

Abstract
Glioblastoma (GBMs) is the most common and aggressive type of primary brain tumor in adults with dismal prognosis despite radical surgical resection coupled with chemo- and radiotherapy. Recent studies have proposed the use of small-molecule inhibitors, including verteporfin (VP), to target oncogenic networks in cancers. Here we report efficient encapsulation of water-insoluble VP in poly(lactic-co-glycolic acid) microparticles (PLGA MP) of ~1.5 μm in diameter that allows tunable, sustained release. Treatment with naked VP and released VP from PLGA MP decreased cell viability of patient-derived primary GBM cells in vitro by ~70%. Moreover, naked VP treatment significantly increased radiosensitivity of GBM cells, thereby enhancing overall tumor cell killing ability by nearly 85%. Our in vivo study demonstrated that two intratumoral administrations of sustained slow-releasing VP-loaded PLGA MPs separated by two weeks significantly attenuated tumor growth by ~67% in tumor volume in a subcutaneous patient-derived GBM xenograft model over 26 d. Additionally, our in vitro data indicate broader utility of VP for treatment for other solid cancers, including chordoma, malignant meningioma, and various noncentral nervous system-derived carcinomas. Collectively, our work suggests that the use of VP-loaded PLGA MP may be an effective local therapeutic strategy for a variety of solid cancers, including unresectable and orphan tumors, which may decrease tumor burden and ultimately improve patient prognosis.
Keywords: verteporfin, poly(lactic-co-glycolic acid), PLGA, microparticles, microspheres, local delivery, cancer therapy, intratumoral treatment, radiosensitization, glioblastoma, chordoma, malignant meningioma, brain cancer
Graphical Abstract
INTRODUCTION
Glioblastoma (GBM) is the most aggressive form of primary brain cancer. GBMs are characterized by their high proliferative nature coupled with their enhanced ability to disseminate into the intricate microenvironment of the human brain, confounding surgical excision and chemo- and radiotherapy.1,2 Despite state-of-the-art standard of care, GBMs account for ~13 000 deaths annually in the U.S with an annual survival rate of 35%.3 Hence, it is crucial to design effective treatment strategies to attenuate growth and recurrence of this malignancy.
In an effort to identify potential genetic vulnerabilities in GBMs, numerous studies have continuously interrogated the complex molecular and genetic landscape of this disease, offering new therapeutic targets.4 Aggressive tumors frequently exhibit chromosomal instability; of which, GBMs harbor amplification of 11q22.5 Interestingly, Yes-Associated-Protein (YAP), a developmentally relevant transcriptional coactivator and effector of the mammalian Hippo pathway, resides in this human amplicon.6,7 Moreover, previous studies report overexpression of YAP and loss of upstream kinases of the Hippo pathway in several cancers including GBMs, suggesting that hyperactive YAP-dependent signaling plays an important role in tumor development and progression.8–10 Therefore, targeting these YAP-driven networks may serve as a potent and effective therapeutic approach for aggressive and lethal cancers.
Recently, verteporfin (VP) has been identified as a potent inhibitor of YAP-driven signaling by disrupting YAP’s interaction with its cognate transcription factor, TEAD4.11 Currently, a lipophilic formulation of VP is clinically approved for photodynamic treatment of age-related macular degeneration.12 In this format, VP accumulates in the retinal vasculature, whereby it is activated upon exposure to a low-energy infrared laser of 693 nm wavelength, generating free radicals that lead to destabilization and destruction of the abnormal vessels. In cancer, which is highly vascularized, free radicals that are generated during VP photodynamic therapy have been proposed to disrupt tumor vascularization and also exert cytotoxic effects on adjacent tumor cells by induction of apoptosis and autophagy. However, studies have demonstrated VP’s ability to attenuate tumor growth in a variety of cancer models without photoactivation.13–19 Yi Liu-Chittenden et al. demonstrated that verteporfin, without light activation, can inhibit TEAD–YAP association, which is hyperactive in malignant cells, and attenuate tumor growth.11 Additionally, it has been reported that VP can decrease the in vitro proliferative capacity of GBM cells.20 However, it is unknown whether VP is efficacious in vivo in GBM xenograft models.
Despite numerous studies identifying, developing, and testing potential therapies, several experimental drugs have failed clinical trials. Several factors may explain this lack of success, including the drug’s inability to penetrate the blood–brain barrier, poor solubility, limited bioavailability, unfavorable pharmacokinetics, and rapid clearance.21 Previous in vivo studies with VP have relied on daily intraperitoneal injections, making its clinical use difficult to envision.13,15,18 Furthermore, VP is a hydrophobic small molecule with poor solubility in aqueous solutions, which makes it difficult to administer without excipients that can potentially cause severe side effects. Thus, for practical translational use of VP as a potential therapy for GBMs and other solid cancers, the issue of solubility must be addressed further. Recently, a number of nanoparticle systems composed of lipids or polymers, such as poly(beta-amino ester) and poloxamers, have been engineered to enhance pharmacokinetics and biodistribution of encapsulated VP for application in systemic delivery to tumor.22–24 However, these nanoscale systems display relatively brief release kinetics (≤72 h) and, thus, offer limited applicability as a sustained, long-lived release depot. For example, for hard-to-reach organs such as the brain, where it is important to limit the number of surgeries and local injections while maintaining a constant therapeutic VP dosage over long periods, such a short-release therapeutic system may not be optimal. These previous formulations, if they were to reach the tumor site either through local or systemic administration, would most likely demonstrate a fast clearance and efflux from the tumor microenvironment, thereby reducing the effective therapeutic dose and ultimately diminishing its efficacy. Thus, to our knowledge, the possibility of creating VP-based biodegradable depots for longer-term chemotherapeutic activity has not been explored. We hypothesized that VP-loaded biodegradable microparticles could be fabricated to meet this challenge and act as a versatile and effective local treatment strategy for GBMs and other solid cancers.
Over the last several decades, polymeric microparticles have been shown to be efficient drug-carrying vehicles for controlled release and delivery of drugs for combatting a variety of diseases.25 The size and structure of microparticles enables large payloads of materials including drugs, proteins, and genes. Microparticles have been most commonly fabricated from poly(lactic-co-glycolic acid) (PLGA), a material that has been generally regarded as safe, and PLGA-based particle devices have been Food and Drug Administration (FDA)-approved. Depending on the solubility of the cargo, a single or double emulsion procedure is usually utilized for encapsulation of these materials into hard particles. The slow hydrolysis of the PLGA polymer can provide a favorable controlled release, including under some conditions zero-order kinetics, of the loaded cargo when administered in vivo. PLGA microparticles (PLGA MP) are better suited than nanoparticles as local, long-term drug depots, since larger micron-scale size increases the time scale needed for polymer degradation to occur throughout the particle and also increases the time scale of diffusion for the encapsulated drug that needs to diffuse through the pores of the particle.26 Thus, the larger size of the micron-scale biodegradable PLGA device extends the duration of release compared to the nanoscale device. Additionally, PLGA can be tuned to modify its release rate either by changing the ratio of lactic to glycolic acid or the molecular weight of the polymer. In this study, we demonstrate the strong therapeutic ability of naked and encapsulated VP in PLGA MP on patient-derived GBM cells, in both in vitro and in vivo models. Additionally, we explore its efficacy in other solid cancers, including chordoma, meningioma, and various noncentral nervous system-derived carcinomas.
EXPERIMENTAL SECTION
Cell culture.
GBM1A cells were derived by Galli, R et al.27 and cultured according to previously described methods.9 Specifically, these cells were cultured on laminin-coated (1ug/cm2) plates in media prepared using Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 (Invitrogen) and supplemented with GEM21 (Gemini) or B27 supplement (Invitrogen), antibiotic/antimycotic (Invitrogen), human EGF (Peprotech), and FGF (Peprotech). IOMM-LEE and KT21-MG1 cells were purchased from ATCC and maintained in DMEM (Invitrogen) with 10% Fetal Bovine Serum (Gibco) and antibiotic/antimycotic (Invitrogen). JHC7 Cells were cultured in MesenPRO basal medium with associated growth supplement (Invitrogen), glutamax (Invitrogen), and antibiotic/antimycotic (Invitrogen). All non-central nervous system (CNS)-derived commercial tumor cell lines were cultured according to manufacturer’s protocol.
In Vitro Cell Viability using MTT Assay.
Verteporfin (Sigma) was solubilized in dimethyl sulfoxide (DMSO) at 2 mg/mL per manufacturer’s recommendation. Concentrated solution was stored in LightSafe centrifuge tubes (Z688339, Sigma). In vitro treatment solutions were prepared by diluting concentrated VP solution in corresponding cell culture medium to appropriate concentrations for each experiment/condition. Cells were seeded and grown to achieve ~25–50% density prior to media removal and bolus treatment of DMSO-media or VP-media solutions. All of these steps were performed following standard sterile, aseptic technique under dark conditions to avoid any light exposure. Cell viability was evaluated using monotetrazolium (MTT) assay in 96-well plates, with each independent time point performed in 3–8 replicates to establish statistical significance. Cells were seeded at density of 12 × 103 cells/well. MTT (Sigma) was prepared at a concentration of 5 mg/mL in PBS (MTT solution). A 10% MTT solution was prepared in the appropriate cell culture media, and cells were incubated at 37 °C for 4 h. After 4 h, each well was carefully aspirated, and the formazan salt was solubilized using 200 μL of isopropyl alcohol. Optical density measurements were read using a spectro-photometric plate reader (Epoch) at 570 nm. Measurements were taken at different time points. Raw data were analyzed as a percentage of surviving cells from vehicle control and normalized to Day 0 when appropriate.
In Vitro Cell Irradiation.
Cells were plated in monolayer in sterile cell culture dishes. After 2 d, cells were treated with single specific doses of radiation (Grays) according to experimental conditions using a GammaCell 40 irradiator (Best Theratronics).28 At specific postradiation time points according to the experiment, cells were dissociated, and viable cells were counted using an automated cell counter (ViCell XR Cell Viability Analyzer, Beckman Coulter) in triplicate. A dose response curve was performed to identify an in vitro IC50 radiation dose to be used in subsequent in vitro experiments. Raw data were analyzed as a percentage of surviving cells from vehicle control or non-irradiated control.
Poly(lactic-co-glycolic acid) Microparticle (PLGA MP) Formulation and Characterization.
PLGA MP encapsulating VP was prepared using the single emulsion method. Two different MPs were formulated using PLGA polymers of varying hydrophobicity and molecular weight. 50/50 PLGA MP was formulated with less hydrophobic PLGA polymer with lactide-to-glycolide ratio of 50:50 and molecular weight of 7–17 kDa (Resomer RG 502H, Sigma), while 85/15 PLGA MP was prepared with more hydrophobic PLGA polymer with lactide-to-glycolide ratio of 85:15 and molecular weight of 190–240 kDa (Resomer RG 858S, Sigma). PLGA (100–200 mg) is first solubilized in dichloromethane at 20 mg/mL. VP dissolved in DMSO at 20 mg/mL was added to the polymer solution at a v/v ratio of 1:20 for 5% drug loading by mass to the polymer, and the solution was mixed by sonication for 20 s with a tip sonicator (Misonix) at an amplitude of 30 A. For vehicle-loaded PLGA MP (Veh PLGA MP), pure DMSO was used in place of VP-dissolved DMSO. The mixture was then added to 50 mL of 1% poly(vinyl alcohol) (PVA; molecular weight (MW) = 25 kDa) and homogenized at 15 krpm for 1 min (Ika T25 digital Ultra-Turrax) to emulsify the drug-polymer organic phase in PVA aqueous phase. The emulsion was transferred to 100 mL of 0.5% PVA solution and stirred for 4 h to allow for solvent evaporation. PLGA MPs were washed three times with deionized H2O by repeating centrifugation at 4 krpm for 5 min, removal of supernatant, and addition of fresh H2O. Lastly, the MPs were lyophilized and stored in −20 °C until use.
VP PLGA MPs were characterized for morphology, size, surface charge, drug loading, and drug release. Scanning electron microscopy (SEM) was used to visualize the particles. Lyophilized MPs were placed on a piece of carbon tape (EMS), sputter-coated with gold/palladium, and imaged using LEO/Zeiss Field Emission SEM (Johns Hopkins University School of Medicine Microscope Facility). SEM images were analyzed with ImageJ to determine particle size distribution. Zeta potential of the VP PLGA MPs was measured using Malvern Zetasizer Nano ZS (Malvern Instruments) by resuspending lyophilized MPs in 10 mM NaCl at 1 mg/mL.
The amount of VP encapsulated in PLGA MP was quantified using the drug’s inherent fluorescence. Briefly, a known mass of both lyophilized VP PLGA MP and naked VP drug were first dissolved in DMSO to 2 mg/mL and 200 ug/mL, respectively. Then, twofold dilution series were prepared with DMSO, and fluorescence was measured at the VP’s excitation (420 nm) and emission (680 nm) wavelengths using Synergy 2 plate reader (Biotek). The amount of drug from dissolved MP was back-calculated from its fluorescence reading using the standard curve generated with naked VP. The loading capacity and encapsulation efficiency are calculated as follows:
The ability of PLGA MP to efficiently encapsulate VP and prevent precipitation of free, unloaded VP in aqueous solution was determined through turbidity of the solution. 1X phosphate-buffered saline (PBS) was added to either naked VP drug or VP-loaded 50/50 PLGA MP at 20 μg/mL VP concentration, and absorbance spectrum was recorded using Synergy 2 plate reader (Biotek) and signal quantified at 550 nm.
The drug release from both 50/50 and 85/15 PLGA MPs was measured in vitro by incubating MPs in PBS buffer. Lyophilized PLGA MP (5 mg) was resuspended in 1 mL of PBS and incubated on a shaker at 37 °C. MP solution was centrifuged at 4 krpm for 5 min, supernatant was removed, and 1 mL of fresh PBS was added for incubation daily until day 7 and on day 14. Supernatant was lyophilized and resuspended in DMSO to measure fluorescence at the VP’s excitation (420 nm) and emission (680 nm) wavelengths to back-calculate the VP mass released at each time period via predetermined standard curve.
In Vitro Cell Treatment with VP Released from PLGA MP.
Five milligrams of VP-loaded 50/50 PLGA MP and 85/15 PLGA MP were incubated in 1 mL of deionized H2O at 37 °C for 72 h to allow the release of VP. Following the incubation, the solution was centrifuged at 4 krpm for 5 min. An aliquot (50 μL) of the supernatant was collected, lyophilized, and dissolved in DMSO to determine the mass of released VP in the supernatant by fluorescence. The rest of the supernatant was lyophilized and dissolved in the calculated volume of cell culture media to obtain 12.5 μM solution. Cells were seeded and grown in a 96-well tissue culture plate to achieve ~25–50% density prior to media removal and bolus treatment of 100 μL of released VP-media solution. Cells were incubated with the drug-containing media for 4 h, and then GBM1A, JHC7, or H460 cells were further incubated in fresh media for 3 and 5 d, and H460 cells for 1 and 2 d. At the given time points, cells were imaged with bright-field microscope, and MTT cell viability/proliferation assay was performed.
In Vivo Tumor Growth Inhibition.
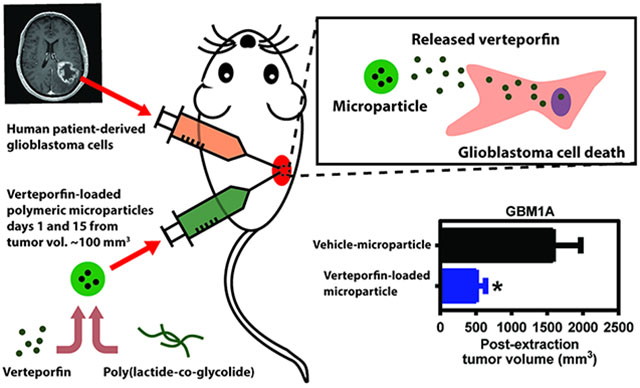
All animal protocols were approved by the Mayo Clinic Animal Care Institutional and Use Committee. Eight-week male nude mice (athymic nude Foxn1nu, Jackson Laboratories) were injected subcutaneously in the right flank with 2 × 106 primary glioblastoma cells (previously transduced with lentiviral particles to constitutively express GFP-Luciferase gene). Cells were resuspended in 200 μL of 1:1 mixture of DMEM F12/Matrigel matrix (Corning). Tumors were allowed to grow to ~100 mm3, and then mice were randomized into individual treatment groups. For the naked verteporfin experiment, animals were treated with naked VP (100 mg/kg, cat: 1711461, USP) or vehicle (5%DMSO-95%PBS) daily for 7 d by intraperitoneal injections (IP). For the verteporfin-loaded PLGA microparticles, animals received two intratumoral injections (on day 1 and day 15) containing 50 ul of VP PLGA MPs (~288ug of VP) diluted in PBS. As a control we used Veh PLGA MPs diluted in PBS.
Tumor growth in each mouse was assessed and recorded weekly by caliper measurements and bioluminescence imaging (see timeline in Figure 2a and Figure 5a). The tumor volume was measured by the modified ellipsoid formula: tumor volume = 1/2(length × width2).
Figure 2. Verteporfin decreases glioblastoma growth in vivo.

(a) Timeline of in vivo experiment. (b) Representative bioluminescence images of free-VP vs Vehicle-treated mice on Day 21. (c) Average GBM1A tumor volume of free VP vs Vehicle-treated mice measured using calipers over a 21 d period. All error bars are standard error of measurement (s.e.m.). * = P < 0.05 as determined by ANOVA, Tukey’s multiple comparison test.
Figure 5. Verteporfin PLGA microparticles attenuate glioblastoma growth in vivo.

(a) Timeline of in vivo experiment. (b) Average bioluminescence readings of GBM1A tumors treated VP-loaded PLGA MP (VP-MP) vs Vehicle control MP (Veh-MP) over a 26 d period. (c) Representative bioluminescence images of GBM1A tumors before (Day 0) and after (Day 26) VP-MP vs Veh-MP intratumoral treatment. (c) Average GBM1A tumor volume of VP-MP vs Veh-MP treated mice measured using calipers over a 26 d period. (e) Average fold change in tumor volume pre- vs post-treatment of VP-MP vs Veh-MP. (f) Macroscopic images of GBM1A tumors post-VP-MP and Veh-MP treatment. (f) Postextraction volume of VP-MP vs Veh-MP treated GBM1A tumors. All error bars are s.e.m. * = P < 0.05 as determined by Student’s t-test or ANOVA, Tukey’s multiple comparison test.
In vivo bioluminescence images of tumor-implanted mice were obtained using the IVIS Spectrum System. Before imaging, D-luciferin (XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate PerkinElmer) was injected IP at a dose of 10 mg/kg and allowed to distribute for 10 min. Mice were imaged once a week after tumor engrafting. Data acquisition and analysis were performed using the Living Image Software. For quantitation of the detected light, regions of interest were drawn, and the light emitted was recorded as the total flux (number of photons per second).
Statistics.
Statistical analysis was performed using GraphPad Prism 6 software package. As indicated in the figure legend for each figure, one-way ANOVA with Dunnett posthoc test was performed to compare multiple conditions against the control group, Tukey posthoc test to compare all pairs, or Student’s t-test to compare two conditions whereby * = p < 0.05.
RESULTS
Verteporfin Decreases Proliferation of Glioblastoma Cells.
Our prior work has shown that YAP is overexpressed and hyperactive in different solid cancers, including GBMs.9 Moreover, we recently showed that YAP-dependent signaling promotes GBM cell proliferation.8 Hence, we hypothesized that VP treatment would decrease the proliferative capacity of GBM cells. To this end, using established patient-derived GBM cell line, GBM1A cells, the effects of naked VP treatment without photoactivation were investigated in vitro. Treatment with increasing doses of naked VP (5–20 μM) significantly decreased cell viability at 24 h compared to our vehicle control of DMSO (Figure 1a). Moreover, a nearly 50% reduction in cell number was observed after 24 h of treatment with 10–20 μM of naked VP compared to Vehicle (Figure 1a). In addition to an initial decrease in cell viability, VP may also cause further decreases in cell viability over time. To measure this, treatment of naked VP for 48 h was also conducted. Increasing doses of naked VP treatment (1.25–20 μM) for 48 h significantly decreased GBM1A cell proliferation compared to Vehicle control (Figure 1b). These results from patient-derived primary cells confirm prior observations made by Al-Moujahed, et al., using commercial GBM cells.20
Figure 1. Verteporfin decreases proliferation and radiosensitizes glioblastoma cells.

In vitro proliferation of GBM1A cells after increasing doses of VP treatment for (a) 24 and (b) 48 h. (c) GBM1A cells pretreated with VP (2.5 μM) for 24 h had higher radiosensitivity at increasing doses of radiation than corresponding vehicle-treated control cells. (d) The radiosensitivity was preserved at a prolonged 4 d time-point in JHGBM1A cells treated with VP and 10 Gray. All gray error bars are standard deviation. * = P < 0.05 as determined by ANOVA, Tukey’s multiple comparison test; compared to dose 0 μM.
Verteporfin Attenuates Proliferation of Other CNS- and Non-CNS Derived Carcinoma Cells.
Next, the effect of naked VP treatment was tested in two other types of central nervous system-derived cancers, meningiomas, and chordoma, which currently lack any FDA-approved chemotherapy. Thus, we evaluated the effects of VP on malignant meningioma cells (IOMM LEE and KT21-MG1) and JHC7 chordoma cells. Short-term (1–3 d) and long-term (5 d) treatment of increasing doses of naked VP significantly reduced the proliferative capacity of IOMM LEE, KT21-MG1, JHC7 cells (Figure S1). A dramatic reduction in cell proliferation of these CNS-derived cell lines was observed at higher doses of naked VP (5–20 μM) at Day 5 (Figure S1b, d, f). These data demonstrate the therapeutic efficacy of VP in a broad variety of CNS-derived cancers.
Given our findings on CNS-derived cancers, we investigated the efficacy of long-term treatment of naked VP in a panel of non-CNS derived carcinoma cells, including metastatic colon (SW480), pancreatic (PANC-1), breast (MDA-MB-231 and MDA-MB-468), and cervical (HELA) cancer cells. A significant reduction in cell proliferation was observed after 5 and 6 d of 12.5 μM treatment of naked VP cross all tumor cell types (Figure S2). These findings further confirm the broader utility of VP as an antiproliferative treatment agent.
Verteprofin Radiosensitizes GBM and Other Brain Tumor Cells.
Radiotherapy remains an integral part of the standard of care for GBMs.29 Previous studies have reported that radiation coupled with surgical resection extends median survival of patients by six to nine months.30 Adjuvant chemotherapy using Temozolomide further augments patient survival by nearly four to six months.29 Thus, it is of interest to determine whether VP treatment radiosensitizes GBM cells given that a population of GBM stem cells are notoriously radioresistant and are likely responsible for tumor recurrence, leading to an overall dismal patient prognosis. Exposure to increasing doses of radiation (2–12 Grays) significantly decreased cell viability of GBM1A cells pretreated with 2.5 μM of naked VP in a dose-dependent manner compared to nontreated cells at 24 h (Figure 1c). Specifically, 50% killing of GBM1A cells was observed at a radiation dose of 3 Gray with 24 h of VP treatment. However, at the same VP dosage of 2.5 μM without radiation, the cellular viability was at 100% and ~75% for the 24 and 48 h treatments, respectively. Moreover, the viability of the cells plateaued at ~15% starting at 8 Gray, suggesting that there is no additional significant therapeutic benefit of increasing the radiation past this dose (Figure 1c). Furthermore, the reduction in cell survival was maintained long-term (Figure 1d). That is, a significant reduction in proliferation of GBM1A cells was observed in VP-treated irradiated cells (2.5 μM VP and 10 Gray) compared to VP or radiation treatment alone after 4 d (Figure 1d). Collectively, these results indicate that VP treatment radiosensitizes GBM cells. Therefore, adjuvant treatment of VP coupled with radiation may improve patient therapy and overall outcome.
Unlike GBMs, other brain tumors like malignant meningiomas are treated with radiation therapy alone and/or surgical resection of the tumor mass. Currently, no consensus chemotherapeutic agent is administered for treatment of this malignant meningiomas.31 Therefore, it is of particular importance to identify those agents that may enhance the effect of radiation in malignant meningiomas. A dose-dependent reduction in cell viability was observed in KT21-MG1 cells pretreated with 2.5 μM of naked VP than Vehicle treated cells when exposed to increasing doses of radiation (5–50 Grays; Figure S3a). In addition, adjuvant treatment of KT21-MG1 cells with 2.5 μM VP and 10 Gray significantly decreased cell proliferation overtime compared to cells treated with naked VP or radiation alone (Figure S3b). Thus, our data propose VP as a novel chemotherapeutic agent for the treatment of malignant meningioma and demonstrate its antiproliferative and radiosensitization capabilities.
Verteporfin Attenuates Glioblastoma Growth in Vivo.
Given our in vitro results demonstrating the antiproliferative properties of VP in GBM cells and other solid tumors, we tested its in vivo efficacy in a subcutaneous GBM xenograft model in mice using GFP-luciferase expressing GBM1A cells. After a subcutaneous tumor volume of 100 mm3 was established, which was verified using caliper measurements and bioluminescence readings, naked VP was administered intraperitoneally daily for a week, and tumor measurements were collected over a course of 21 d (Figure 2a,b). A significant, but modest decrease in GBM1A tumor volume was observed in mice treated with daily injections of naked VP than Vehicle-treated mice (Figure 2c). Specifically, tumor volumes were significantly smaller in VP-treated mice from d 8 through 21 (Figure 2c). These results show that VP attenuates growth of GBMs in vivo and establishes its in vivo efficacy for treatment of GBMs for the first time, to our knowledge.
PLGA Microparticle Characterization.
While naked VP shows significant antitumor efficacy both in vitro and in vivo, its low aqueous solubility limits the clinical use and requires further modification to increase its translational potential. A generally regarded as safe (GRAS), biodegradable PLGA polymer was used to formulate microparticles with encapsulated verteporfin. SEM images showed two types of MPs composed of PLGA polymers of different lactic to glycolic acid ratios (50:50 and 85:15), molecular weights (7–17 and 190–240 kDa), and polymer end group (carboxylic acid and ester). Both formed spherical PLGA MPs with smooth surface topology and similar particle sizes of 1.2 ± 0.3 and 1.6 ± 0.5 μm, respectively (Figure 3a,b). Surface charge of the PLGA MPs were both nearly neutral (Figure 3c). 85/15 PLGA MP had slightly positive and 50/50 PLGA MP slightly negative zeta potential due to the PLGA polymers terminating with an ester or a carboxylic acid moiety, respectively. These MPs enable hydrophobic VP to remain in the same state when added to physiological solution (PBS), unlike naked encapsulated VP that precipitates out at low concentration when added to physiological solutions (13.6 μg/mL solubility in water). Both 50/50 and 85/15 PLGA MPs were loaded with ~6% by mass of VP, which corresponds to 47.3% and 39.5% loading efficiency, respectively (Table 1). As shown in Figure 3d, turbidity of VP-loaded 50/50 PLGA MP in PBS is decreased by fivefold in comparison to naked VP, as determined by the amount of nonspecific absorption of light at 550 nm wavelength, indicating the VP is efficiently encapsulated inside microparticles and not precipitating out into aqueous solution.
Figure 3. PLGA microparticle characterization.

(a) Histogram of 85/15 and 50/50 PLGA MP size distribution measured via ImageJ analysis of (b) representative SEM images. (c) Zeta potential of 85/15 and 50/50 PLGA MP measured in 10 mM NaCl with Zetasizer (n = 3). (d) Turbidity of PBS solution of naked VP vs VP-loaded 50/50 PLGA MP with equivalent VP concentration of 20 μg/mL, determined by absorbance at 550 nm (n = 3). All error bars are s.e.m. * = P < 0.05 as determined by unpaired Student’s t-test comparison.
Table 1.
Verteporfin Loading in PLGA Microparticlesa
| MP | loading capacity (%) | encapsulation efficiency (%) |
|---|---|---|
| 50/50 PLGA | 6.03 ± 0.26 | 47.26 |
| 85/15 PLGA | 5.76 ± 0.12 | 39.46 |
Loading capacity and encapsulation efficiency of verteporfin in 50/50 and 85/15 PLGA MP, calculated from fluorescence measurement of DMSO solution of dissolved PLGA MP using VP standard curve. Data given as mean ± s.e.m. of triplicates.
The drug-release kinetics of 50/50 and 85/15 PLGA MPs were characterized as the MPs are aimed to be delivered locally, reside in the local tumor ECM matrix, and potentially induce prolonged inhibition of tumor growth through sustained release of VP. Drug release from PLGA MPs has been previously reported to come in several phases.32 Initial burst release occurs primarily due to the diffusion of drugs near the surface through the polymer matrix, followed by a sustained release of drugs due to bulk erosion of the polymer. As expected, both 50/50 and 85/15 PLGA MPs showed a burst release of VP during the first 2 d before a slow sustained release, but there was an approximately fourfold greater amount of VP released from 50/50 in comparison to 85/15 PLGA MP (Figure 4a). This difference is potentially due to the stronger hydrophobic interaction between the hydrophobic VP drug and 85/15 PLGA compared to 50/50 PLGA, slowing the encapsulated drug’s diffusion through the polymer matrix. The slower, controlled release of VP from 85/15 PLGA MP can potentially lead to extended therapeutic efficacy.
Figure 4. Released verteporfin from PLGA microparticles decreases proliferation of glioblastoma cells.

(a) Percent cumulative release of VP from 85/15 and 50/50 PLGA MP in 1X PBS incubated at 37 °C (n = 3). VP amount was calculated from the fluorescence measurement of release medium using VP standard curve. (b) Cell killing efficacy of released medium from DMSO- (rVeh) and VP-loaded (rVP) 85/15 and 50/50 PLGA MPs normalized to untreated control (n = 4). Released medium was lyophilized and reconstituted to 12.5 μM VP concentration for cell treatment. (c) Representative bright-field microscope images of GBM1A cells treated with released medium from DMSO and VP-loaded 85/15 and 50/50 PLGA MPs (scale bar = 100 μm). All error bars are s.e.m. * = P < 0.05 as determined by ANOVA, Dunnett’s multiple comparison test with untreated as control.
Verteporfin Released from PLGA Microparticles Decreases CNS- and non-CNS-Derived Tumor Cell Proliferation.
To test evaluate if VP following particle encapsulation and subsequent release has activity, GBM1A cells were incubated with a bolus treatment of VP released from 50/50 and 85/15 PLGA MPs. Released VP (rVP) diluted to 12.5 μM induced significant cell death at day 3, killing ~80% of the GBM1A cells (Figure 4b,c). On day 5, cell death was 64% and 60% for rVP from 50/50 and 85/15 PLGA MP, respectively. A moderate increase in cell viability was observed between days 3 and 5 after rVP treatment similar to our results with naked VP in from Figure 1a,b, potentially from reproliferation of the surviving cells from VP treatment. In addition, we tested rVP on JHC7 chordoma and H460 lung carcinoma cells for 3–5 or 1–2 d, respectively, whereby we observed a dramatic reduction in cell viability that time period (Figure S4). Sustained release from MP in vivo can potentially extend the duration of cell killing. As expected, rVP from 50/50 and 85/15 PLGA MP showed no significant difference in its efficacy, further indicating the particle formulation and the type of polymer used tunes the release of, but does not affect the function of, the encapsulated VP drug itself.
Intratumoral Injection of Verteporfin-Loaded PLGA Microparticles Attenuates Glioblastoma Growth in Vivo.
Our findings thus far demonstrate that VP is a promising treatment strategy for GBMs. However, it is unclear where intratumoral delivery of VP-loaded PLGA MP is an efficacious local therapeutic modality for treatment of GBMs. To address this question, subcutaneous flank tumors were established in mice using GFP-luciferase expressing GBM1A (Figure 5a). Tumor size was tracked and recorded for each mice over a 26 d period using caliper measurements and bioluminescence readings (Figure 5a). Two intratumoral injections of VP-loaded 85/15 PLGA MP (VP-MP) or Vehicle control MP (Veh-MP) were administered at Days 1 and 15 (Figure 5a). On the basis of bioluminesce readings, VP-MP treatment attenuated tumor growth (Figure 5b). While tumor signal from tumor treated with Veh-MP increased after the treatment, there was no significant increase of tumor signal in the VP-MP treated tumors, confirming the potent effect of VP treatment (Figure 5c). Moreover, caliper measurements show a significant decrease in tumor growth in VP-MP compared to Veh-MP treated mice (Figure 5d) with an approximate 20-fold versus 10-fold tumor growth over time, respectively (Figure 5e). These findings are further corroborated by tumor measurement poststudy (Figure 5f), whereby Veh-MP treated tumors were nearly twice the size than VP-MP treated tumors (Figure 5g). Further confirmation of VP-release intratumorally from the PLGA MP is evidenced by the dark coloration, reminiscent of soluble/free/naked VP, observed in the VP-MP treated tumors postresection (Figure 5f). Collectively, these results demonstrate the utility and efficacy of local intratumoral use of VP-loaded PLGA MP for treatment of GBMs.
DISCUSSION
Our findings highlight the potential of VP as a therapy for GBM, in addition to demonstrating the utility of encapsulating VP inside a delivery vehicle versus free drug administration. The benefits of a sustained slow VP-releasing conduit are highlighted in this work as a means to prevent frequent administrations of VP and maximize the clinical potential of the drug. In pursuing this study, we (1) demonstrated the chemotherapeutic potential of VP for GBMs and other CNS- and non-CNS-derived tumors, (2) encapsulated VP inside a PLGA MP to overcome the poor aqueous solubility of VP alone, and (3) evaluated overall therapeutic potential of free VP and VP-loaded PLGA MP in vivo in subcutaneous GBM xenograft mouse model.
PLGA MP may prove to be an advantageous delivery system for local treatment of GBMs, since in general sustained release of encapsulated drug can provide a prolonged therapeutic effect at the target site from a single dose. We used PLGA polymer, since it is GRAS and has been demonstrated previously as an effective carrier for other types of small molecules.33,34 The specific 85:15 (lactide/glycolide) ratio PLGA confers a more hydrophobic polymer structure, which prolongs the release kinetics of the drug. In the case of PLGA MP, the polymer undergoes hydrolysis from the surrounding water that induces bulk erosion and initial burst release of the cargo. Additionally, given their size, microparticles typically remain localized at the site of injection, rather than diffusing away, benefiting local sustained delivery. A long-term locally administered and acting drug delivery system is desirable to treat GBMs, because the brain is a very precarious organ to expose surgically, and it is important to prevent unnecessary trauma for the patient in as few surgeries as possible. Biodegradable microparticles could be an excellent therapeutic device by not only providing advantageous release kinetics but also enabling encapsulation of a high enough dosage to ensure prolonged exposure of a therapeutic dose. While microparticles require local administration, surgical resection is currently the initial standard of care for management and treatment of GBMs,30 so injection of microparticles could be administered as an adjuvant therapy to prevent secondary tumorigenesis and recurrence at the resection cavity.
While numerous experimental therapies are continuously being tested, no potent chemotherapeutic agent has been proposed to significantly augment GBM patient prognosis beyond the short-term benefits of Temozolomide or BCNU.4 Additionally, currently, both malignant meningiomas and chordoma lack any FDA-approved chemotherapy, leading to their dismal overall patient outcome.31,35 Although most meningiomas are benign, malignant meningiomas account for 1–3% of all meningiomas with a 10-year survival rate of ~34.5%.31 While meningiomas are one of the most common type of brain cancer to develop in adults, chordomas are rather rare.9,36–39 Despite their slow growth rate, chordomas are fatal with a median survival of approximately six years.35 Since both types of cancers lack any available FDA-approved chemotherapy, it is important to test and identify potentially effective chemo-agents for management and treatment of these malignancies. Recent advances in molecular profiling of these cancers have continued to identify potential vulnerabilities for therapeutic targeting, of which YAP has emerged as an attractive candidate given its near-ubiquitous expression and hyperactivity in cancer.10 Our study demonstrates the use of VP, an inhibitor of YAP signaling, as an effective antitumor agent in a variety of different CNS- and non-CNS-derived cancers. It has been previously shown that VP prevents YAP’s association with its cognate transcription factor, TEAD4, thereby diminishing its transcriptional activity. Our data also show that VP radiosensitizes GBM cells, which may also be especially beneficial for those tumors such as malignant meningiomas, where radiation alone is currently the standard of care. Previous studies by Andrade et al.40 suggests that YAP inhibition blocks the repair of radiation induced double-stranded DNA breaks. Specifically, an increase in phosphorylated-ATMS1981 and its effector target phosphorylated-Chk2T68 were observed upon YAP inhibition, an event reflecting the activation of ATM by DNA damage. Thus, in the case of VP, which inhibits YAP signaling, it likely suppresses the cell’s DNA damage response and consequently renders the GBM cells sensitive to radiation. Collectively, these results suggest an additive effect whereby VP inhibits proliferation, elicits DNA damage, and inhibits DNA repair mechanisms, which, ultimately, radiosensitizes the cells. Therefore, our results further lend support to use of VP in conjunction with radiotherapy for the treatment of cancer. In addition, for orphan cancers such as chordomas that currently lack any defined FDA-approved chemotherapeutic,35 VP may serve as a promising treatment option. Given our promising results in chordoma cells, our recent orthotopic murine model of human patient-derived chordoma41 provides another preclinical platform to test the intratumoral delivery of VP-loaded PLGA MP for long-term management and treatment of this rare cancer. Recent studies have also demonstrated VP’s efficacy in non-YAP-driven cancers, therefore highlighting its broad therapeutic utility in a diversity of cancers.42,43 Apart from its poor solubility, another issue with VP is it requires daily administration. However, repeated treatment is unfavorable in a clinical setting, which may create a burden for cancer patients and ultimately lead to poor patient compliance. Therefore, local delivery postresection using microparticles might be a viable option, because it abrogates further trauma for the patient, minimizes potential systemic side effects, and overcomes any potential blood–brain barrier issues.21 Likewise, for unresectable tumors, VP-packaged MPs may offer a plausible and effective local treatment modality.
CONCLUSION
In this study, we propose the use of VP-packaged MPs as potent and effective chemotherapeutic for long-term management and treatment of a variety of CNS- and non-CNS-derived cancers. Overall, this work discusses two important breakthroughs for cancer: (1) identifying VP as a potent molecule for the killing and radiosensitizing primary patient-derived GBM and other CNS-and non-CNS-derived tumor cells and (2) packaging VP in a vehicle to enable efficacy in vivo for long-term management and treatment of these malignancies. Specifically, we show that naked VP decreases cell viability and proliferation and radiosensitizes GBM cells in vitro. Released VP from PLGA MP also induces cell death of GBM cells in vitro, showing PLGA MP as a suitable platform for VP delivery. Furthermore, local intratumoral delivery of VP-loaded PLGA MP attenuates tumor growth in our patient-derived GBM xenograft model. Additionally, our data indicate the efficacy of VP for treatment of other solid cancers, including chordoma, meningioma, and various noncentral nervous system-derived carcinomas. Therefore, our work suggests that the use of PLGA MP as a VP delivery system may be an effective therapeutic strategy for variety of solid cancers, which may decrease tumor growth and ultimately lead to improved patient outcomes.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. J. Laterra and A. Vescovi for access to GBM1A cells. We thank K. RaFaey for assistance with patient MRI collection. We also thank A.Q.H. and Green lab members for their critical feedback. A.Q.H. was supported by the Mayo Clinic Professorship and a Clinician Investigator award. J.J.G. was supported by the Bloomberg–Kimmel Institute for Cancer Immunotherapy. The authors thank the NIH for support (R43CA221490, R01CA200399, R01EB016721, R01CA183827, R01CA195503, R01CA216855). J.K. received fellowship support from Samsung Scholarship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00959.
Plots of verteporfin concentration versus proliferation (percentage of vehicle), plots showing verteporfin attenuates proliferation of several non-CNS derived carcinoma cells, plots showing verteporfin radiosensitizes meningioma cells (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Louis DN; Perry A; Reifenberger G; von Deimling A; Figarella-Branger D; Cavenee WK; Ohgaki H; Wiestler OD; Kleihues P; Ellison DW The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016, 131 (6), 803–20. [DOI] [PubMed] [Google Scholar]
- (2).Lara-Velazquez M; Al-Kharboosh R; Jeanneret S; Vazquez-Ramos C; Mahato D; Tavanaiepour D; Rahmathulla G; Quinones-Hinojosa A Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci. 2017, 7 (12), 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ostrom QT; Gittleman H; Fulop J; Liu M; Blanda R; Kromer C; Wolinsky Y; Kruchko C; Barnholtz-Sloan JS CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol 2015, 17, iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Reifenberger G; Wirsching HG; Knobbe-Thomsen CB; Weller M Advances in the molecular genetics of gliomas - implications for classification and therapy. Nat. Rev. Clin. Oncol 2016, 14 (7), 434–452. [DOI] [PubMed] [Google Scholar]
- (5).Overholtzer M; Zhang J; Smolen GA; Muir B; Li W; Sgroi DC; Deng CX; Brugge JS; Haber DA Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (33), 12405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lorenzetto E; Brenca M; Boeri M; Verri C; Piccinin E; Gasparini P; Facchinetti F; Rossi S; Salvatore G; Massimino M; Sozzi G; Maestro R; Modena P YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014, 5 (9), 2608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dong J; Feldmann G; Huang J; Wu S; Zhang N; Comerford SA; Gayyed MF; Anders RA; Maitra A; Pan D Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130 (6), 1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wei S; Wang J; Oyinlade O; Ma D; Wang S; Kratz L; Lal B; Xu Q; Liu S; Shah SR; Zhang H; Li Y; Quinones-Hinojosa A; Zhu H; Huang ZY; Cheng L; Qian J; Xia S Heterozygous IDH1(R132H/WT) created by “single base editing” inhibits human astroglial cell growth by downregulating YAP. Oncogene 2018, 37 (38), 5160–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shah SR; David JM; Tippens ND; Mohyeldin A; Martinez-Gutierrez JC; Ganaha S; Schiapparelli P; Hamilton DH; Palena C; Levchenko A; Quinones-Hinojosa A Brachyury-YAP Regulatory Axis Drives Stemness and Growth in Cancer. Cell Rep.2017, 21 (2), 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zanconato F; Cordenonsi M; Piccolo S YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29 (6), 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Liu-Chittenden Y; Huang B; Shim JS; Chen Q; Lee SJ; Anders RA; Liu JO; Pan D Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26 (12), 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bressler NM; Bressler SB Photodynamic therapy with verteporfin (Visudyne): impact on ophthalmology and visual sciences. Invest Ophthalmol Vis Sci. 2000, 41 (3), 624–8. [PubMed] [Google Scholar]
- (13).Yu FX; Luo J; Mo JS; Liu G; Kim YC; Meng Z; Zhao L; Peyman G; Ouyang H; Jiang W; Zhao J; Chen X; Zhang L; Wang CY; Bastian BC; Zhang K; Guan KL Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25 (6), 822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Song S; Ajani JA; Honjo S; Maru DM; Chen Q; Scott AW; Heallen TR; Xiao L; Hofstetter WL; Weston B; Lee JH; Wadhwa R; Sudo K; Stroehlein JR; Martin JF; Hung MB; Johnson RL Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74 (15), 4170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Perra A; Kowalik MA; Ghiso E; Ledda-Columbano GM; Di Tommaso L; Angioni MM; Raschioni C; Testore E; Roncalli M; Giordano S; Columbano A YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol 2014, 61 (5), 1088–96. [DOI] [PubMed] [Google Scholar]
- (16).Li H; Huang Z; Gao M; Huang N; Luo Z; Shen H; Wang X; Wang T; Hu J; Feng W Inhibition of YAP suppresses CML cell proliferation and enhances efficacy of imatinib in vitro and in vivo. J. Exp. Clin. Cancer Res 2016, 35 (1), 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Brodowska K; Al-Moujahed A; Marmalidou A; Meyer Zu Horste M; Cichy J; Miller JW; Gragoudas E; Vavvas DG The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp. Eye Res 2014, 124, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wei H; Wang F; Wang Y; Li T; Xiu P; Zhong J; Sun X; Li J Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017, 108 (3), 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Martinez-Gutierrez JC; Ruiz-Valls A; Shah SR; Riggins GJ; Quinones-Hinojosa A Meningioma growth inhibition and radiosensitization by the small molecular YAP inhibitor verteportin. Neuro-Oncology 2015, 17 (5), v131.4. [Google Scholar]
- (20).Al-Moujahed A; Brodowska K; Stryjewski TP; Efstathiou NE; Vasilikos I; Cichy J; Miller JW; Gragoudas E; Vavvas DG Verteporfin inhibits growth of human glioma in vitro without light activation. Sci. Rep 2017, 7 (1), 7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).van Tellingen O; Yetkin-Arik B; de Gooijer MC; Wesseling P; Wurdinger T; de Vries HE Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [DOI] [PubMed] [Google Scholar]
- (22).Kim J; Shamul JG; Shah SR; Shin A; Lee BJ; Quinones-Hinojosa A; Green JJ Verteporfin-Loaded Poly(ethylene glycol)-Poly(beta-amino ester)-Poly(ethylene glycol) Triblock Micelles for Cancer Therapy. Biomacromolecules 2018, 19 (8), 3361–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pellosi DS; Moret F; Fraix A; Marino N; Maiolino S; Gaio E; Hioka N; Reddi E; Sortino S; Quaglia F Pluronic((R)) P123/F127 mixed micelles delivering sorafenib and its combination with verteporfin in cancer cells. Int. J. Nanomed 2016, 11, 4479–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang JX; Hansen CB; Allen TM; Boey A; Boch R Lipid-derivatized poly(ethylene glycol) micellar formulations of benzoporphyrin derivatives. J. Controlled Release 2003, 86 (2–3), 323–38. [DOI] [PubMed] [Google Scholar]
- (25).Makadia HK; Siegel SJ Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel, Switz.) 2011, 3 (3), 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen W; Palazzo A; Hennink WE; Kok RJ Effect of Particle Size on Drug Loading and Release Kinetics of Gefitinib-Loaded PLGA Microspheres. Mol. Pharmaceutics 2017, 14 (2), 459–467. [DOI] [PubMed] [Google Scholar]
- (27).Galli R; Binda E; Orfanelli U; Cipelletti B; Gritti A; De Vitis S; Fiocco R; Foroni C; Dimeco F; Vescovi A Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64 (19), 7011–21. [DOI] [PubMed] [Google Scholar]
- (28).Ford EC; Achanta P; Purger D; Armour M; Reyes J; Fong J; Kleinberg L; Redmond K; Wong J; Jang MH; Jun H; Song HJ; Quinones-Hinojosa A Localized CT-guided irradiation inhibits neurogenesis in specific regions of the adult mouse brain. Radiat. Res 2011, 175 (6), 774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stupp R; Mason WP; van den Bent MJ; Weller M; Fisher B; Taphoorn MJ; Belanger K; Brandes AA; Marosi C; Bogdahn U; Curschmann J; Janzer RC; Ludwin SK; Gorlia T; Allgeier A; Lacombe D; Cairncross JG; Eisenhauer E; Mirimanoff RO Radiotherapy plus concomitant and adjuvant Temozolomide for glioblastoma. N. Engl. J. Med 2005, 352 (10), 987–96. [DOI] [PubMed] [Google Scholar]
- (30).Kreth FW; Thon N; Simon M; Westphal M; Schackert G; Nikkhah G; Hentschel B; Reifenberger G; Pietsch T; Weller M; Tonn JC Gross total but not incomplete resection of glioblastoma prolongs survival in the era of radiochemotherapy. Ann. Oncol 2013, 24 (12), 3117–3123. [DOI] [PubMed] [Google Scholar]
- (31).Preusser M; Brastianos PK; Mawrin C Advances in meningioma genetics: novel therapeutic opportunities. Nat. Rev. Neurol 2018, 14 (2), 106–115. [DOI] [PubMed] [Google Scholar]
- (32).Kamaly N; Yameen B; Wu J; Farokhzad OC Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev 2016, 116 (4), 2602–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Grill AE; Shahani K; Koniar B; Panyam J Chemopreventive efficacy of curcumin-loaded PLGA microparticles in a transgenic mouse model of HER-2-positive breast cancer. Drug Delivery Transl Res. 2018, 8 (2), 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang H; Zhang G; Ma X; Liu Y; Feng J; Park K; Wang W Enhanced encapsulation and bioavailability of breviscapine in PLGA microparticles by nanocrystal and water-soluble polymer template techniques. Eur. J. Pharm. Biopharm 2017, 115, 177–185. [DOI] [PubMed] [Google Scholar]
- (35).Walcott BP; Nahed BV; Mohyeldin A; Coumans JV; Kahle KT; Ferreira MJ Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012, 13 (2), e69–76. [DOI] [PubMed] [Google Scholar]
- (36).Hsu W; Mohyeldin A; Shah SR; ap Rhys CM; Johnson LF; Sedora-Roman NI; Kosztowski TA; Awad OA; McCarthy EF; Loeb DM; Wolinsky JP; Gokaslan ZL; Quinones-Hinojosa A Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. J. Neurosurg 2011, 115 (4), 760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hsu W; Mohyeldin A; Shah SR; Gokaslan ZL; Quinones-Hinojosa A Role of cancer stem cells in spine tumors: review of current literature. Neurosurgery 2012, 71 (1), 117–25. [DOI] [PubMed] [Google Scholar]
- (38).Hu Y; Mintz A; Shah SR; Quinones-Hinojosa A; Hsu W The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis 2014, 35 (7), 1491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mathios D; Ruzevick J; Jackson CM; Xu H; Shah SR; Taube JM; Burger PC; McCarthy EF; Quinones-Hinojosa A; Pardoll DM; Lim M PD-1, PD-L1, PD-L2 expression in the chordoma microenvironment. J. Neuro-Oncol 2015, 121 (2), 251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Andrade D; Mehta M; Griffith J; Panneerselvam J; Srivastava A; Kim TD; Janknecht R; Herman T; Ramesh R; Munshi A YAP1 inhibition radiosensitizes triple negative breast cancer cells by targeting the DNA damage response and cell survival pathways. Oncotarget 2017, 8 (58), 98495–98508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sarabia-Estrada R; Ruiz-Valls A; Shah SR; Ahmed AK; Ordonez AA; Rodriguez FJ; Guerrero-Cazares H; Jimenez-Estrada I; Velarde E; Tyler B; Li Y; Phillips NA; Goodwin CR; Petteys RJ; Jain SK; Gallia GL; Gokaslan ZL; Quinones-Hinojosa A; Sciubba DM Effects of primary and recurrent sacral chordoma on the motor and nociceptive function of hindlimbs in rats: an orthotopic spine model. J. Neurosurg Spine 2017, 27 (2), 215–226. [DOI] [PubMed] [Google Scholar]
- (42).Dasari VR; Mazack V; Feng W; Nash J; Carey DJ; Gogoi R Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 2017, 8 (17), 28628–28640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang H; Ramakrishnan SK; Triner D; Centofanti B; Maitra D; Gyorffy B; Sebolt-Leopold JS; Dame MK; Varani J; Brenner DE; Fearon ER; Omary MB; Shah YM Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci. Signaling 2015, 8 (397), ra98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.