur F. (2015). Pituitary dysfunction after traumatic brain injury: a clinical and pathophysiological approach. Endocr. Rev. 36, 305–342 [DOI] [PubMed] [Google Scholar]
- 6. Schneider H.J., Sämann P.G., Schneider M., Croce C.G., Corneli G., Sievers C., Ghigo E., Stalla G.K., and Aimaretti G. (2007). Pituitary imaging abnormalities in patients with and without hypopituitarism after traumatic brain injury. J. Endocrinol. Invest. 30, RC9–RC12 [DOI] [PubMed] [Google Scholar]
- 7. Zgaljardic D.J., Durham W.J., Mossberg K.A., Foreman J., Joshipura K., Masel B.E., Urban R., and Sheffield-Moore M. (2014). Neuropsychological and physiological correlates of fatigue following traumatic brain injury. Brain Inj. 28, 389–397 [DOI] [PubMed] [Google Scholar]
- 8. Cuesta M., Hannon M.J., Crowley R.K., Behan L.A., Tormey W., Rawluk D., Delargy M., Agha A., and Thompson C.J. (2016). Symptoms of gonadal dysfunction are more predictive of hypopituitarism than nonspecific symptoms in screening for pituitary dysfunction following moderate or severe traumatic brain injury. Clin. Endocrinol. (Oxf.) 84, 92–98 [DOI] [PubMed] [Google Scholar]
- 9. Barton D.J., Kumar R.G., McCullough E.H., Galang G., Arenth P.M., Berga S.L., and Wagner A.K. (2016). Persistent hypogonadotropic hypogonadism in men after severe traumatic brain injury: temporal hormone profiles and outcome prediction. J. Head Trauma Rehabil. 31, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bondanelli M., Ambrosio M.R., Cavazzini L., Bertocchi A., Zatelli M.C., Carli A., Valle D., Basaglia N., and Uberti E.C.D. (2007). Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J. Neurotrauma 24, 1687–1697 [DOI] [PubMed] [Google Scholar]
- 11. Carlson N.E., Brenner L.A., Wierman M.E., Harrison-Felix C., Morey C., Gallagher S., and Ripley D. (2009). Hypogonadism on admission to acute rehabilitation is correlated with lower functional status at admission and discharge. Brain Inj. 23, 336–344 [DOI] [PubMed] [Google Scholar]
- 12. Popovic V., Pekic S., Pavlovic D., Maric N., Jasovic-Gasic M., Djurovic B., Medic Stojanoska M., Zivkovic V., Stojanovic M., Doknic M., Milic N., Djurovic M., Dieguez C., and Casanueva F.F. (2004). Hypopituitarism as a consequence of traumatic brain injury (TBI) and its possible relation with cognitive disabilities and mental distress. J. Endocrinol. Invest. 27, 1048–1054 [DOI] [PubMed] [Google Scholar]
- 13. Wagner A.K., Brett C.A., McCullough E.H., Niyonkuru C., Loucks T.L., Dixon C.E., Ricker J., Arenth P., and Berga S.L. (2012). Persistent hypogonadism influences estradiol synthesis, cognition and outcome in males after severe TBI. Brain Inj. 26, 1226–1242 [DOI] [PubMed] [Google Scholar]
- 14. Crompton M.R. (1971). Hypothalamic lesions following closed head injury. Brain J. Neurol. 94, 165–172 [DOI] [PubMed] [Google Scholar]
- 15. Daniel P.M., Prichard M.M., and Treip C.S. (1959). Traumatic infarction of the anterior lobe of the pituitary gland. Lancet 2, 927–931 [DOI] [PubMed] [Google Scholar]
- 16. Aimaretti G., Ambrosio M.R., Di Somma C., Gasperi M., Cannavò S., Scaroni C., Fusco A., Del Monte P., De Menis E., Faustini-Fustini M., Grimaldi F., Logoluso F., Razzore P., Rovere S., Benvenga S., Degli Uberti E.C., De Marinis L., Lombardi G., Mantero F., Martino E., Giordano G., and Ghigo E. (2005). Residual pituitary function after brain injury-induced hypopituitarism: a prospective 12-month study. J. Clin. Endocrinol. Metab. 90, 6085–6092 [DOI] [PubMed] [Google Scholar]
- 17. O'Neil M.E., Carlson K., Storzbach D., Brenner L., Freeman M., Quiñones A., Motu'apuaka M., Ensley M., and Kansagara D. (2013). Complications of Mild Traumatic Brain Injury in Veterans and Military Personnel: A Systematic Review. Department of Veterans Affairs (US): Washington, DC; [PubMed] [Google Scholar]
- 18. Kasturi B.S., and Stein D.G. (2009). Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. J. Neurotrauma 26, 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanriverdi F., Unluhizarci K., and Kelestrimur F. (2010). Persistent neuroinflammation may be involved in the pathogenesis of traumatic brain injury (TBI)-induced hypopituitarism: potential genetic and autoimmune factors. J. Neurotrauma 27, 301–302 [DOI] [PubMed] [Google Scholar]
- 20. Tanriverdi F., De Bellis A., Bizzarro A., Sinisi A.A., Bellastella G., Pane E., Bellastella A., Unluhizarci K., Selcuklu A., Casanueva F.F., and Kelestimur F. (2008). Antipituitary antibodies after traumatic brain injury: is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 159, 7–13 [DOI] [PubMed] [Google Scholar]
- 21. Wang K.K., Yang Z., Zhu T., Shi Y., Rubenstein R., Tyndall J.A., and Manley G.T. (2018). An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev. Mol. Diagn. 18, 165–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Z., Zhu T., Weissman A.S., Jaalouk E., Rathore D.S., Romo P., Shi Y., Wagner A.K., and Wang K.K.W. (2017). Autoimmunity and traumatic brain injury. Curr. Phys. Med. Rehabil. Rep. 5, 22–29 [Google Scholar]
- 23. Guaraldi F., Grottoli S., Arvat E., and Ghigo E. (2015). Hypothalamic-pituitary autoimmunity and traumatic brain injury. J. Clin. Med. 4, 1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chodobski A., Zink B.J., and Szmydynger-Chodobska J. (2011). Blood–brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2, 492–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hay J.R., Johnson V.E., Young A.M.H., Smith D.H., and Stewart W. (2015). Blood–brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 74, 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanriverdi F., De Bellis A., Ulutabanca H., Bizzarro A., Sinisi A.A., Bellastella G., Amoresano Paglionico V., Dalla Mora L., Selcuklu A., Unluhizarci K., Casanueva F.F., and Kelestimur F. (2013). A five year prospective investigation of anterior pituitary function after traumatic brain injury: is hypopituitarism long-term after head trauma associated with autoimmunity? J. Neurotrauma 30, 1426–1433 [DOI] [PubMed] [Google Scholar]
- 27. Notley C.A., Brown M.A., Wright G.P., and Ehrenstein M.R. (2011). Natural IgM is required for suppression of inflammatory arthritis by apoptotic cells. J. Immunol. 186, 4967–4972 [DOI] [PubMed] [Google Scholar]
- 28. Boes M., Prodeus A.P., Schmidt T., Carroll M.C., and Chen J. (1998). A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 188, 2381–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haas K.M., Poe J.C., Steeber D.A., and Tedder T.F. (2005). B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23, 7–18 [DOI] [PubMed] [Google Scholar]
- 30. Schwartz M., and Raposo C. (2014). Protective autoimmunity: a unifying model for the immune network involved in CNS repair. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 20, 343–358 [DOI] [PubMed] [Google Scholar]
- 31. Kobeissy F., Mondello S., Tümer N., Toklu H.Z., Whidden M.A., Kirichenko N., Zhang Z., Prima V., Yassin W., Anagli J., Chandra N., Svetlov S., and Wang K.K.W. (2013). Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front. Neurol. 4, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z., Zoltewicz J.S., Mondello S., Newsom K.J., Yang Z., Yang B., Kobeissy F., Guingab J., Glushakova O., Robicsek S., Heaton S., Buki A., Hannay J., Gold M.S., Rubenstein R., Lu X.-C.M., Dave J.R., Schmid K., Tortella F., Robertson C.S., and Wang K.K.W. (2014). Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PloS One 9, e92698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holmin S., Söderlund J., Biberfeld P., and Mathiesen T. (1998). Intracerebral inflammation after human brain contusion. Neurosurgery 42, 291–299 [DOI] [PubMed] [Google Scholar]
- 34. Kumar R.G., Boles J.A., and Wagner A.K. (2015). Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J. Head Trauma Rehabil. 30, 369–381 [DOI] [PubMed] [Google Scholar]
- 35. Teasdale G., Murray G., Parker L., and Jennett B. (1979). Adding up the Glasgow Coma Score. Acta Neurochir. Suppl. (Wien) 28, 13–16 [DOI] [PubMed] [Google Scholar]
- 36. Baker S.P., O'Neill B., Haddon W., and Long W.B. (1974). The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. J. Trauma 14, 187–196 [PubMed] [Google Scholar]
- 37. Smith C.G. (1982). Drug effects on male sexual function. Clin. Obstet. Gynecol. 25, 525–531 [DOI] [PubMed] [Google Scholar]
- 38. Smith C.G. (1983). Reproductive toxicity: hypothalamic-pituitary mechanisms. Am. J. Ind. Med. 4, 107–112 [PubMed] [Google Scholar]
- 39. Gunnet J.W., and Moore K.E. (1988). Neuroleptics and neuroendocrine function. Annu. Rev. Pharmacol. Toxicol. 28, 347–366 [DOI] [PubMed] [Google Scholar]
- 40. Meltzer H.Y., Fang V.S., Tricou B.J., and Robertson A. (1982). Effect of antidepressants on neuroendocrine axis in humans. Adv. Biochem. Psychopharmacol. 32, 303–316 [PubMed] [Google Scholar]
- 41. Torre D.L., and Falorni A. (2007). Pharmacological causes of hyperprolactinemia. Ther. Clin. Risk Manag. 3, 929–951 [PMC free article] [PubMed] [Google Scholar]
- 42. NAMI: National Alliance on Mental Illness (2020). Mental health medications | https://www.nami.org/learn-more/treatment/mental-health-medications (last accessed December30, 2019)
- 43. Niyonkuru C., Wagner A.K., Ozawa H., Amin K., Goyal A., and Fabio A. (2013). Group-based trajectory analysis applications for prognostic biomarker model development in severe TBI: a practical example. J. Neurotrauma 30, 938–945 [DOI] [PubMed] [Google Scholar]
- 44. Goyal A., Failla M.D., Niyonkuru C., Amin K., Fabio A., Berger R.P., and Wagner A.K. (2013). S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J. Neurotrauma 30, 946–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salonia R., Empey P.E., Poloyac S.M., Wisniewski S.R., Klamerus M., Ozawa H., Wagner A.K., Ruppel R., Bell M.J., Feldman K., Adelson P.D., Clark R.S.B., and Kochanek P.M. (2010). Endothelin-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes in children after severe traumatic brain injury. J. Neurotrauma 27, 1819–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santarsieri M., Kumar R.G., Kochanek P.M., Berga S., and Wagner A.K. (2015). Variable neuroendocrine-immune dysfunction in individuals with unfavorable outcome after severe traumatic brain injury. Brain. Behav. Immun. 45, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santarsieri M., Niyonkuru C., McCullough E.H., Dobos J.A., Dixon C.E., Berga S.L., and Wagner A.K. (2014). Cerebrospinal fluid cortisol and progesterone profiles and outcomes prognostication after severe traumatic brain injury. J. Neurotrauma 31, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wagner A.K., Amin K.B., Niyonkuru C., Postal B.A., McCullough E.H., Ozawa H., Dixon C.E., Bayir H., Clark R.S., Kochanek P.M., and Fabio A. (2011). CSF Bcl-2 and cytochrome C temporal profiles in outcome prediction for adults with severe TBI. J. Cereb. Blood Flow Metab. 31, 1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wagner A.K., McCullough E.H., Niyonkuru C., Ozawa H., Loucks T.L., Dobos J.A., Brett C.A., Santarsieri M., Dixon C.E., Berga S.L., and Fabio A. (2011). Acute serum hormone levels: characterization and prognosis after severe traumatic brain injury. J. Neurotrauma 28, 871–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baron R.M., and Kenny D.A. (1986). The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 51, 1173–1182 [DOI] [PubMed] [Google Scholar]
- 51. Rijnhart J.J.M., Twisk J.W.R., Eekhout I., and Heymans M.W. (2019). Comparison of logistic-regression based methods for simple mediation analysis with a dichotomous outcome variable. BMC Med. Res. Methodol. 19, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. American College of Physicians (n.d.).Normal laboratory values. http://idgateway.wustl.edu/Normal%20lab%20values.pdf (last accessed July23, 2019)
- 53. Decaroli M.C., and Rochira V. (2017). Aging and sex hormones in males. Virulence 8, 545–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schubert M., and Jockenhövel F. (2005). Late-onset hypogonadism in the aging male (LOH): definition, diagnostic and clinical aspects. J. Endocrinol. Invest. 28, 23–27 [PubMed] [Google Scholar]
- 55. Paganelli R., Quinti I., Fagiolo U., Cossarizza A., Ortolani C., Guerra E., Sansoni P., Pucillo L.P., Scala E., and Cozzi E. (1992). Changes in circulating B cells and immunoglobulin classes and subclasses in a healthy aged population. Clin. Exp. Immunol. 90, 351–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weiskopf D., Weinberger B., and Grubeck-Loebenstein B. (2009). The aging of the immune system. Transpl. Int. 22, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 57. Ghigo E., Masel B., Aimaretti G., Léon-Carrión J., Casanueva F.F., Dominguez-Morales M.R., Elovic E., Perrone K., Stalla G., Thompson C., and Urban R. (2005). Consensus guidelines on screening for hypopituitarism following traumatic brain injury. Brain Inj. 19, 711–724 [DOI] [PubMed] [Google Scholar]
- 58. Feldman H.A., Longcope C., Derby C.A., Johannes C.B., Araujo A.B., Coviello A.D., Bremner W.J., and McKinlay J.B. (2002). Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts Male Aging Study. J. Clin. Endocrinol. Metab. 87, 589–598 [DOI] [PubMed] [Google Scholar]
- 59. Harman S.M., Metter E.J., Tobin J.D., Pearson J., Blackman M.R., and Baltimore Longitudinal Study of Aging (2001). Longitudinal effects of aging on serum total and free testosterone levels in healthy men. Baltimore Longitudinal Study of Aging. J. Clin. Endocrinol. Metab. 86, 724–731 [DOI] [PubMed] [Google Scholar]
- 60. Tanriverdi F., De Bellis A., Battaglia M., Bellastella G., Bizzarro A., Sinisi A.A., Bellastella A., Unluhizarci K., Selcuklu A., Casanueva F.F., and Kelestimur F. (2010). Investigation of antihypothalamus and antipituitary antibodies in amateur boxers: is chronic repetitive head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 162, 861–867 [DOI] [PubMed] [Google Scholar]
- 61. Ranganathan P., Kumar R.G., Davis K., McCullough E.H., Berga S.L., and Wagner A.K. (2016). Longitudinal sex and stress hormone profiles among reproductive age and post-menopausal women after severe TBI: a case series analysis. Brain Inj. 30, 452–461 [DOI] [PubMed] [Google Scholar]
- 62. Ripley D.L., Harrison-Felix C., Sendroy-Terrill M., Cusick C.P., annels-McClure A., and Morey C. (2008). The impact of female reproductive function on outcomes after traumatic brain injury. Arch. Phys. Med. Rehabil. 89, 1090–1096 [DOI] [PubMed] [Google Scholar]
- 63. Tanriverdi F., and Kelestimur F. (2017). Classical and non-classical causes of GH deficiency in adults. Best Pract. Res. Clin. Endocrinol. Metab. 31, 3–11 [DOI] [PubMed] [Google Scholar]
- 64. Cuesta M., Hannon M.J., Crowley R.K., Behan L.A., Tormey W., Rawluk D., Delargy M., Agha A., and Thompson C.J. (2016). Symptoms of gonadal dysfunction are more predictive of hypopituitarism than nonspecific symptoms in screening for pituitary dysfunction following moderate or severe traumatic brain injury. Clin. Endocrinol. (Oxf.) 84, 92–98 [DOI] [PubMed] [Google Scholar]
- 65. Hedegaard C.J., Chen N., Sellebjerg F., Sørensen P.S., Leslie R.G.Q., Bendtzen K., and Nielsen C.H. (2009). Autoantibodies to myelin basic protein (MBP) in healthy individuals and in patients with multiple sclerosis: a role in regulating cytokine responses to MBP. Immunology 128, e451–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nielsen C.H., Leslie R.G., Jepsen B.S., Kazatchkine M.D., Kaveri S.V., and Fischer E. (2001). Natural autoantibodies and complement promote the uptake of a self antigen, human thyroglobulin, by B cells and the proliferation of thyroglobulin-reactive CD4(+) T cells in healthy individuals. Eur. J. Immunol. 31, 2660–2668 [DOI] [PubMed] [Google Scholar]
- 67. O'Connor K.C., Chitnis T., Griffin D.E., Piyasirisilp S., Bar-Or A., Khoury S., Wucherpfennig K.W., and Hafler D.A. (2003). Myelin basic protein-reactive autoantibodies in the serum and cerebrospinal fluid of multiple sclerosis patients are characterized by low-affinity interactions. J. Neuroimmunol. 136, 140–148 [DOI] [PubMed] [Google Scholar]
- 68. Grönwall C., Vas J., and Silverman G.J. (2012). Protective roles of natural IgM antibodies. Front. Immunol. 3, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vas J., Grönwall C., and Silverman G.J. (2013). Fundamental roles of the innate-like repertoire of natural antibodies in immune homeostasis. Front. Immunol. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lobo P.I., Schlegal K.H., Vengal J., Okusa M.D., and Pei H. (2010). Naturally occurring IgM anti-leukocyte autoantibodies inhibit T-cell activation and chemotaxis. J. Clin. Immunol. 30, Suppl. 1, S31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baumgarth N., Tung J.W., and Herzenberg L.A. (2005). Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin. Immunopathol. 26, 347–362 [DOI] [PubMed] [Google Scholar]
- 72. Chen Y., Khanna S., Goodyear C.S., Park Y.B., Raz E., Thiel S., Grönwall C., Vas J., Boyle D.L., Corr M., Kono D.H., and Silverman G.J. (2009). Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J. Immunol. 83, 1346–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen Y., Park Y.-B., Patel E., and Silverman G.J. (2009). IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J. Immunol. 182, 6031–6043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. deCathelineau A.M., and Henson P.M. (2003). The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 39, 105–117 [DOI] [PubMed] [Google Scholar]
- 75. Gray M., Miles K., Salter D., Gray D., and Savill J. (2007). Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc. Natl. Acad. Sci. U. S. A. 104, 14,080–14,085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Huynh M.-L.N., Fadok V.A., and Henson P.M. (2002). Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kjaer T.R., Thiel S., and Andersen G.R. (2013). Toward a structure-based comprehension of the lectin pathway of complement. Mol. Immunol. 56, 413–422 [DOI] [PubMed] [Google Scholar]
- 78. Schluns K.S., Kieper W.C., Jameson S.C., and Lefrançois L. (2000). Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 1, 426–432 [DOI] [PubMed] [Google Scholar]
- 79. Goldrath A.W., and Bevan M.J. (1999). Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 11, 183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Louveau A., Harris T.H., and Kipnis J. (2015). Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Louveau A., Smirnov I., Keyes T.J., Eccles J.D., Rouhani S.J., Peske J.D., Derecki N.C., Castle D., Mandell J.W., Lee K.S., Harris T.H., and Kipnis J. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Louveau A., Smirnov I., Keyes T.J., Eccles J.D., Rouhani S.J., Peske J.D., Derecki N.C., Castle D., Mandell J.W., Lee K.S., Harris T.H., and Kipnis J. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Engelhardt B., Vajkoczy P., and Weller R.O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131 [DOI] [PubMed] [Google Scholar]
- 84. Brait V.H., Arumugam T.V., Drummond G.R., and Sobey C.G. (2012). Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J. Cereb. Blood Flow Metab. 32, 598–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Graber J.J., and Dhib-Jalbut S. (2009). Protective autoimmunity in the nervous system. Pharmacol. Ther. 121, 147–159 [DOI] [PubMed] [Google Scholar]
- 86. Saltzman J.W., Battaglino R., Stott H., and Morse L.R. (2013). Neurotoxic or neuroprotective? Current controversies in sci-induced autoimmunity. Curr. Phys. Med. Rehabil. Rep. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schwartz M., and Baruch K. (2014). Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J. Autoimmun. 54, 8–14 [DOI] [PubMed] [Google Scholar]
- 88. McAlister C.C., Gao Z.-H., McAlister V.C., Gupta R., Wright J.R., MacDonald A.S., and Peltekian K. (2004). Protective anti-donor IgM production after crossmatch positive liver-kidney transplantation. Liver Transpl. 10, 315–319 [DOI] [PubMed] [Google Scholar]
- 89. Eriksson U.K., Sjöberg B.G., Bennet A.M., de Faire U., Pedersen N.L., and Frostegård J. (2010). Low levels of antibodies against phosphorylcholine in Alzheimer's disease. J. Alzheimers Dis. 21, 577–584 [DOI] [PubMed] [Google Scholar]
- 90. Fiskesund R., Stegmayr B., Hallmans G., Vikström M., Weinehall L., de Faire U., and Frostegård J. (2010). Low levels of antibodies against phosphorylcholine predict development of stroke in a population-based study from northern Sweden. Stroke 41, 607–612 [DOI] [PubMed] [Google Scholar]
- 91. Stein T.D., Fedynyshyn J.P., and Kalil R.E. (2002). Circulating autoantibodies recognize and bind dying neurons following injury to the brain. J. Neuropathol. Exp. Neurol. 61, 1100–1108 [DOI] [PubMed] [Google Scholar]
- 92. Hazeldine J., Lord J.M., and Belli A. (2015). Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front. Neurol. 6, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Riegger T., Conrad S., Schluesener H.J., Kaps H.-P., Badke A., Baron C., Gerstein J., Dietz K., Abdizahdeh M., and Schwab J.M. (2009). Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience 158, 1194–1199 [DOI] [PubMed] [Google Scholar]
- 94. Brait V.H., Jackman K.A., Walduck A.K., Selemidis S., Diep H., Mast A.E., Guida E., Broughton B.R.S., Drummond G.R., and Sobey C.G. (2010). Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J. Cereb. Blood Flow Metab. 30, 1306–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ernst B., Lee D.S., Chang J.M., Sprent J., and Surh C.D. (1999). The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 11, 173–181 [DOI] [PubMed] [Google Scholar]
- 96. Katzman S.D., Hoyer K.K., Dooms H., Gratz I.K., Rosenblum M.D., Paw J.S., Isakson S.H., and Abbas A.K. (2011). Opposing functions of IL-2 and IL-7 in the regulation of immune responses. Cytokine 56, 116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Harrison J.K., Jiang Y., Chen S., Xia Y., Maciejewski D., McNamara R.K., Streit W.J., Salafranca M.N., Adhikari S., Thompson D.A., Botti P., Bacon K.B., and Feng L. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U. S. A. 95, 10,896–10,901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Imai T., Hieshima K., Haskell C., Baba M., Nagira M., Nishimura M., Kakizaki M., Takagi S., Nomiyama H., Schall T.J., and Yoshie O. (1997). Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530 [DOI] [PubMed] [Google Scholar]
- 99. Bazan J.F., Bacon K.B., Hardiman G., Wang W., Soo K., Rossi D., Greaves D.R., Zlotnik A., and Schall T.J. (1997). A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644 [DOI] [PubMed] [Google Scholar]
- 100. Shin M.S., You S., Kang Y., Lee N., Yoo S.-A., Park K., Kang K.S., Kim S.H., Mohanty S., Shaw A.C., Montgomery R.R., Hwang D., and Kang I. (2015). DNA methylation regulates the differential expression of CX3CR1 on human IL-7Rαlow and high effector memory CD8+ T cells with distinct migratory capacities to fractalkine. J. Immunol. 195, 2861–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Spolski R., and Leonard W.J. (2014). Interleukin-21: a double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 13, 379–395 [DOI] [PubMed] [Google Scholar]
- 102. Trinchieri G. (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3, 133–146 [DOI] [PubMed] [Google Scholar]
- 103. Saraiva M., and O'Garra A. (2010). The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181 [DOI] [PubMed] [Google Scholar]
- 104. Choi P., and Reiser H. (1998). IL-4: role in disease and regulation of production. Clin. Exp. Immunol. 113, 317–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Campbell I.K., Rich M.J., Bischof R.J., Dunn A.R., Grail D., and Hamilton J.A. (1998). Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J. Immunol. 161, 3639–3644 [PubMed] [Google Scholar]
- 106. Gonzalez-Juarrero M., Hattle J.M., Izzo A., Junqueira-Kipnis A.P., Shim T.S., Trapnell B.C., Cooper A.M., and Orme I.M. (2005). Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J. Leukoc. Biol. 77, 914–922 [DOI] [PubMed] [Google Scholar]
- 107. Shi Y., Liu C.H., Roberts A.I., Das J., Xu G., Ren G., Zhang Y., Zhang L., Yuan Z.R., Tan H.S.W., Das G., and Devadas S. (2006). Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. 16, 126. [DOI] [PubMed] [Google Scholar]
- 108. Malek T.R., and Castro I. (2010). Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity 33, 153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Morgan D.A., Ruscetti F.W., and Gallo R. (1976). Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 193, 1007–1008 [DOI] [PubMed] [Google Scholar]
- 110. Mikolajczyk T.P., Nosalski R., Szczepaniak P., Budzyn K., Osmenda G., Skiba D., Sagan A., Wu J., Vinh A., Marvar P.J., Guzik B., Podolec J., Drummond G., Lob H.E., Harrison D.G., and Guzik T.J. (2016). Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 30, 1987–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pandey M., Tuncman G., Hotamisligil G.S., and Samad F. (2003). Divergent roles for p55 and p75 TNF-α receptors in the induction of plasminogen activator inhibitor-1. Am. J. Pathol. 162, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sedger L.M., and McDermott M.F. (2014). TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev. 25, 453–472 [DOI] [PubMed] [Google Scholar]
- 113. Cope A.P., Aderka D., Wallach D., Kahan M., Chu N.R., Brennan F.M., and Feldmann M. (1995). Soluble TNF receptor production by activated T lymphocytes: differential effects of acute and chronic exposure to TNF. Immunology 84, 21–30 [PMC free article] [PubMed] [Google Scholar]
- 114. Waage A., and Espevik T. (1994). TNF receptors in chronic lymphocytic leukemia. Leuk. Lymphoma 13, 41–46 [DOI] [PubMed] [Google Scholar]
- 115. Suvannavejh G.C., Lee H.O., Padilla J., Dal Canto M.C., Barrett T.A., and Miller S.D. (2000). Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 205, 24–33 [DOI] [PubMed] [Google Scholar]
- 116. Aspinall R., Lapenna A., B-Lynch C., and Lang P.O. (2014). Cellular signalling pathways in immune aging and regeneration. Biochem. Soc. Trans. 42, 651–656 [DOI] [PubMed] [Google Scholar]
- 117. Frasca D., Riley R.L., and Blomberg B.B. (2005). Humoral immune response and B-cell functions including immunoglobulin class switch are downregulated in aged mice and humans. Semin. Immunol. 17, 378–384 [DOI] [PubMed] [Google Scholar]
- 118. Pereira B.I., and Akbar A.N. (2016). Convergence of innate and adaptive immunity during human aging. Front. Immunol. 7, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wagner A.K. (2010). TBI translational rehabilitation research in the 21st Century: exploring a Rehabilomics research model. Eur. J. Phys. Rehabil. Med. 46, 549–556 [PubMed] [Google Scholar]
- 120. Wagner A.K., and Sowa G. (2014). Rehabilomics research: a model for translational rehabilitation and comparative effectiveness rehabilitation research. Am. J. Phys. Med. Rehabil. 93, 913–916 [DOI] [PubMed] [Google Scholar]
- 121. Wagner A.K., and Zitelli K.T. (2013). A Rehabilomics focused perspective on molecular mechanisms underlying neurological injury, complications, and recovery after severe TBI. Pathophysiology 20, 39–48 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.