

Abstract
Excessive alcohol consumption results in significant changes in gene expression and isoforms due to altered mRNA splicing. As such, an intriguing possibility is that disturbances in alternative splicing are involved in key pathological pathways triggered by alcohol exposure. However, no resources have been available to systematically analyze this possibility at a genome-wide scale. Here, we performed RNA sequencing of human fetal cortical slices that were obtained at the late first trimester and exposed to ethanol or in control medium. We report 382 events that were identified as changes affecting the ratio of splicing isoforms in the ethanol-exposed fetal human cortex. Additionally, previously unreported novel isoforms of several genes were also identified. These results provide a broad perspective on the post-transcriptional regulatory network underlying ethanol-induced pathogenesis in the developing human cortex.
Keywords: Alternative splicing, RNA sequencing, human fetal cerebral cortex, prenatal alcohol exposure
Introduction:
Excessive alcohol consumption results in devastating consequences for newborns of women who have abused this substance (Mattson & Riley, 1998; Riley et al., 1995; Riley & McGee, 2005; Roebuck, Mattson, & Riley, 1998). Alcohol misuse results in severe impairments of fetal development, including the formation of complex neural circuitry of the cerebral cortex (Fryer et al., 2007; Gautam et al., 2014; McGee, Fryer, Bjorkquist, Mattson, & Riley, 2008; McGee, Schonfeld, Roebuck-Spencer, Riley, & Mattson, 2008; Sowell et al., 2008; Yang et al., 2012; Zhou et al.). Permanent birth deficits caused by fetal alcohol exposure are referred to as Fetal Alcohol Spectrum Disorder (FASD) (Calhoun & Warren, 2007; Clarren, Alvord, Sumi, Streissguth, & Smith, 1978; Warren & Bast, 1988; Warren et al., 2001).
The human cerebral cortex is highly vulnerable to alcohol exposure during the mid-gestational period, which is associated with extensive neurogenesis and neuronal migration (Ben-Ari, 2008; Miranda et al., 2010; Thompson, Levitt, & Stanwood, 2009). This period is also the most crucial in terms of the development of mental dysfunctions in FASD (Riley & McGee, 2005). Our previous study had focused on the impact of alcohol exposure on human cortical development in the mid-gestational period, more precisely, between 15 and 18 gestational weeks (GW) (Hashimoto-Torii, Kawasawa, Kuhn, & Rakic, 2010). Using microarray gene analysis, we observed the expression of stress-responsive genes such as Heat Shock Proteins and GADD45 to be highly upregulated in the alcohol-exposed fetal cortical slices, and the expression of many genes involved in specification of the neuronal subtypes to be downregulated (Hashimoto-Torii et al., 2010). In our subsequent study, we showed that the increased expression of stress-responsive genes was required for the protection of the fetal cerebral cortex against alcohol-inducible damage (Hashimoto-Torii et al., 2014; Ishii & Hashimoto-Torii, 2015).
Using RNA sequencing, the present study revealed an altered mRNA splicing signature in the alcohol-exposed fetal cortex. We also identified a number of unreported novel isoforms in several genes.
Materials and Methods:
Human tissue culture
Two GW15-18 female embryonic cortices from aborted donors were obtained from the Human Fetal Tissue Repository at Albert Einstein College of Medicine. The research protocol was approved by the Human Investigation Committees of Yale University and Children’s National Medical Center. None of the samples had histories of alcohol exposure during pregnancy. Tissues were stored in L-15 Leibovitz medium on ice and were cut at 300 μm using a tissue chopper within 3 h after the abortion. Each cortical slice was cultured on a membrane floating in neurobasal medium containing 50 mM ethanol or PBS (control) with supplements for 24 h in a 6-well tissue culture plate with a low evaporation lid (353046; Falcon). Sequential slices from each brain were placed in an alternating manner into plates containing exclusively either ethanol-containing or control culture medium.
Library preparation and sequencing for mRNA
Total RNA was extracted using an RNeasy mini kit (Qiagen) and kept at −80 °C until further use. Optical density values of extracted RNA were measured using NanoDrop (Thermo Scientific) to confirm an A260:A280 ratio above 1.9. RNA integrity number (RIN) was measured using BioAnalyzer RNA 6000 Pico Kit (Agilent Technologies) to confirm RIN above 8. The cDNA libraries were prepared using KAPA Stranded RNA-Seq Kit with RiboErase (Kapa Biosystems) as per the manufacturer’s instructions. The unique barcode sequences were incorporated in the adaptors for multiplexed sequencing. The final product was assessed for its size distribution and concentration using BioAnalyzer High Sensitivity DNA Kit (Agilent) and Kapa Library Quantification Kit (Kapa Biosystems). The libraries were pooled and diluted to 2 nM in EB buffer (Qiagen) and then denatured using the Illumina protocol. The denatured libraries were diluted to 10 pM by pre-chilled hybridization buffer and loaded onto TruSeq SR v3 flow cells on an Illumina HiSeq 2000 (Illumina) and run for 51 cycles using a single-read recipe (TruSeq SBS Kit v3, Illumina) according to the manufacturer’s instructions. Illumina CASAVA pipeline (released version 1.8, Illumina) was used to obtain de-multiplexed sequencing reads (fastq files) passed the default purify filter. Additional quality filtering used FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) to keep only reads that have at least 80% of bases with a quality score of 20 or more (conducted by fastq_quality_filter function) and reads left with minimum 10 bases after being trimmed with reads with a quality score of 20 (conducted by fastq_quality_trimmer function).
RNA-seq alignment and differential expression analysis
A bowtie2 index was built for the GRCh37 genome assembly using bowtie version 2.2.3. The RNA-seq reads of each of the 4 samples were mapped using Tophat version 2.0.9 (Kim et al., 2013; Trapnell, Pachter, & Salzberg, 2009) supplied by Ensembl annotation file; GRCh37.67.gtf. Log2-transformed RPKM (Reads Per Kilobase per Million mapped reads) values were calculated using HTSeq-count (Anders, Pyl, & Huber, 2015), followed by GC-correction provided with the Ensemble gene annotation using cqn R package (Hansen, Irizarry, & Wu, 2012). The density plots of RPKM values were generated by ggplot2 version 1.0.1 R package after filtering out genes that were not expressed in all samples. RnaSeqMetrics function under Picard tools (v.1.102; http://picard.sourceforge.net) was used to compute the 5′–3′ coverage bias along gene body as well as the number of bases assigned to various classes of RNA. The Weighted Average Difference (WAD) method (Hashimoto-Torii et al., 2010; Kadota, Nakai, & Shimizu, 2008) was used to identify differentially expressed genes.
Alternative splicing analysis
juncBASE (Junction-Based Analysis of Splicing Events) version 0.9 software (Brooks et al., 2011) was used to identify and classify exon-centric alternative splicing events based on splice junction reads from Cufflinks and Cuffmerge, and exon coordinates from GRCh37.67.gtf. For the identification of alternative splicing events and quantifying of events from each sample (step 5 of juncBASE) the following parameters were used: an SQLite de novo transcript database created from the Cuffmerge output (step 0 of juncBASE) for the “txt_db1” and “txt_db2” parameters and an SQLite transcript database created from the exons in GRCh37.67.gtf for the “txt_db3” parameter. Differentially spliced events were calculated for each event category: alternative acceptor, alternative donor, alternative first exon, alternative last exon, cassette, intron retention, alternative acceptor (AA) only junction, alternative donor (AD) only junction and mutually exclusive spliced events.
As an alternative approach for splicing analysis, FineSplice pipeline version 0.2.2 (Gatto et al., 2014) was used to confirm vignette alternative splicing events from the juncBASE analysis. Following alignment with TopHat2 (using known transcript annotations), FineSplice takes the resulting BAM file as input, and provides a confident set of expressed junctions with the corresponding read counts. Potential false positives arising from spurious alignments were filtered out through a semi-supervised anomaly detection strategy based on logistic regression. Multiple mapping reads with a unique hit after filtering are rescued and reallocated to the most reliable candidate location. The resulting list of splice junctions was filtered at 0.1> probability score (i.e., junction reliability) and compared with that of juncBASE results.
Semi-quantitative RT-PCR
A 0.1 ng aliquot from each cDNA sample used in the RNA-seq analysis was used for semi-quantitative RT-PCR. Forward: 5’-CATCCCTCGAGGTACGATGC-3’ and Reverse: 5’-CAGCATTGGAGGAGCCAGAA-3’ primers for SHANK2 and Forward: 5’-GGTACACCAATAAGAGGAGCCC-3’ and Reverse: 5’-ATCTTCTGCCCCCAACATCC-3’ primers for PTPRD, respectively, were designed to span the alternatively spliced exon-exon junctions. Semi-quantitative PCR was performed for SHANK2 [94 °C for 2 min, 30 cycles of (94 °C for 30 sec, 55 °C for 30 sec and 72 °C for 60 s), and 72 °C for 60 sec] and PTPRD [94 °C for 2 min, 30 cycles of (94 °C for 30 sec, 65 °C for 30 sec and 72 °C for 60 sec), and 72 °C for 60 sec) genes, respectively using GoTaq Master Mix (Promega). The cycle number was determined appropriate by comparing 5 cycles more or less to ensure exponential (semi-quantitative) amplification. The 103-bp and 82-bp products for SHANK2 and the 273-bp and 261-bp products for PTPRD were applied to a BioAnalyzer High Sensitivity chip (Agilent Technologies) for quantification of each band that is specific to either inclusion or exclusion of an alternative exon.
Protein structure and sequence analysis
The X-ray crystallographic structure 4RCA (Protein Data Bank (RCSB.org) contains PTPRD and SLITK1. VMD was used to visualize the structures (Humphrey, Dalke, & Schulten, 1996).
Data deposition
The data reported in this paper has been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE86006).
Results:
Quality control of RNA sequencing data.
To obtain a comprehensive transcriptome of the ethanol-exposed human fetal cortex, total RNAs were isolated from human cortical slices cultured in a medium containing 50 mM ethanol or the same volume of PBS (control) for 24 hours (two female samples per condition - control: C1, C2, and experimental: E1, E2)(Fig. 1A). All the RNA and sequencing libraries were prepared at the same time and sequenced in the same flow cell to minimize confounding factors.
Figure 1. RNA sequencing of human fetal cortical tissue exposed to ethanol.
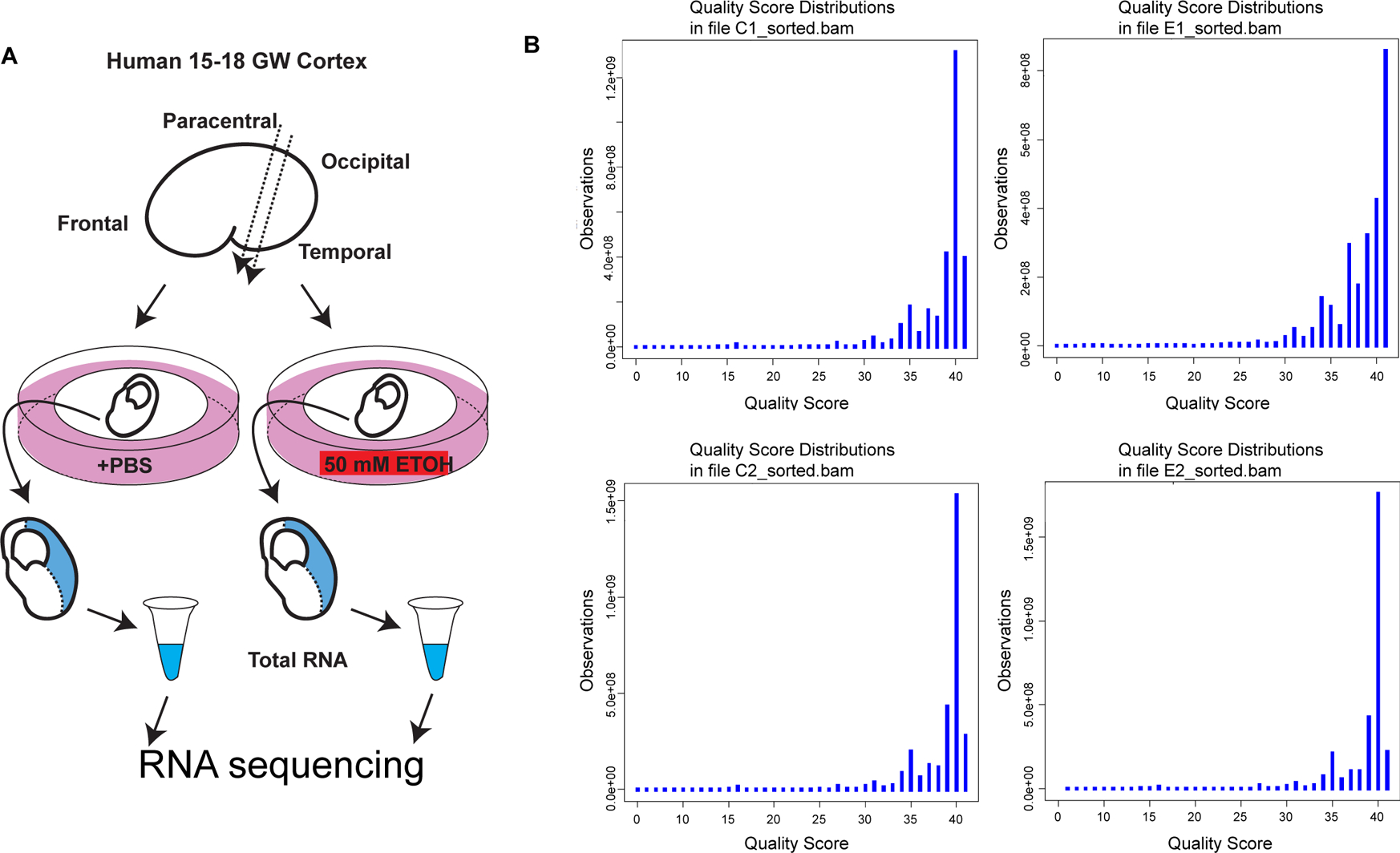
(A) Schematic drawing showing the sample preparation for RNA sequencing. (B) Quality score distribution for each of the human samples. Base calling accuracy was measured by the Phred quality score (Q score).
The quality of the tissue samples has been evaluated previously based on the expression of molecular markers for cellular stress (e.g., HSP70 and FOS) and analysis of cell death prior to the culture (Hashimoto-Torii et al., 2010). Gender-specific gene expression (Reinius & Jazin, 2009; Vawter et al., 2004) in each sample was confirmed with the RNA sequencing data (Table S1). RNA integrity number (RIN) was measured using BioAnalyzer RNA 6000 Pico Kit (Agilent Technologies) to confirm RIN above 8. We obtained a minimum of 61.7 million high quality reads at 1 × 51 bp per sample. Tophat2 alignment utilized the Ensembl reference genome and annotation file (GRCh37, release 67) which, while not the most recent, is the required genome build for computing juncBASE analysis.
We first monitored sequencing quality and robustness. Base calling accuracy was measured by the Phred quality score (Q score). More than 85% bases were above Q30, confirming the high quality of the sequencing (Fig. 1B). The aligned reads contained minimal ribosomal RNA contamination (approximately 0.0006%), highly correct strandedness (>97.9%), and minimal 5′ to 3′ bias (Fig. 2), minimizing false-positive differential gene expression callings. The RPKM distributions of the four samples show a high degree of similarity (Figure S1), confirming the quality of the data. The transcription occurred not only within the coding region (~31.9%), but also within untranslated (UTRs, ~32.4%) and intronic regions of known protein-coding genes (~30.8%), which suggests the existence of novel alternative splicing isoforms. The remaining ~4.9% of reads aligned within intergenic regions, suggesting the expression of novel transcripts.
Figure 2. Distribution plots of aligned reads.
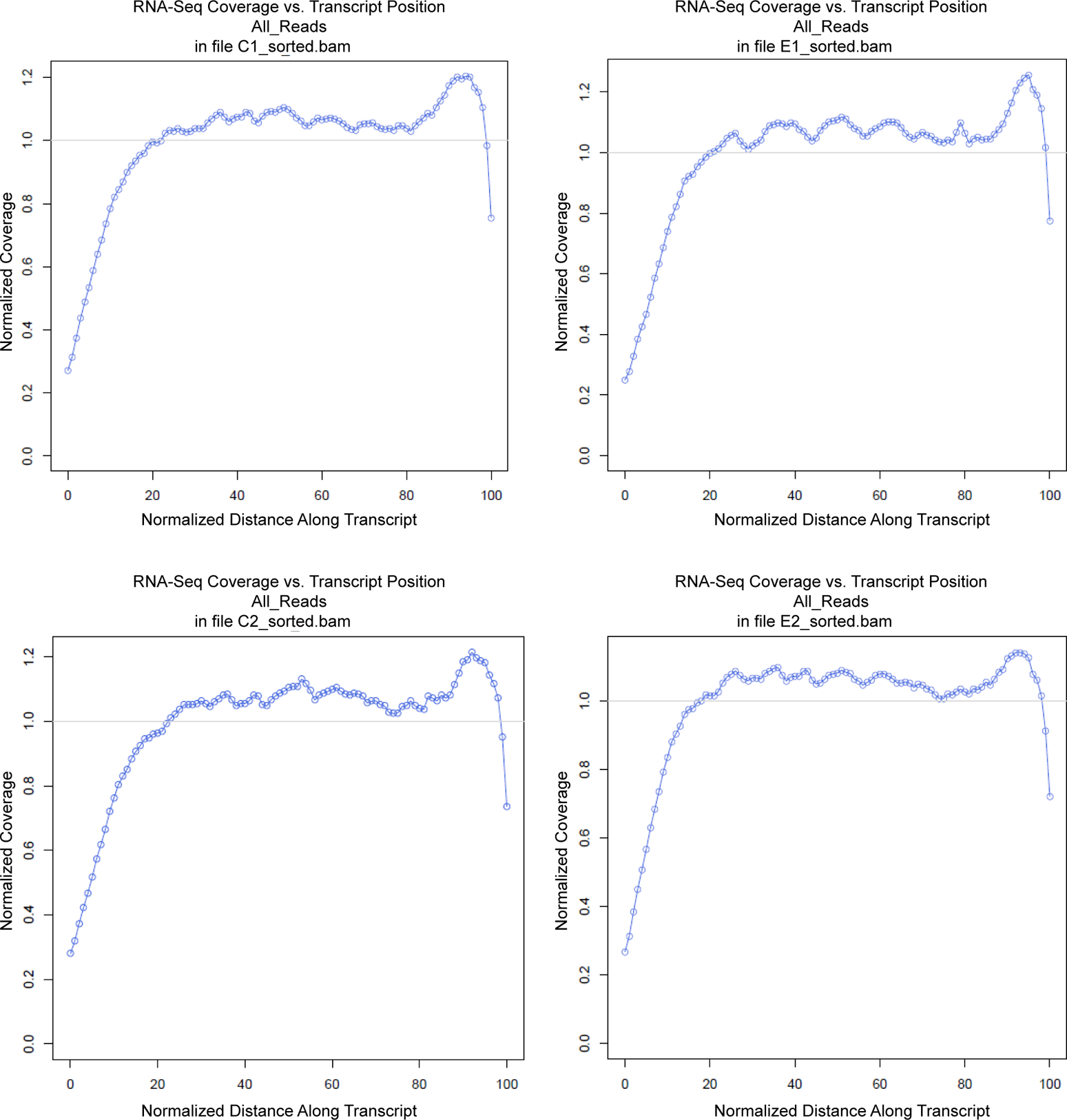
The read coverage in RNA sequencing was plotted across normalized (binned) transcript length for each sample. We confirmed similar distribution of the reads across all transcripts.
For identification of differentially expressed genes (DEGs), we utilized the weighted average difference (WAD) method as it has previously shown superior performance (Hashimoto-Torii et al., 2010; Kadota et al., 2008). As WAD weighs the absolute gene expression levels (Kadota et al., 2008), we did not apply a cutoff based on the RPKM levels. With the WAD value cut off at 0.1, we obtained 9,281 and 9,443 genes as upregulated and downregulated genes in the ethanol-exposed fetal cortex, respectively (Fig. 3, Table S2). qRT-PCR for representative DEGs including HSPA1A, HSPA1B, GADD45B, VEGFA and SATB2 with the same samples showed consistent results (Hashimoto-Torii et al., 2010) confirming the quality and reproducibility of the transcriptome of alcohol-exposed human fetal cerebral cortex.
Figure 3. WAD cutoff and frequency of genes.
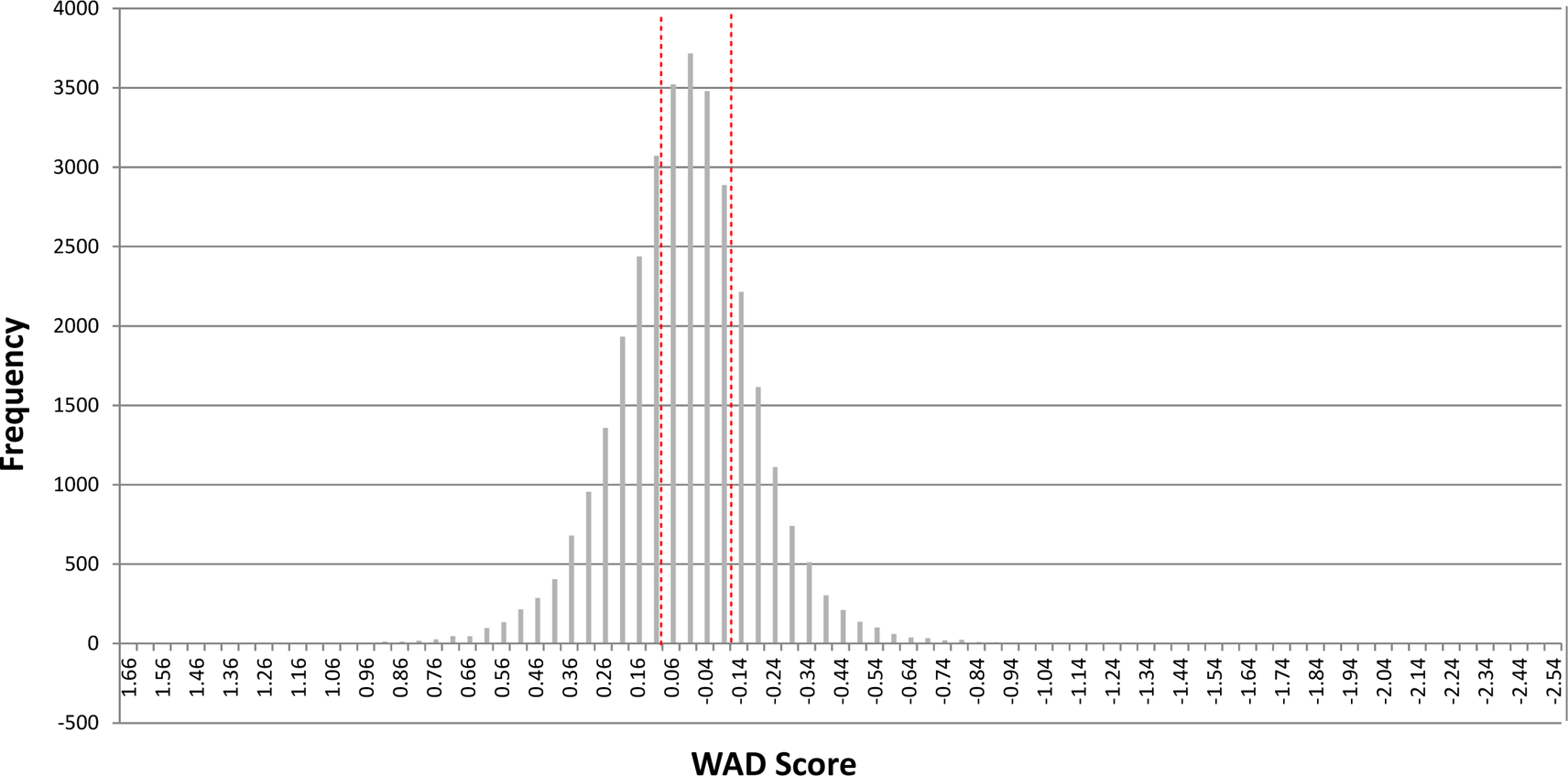
Histograms showing the frequency of genes corresponding to the WAD score. The vertical broken red lines demarcate the WAD cut off points (0.1).
Alternative splicing events affected in the ethanol-exposed fetal cortex.
We next explored the impact of alcohol exposure on alternative splicing events using RNA sequencing data. Analysis of alternative splicing can be performed using two different approaches; the isoform-centric approach or the exon-centric approach (Hooper, 2014). We chose an exon-centric approach, namely juncBASE (Brooks et al., 2011), that considers the inclusion ratio of each individual exon, which can be included in multiple transcript isoforms while excluded in other isoforms. This approach is not dependent on, or biased by, the number of exons within a gene and is more tolerant of the shortness of sequencing reads. Using juncBASE, we analyzed 8 basic splicing modalities: cassette exon, mutually exclusive exon, coordinate cassette exons, alternative 5′ or 3′ splice site, alternative first or last exon, and retained intron (Fig. 4A, Table S3). Using 0.40 as a cut-off of the delta value, which is the ratio of altered splicing index compared to the control (Fig. 4B), we defined a total of 382 alternative splicing events that are differentially regulated in alcohol-exposed cortical samples comparing to the controls. For those events, retained introns were the most frequently affected modality. Of the 382 alternative splicing events, 208 and 174 were known and novel events, respectively (Fig. 4A). The genes that showed significant isoform shifts by ethanol exposure were enriched in those associated with cell death and cell junction (Table S4).
Figure 4. Alternative splicing events enriched in the ethanol-exposed cortical tissues.
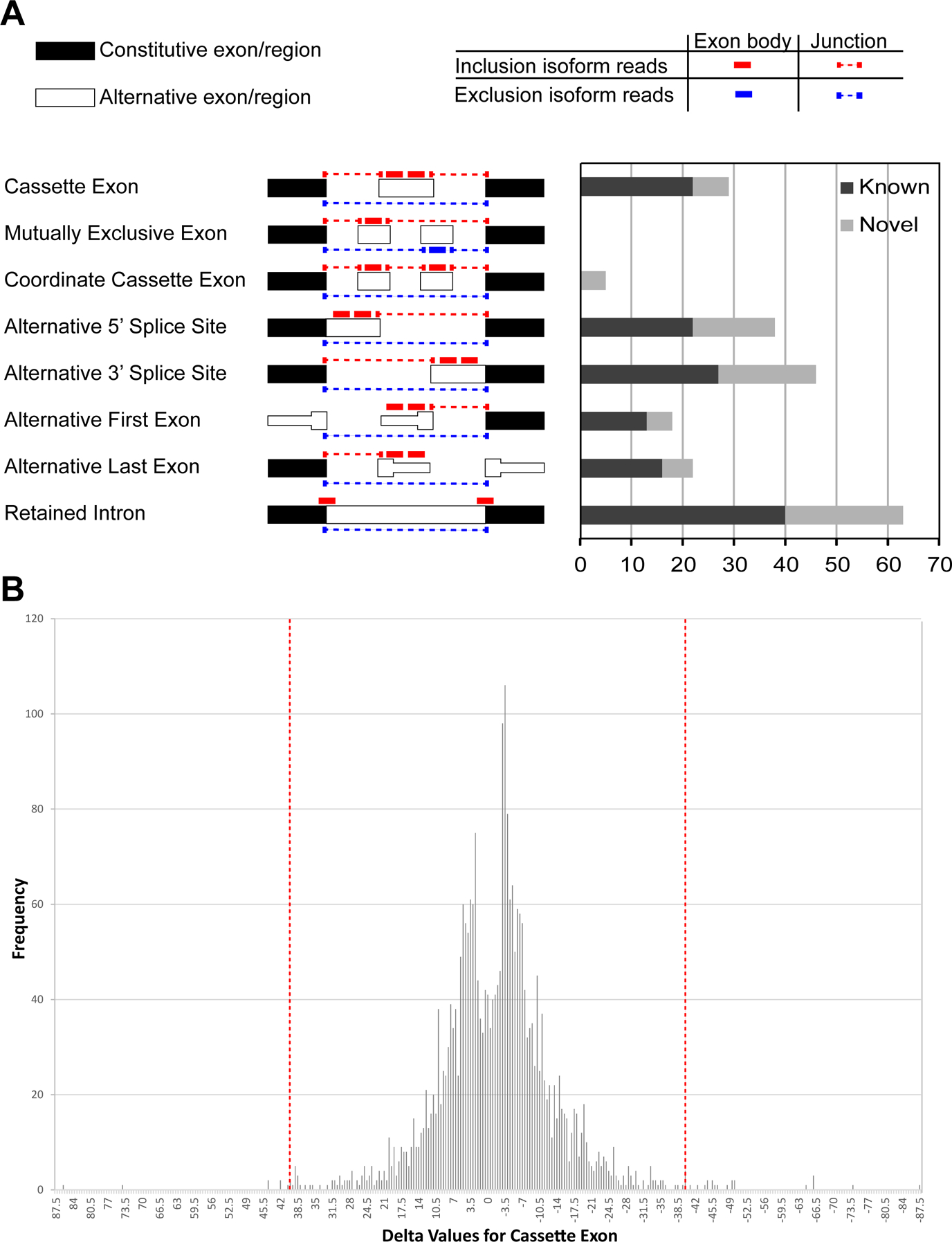
(A) Numbers of known (blue) and novel (red) ethanol exposure-enriched splicing events identified by juncBASE analysis. Diagram of each event is adapted from the manual of juncBASE. (B) Delta cut-off and frequency of genes. Histograms show the frequency of probe sets corresponding to the delta score. The vertical broken red lines demarcate the delta cut off points.
Novel splicing events occur in the ethanol-exposed fetal cortex.
One of the 174 novel events predicted by juncBASE is a novel cassette exon of the SHANK2 (SH3 and multiple ankyrin repeat domains 2) gene (Fig. 5A). There are 3 transcripts and 6 isoforms registered for human SHANK2. The new isoform, which skips an exon that contains 21 base pairs and therefore lacking a 7 amino acid sequence, was more abundantly detected in alcohol-exposed samples, in which the level of total SHANK2 mRNA expression was not significantly changed (Table S2).
Figure 5. Splicing isoforms of SHANK2 and PTPRD genes specific in ethanol exposure.
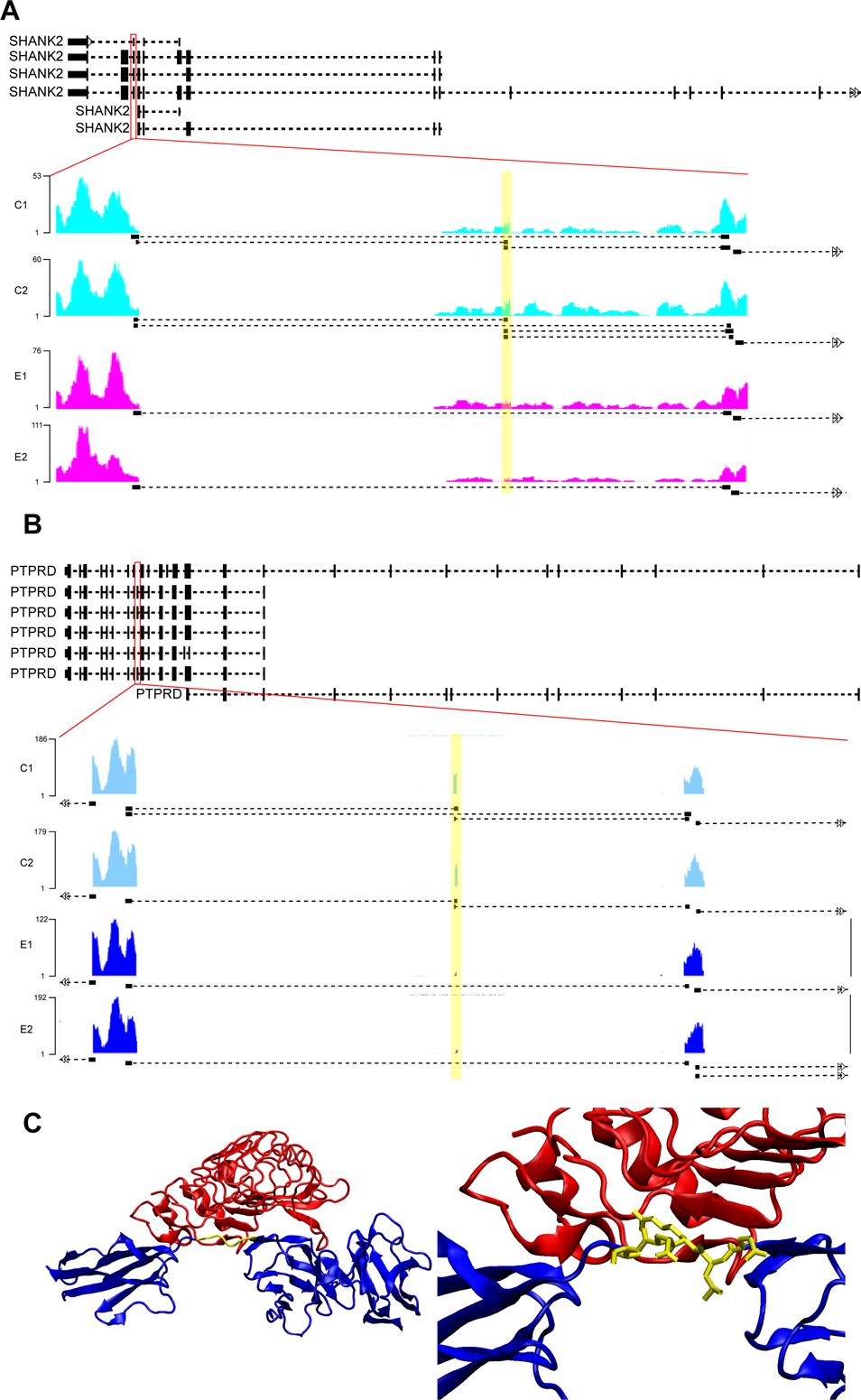
(A, B) The reads coverage in SHANK2 (novel, A) and PTPRD (known, B) gene regions showing the isoforms that are differentially spliced in ethanol-exposed cortices (highlight). Black bars/boxes underneath the exonic read distribution indicate junction reads. (C) The interaction of PTPRD (blue) and SLIRTK (red) is through the amino acid sequence of PTPRD skipped by ethanol exposure (yellow).
PTPRD (Protein Tyrosine Phosphatase, Receptor Type D) is one of the genes of which a specific known isoform is enriched in alcohol-exposed group (Fig. 5B). 7 isoforms of human PTPRD have been registered; in the differentially spliced isoform, the exon that consists of 15 base pairs for 5 amino acids was highly skipped by alcohol exposure. This region corresponds to an interdomain linker that has been identified as part of the minimal interaction region for PTPRD and SLIRTK (Fig. 5C) (Um et al., 2014; Yamagata et al., 2015). SLIRTK (Slit-and Trk-like family proteins) is implicated in the orchestration of synaptogenesis.
Validation of the splicing events in the ethanol-exposed fetal cortex.
Recently, Gatto et al. (Gatto et al., 2014) have developed a novel analysis pipeline based on TopHat2, combined with a splice junction detection algorithm named as FineSplice. FineSplice allows effective elimination of spurious junction hits arising from artefactual alignments, achieving up to 99% precision in both real and simulated data sets. They also showed that only 20 million single-end reads can be sufficient to sensitively and reproducibly detect exon-exon junctions. By using FineSplice (Gatto et al., 2014), we further confirmed the exon skipping of PTPRD and SHANK2 (Fig.S2, Table S5). We selected 0.1 as the cutoff probability score. In addition, qRT-PCR also validated the specific isoforms in the ethanol-exposed cortical tissue (Fig.6).
Figure 6. RT-PCR confirms splicing isoforms of SHANK2 and PTPRD genes specific to ethanol exposure.
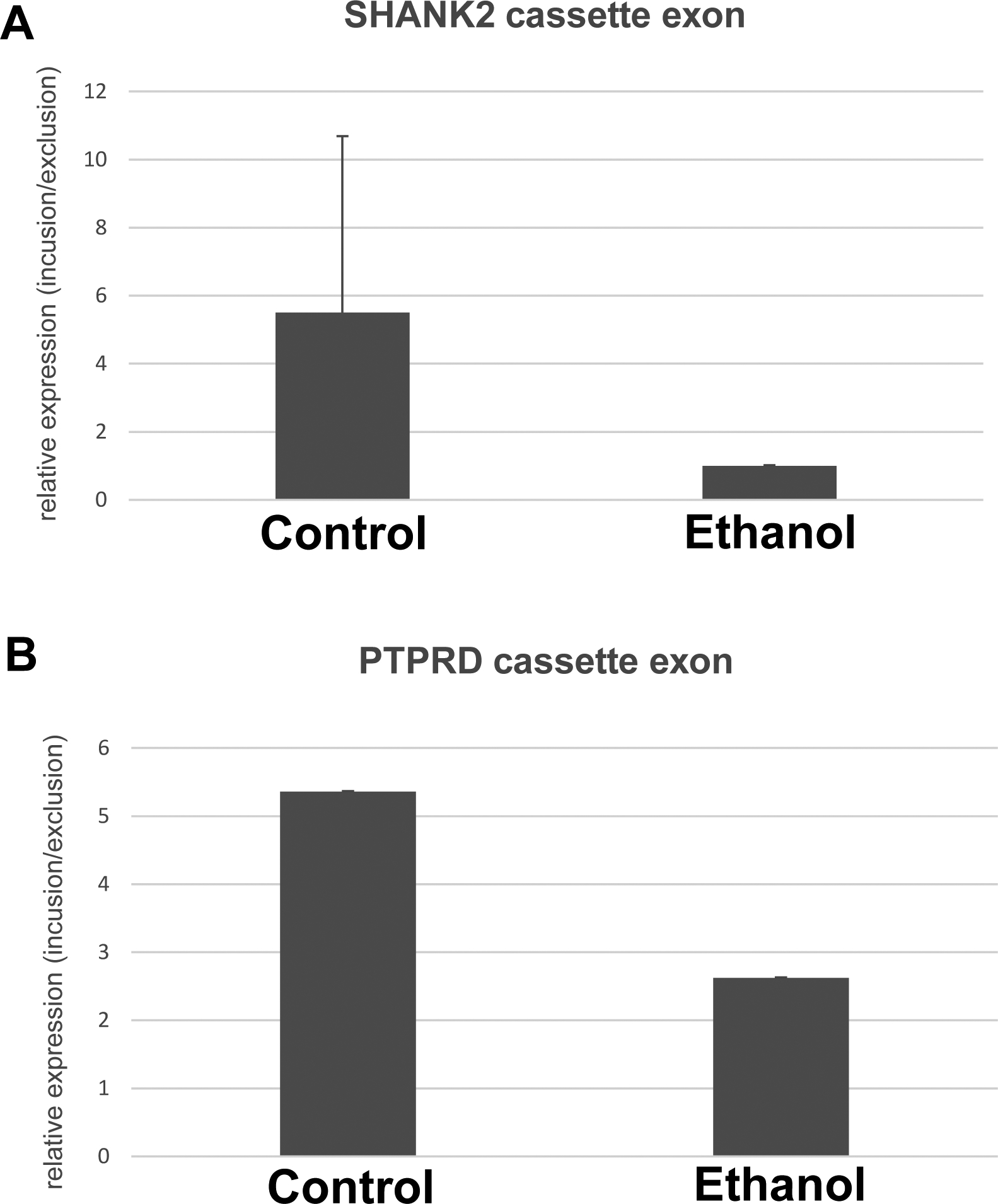
Semi-quantitative RT-PCR was performed using primers that were designed to span the alternatively spliced exon-exon junctions for SHANK2 (A) and PTPRD (B). The mole ratio of short amplicons (derived from exclusion cassette exon events) over long amplicons (derived from inclusion cassette exon events) was calculated and statistically analyzed between the control and ethanol-exposure groups. Comparisons between control and ethanol by t-test yield p<0.05 in both genes. Mean +/− SEM.
Discussion:
In this study, we utilized RNA sequencing to identify more than 18,000 genes that are differentially expressed between alcohol- and control-exposed human fetal cortical samples. Compared to our previous microarray study which had identified 9,000 genes that show altered expression with the same samples using the same WAD cut off (0.1, Hashimoto-Torii et al., 2010), the technical advances of our current study demonstrates the higher sensitivity of RNA sequencing than microarray.
Our analysis found significant changes in alternative splicing events in the ethanol-exposed fetal cortex. The Gene Ontology terms enriched in differentially spliced genes include cell death, apoptosis, and cell junctions (Table S4), all of which are known to be altered by ethanol exposure (Cheema, West, & Miranda, 2000; Goldowitz et al., 2014; Mooney & Miller, 2003; Ramanathan, Wilkemeyer, Mittal, Perides, & Charness, 1996). In addition, we found previously unknown isoforms of several genes in our analysis, including SHANK2 and TIA1. TIA1, which encodes an RNA binding protein, is involved in the control of cell death in response to the endoplasmic reticulum stress through the post-transcriptional control of stress granules (Arimoto-Matsuzaki, Saito, & Takekawa, 2016).
Furthermore, the Ingenuity Pathway Analysis has revealed that the differentially alternatively spliced genes are enriched with disease pathways implicated in pervasive neurodevelopmental conditions such as autism and intellectual disability. These include SHANK2 and PTPRD, genes known to regulate synaptic formation and function (Berkel et al., 2010; Boeckers et al., 2005; Choucair et al., 2015; Kumar, 2010; Lim et al., 2016). Therefore, abnormalities in alternative splicing events may be critically involved in the pathogenic effects of alcohol on cortical development.
Our findings provide a landscape of RNA splicing changes by ethanol. Further defining the cell type-specific changes will enable us to evaluate the biological impacts of such gene isoform changes. Since recent studies have demonstrated significant effects of ethanol at lower exposure levels (Cullen, Burne, Lavidis, & Moritz, 2013), it is important to examine whether the defined splicing events also occur at lower doses of ethanol. Another interesting topic would be the regulatory mechanisms of such specific splicing. Although our GO analysis of the top 3,000 DEGs (Table S6) found no particular enrichment of GOs related to RNA splicing (FDR non-controlled p value>0.05), several genes relevant to RNA splicing are included in the DEG list, suggesting the possibility that the altered expression of these genes may play critical roles in the ethanol-induced specific splicing events. As small RNAs such as snRNA and miRNAs are known to regulate RNA splicing (Boutz, Chawla, Stoilov, & Black, 2007; Wu, Chiou, Chiu, & Yang, 2013), profiling of such small RNAs to examine their potential involvement in ethanol-induced splicing changes would be of great interest.
Conclusion:
The RNA sequencing of early mid-gestation human cortical tissue exposed to alcohol revealed a number of differential alternative splicing events induced by ethanol exposure. Those genes are involved in the control of cell death, cell junctions, and synapse formation, suggesting that these biological events are disrupted epigenetically by ethanol exposure. In particular, the alternatively spliced PTPRD lacks part of the minimal essential interaction domain with SLIRTK, suggesting one mechanism for epigenetic disruptions in the alcohol-exposed fetal brain.
Supplementary Material
Figure S1 Validation of sequencing quality. (A) Each box plot show quantile-normalized and log2-transformed RPKM for all annotated genes that were expressed at log2RPKM >=0 in at least two samples in entire four samples. The RPKM distributions are similar between samples. (B, C) Scatter plots (B) and correlation plots (C), confirm the overall similarity in the RPKM distribution between samples, especially of highly expressed genes (B). Pearson correlation coefficients were shown with scaled-colored circles as well as numbers (C).
Figure S2 Output of SHANK2 and PTPRD in FineSplice. Schematic illustration of alternative transcripts for SHANK2 (A) and PTPRD (B) are depicted. Black rectangles and dashed lines represent exons and intronic regions that are spliced out, respectively. Zoomed images depict junction reads from RNA-seq. Ethanol-treated samples (E1 and E2) were predicted to express transcripts that skip the middle exon (which are illustrated as red dashed arrows labeled as “both”) exclusively and lack the expression of transcripts that include this middle exon (which yield “left” and “right” junction reads). The number of junction reads for non-skipping (“left”, “right”) and skipping (“both”) junctions in each samples (C1, C2 (control), E1, E2 (ethanol)) were calculated by FineSplice and summarized in the tables.
Table S1 Expression of gender specific genes. https://www.dropbox.com/s/5wxlpia7wpm8awm/Supplemental%20Table%201.xls?dl=0
Table S2 WAD ranking of RNA sequencing data of the ethanol-exposed cortex. All are listed without cut off. https://www.dropbox.com/s/tq1imzjlrb8zzlr/Supplementary%20Table1.xlsx?dl=0
Table S3 Splicing events defined by juncBASE. https://www.dropbox.com/s/zjr6zxos8p9h0jm/Supplementary%20Table%202.xlsx?dl=0
Table S4 Gene Ontology analysis output of changed splicing events. All are listed without cut off. https://www.dropbox.com/s/c9tpzf2daw1urre/Supplementary%20Table3.xlsx?dl=0
Table S6 Gene Ontology analysis output of differentially expressed genes. https://www.dropbox.com/s/lhe349tvym2cj1n/Supplementary%20Table%206.xls?dl=0
Table S5 Splicing events defined by FineSplice. All are listed without cut off. https://www.dropbox.com/s/vl9vhpj1afy820k/Supplementary%20Table%205.xlsx?dl=0
Acknowledgements:
This work was supported by National Institute of Health (R21AA024882, R01AA025215), ABMRF/The Foundation for Alcohol Research (K.H-T.), Brain & Behavior Research Foundation, Scott-Gentle Foundation (K.H-T. and M.T.) and Avery Translational Research Career Development Program Award (M.T.).
References:
- Anders S, Pyl PT, & Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto-Matsuzaki K, Saito H, & Takekawa M (2016). TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun, 7, 10252. doi: 10.1038/ncomms10252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y (2008). Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci, 31(12), 626–636. doi: 10.1016/j.tins.2008.09.002[pii] S0166-2236(08)00211-7 [DOI] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, … Rappold GA (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet, 42(6), 489–491. doi: 10.1038/ng.589 [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, & Gundelfinger ED (2005). C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. J Neurochem, 92(3), 519–524. doi: 10.1111/j.1471-4159.2004.02910.x [DOI] [PubMed] [Google Scholar]
- Boutz PL, Chawla G, Stoilov P, & Black DL (2007). MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev, 21(1), 71–84. doi: 10.1101/gad.1500707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, Dudoit S, … Graveley BR (2011). Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res, 21(2), 193–202. doi: 10.1101/gr.108662.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun F, & Warren K (2007). Fetal alcohol syndrome: historical perspectives. Neurosci Biobehav Rev, 31(2), 168–171. [DOI] [PubMed] [Google Scholar]
- Cheema ZF, West JR, & Miranda RC (2000). Ethanol induces Fas/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol Clin Exp Res, 24(4), 535–543. [PubMed] [Google Scholar]
- Choucair N, Mignon-Ravix C, Cacciagli P, Abou Ghoch J, Fawaz A, Megarbane A, … Chouery E (2015). Evidence that homozygous PTPRD gene microdeletion causes trigonocephaly, hearing loss, and intellectual disability. Mol Cytogenet, 8, 39. doi: 10.1186/s13039-015-0149-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarren SK, Alvord EC Jr., Sumi SM, Streissguth AP, & Smith DW (1978). Brain malformations related to prenatal exposure to ethanol. J Pediatr, 92(1), 64–67. [DOI] [PubMed] [Google Scholar]
- Cullen CL, Burne TH, Lavidis NA, & Moritz KM (2013). Low dose prenatal ethanol exposure induces anxiety-like behaviour and alters dendritic morphology in the basolateral amygdala of rat offspring. PLoS One, 8(1), e54924. doi: 10.1371/journal.pone.0054924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer SL, Tapert SF, Mattson SN, Paulus MP, Spadoni AD, & Riley EP (2007). Prenatal alcohol exposure affects frontal-striatal BOLD response during inhibitory control. Alcohol Clin Exp Res, 31(8), 1415–1424. doi: ACER443 [pii] 10.1111/j.1530-0277.2007.00443.x [DOI] [PubMed] [Google Scholar]
- Gatto A, Torroja-Fungairino C, Mazzarotto F, Cook SA, Barton PJ, Sanchez-Cabo F, & Lara-Pezzi E (2014). FineSplice, enhanced splice junction detection and quantification: a novel pipeline based on the assessment of diverse RNA-Seq alignment solutions. Nucleic Acids Res, 42(8), e71. doi: 10.1093/nar/gku166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P, Nunez SC, Narr KL, Mattson SN, May PA, Adnams CM, … Sowell ER (2014). Developmental Trajectories for Visuo-Spatial Attention are Altered by Prenatal Alcohol Exposure: A Longitudinal FMRI Study. Cereb Cortex. doi: 10.1093/cercor/bhu162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldowitz D, Lussier AA, Boyle JK, Wong K, Lattimer SL, Dubose C, … Hamre KM (2014). Molecular pathways underpinning ethanol-induced neurodegeneration. Front Genet, 5, 203. doi: 10.3389/fgene.2014.00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Irizarry RA, & Wu Z (2012). Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics, 13(2), 204–216. doi: 10.1093/biostatistics/kxr054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Kawasawa YI, Kuhn A, & Rakic P (2010). Combined transcriptome analysis of fetal human and mouse cerebral cortex exposed to alcohol. Proc Natl Acad Sci U S A, 108(10), 4212–4217. doi: 1100903108 [pii] 10.1073/pnas.1100903108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Torii M, Fujimoto M, Nakai A, El Fatimy R, Mezger V, … Rakic P (2014). Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron, 82(3), 560–572. doi: 10.1016/j.neuron.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JE (2014). A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum Genomics, 8, 3. doi: 10.1186/1479-7364-8-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, & Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph, 14(1), 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Ishii S, & Hashimoto-Torii K (2015). Impact of prenatal environmental stress on cortical development. Front Cell Neurosci, 9, 207. doi: 10.3389/fncel.2015.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota K, Nakai Y, & Shimizu K (2008). A weighted average difference method for detecting differentially expressed genes from microarray data. Algorithms Mol Biol, 3, 8. doi: 10.1186/1748-7188-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, & Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol, 14(4), R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA (2010). SHANK2 redemption: another synaptic protein for mental retardation and autism. Clin Genet, 78(6), 519–521. doi: 10.1111/j.1399-0004.2010.01530_2.x [DOI] [PubMed] [Google Scholar]
- Lim CS, Kim H, Yu NK, Kang SJ, Kim T, Ko HG, … Kaang BK (2016). Enhancing inhibitory synaptic function reverses spatial memory deficits in Shank2 mutant mice. Neuropharmacology. doi: 10.1016/j.neuropharm.2016.08.016 [DOI] [PubMed] [Google Scholar]
- Mattson SN, & Riley EP (1998). A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res, 22(2), 279–294. [DOI] [PubMed] [Google Scholar]
- McGee CL, Fryer SL, Bjorkquist OA, Mattson SN, & Riley EP (2008). Deficits in social problem solving in adolescents with prenatal exposure to alcohol. Am J Drug Alcohol Abuse, 34(4), 423–431. [DOI] [PubMed] [Google Scholar]
- McGee CL, Schonfeld AM, Roebuck-Spencer TM, Riley EP, & Mattson SN (2008). Children with heavy prenatal alcohol exposure demonstrate deficits on multiple measures of concept formation. Alcohol Clin Exp Res, 32(8), 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda RC, Pietrzykowski AZ, Tang Y, Sathyan P, Mayfield D, Keshavarzian A, … Hereld D (2010). MicroRNAs: master regulators of ethanol abuse and toxicity? Alcohol Clin Exp Res, 34(4), 575–587. doi: 10.1111/j.1530-0277.2009.01126.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney SM, & Miller MW (2003). Ethanol-induced neuronal death in organotypic cultures of rat cerebral cortex. Brain Res Dev Brain Res, 147(1–2), 135–141. [DOI] [PubMed] [Google Scholar]
- Ramanathan R, Wilkemeyer MF, Mittal B, Perides G, & Charness ME (1996). Alcohol inhibits cell-cell adhesion mediated by human L1. J Cell Biol, 133(2), 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B, & Jazin E (2009). Prenatal sex differences in the human brain. Mol Psychiatry, 14(11), 987,988–989. doi: 10.1038/mp.2009.79 [DOI] [PubMed] [Google Scholar]
- Riley EP, Mattson SN, Sowell ER, Jernigan TL, Sobel DF, & Jones KL (1995). Abnormalities of the corpus callosum in children prenatally exposed to alcohol. Alcohol Clin Exp Res, 19(5), 1198–1202. [DOI] [PubMed] [Google Scholar]
- Riley EP, & McGee CL (2005). Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood), 230(6), 357–365. doi: 230/6/357 [pii] [DOI] [PubMed] [Google Scholar]
- Roebuck TM, Mattson SN, & Riley EP (1998). A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res, 22(2), 339–344. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Mattson SN, Kan E, Thompson PM, Riley EP, & Toga AW (2008). Abnormal cortical thickness and brain-behavior correlation patterns in individuals with heavy prenatal alcohol exposure. Cereb Cortex, 18(1), 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BL, Levitt P, & Stanwood GD (2009). Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci, 10(4), 303–312. doi: nrn2598 [pii] 10.1038/nrn2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, & Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25(9), 1105–1111. doi: 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Kim KH, Park BS, Choi Y, Kim D, Kim CY, … Kim HM (2014). Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat Commun, 5, 5423. doi: 10.1038/ncomms6423 [DOI] [PubMed] [Google Scholar]
- Vawter MP, Evans S, Choudary P, Tomita H, Meador-Woodruff J, Molnar M, … Bunney WE (2004). Gender-specific gene expression in post-mortem human brain: localization to sex chromosomes. Neuropsychopharmacology, 29(2), 373–384. doi: 10.1038/sj.npp.1300337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KR, & Bast RJ (1988). Alcohol-related birth defects: an update. Public Health Rep, 103(6), 638–642. [PMC free article] [PubMed] [Google Scholar]
- Warren KR, Calhoun FJ, May PA, Viljoen DL, Li TK, Tanaka H, … Mundle G (2001). Fetal alcohol syndrome: an international perspective. Alcohol Clin Exp Res, 25(5 Suppl ISBRA), 202S–206S. [DOI] [PubMed] [Google Scholar]
- Wu CT, Chiou CY, Chiu HC, & Yang UC (2013). Fine-tuning of microRNA-mediated repression of mRNA by splicing-regulated and highly repressive microRNA recognition element. BMC Genomics, 14, 438. doi: 10.1186/1471-2164-14-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata A, Sato Y, Goto-Ito S, Uemura T, Maeda A, Shiroshima T, … Fukai S (2015). Structure of Slitrk2-PTPdelta complex reveals mechanisms for splicing-dependent trans-synaptic adhesion. Sci Rep, 5, 9686. doi: 10.1038/srep09686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Roussotte F, Kan E, Sulik KK, Mattson SN, Riley EP, … Sowell ER (2012). Abnormal cortical thickness alterations in fetal alcohol spectrum disorders and their relationships with facial dysmorphology. Cereb Cortex, 22(5), 1170–1179. doi: 10.1093/cercor/bhr193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Lebel C, Lepage C, Rasmussen C, Evans A, Wyper K, … Beaulieu C Developmental cortical thinning in fetal alcohol spectrum disorders. Neuroimage, 58(1), 16–25. doi: S1053-8119(11)00644-6 [pii] 10.1016/j.neuroimage.2011.06.026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Validation of sequencing quality. (A) Each box plot show quantile-normalized and log2-transformed RPKM for all annotated genes that were expressed at log2RPKM >=0 in at least two samples in entire four samples. The RPKM distributions are similar between samples. (B, C) Scatter plots (B) and correlation plots (C), confirm the overall similarity in the RPKM distribution between samples, especially of highly expressed genes (B). Pearson correlation coefficients were shown with scaled-colored circles as well as numbers (C).
Figure S2 Output of SHANK2 and PTPRD in FineSplice. Schematic illustration of alternative transcripts for SHANK2 (A) and PTPRD (B) are depicted. Black rectangles and dashed lines represent exons and intronic regions that are spliced out, respectively. Zoomed images depict junction reads from RNA-seq. Ethanol-treated samples (E1 and E2) were predicted to express transcripts that skip the middle exon (which are illustrated as red dashed arrows labeled as “both”) exclusively and lack the expression of transcripts that include this middle exon (which yield “left” and “right” junction reads). The number of junction reads for non-skipping (“left”, “right”) and skipping (“both”) junctions in each samples (C1, C2 (control), E1, E2 (ethanol)) were calculated by FineSplice and summarized in the tables.
Table S1 Expression of gender specific genes. https://www.dropbox.com/s/5wxlpia7wpm8awm/Supplemental%20Table%201.xls?dl=0
Table S2 WAD ranking of RNA sequencing data of the ethanol-exposed cortex. All are listed without cut off. https://www.dropbox.com/s/tq1imzjlrb8zzlr/Supplementary%20Table1.xlsx?dl=0
Table S3 Splicing events defined by juncBASE. https://www.dropbox.com/s/zjr6zxos8p9h0jm/Supplementary%20Table%202.xlsx?dl=0
Table S4 Gene Ontology analysis output of changed splicing events. All are listed without cut off. https://www.dropbox.com/s/c9tpzf2daw1urre/Supplementary%20Table3.xlsx?dl=0
Table S6 Gene Ontology analysis output of differentially expressed genes. https://www.dropbox.com/s/lhe349tvym2cj1n/Supplementary%20Table%206.xls?dl=0
Table S5 Splicing events defined by FineSplice. All are listed without cut off. https://www.dropbox.com/s/vl9vhpj1afy820k/Supplementary%20Table%205.xlsx?dl=0