

Abstract
Coronaviruses are responsible on respiratory diseases in animal and human. The combination of numerical encoding techniques and digital signal processing methods are becoming increasingly important in handling large genomic data. In this paper, we propose to analyze the SARS-CoV-2 genomic signature using the combination of different nucleotide representations and signal processing tools in the aim to identify its genetic origin. The sequence of SARS-CoV-2 was compared with 21 relevant sequences including Bat, Yak and Pangolin coronavirus sequences. In addition, we developed a new algorithm to locate the nucleotide modifications. The results show that the Bat and Pangolin coronaviruses were the most related to SARS-CoV-2 with 96% and 86% of identity all along the genome. Within the S gene sequence, the Pangolin sequence presents local highest nucleotide identity. Those findings suggest genesis of SARS-Cov-2 through evolution from Bat and Pangolin strains. This study offers new ways to automatically characterize viruses.
Keywords: SARS-CoV-2, Bat, Yak, Pangolin, Genome signature, COVID19
Highlights
-
•
We propose to analyze the SARS-CoV-2 genomic signature using the combination of different nucleotide representations in the aim to identify its genetic origin.
-
•
The SARS-CoV-2 sequence was compared with 21 relevant sequences including Bat, Yak and Pangolin coronavirus sequences.
-
•
the Bat and Pangolin coronaviruses were the most related to SARS-CoV-2
-
•
Within the S gene sequence, the Pangolin sequence presents local highest nucleotide identity.
-
•
This study suggests genesis of SARS-Cov-2 through evolution from bat and pangolin strains.
1. Introduction
Recently, unidentified human pneumonia has started from a local fresh seafood market in Wuhan, in December 2019. This emerging virus was later identified as coronavirus called SARS-CoV-2 responsible on Coronavirus Disease 19 (COVID-19) [1,2]. It spreads to more than 216 countries all around the world causing a pandemic [3]. It is considered as the third major zoonotic human coronavirus outbreak of this century after the prominence of the two coronavirus pandemics; Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV) [4,5].
Despite the fact that COVID-19 has a death rate of 2.8% as of April, the 8,844,171 confirmed cases with 465,460 confirmed deaths in a few months (December 8, 2019 to June 22, 2020) are terrifying. Indeed, this virus is highly contagious and the number of infected people can be doubled in less than seven days with a basic reproductive number (R0) of 2.2–2.7 [6].
The SARS-CoV-2 is an enveloped, positive-sense single-stranded RNA virus of almost 29,700 nucleotides. It belongs to the family of Coronaviridae and the subfamily of Orthocoronavirinae which contains 4 genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and the recently defined Deltacoronavirus. SARS-CoV-2 is a member of the genera of Betacoronavirus and the SARSr-CoV specie responsible on severe lower respiratory tract infection in human as SARS and MERS infections [7].
Phylogenetic studies proved a complex coronaviruses evolutionary history originated from an ancient common ancestor (around 10,000 years ago) [8]. Indeed, these viruses present a high rate of mutation and recombination that enable great plasticity and high dynamic genome evolution. The mutation rate of coronaviruses varies from 1 /1000 to 1 /10,000 nucleotides during the replication of RNA-dependent RNA polymerases (RdRP) [9]. Also, coronaviruses are known to use template switching mechanism resulting in high rate of homologous RNA recombination among different viral strain genomes [4]. Furthermore, the existence of different virus hosts favors cross species infection resulting in adaptation of viruses and the emergence of new ones [8,9]. For instance, Bats can harbor different types of coronavirus creating a favorable environment for the emergence of new viruses [10].
Till now, scientists are trying to know how SARS-CoV-2 was emerged and infected Humans. Different hypotheses have been proposed. Recently, analysis of the whole genome of two viruses (HKU-SZ-002a and HKU-SZ-005b) confirmed that SARS-CoV-2 belongs to lineage B (Sarbecovirus) of Betacoronavirus and demonstrated the existence of a novel coronavirus genetically closer to the bat SARS-like coronavirus bat-SL-CoVZXC21 (MG772934) and bat-SL-CoVZC45 (MG772933) [10,11]. Another research in [12] proved that SARS-CoV-2 has the highest similarity (96.3%) with the Bat coronavirus RaTG13 all along the genome, using phylogenetic and similarity plot analysis. Other ones suggest that the human SARS-CoV-2, could also evaluate from Yak coronaviruses [13] and also through recombination with Pangolin coronaviruses [14,15].
The organism's genomic signature is a very important graphical representation that allows understanding of the intragenic variations [[16], [17], [18]], especially for handling large genomic data. This assay is based on the Chaos Game Representation (CGR) derived from the chaos theory of Jeffrey et al. (1990) and it was considered as a mapping method of large genome sequences [19]. In this study, we try to identify the SARS-CoV-2 genetic origin using a combination of different DNA representation and signal processing tools. We compared full genome sequence of SARS-CoV-2 to relevant viral genomes: sequences of the four Orthocoronavirinae genus, the 15th species included in the Betacoronavirus genus and also Yak and Pangolin coronaviruses.
2. Material and methods
First, we start by extracting the genomic sequences from the NCBI database (http://www.ncbi.nlm.nih.gov/Genbank/). Secondly, we applied to each sequence a CGR image to capture the information of the whole genome sequence. This considered step is followed by computing the centroid points (Chaos-Centroid) of M subsquare CGR images. The third step consists of applying the Smoothed Discrete Fourier Transform (SDFT) to the Frequency Chaos Game Signal (FCGS) order two, corresponding to genomic sequences with the goal of seeing the correlation between the genomes.
2.1. Genomic database
The complete genomes were downloaded from the NCBI database. Sequences investigated in this study are presented in Table 1 . Our genomic database contains in totally 22 species (Table 1).
Table 1.
Genomics sequences database and their correspond length extracted from NCBI platform.
| Order | Species | Accession number | Length (nt) |
|---|---|---|---|
| A | SARS-CoV-2 | NC_045512 | 29,903 |
| B | Betacoronavirus RaTG13 | MN996532 | 29,855 |
| C | Betacoronavirus CoVZC45 | MG772933 | 29,802 |
| D | Betacoronavirus CoVZXC21 | MG772934.1 | 29,732 |
| E | Alphacoronavirus | DQ811787 | 27,533 |
| F | Gammacoronavirus A116E7 | FN430415 | 27,593 |
| G | Deltacoronavirus | KJ481931 | 25,406 |
| H | Bat Hp-betacoronavirus/ | NC_025217 | 31,491 |
| I | Bovine coronavirus Mebus | BCU00735 | 31,032 |
| J | Human coronavirus OC43 | KX344031 | 30,713 |
| K | Human coronavirus OC43 ATCC VR-759 | AY585228 | 30,741 |
| L | Porcine hemagglutinating encephalomyelitis virus VW572 | DQ011855 | 30,480 |
| M | Murine hepatitis virus JHM | AC_000192 | 31,526 |
| N | Rat coronavirus Parker | FJ938068.1 | 31,250 |
| O | Pipistrellus bat coronavirus HKU5/HK/03/2005 | NC_009020 | 30,482 |
| P | Rousettus bat coronavirus HKU9/GD/005/2005 | NC_009021 | 29,114 |
| Q | SARS-related Rhinolophus bat coronavirus Rf1/2004 | NC_004718.3 | 29,751 |
| R | SARS-related palm civet coronavirus SZ3/2003 | AY304486.1 | 29,741 |
| S | SARS-related chinese ferret badger coronavirus CFB/SZ/94/03 | AY545919 | 29,739 |
| T | Tylonycteris bat coronavirus HKU4/HK/04/2005 | NC_009019 | 30,286 |
| U | Yak coronavirus strain YAK/HY24/CH/2017 | MH810163 | 31,032 |
| V | Pangolin coronavirus isolate MP789 | MT121216 | 29,521 |
2.2. CGR technique
CGR technique was proposed by Jeffrey as a unique and scale-independent representation for DNA sequences [19]. Mapping the genome sequence using the frequency chaos game representation (FCGR) produces fractal landscapes. This iterative mapping technique assigns, for each nucleotide in a DNA or amino acid in a protein, a unique coordinate in a 2-dimensional space (x, y). This 2-D image contains the distribution of the dots captured in a form of 0–1 square matrix, where 0 represents an empty coordinate and 1 represents a dot. Thus, an element occupying the nth position of the DNA sequence (Seq = S1, S2,.., SL) composed by L nucleotides (A, T, C, or G) is represented into a square by a point CGR n. This point CGR n is repeatedly placed halfway between the previous plotted point CGR n−1 and the segment joining the vertex corresponding to the read letter Sn [19]. This value is chosen according to previous research [16,17,19]. The proposed CGR steps are shown in the following Algorithm 1.

Fig. 1.a shows the CGR representation plot of ‘AGCTGC’ sequence. The obtained point set shows a fractal pattern. The CGR algorithm applied to various species has produced specific and structured images that can differentiate each genome between the studied genomes. CGR-Centroid is the result of calculating the centroid point of each sub-image of CGR image after dividing the image into M sub-images. The number M is equal to 4m, and m presents the oligonucleotide length considered for the study that should be superior to zero. The idea is to divide the CGR image into sub-images to get the genomic signature for each region. Essentially, each pixel in a CGR image is associated to a specific position word in a given genomic sequence. Therefore, each visible pattern in the CGR corresponds to some specific pattern of the genomic sequence. For this, the CGR representation shows the global information of the nucleotide sequence. Each partition of this image contains a local information.
Fig. 1.



CGR process and CGR-Centroid examples.
For example, if we divide the image to 4 images and calculate the center, we can find the CGR centroid corresponding to each nucleotide (A, T, C or G). When the two-point are within the same quadrant, they correspond to a succession of nucleotides in the sequences with the same last mononucleotide; and when they are within the same sub-quadrant, the sequences have the same last dinucleotides; and so on. Therefore, the coordinate of the centroid which corresponds to local information of the sub-region can differentiate the sequences and can be used to find the relationship between nucleotide sequences. To obtain the CGR Centroid, a mandatory step is to divide the CGR image into 4 squares. For CGR Centroid, the CGR image is partitioned into (n/M) × (n/M) equate sub-images, where n is the size of CGR image. Then, for each sub-region, all pairs of distances between the Covid-19 centroids and the others centroids sequences are extracted. After that, these distances can indicate the relation between the DNA sequences. The following algorithm 2 presents the steps GGR Centroid.

Fig. 1.b shows an example of the CGR-centroid plot of each sub-region and the center of CGR (X) for COVID 19 after being partitioned into sub-regions of size 2 × 2. These distance values between each sub-region (A-centroid, T-centroid, C-centroid, and G-centroid) and the CGR-center (X) can indicate the existence of the similarities or not between the nucleotide sequences.
2.3. Time-frequency analysis technique
The numerical genomic representation using coding methods is an important step to visualize and characterize the hidden information that can be contain in it, especially in this case where the nucleotide sequences do not have any continuously or homologous between them. Different coding techniques exist: the binary [20], the structural bending trinucleotide (PNUC) [21], the electron-ion interaction pseudo-potential (EIIP) mapping [22], the FCGS [[23], [24], [25]], and so on. In addition, several signal processing techniques were applied with success to detect the relationship between sequences and detect some biological repetitive sequences, and so on.
In this paper we use the Frequency Chaos Game Signal (FCGS) as a coding technique (first step) and the Smoothed Discrete Fourier Transform (SDFT) and the Wavelet Transform as an analysis techniques (second step).
2.3.1. Genomic signal representation
Nucleotide sequences are converted into a numerical sequence (1D signals) before processing from our database extracted from NCBI platform. Then, 1-D signals are generated by applying the FCGS order 2: FCGS2. This type of coding technique is based on the apparition's probability of two successive nucleotides in an entry sequence [[23], [24], [25]]. The probability (P2_nuc) of given L nucleotides in the sequence is as follows:
| (1) |
N2_nuc represents the apparition number two successive nucleotides in the sequence and L represents the length in base pairs of the sequence. After that, in position (k), the oligomer (i), which consists of 2 nucleotides, is replaced by the corresponding occurrence probability:
| (2) |
2.3.2. Smoothed discrete Fourier transform: SDFT
We choose SDFT, a space-scale analysis, to be applied on nucleotide signals which is based on:
-
-
Dividing the helitron signal S[n] into R portions with an overlap ΔR.
-
-
Dividing each portion into N frames by multiplication with a sliding window W[n]:
| (3) |
where Δn represents the overlap value and l the window index. Here, W (the width of the window) must be chosen in such a way that the samples number of S[n] provides best frequency resolution. The sequence size and the type of window influence the frequency parameters values. N and R couple are taken as a power of two, this is recommended by the Fast Fourier Transform (FFT) algorithm [[20], [21], [22], [23]].
-
-
Applying the Discrete Fourier Transform (DFT) on each weighted block S w[n, l]. In the spectral domain, S w[ω] is given by:
| (4) |
Here, ω represents the frequency index; ω ∈ [0 : N − 1]
-
-
Calculating the mean value corresponding to each N frame within the R segments; then carrying out the DFT mean value for all R frames. The following equation (Eq. 5) gives the mean Smoothed Spectrum:
| (5) |
Here, l represents the frame index of the N frames (l ∈ [0 : N − 1]) and i, the frame index of the R frames (i ∈ [0 : R − 1]).
To ensure the best accurate smoothed spectrum, the Blackman window was chosen as window type. In addition we can follow the instantaneous frequencies evolution by considering the 2-D spectrogram representation resulting from the following mathematical equation:
| (6) |
The final obtained matrix Mat Hel(i,w) contains the time-frequency information corresponding to the studied sequence. This is an efficient representation to visualize the evolution of periodicities along a nucleotide sequence.
2.3.3. Wavelet transform
The time-frequency nucleotide image with three color channels (Red, Green, Blue) is the best way to visualize the different patterns that can differentiate the sequences. If the pixels luminance in nucleotide image changes between two images, we can determine the modified patterns. To reach this goal, we choose the wavelet transform as an analyzing technique which present the nucleotide signal into a nucleotide image. Our choice is based on the performance of the wavelet transform which have been especially used in data related to the biological domain [22,24,26,27].
In this paper, the complex Morlet wavelet (CWT) has been used as a wavelet type which is the best one in terms of time-frequency domains localization. The method's principle consists of decomposing a nucleotide signal into a sum of basic functions called wavelets. These wavelets are issued from the mother wavelet by expansion and translation operations. These wavelets analysis technique applied to a given signal takes into account both time and frequency variations. Unlike the mother wavelet that only has a time variation parameter expressed by the function ψ(t), the daughter wavelet depends on time (a) and scale parameters (b) and it is generated by the expression given by the following equations, where * indicates the conjugate complex and ɷ0 is the oscillation's number that must be greater than 5 (admissibility condition) [[28], [29], [30]]:
| (7) |
The CWT of a DNA signal x(t) is a matrix W (a,b)which contains the continuous wavelet coefficients. The DNA scalogram (2D) is a representation of the modulus |W (a,b)|.
After obtaining the matrix W (a,b)of each sequence, for example SARS-COV-2 and RATG13, we develop a new algorithm which detects the nucleotide variation that exists between these sequences. The new algorithm we have developed is based on computing the correlation value between two matrices corresponding to two different genomes with a variable window. The following figure (Fig. 2 ) shows a flowchart methodology to extract similar nucleotide sequences in two genomes, example of SARS-COV-2 and betacoronavirus RATG13. The aim here is to find similar sequences and the modified nucleotides in two genomes by computing the correlation values and shifting one position to the next base pair if not equal to 1 until obtaining similar matrices.
Fig. 2.

Flowchart diagram of the adopted localization methodology to extract similar nucleotide sequences between two genomes.
2.3.4. Recombination analysis
To detect recombination event, complete genomic sequences of Sars-Cov-2 with other coronavirus sequences were investigated. Sequences were first aligned using Clustal X program and then analyzed by Simplot program [31]. The default settings were used. These included window size = 200, a step size = 20, replicate used = 100, gap stripping = “on”, distance model=”Kimura”, tree model=”Neighbor Joining”.
3. Results
3.1. CGR analysis
The CGR image (2-D) graphical representations are the results of converting the nucleotides succession in a nucleotide sequence to a visual image. The CGR plots of all investigated sequences are presented in “Supplementary Fig. 1” file. Fig. 3 shows the correlation value between SARS-Cov-2 and others species. In general, our results shows that SARS-Cov-2 is close to betacoronavirus genomes (B, V, C, D, S, T, R, Q), more precisely, the higher correlation value (0.9) corresponds to SARS-CoV-2 Vs. Beta Cov-RaTG13 (B) and Pangolin coronavirus isolate MP789 (V) genomes, followed by the bat-SL-CoVZC45 (C) and bat-SL-CoVZXC21 (D). The Yak coronavirus strain YAK/HY24/CH/2017 (U) genome seems to be different (0.55 correlation value).
Fig. 3.

CGR image correlation between SARS-CoV-2 and others genome; RaTG13 (B), Betacoronavirus CoVZC45, Betacoronavirus CoVZXC21(D), Alphacoronavirus DQ811787 (E), Gammacoronavirus A116E7 (F), Deltacoronavirus KJ481931 (G), Bat Hp-betacoronavirus (H), Bovine coronavirus Mebus(I), Human coronavirus OC43 (J), Human coronavirus OC43 ATCC (K), Porcine hemagglutinating encephalomyelitis virus VW572 (L), Murine hepatitis virus JHM (M), Rat coronavirus Parker (N), Pipistrellus bat coronavirus HKU5 (O), Rousettus bat coronavirus HK 9 (P), SARS-related Rhinolophus bat coronavirus (Q), SARS-related palm civet coronavirus (R), SARS-related chinese ferret badger coronavirus (S), Tylonycteris bat coronavirus HKU4 (T) Yak coronavirus strain YAK/HY24/CH/2017(U), and Pangolin coronavirus isolate MP789(V).
To confirm these results, we calculated the CGR Centroid between SARS-CoV-2 and other investigated sequences. Fig. 4 presents CGR Centroid points of each sequence. It shows the plot of CGR Centroid points of 4 regions (A, T, C, and G nucleotides) of investigated sequences (Fig. 4.a). Red point A presents the centroid of SARS-CoV-2 sequence. The Fig. 4.b shows the plot of CGR Centroid points of 16 regions (AA, AT, AC….GC, and GG dinucleotides) of each sequence.
Fig. 4.


CGR centroids plots where the points; where the first subfigure (a) show the nucleotides centroid plot of 20 species; SARS-CoV-2 (A) Betacoronavirus RaTG13 (B), Betacoronavirus CoVZC45, Betacoronavirus CoVZXC21(D), Alphacoronavirus DQ811787 (E), Gammacoronavirus A116E7 (F), Deltacoronavirus KJ481931 (G), Bat Hp-betacoronavirus (H), Bovine coronavirus Mebus(I), Human coronavirus OC43(J), Human coronavirus OC43 ATCC (K), Porcine hemagglutinating encephalomyelitis virus VW572 (L), Murine hepatitis virus JHM (M), Rat coronavirus Parker(N), Pipistrellus bat coronavirus HKU5(O), Rousettus bat coronavirus HKU9(P), SARS-related Rhinolophus bat coronavirus (Q), SARS-related palm civet coronavirus (R), SARS-related chinese ferret badger coronavirus (S), Tylonycteris bat coronavirus HKU4(T), Yak coronavirus strain YAK/HY24/CH/2017(U), and Pangolin coronavirus isolate MP789(V), and the second figure (b) show the dinucleotides centroid plots of the more similar species.
The degree of correlation is displayed by the distance variation between the points. Fig. 5 shows that points B (Betacoronavirus RaTG13), V (Pangolin coronavirus isolate MP789), C (Bat Betacoronavirus CoVZC45) and D (Bat coronavirus CoVZXC21) are more strongly correlated with point A (SARS-CoV-2) and we can see that distances between points A, B and V are the shortest one. This obtained result confirms the similarities obtained by CGR analysis.
Fig. 5.

Graphical representation of distance values between each nucleotide region (A, T, C and G) CGR centroids of SARS-Cov-2 (A) and other investigated viruses.
The distance values of CGR centroid between SARS-CoV-2 (A) and other investigated virus sequences are presented in Fig. 5. The nearest to zero the value is, the greater is the similarity between SARS-CoV-2 genome and other genome which is compared to. It appears that the most similar genome corresponds to genome B (Betacoronavirus RaTG13) and V (Pangolin coronavirus isolate MP789).
3.2. SDFT time frequency analysis
The application of the SDFT spectral analysis to the genomic signal gives us the opportunity to detect any latent or hidden periodic signal in the original sequences. Here, the idea is to characterize each genome independently of their length with a specific specter (1D signature) and spectrum (2D signature) which indicate the variation region if it exist between more of genomes. Exploring the latent periodicities of global genomes using SDFT method can play a key role in the homology detection between these viruses' classes.
For more investigation, we visualize the sequences in 1D spectrum and 2D spectrogram representations by applying the SDFT to the FCGS2 signals. These representations reflect the time-frequency signatures of each sequence which may differentiate each genome or indicate the similarities between them by highlighting its periodicities. In this work, the “Blackman” window was chosen as window type and we consider 1024 as the R frames length, with a shift index Δr = 512, for the sub-frames, we take N = 256, with Δn = 64. The major effect of windowing is guarantee the converting the frequency response discontinuities into transition bands between values on either side of the discontinuity. The spectral representation (1D) of different genomes and the 2-D spectrum representation are presented in Fig. 6 and Supplementary Fig. 2.
Fig. 6.

Spectra superposition of Sequences (20 viruses) investigated in this study (a) and of the more correlated genome Cov-RATG13 to SARS-Cov2 (A) genome (b), where these viruses are Betacoronavirus RaTG13 (B), Betacoronavirus CoVZC45, Betacoronavirus CoVZXC21(D), Alphacoronavirus DQ811787 (E), Gammacoronavirus A116E7 (F), Deltacoronavirus KJ481931 (G), Bat Hp-betacoronavirus (H), Bovine coronavirus Mebus(I), Human coronavirus OC43(J), Human coronavirus OC43 ATCC (K), Porcine hemagglutinating encephalomyelitis virus VW572 (L), Murine hepatitis virus JHM (M), Rat coronavirus Parker(N), Pipistrellus bat coronavirus HKU5(O), Rousettus bat coronavirus HKU9(P), SARS-related Rhinolophus bat coronavirus (Q), SARS-related palm civet coronavirus (R), SARS-related chinese ferret badger coronavirus (S), Tylonycteris bat coronavirus HKU4(T), Yak coronavirus strain YAK/HY24/CH/2017(U), and Pangolin coronavirus isolate MP789 (V).
The superposed spectra presented in Fig. 6 reflect the existence of similarities between all investigated sequences and highlight the high correlation between SARS-Cov-2 genome and Betacoronavirus RaTG13 Bat (A) and Pangolin coronavirus isolate MP789 (V) genomes followed by two Betacoronavirus CoVZC45 (B) and Betacoronavirus CoVZXC21 (C) genomes. The spectrum correlation values between SARS-CoV-2 and Betacoronavirus RaTG13 and Pangolin coronavirus isolate MP789 genomes are about 0.995 and 0.9889 respectively, the highest value among investigated sequences. In the other hand it's about 0.5518 for Yak coronavirus strain YAK/HY24/CH/2017 genome (Supplementary Fig. 2).
For more interpretation, we can use the Neighbor joining (NJ) clustering which is an alternative method for hierarchical cluster analysis [32]. Here, we can draw phylogenetic trees of the SARS-Cov-2 and other investigated viruses using the calculated spectra correlation values between SRS-Cov2 and other viruses (B to T) mentioned in “Supplementary Fig. 3”. The dendrogram in Fig. 7 was developed using the Past PAleontological Statistics program version 3.23 [33]. It shows the homology viruses degree to SARA-Cov2 generated from phylogenetic relationships using the spectra from SDFT method SARS-Cov-2 (A) is close to betacoronavirus genomes type: B, V, C, D, S, T, U, R, and Q successively. This phylogenetic tree confirms our previous results that indicate a high correlation between the SARS-Cov-2 genome and Betacoronavirus RaTG13 (B) Bat and Pangolin coronavirus isolate MP789 (V) genomes followed by two Betacoronavirus CoVZC45 (C) and Betacoronavirus CoVZXC21 (D) genomes. (See Fig. 7.)
Fig. 7.

Phylogenetic tree of SARS-COV-2 and the investigated viruses (21 viruses) using the spectra correlation vector and Neighbor-Joining method. The most related sequences to SARS-Cov-2 are highlighted with a red circle. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. CWT time frequency analysis
In this work, we developed a new algorithm to extract the zone of similarities between two sequences using signal processing tools. The scalograms signatures (2-D images) of all investigated sequences are presented in “Supplementary Fig. 4” file.
As an example, the Fig. 8 shows a scalogram representation of a limited nucleotide sequences of the two coronavirus genomes; SARS-Cov-2 (26,004 to 26,503 nucleotide position) and Betacoronavirus RATG13 (25,973 to 26,473 nucleotide position). These two scalograms covering 500 nucleotides, present the time-frequency representation of the sequences. After applying our method we find 4 nucleotides modifications between two sequences; one C➔T in 26,023 bp and T➔C in [26,053 bp; 26,169 bp; 26,332 bp] positions (Fig. 9 ).
Fig. 8.

2-D representations (scalograms) of two nucleotide sequences with size equal to 500 bp show the high homology between these sequences.
Fig. 9.
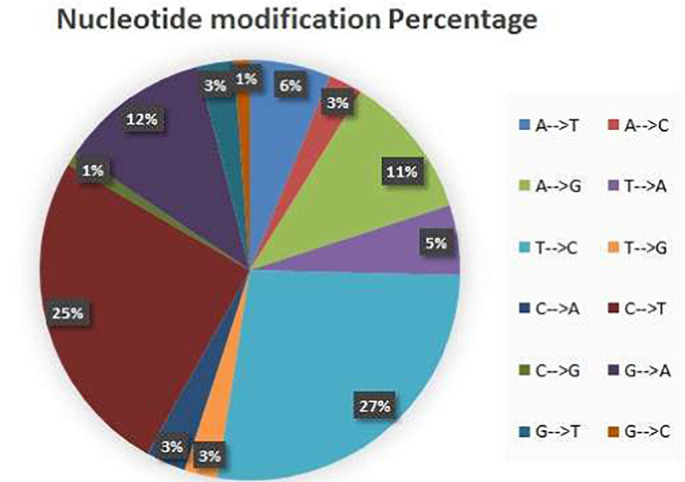
Dispatching of mutation between SARS-COV-2 and betacoronavirus RATG13 genomes ratios found using our methods.
The combination of bioinformatics and signal processing tools applied to genetic sequences show that SARS-Cov-2 is highly related to RATG13 Betacoronavirus and Pangolin coronavirus isolate MP789 with 96% and 86% identity along the whole genome. This global result is similar to result obtained by BLAST platform. To highlight these results, we analyzed nucleotide modification positions of both viruses in comparison with SARS-Cov-2 genome. We developed a new algorithm based on the scalogram images resulting from wavelet transform applied to the genomic signal. Table 2 summarizes nucleotide modification positions according to SARS-Cov-2 genome and identity percent for each genomic region. The total modification number is about 1173 and 2780 nucleotides for RATG13 Betacoronavirus and Pangolin coronavirus isolate MP789 respectively. For all the genes RATG13 sequences are most related to SARS-Cov-2 than Pangolin coronavirus sequences.
Table 2.
Nucleotide modifications distribution along the SARS-COV 2 genome comparing to the Cov-RATG13 genome.
| Region | Begin | End | Length | Protein | Nucleotide modification RATG13 |
Nucleotide modification Pangolin |
|
|---|---|---|---|---|---|---|---|
| Number (%) | Number (%) | ||||||
| 1 | 5′UTR | 16 | 265 | 250 | – | 6 (2.4%) | 2 (0.8%) |
| 2 | Gene = “orf1ab” | 266 | 21,555 | 21,290 | GU280_gp01 | 744 (3.494%) | 1924 (9%) |
| 3 | Gene = “S” | 21,563 | 25,384 | 3822 | GU280_gp02 | 271 (7.09%) | 323 (8.45%) |
| 4 | Gene = “ORF3a” | 25,393 | 26,220 | 828 | GU280_gp03 | 31 (3.743%) | 34 (4.106%) |
| 5 | Gene = “E” | 26,245 | 26,472 | 228 | GU280_gp04 | 1 (0.438%) | 2 (0.877%) |
| 6 | Gene = “M” | 26,532 | 27,191 | 660 | GU280_gp05 | 30 (4.545%) | 38 (5.75%) |
| 7 | Gene = “ORF6” | 27,202 | 27,387 | 186 | GU280_gp06 | 3 (1.612%) | 3 (1.61%) |
| 8 | Gene = “ORF7a” | 27,394 | 27,759 | 366 | GU280_gp07 | 16 (4.371%) | 24 (6.557%) |
| 9 | Gene = “ORF7b” | 27,756 | 27,887 | 132 | GU280_gp08 | 1 (0.757%) | 132 (100%) |
| 10 | Gene = “ORF8” | 27,894 | 28,259 | 366 | GU280_gp09 | 11 (3.005%) | 229 (62.5%) |
| 11 | Gene = “N” | 28,274 | 29,533 | 1260 | GU280_gp10 | 39 (3095%) | 48 (3.809%) |
| 12 | Gene = “ORF10” | 29,558 | 29,674 | 117 | GU280_gp11 | 1 (0,854%) | 1 (0.854%) |
| 13 | 3′UTR | 29,675 | 29,889 | 229 | – | 3 (1.39%) | 0 (0%) |
We can clearly see the great modification number in S gene which with 271 (7.09%) for RATG13 genome and 323 (8.45%) for Pangolin coronavirus genome.
Comparing the scalogram images of S gene of SARS-Cov2 to RATG13 and Pangolin coronavirus genomes, we find that the homology changes depending on selected regions in this gene. For this reason, we adopted our analysis algorithm according to the region where the similarities in the alignment are strong (Table 3 ). Between the nucleotide positions [1251–1600] pangolin sequence showed the lower nucleotide modification than RATG13. These result suggest a possible recombination event in the S gene.
Table 3.
Nucleotide modifications distribution of S gene genome comparing to the S genes of Cov-RATG13 and Pangolin genomes.
| Position of selected region in S gene | Length | Nucleotide modification Pangolin |
Nucleotide modification |
|---|---|---|---|
| Number (%) | RATG13 |
||
| Number (%) | |||
| 1–769 | 769 | All (100%) | 38 (4.094%) |
| 770–1250 | 481 | 81 (16.83%) | 16 (3.326%) |
| 1251–1600 | 350 | 50 (14.28%) | 125 (35.7%) |
| 1601–2500 | 900 | 36 (4%) | 30 (3.33%) |
| 2501–2550 | 50 | 4 (8%) | 9 (18%) |
| 2551–3822 | 1272 | 132 (10.3%) | 52 (4.088) |
Our results of the comparison between SARS-CoV-2 and Cov-RATG13 along the genome, showed 1173 nucleotide modifications dispatched as shown in the following Fig. 9.
The minimum mutation ratio is for the nucleotide G that becomes C with 1.22% ratio, corresponding to numbers of 6 mutations. The minimum mutation ratio is for the nucleotide T that becomes C with 1.22% ratio. The global results are presented in the “Supplementary results” file.
3.4. Recombination analysis result
To confirm occurrence of recombination between RATG 13 and pangolin MP789 sequences, we assessed Simplot analysis. Fig. 10 shows evidence of possible recombination event between the nucleotide positions 1250 and 1575 of the S gene.
Fig. 10.

Simplot analysis of SARS-cov-2 genome in comparison with RATG13 and Pangolin coronavirus genomes: (a) Simplot analysis of complete genome and (b) S gene Simplot analysis of S gene.
4. Discussion
The world health organization informed in December 31, 2019 that the unexplained respiratory disease called COVID-19 is caused by a new coronavirus called SARS-CoV-2. This virus caught the attention of scientists, who set out to analyze the virus, the disease it causes, and how it spreads. Finding the evolutionary relationship between these coronaviruses, and their genomic characterization is a crucial task. It can improve our understanding about the evolution and generation of new viruses and help in the future the prevention against new emergence. In this paper, we used combination of different DNA representation and signal processing tools to compare full genome sequence of SARS-CoV-2 to relevant viral genomes including the most related ones: Bat, Yak and Pangolin coronaviruses.
The image genomic signature provided a rapid identification of relationship similarity between SARS-CoV-2 and other coronaviruses genomes. We used three different representations and we developed a new algorithm allowing global and local comparisons of genomic sequences and thus informing on possible recombination event occurrence. The obtained results using different representations were confirmed by each other. Furthermore, when compared to results based on alignment methods (Blast and Simplot) same findings were obtained.
The used methods are an alignment free method, requesting only a sequence database and a bioinformatics and signal processing knowledges [[16], [17], [18]]. Those methods transformed every nucleotide sequence in numerical form. CGR technique has been used because it has a remarkable capacity to differentiate between genetic sequences belonging to different species [17]. Comparing to alignment-based methods, it uses an image-based approach generated by different techniques to elaborate genomic signatures. These signatures can be processed by mathematical computations and signal processing tools to extract remarkable features as CGR centroid points and time-frequency representation (specter, spectrogram, and scalogram). The CGR centroid plots is a very efficient method to know the relationship between DNA sequences in each square of image. In addition, the spectral representations reflect the frequency repartition of the nucleotides in the genomic sequences which can indicate the position or zone of the great change between the genome signals. It also allows construction of the phylogenetic tree of SARS-Cov-2 in comparison to other coronaviruses. Furthermore, we developed a new algorithm that allows identification of nucleotide modification between SARS-Cov-2 and their closer genomes without any need to previous alignment. The main advantage of our algorithm consists in the generation of a table output with local identity percent and position allowing suspicion of possible recombination events as demonstrated in our study.
This study shows that based on genomic signatures representations, we can easily assess automatic homology identification between genomes and thus they allow rapid genomic characterization when we are compressed by time, such as this critical period during the outbreak of this novel viral. It permits to rapidly mobilize appropriate methods for diagnostic and medical care specific to the identified viruses. Furthermore, it's also crucial to the choice of appropriate preventive methods and contingency plans. In the future, alignment free methods can also be advanced and adapted to specific biological tasks and biologist needs in different fields.
Over all obtained results, signal processing tools highlight the high homology of SARS-CoV-2 and other betacoronavirus than other families and more precisely with Cov-RATG13 and Pangolin coronavirus isolate MP789 genomes. However, the Yak coronavirus strain YAK/HY24/CH/2017 seems to be different [13]. These results show evolutionary evidence of the SARS-CoV-2- from Bat and Pangolin coronaviruses as a consequence of accumulation of mutations and also acquisition of new genomic region by recombination events.
Prior scientists searched the evolutionary history of the Wuhan COVID-19 virus [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15],34,35]. They discovered that three bat SARS-like coronaviruses, Betacoronavirus RaTG13, bat-SL-CoVZC45 and bat-SL-CoVZXC21, were closely related to SARS-CoV-2 [35].
Nevertheless, it was not clear, till now, if the COVID-19 arose from a recombination event or no between those viruses [12]. Others scientists suggested possible genesis of COVID-19 virus through recombination between Bat [10,11] and Pangolin coronaviruses [14,15]. Our results support this possibility of COVID-19 virus genesis though evolution of Bat and Pangolin coronaviruses by accumulation of point mutations and recombination. Close contact between humans and wild animals and consumption of bat and Pangolin meat can creat favorable conditions for evolution of viruses of both hosts.
5. Conclusion
This study offers new and rapid ways to automatically identify the homology between known viruses and emergent ones, given the opportunity for rapid classification and identification of virus origin.
Using bioinformatics combined with signal processing tools, we confirmed the high homology of SARS-CoV-2 with bat BetaCov-RaTG13 and Pangolin coronavirus isolate MP789 genomes.
Thus, our technique can be used to extract numerical features to classify the viruses and to perform evolutionary study of viruses.
Declaration of competing interest
None.
Acknowledgment
This study was funded by the Ministry of Higher Education and Research, LR99ES10 Human Genetics Laboratory.
Biographies

Rabeb Touati: PhD in electrical engineering from the National Engineering School of Tunisia (ENIT). Currently, she has a Post-Doctoral position at the Laboratory of Human Genetics (LR99ES10) at the Faculty of Medicine of Tunis (FMT). Her research interest includes genomic signal processing, bioinformatics, pattern recognition and machine learning.

Sondes Haddad-Boubaker: PhD in Virology from the Faculty of Sciences of Tunis. She is an Assistant Professor in the Laboratory of Clinical Virology, at Pasteur Institute of Tunis, which acts as the WHO Regional Reference Laboratory for Poliomyelitis and Measles in the EMRO region. Her research interest includes molecular characterization and omics data analysis of Human viruses, especially poliovirus and other enteric viruses.

Imen Ferchichi: PhD in molecular biology from Faculty of Sciences of Tunis (FST), University of Tunis El Manar, Tunisia. Currently, she has a Post-Doctoral position at the Genetic Human Laboratory (LR99ES10) at the Faculty of Medicine of Tunis (FMT). Her research interests include Human Genetics.

Imen Messaoudi: Received her PhD degree in electrical engineering from the National Engineering School of Tunisia. She is Assistant professor at the Higher Institute of Information Technologies and Communications (ISTIC) from Carthage University. Her research interest includes biomedical and genomic signal processing.

Afef Elloumi Ouesleti: PhD in electrical engineering from the National Engineering School of Tunisia (ENIT). She is Associate Professor at the National School of Engineers of Carthage (ENICarthage). Her research interest includes issues related to signal and image processing applied on biomedical and genomic fields.

Henda Triki: MD: She is Professor in Virology in 2006 at the Faculty of Medicine of Tunis (FMT). She is the head of laboratory of Clinical Virology, which acts as the WHO Regional Reference Laboratory for Poliomyelitis and Measles in the EMRO region. In 2018, she was assigned as Director of the Clinical Investigation Center entitled: “Transmissible diseases: Natural history and innovated tools for diagnostic, prevention and treatment” in Pasteur Institute of Tunis.

Zied Lachiri: PhD in electrical engineering from the National Engineering School of Tunisia (ENIT). He is Professor and Research Director In Signal, Image and Information Technology laboratory (LR-SITI). His research interests include pattern recognition, signal and image processing in biomedical, multimedia and machine communication.

Maher Kharrat: PhD in Human Genetics from the Faculty of Medicine of Tunis (FMT). He is Associate Professor and Research Director In Genetic Human laboratory (LR99ES10) at the Faculty of Medicine of Tunis (FMT). He is currently works at the Faculty of Medicine, University of Tunis El Manar. His research interests include Human Genetics.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygeno.2020.07.003.
Appendix A. Supplementary data
Supplementary Fig
Supplementary results
References
- 1.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hassan S.S., Choudhury P.P., Basu P., Jana S.S. Molecular conservation and differential mutation on ORF3a gene in Indian SARS-CoV2 genomes. Genomics. 2020:3226–3237. doi: 10.1016/j.ygeno.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luk H.K.H., Li X., Fung J., Lau S.K.P., Woo P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019;71:21–30. doi: 10.1016/j.meegid.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaki A.M., Van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (Accessed 06 may 2020)
- 6.Sanche S., Lin Y.T., Xu C., Romero-Severson E., Hengartner N., Ke R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020:26–27. doi: 10.3201/eid2607.200282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King A.M.Q., Lefkowitz E.J., Adams M.J., Carstens E.B. Elsevier Academic Press; San Diego, CA: 2011. Ninth Report of the International Committee on Taxonomy of Viruses. [Google Scholar]
- 8.Vijaykrishna D., Smith G.J., Zhang J.X., Peiris J.S.M., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007;81(8):4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo P.C., Lau S.K., Huang Y., Yuen K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009;234(10):1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A., Kulcsar K., Misra V., Frieman M., Mossman K. Bats and coronaviruses. Viruses. 2019;11(1):41. doi: 10.3390/v11010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan J.F.W., Yuan S., Kok K.H., To K K W, Chu H., Yang J. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paraskevis D., Kostaki E.G., Magiorkinis G., Panayiotakopoulos G., Sourvinos G., Tsiodras S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 2020;79:104212. doi: 10.1016/j.meegid.2020.104212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dabravolski S.A., Kavalionak Y.K. SARS-CoV-2: structural diversity, phylogeny, and potential animal host identification of spike glycoprotein. J. Med. Virol. 2020:1–5. doi: 10.1002/jmv.25976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X., Giorgi E.E., Marichannegowda M.H., Foley B., Xiao C., Kong X.P. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020:1–14. doi: 10.1126/sciadv.abb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam T.T.Y., Jia N., Zhang Y.W., Shum M.H.H., Jiang J.F., Zhu C. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020:1–4. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- 16.Hoang T., Yin C., Yau S.T. Numerical encoding of DNA sequences by chaos game representation with application in similarity comparison. Genomics. 2016;108(3–4):134–142. doi: 10.1016/j.ygeno.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Touati R., Messaoudi I., Oueslati A.E., Lachiri Z., Kharrat M. New intraclass helitrons classification using DNA-image sequences and machine learning approaches. IRBM. 2020 (In press) [Google Scholar]
- 18.Yousef M., Khalifa W., Acar İ.E., Allmer J. MicroRNA categorization using sequence motifs and k-mers. BMC Bioinform. 2017;18(1):170. doi: 10.1186/s12859-017-1584-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeffrey H.J. Chaos game representation of gene structure. Nucleic Acids Res. 1990;18(8):2163–2170. doi: 10.1093/nar/18.8.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oueslati A.E., Ellouze N., Lachiri Z. 3D spectrum analysis of DNA sequence: application to Caenorhabditis elegans genome. Bioinform. Bioeng. 2007:864–871. [Google Scholar]
- 21.Oueslati A.E., Messaoudi I., Ellouze N., Lachiri Z. Spectral analysis of global behaviour of C. elegans chromosomes. INTECH. 2012;8:205–228. [Google Scholar]
- 22.Chakraborty S., Gupta V. 2016 Second International Conference on Computational Intelligence & Communication Technology (CICT) IEEE. 2016. Dwt based cancer identification using EIIP; pp. 718–723. [Google Scholar]
- 23.Touati R., Oueslati A.E., Messaoudi I., Lachiri Z. The Helitron family classification using SVM based on Fourier transform features applied on an unbalanced dataset. Med. Biol. Eng. Comput. 2019;57(10):2289–2304. doi: 10.1007/s11517-019-02027-5. [DOI] [PubMed] [Google Scholar]
- 24.Touati R., Messaoudi I., Oueslati A.E., Lachiri Z. Distinguishing between intra-genomic helitron families using time-frequency features and random forest approaches. Biomed. Signal Process. Control. 2019;54:101579. [Google Scholar]
- 25.Touati R., Messaoudi I., Oueslati A.E., Lachiri Z., Kharrat M. Classification of intra-genomic helitrons based on features extracted from different orders of FCGS. Inform. Med. Unlock. 2020;18:100271. [Google Scholar]
- 26.Kumar M.R., Vaegae N.K. A new numerical approach for DNA representation using modified Gabor wavelet transform for the identification of protein coding regions. Biocybernet. Biomed. Eng. 2020;40(2):836–848. [Google Scholar]
- 27.Fernández L., Pérez M., Orduña J.M. Visualization of DNA methylation results through a GPU-based parallelization of the wavelet transform. J. Supercomput. 2019;75(3):1496–1509. [Google Scholar]
- 28.Grossmann A., Morlet J. Decomposition of Hardy functions into square integrable wavelets of constant shape. SIAM J. Math. Anal. 1984;15(4):723–736. [Google Scholar]
- 29.Merry R.J.E. DCT Rapporten; 2005. Wavelet Theory and Applications: A Literature Study; p. 2005. [Google Scholar]
- 30.Najmi A.H., Sadowsky J. The continuous wavelet transform and variable resolution time-frequency analysis. Johns Hopkins APL Tech. Digest. 1997;18(1):134–140. [Google Scholar]
- 31.Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G.…Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of virology. 1999;73(1):152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 33.Harper Hammer D.A., Ryan P.D. PAST: paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001;4:9. [Google Scholar]
- 34.Dong N., Yang X., Ye L., Chen K., Chan E.W.C., Yang M., Chen S. Genomic and protein structure modelling analysis depicts the origin and infectivity of 2019-nCoV, a new coronavirus which caused a pneumonia outbreak in Wuhan, China. bioRxiv. 2020:2020. [Google Scholar]
- 35.Hu B., Zeng L.P., Yang X.L., Ge X.Y., Zhang W., Li B. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017:13–36. doi: 10.1371/journal.ppat.1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig
Supplementary results