


Abstract
OGG1 initiated base excision repair (BER) is the major pathway for repair of oxidative DNA base damage 8-oxoguanine (8-oxoG). Here, we report that RECQL4 DNA helicase, deficient in the cancer-prone and premature aging Rothmund-Thomson syndrome, physically and functionally interacts with OGG1. RECQL4 promotes catalytic activity of OGG1 and RECQL4 deficiency results in defective 8-oxoG repair and increased genomic 8-oxoG. Furthermore, we show that acute oxidative stress leads to increased RECQL4 acetylation and its interaction with OGG1. The NAD+-dependent protein SIRT1 deacetylates RECQL4 in vitro and in cells thereby controlling the interaction between OGG1 and RECQL4 after DNA repair and maintaining RECQL4 in a low acetylated state. Collectively, we find that RECQL4 is involved in 8-oxoG repair through interaction with OGG1, and that SIRT1 indirectly modulates BER of 8-oxoG by controlling RECQL4–OGG1 interaction.
INTRODUCTION
RECQL4, one of the five RecQ helicases in mammalian cells, is associated with premature aging and cancer prone syndromes (1–4). Mutations in human RECQL4 contribute to three autosomal recessive disorders: Rothmund–Thomson Syndrome (RTS), RAPADILINO syndrome and Baller–Gerold Syndrome (2). Skeletal abnormalities, growth retardation, and in case of RTS and RAPADILINO, predisposition to cancer, are the common clinical features of RECQL4 deficiency (5). Increased cellular senescence because of the accumulation of DNA damage was also observed in a mouse model of RTS deficient in RECQL4 (6). Numerous studies have shown that RECQL4 functions in multiple cellular processes, including DNA replication (7–10), non-homologous end joining (NHEJ) (11,12) and homologous recombination (HR) (12,13) as well as telomere and mitochondrial DNA maintenance (14–19). However, much less is known about the function of RECQL4 in base excision repair (BER).
Accumulation of oxidative DNA damage has been implicated in cancer and aging (20). Reactive oxygen species (ROS) generated during normal cellular metabolism and from exogenous sources such as ionizing radiation (IR), can generate various types of DNA base lesions, including 7,8-dihydro-8-oxoguanine (8-oxoG) which is thought to be the most common oxidative DNA base damage (21). 8-oxoG is potentially mutagenic. During DNA replication, 8-oxoG may mispair with adenine (A) causing G:C to T:A transversion mutations (22,23). In mammals, the base excision repair (BER) pathway repairs 8-oxoG and a key protein for repair of 8-oxoG is DNA glycosylase 1 (OGG1) (24). OGG1 variants including S326C, which has a lower base excision activity, are associated with increased risk of developing cancer (25–27). OGG1-deficient mice accumulate high levels of 8-oxoG lesions and increased mutations rates (28,29). This data highlights the importance of 8-oxoG repair and OGG1 in maintaining genomic integrity and preventing tumorigenesis.
Sirtuins are evolutionarily conserved protein deacetylases. Seven mammalian sirtuins (1–7) have been identified (30,31). SIRT1 is the mammalian orthologue of yeast Sir2 (silent information regulator 2), which has emerged as an important regulator of aging (32,33). SIRT1 participates in various cellular functions including gene silencing, stress resistance, apoptosis, senescence, metabolism, and tumorigenesis (30,31). Targets of SIRT1 include histones and many DNA repair proteins (34–39). Evidence suggests that SIRT1 plays an important role in DNA repair. It deacetylates Ku70 and promotes non-homologous end-joining (NHEJ) following exposure to ionizing radiation (IR) (35). SIRT1 regulates the enzymatic activity and subcellular localization of Werner helicase (WRN) after DNA damage through deacetylation of WRN (36). SIRT1 promotes homologous recombination (HR) repair of double-strand breaks (DSBs) through activation of NBS1 by deacetylation (37). In response to oxidative stress and IR, SIRT1 redistributes from repetitive DNA foci to DNA breaks to promote DNA repair (40). Besides its role in DSB repair and oxidative stress response, SIRT1 has been shown to participate in the repair of UV-light generated DNA damage through deacetylation of xeroderma pigmentosum group A (XPA) (38). More recently, SIRT1 has been shown to modulate BER activity through deacetylation of OGG1 and apurinic/apyrimidinic endonuclease-1 (APE1) (39,41).
A recent study revealed that 8-oxoG lesions accumulate genome-wide at DNA replication origins within transcribed long genes (42). Intriguingly, 8-oxoG and γH2AX, a sensitive marker for DNA double-strand breaks (43), co-localize at these DNA replication origins within the transcribed long genes (42). Given the role of RECQL4 in replication and DNA double-strand breaks repair, we hypothesize that RECQL4 is involved in 8-oxoG repair, particularly in such regions. Further, a previous study found that RECQL4 is an acetylated protein (44), indicating that the function of RECQL4 may be regulated by acetylation/deacetylation. Because of the involvement of SIRT1 and RECQL4 in DNA repair and since RECQL4 is an acetylated protein, we asked whether SIRT1 could regulate RECQL4 function in DNA repair by deacetylation.
Here, we show that RECQL4 is required for efficient BER of 8-oxoG. RECQL4 stimulates 8-oxoG repair by physical and functional interactions with OGG1. Further, we show that SIRT1 interacts with RECQL4, deacetylates it, and maintains it in a hypoacetylated state. The acetylation of RECQL4 and its binding ability to OGG1 is enhanced after oxidative stress, which is counteracted by SIRT1. Moreover, the acetylation status of RECQL4 is tightly regulated by SIRT1 in response to oxidative stress. Taken together, our study reveals a hitherto uncharacterized function of RECQL4 as a regulator of BER that is further regulated by SIRT1.
MATERIALS AND METHODS
Reagents
Synthetic oligonucleotides were from TAG Copenhagen. Menadione sodium bisulfite (74036), nicotinamide (N0636), Trichostatin A (T8552) and piperidine (411027) were from Sigma-Aldrich. Recombinant RECQL4 was used as previously described unless otherwise indicated (13).
Site-directed point mutation and plasmid construction
Substitution of lysines (376, 380, 382, 385 and 386) of RECQL4 with glutamine or arginine was carried out by PCR using pCMV-Tag4A-3xFLAG-RECQL4 as template using QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Inc., #210515). The primer for RECQL4 (KQ) mutant with all five lysines mutated to glutamine: CAGGCTCCTCCGCCAGCAGGCATGGCAGCAGCAGTGGCGGCAGCAGGGGGAGTGTTTTGG; and the primer for RECQL4 (KR) mutant with all five lysines mutated to arginine: CAGGCTCCTCCGCAGGCAGGCATGGAGGCAGAGGTGGCGGAGGAGAGGGGAGTGTTTTGG. All mutants were verified by sequencing.
Cell culture and generation of stable cell lines
U2OS and HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified incubator with 5% CO2. To generate RECQL4 knock out cells, a double nicking strategy using CRISPR/Cas9n with paired guide RNAs was applied (45). The two paired single guide RNA (sgRNA) sequences were as follows: (i) sgRNA sense: 5 ′-AAGCAGAAACACTACGTGCG-3′; (ii) sgRNA anti-sense: 5′-GTACGTAATTGCCCCTGTCA-3′ (sc-403224-NIC, Santa Cruz). U2OS cells were transiently transfected with 1 μg of RECQL4 double-nickase plasmids at 80% cell density in a six-well culture plate using PolyJet™ transfection reagent (SL100688, SignaGen Laboratories). Transfected cells were selected using 2 μg/ml puromycin for one week. Subsequently, single cells were seeded on 150 mm dishes at 80 cells per dish to obtain single clones and verified for knockout by genotyping and western blot analysis. Stable shRNA-mediated knockdown cell lines were generated by lentivirus transduction. All cell lines were tested for mycoplasma by MycoAlert Mycoplasma Detection Kit (Lonza).
Lentivirus production and infection
Lentivirus production was performed as previously described with some modifications (46). Briefly, for lentiviral packaging, HEK293T cells were cotransfected with packaging plasmids psPAX2 and pMD2.G, and lentiviral plasmids targeting indicated genes. The virus-containing supernatant was collected 48 h after transfection and used for cell infection. Twenty four hours after infection, medium was changed to 5 μg/ml puromycin-containing medium, and cells were incubated for 1 week to select for infected cells. Lentivirus based construct encoding short hairpin RNAs targeting human SIRT1 (TRCN0000218734) was obtained from Sigma-Aldrich.
Western blot and immunoprecipitation
For western blot analyses, cells were lysed in RIPA buffer (89900, Thermo Fisher Scientific) supplemented with protease inhibitor (4693159001, Sigma-Aldrich). Protein concentration in the cell lysates was determined using the protein assay dye reagent (#5000006, Bio-Rad). Proteins were separated in NuPAGE 4–12% Bis–Tris gradient gels (Thermo Fisher Scientific), transferred to 0.2 μm pore-sized PVDF membranes (GE10600021, Sigma-Aldrich) and the blots were detected with the indicated antibodies. Immunoprecipitations were performed as previously described (12,13). Briefly, cells or cells transfected with the indicated expression constructs were lysed in IP lysis buffer containing 40 mM Tris–HCl pH 7.4, 2 mM MgCl2, 150 mM NaCl, 0.2% NP-40, 0.4% Triton X-100, supplemented with protease inhibitor (4693159001, Sigma-Aldrich) and 20 U/ml benzonase (E1014, Sigma-Aldrich). Lysates were incubated with Protein A/G agarose beads (88802, Thermo Fisher Scientific) or GFP-TRAP beads (gtma-20, ChromoTek) or FLAG-magnetic beads (Sigma-Aldrich) overnight at 4°C. The beads were washed with washing buffer (20 mM Tris–HCl, pH7.4, 150 mM NaCl, 0.2% Triton X-100) for six times. The washed Protein A/G agarose beads or GFP-TRAP beads were resuspended in 2 × SDS sample buffer, boiled at 95°C, and then subjected to Western blotting. The washed FLAG-magnetic beads were eluted with the above washing buffer containing 300 μg/ml 3xFLAG peptide (F4799, Sigma-Aldrich). The following antibodies and dilutions were used: anti-RECQL4 (2814; 1:1,000), anti-SIRT1 (8469, 1:1000) antibodies were from Cell Signaling Technology; anti-OGG1 (ab124741, 1:3000), anti-acetyl-OGG1 (acK338 + acK341, ab93670, 1:500), anti-NEIL1 (ab21337, 1:1000), anti-GFP (ab290, 1:5000), anti-Histone H3 (ab1791, 1:5000), anti-APE1 (ab194,1:1000) antibodies were from Abcam; anti-Lamin B1 (sc-374015, 1:3000) was from Santa Cruz Biotechnology; anti-Actin (A5441, 1:10 000); anti-FLAG (F1804, 1:10 000) antibodies were from Sigma-Aldrich. For secondary antibodies, mouse anti-goat IgG-HRP (sc-2354, Santa Cruz), rabbit anti-mouse IgG-HRP (A9044, Sigma-Aldrich), goat anti-rabbit IgG-HRP (A6667, Sigma-Aldrich) were used.
Immunofluorescence analysis
For staining of 8-oxoG in DNA, a previously published protocol was used with slight modifications (47,48). In brief, 2 × 104 cells were seeded on 12 mm diameter coverslip in 24-well plates. After 24 h, cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde (PFA) in PBS for 10 min. Cells were washed with PBS three times for 5 min each with agitation. Cells were then permeabilized with 0.4% Triton-X-100 in PBS, followed by three times washes for 5 min each. Cells were then treated with 2.5 mg/ml RNase A in PBS for 1 h at 37°C, followed by three PBS washes. Nuclear DNA was denatured by treating with freshly prepared 2 N HCl in distilled water for 10 min at room temperature, followed by three PBS washes. Cells were then incubated with mouse anti-8-oxo-dG antibody (ab48508, Abcam). Alexa Fluor 555 anti-mouse secondary antibodies were used. All images were acquired with Carl Zeiss confocal microscope LSM 780 at the Core Facility for Integrated Microscopy at University of Copenhagen. Fluorescence intensity was measured using Fiji software.
Measurement of mitochondrial DNA damage by long-range PCR
This assay was performed as previously described with minor modifications (49–51). Total DNA was isolated from cell pellets using the QIAamp DNA Mini kit (51304, QIAGEN). DNA concentration was measured by an Epoch Microplate Spectrophotometer (BioTek). Amplification of an 8.9 kb segment and a 117 bp segment of mitochondrial DNA was performed using the LongAmp Taq DNA Polymerase (M0323L, New England Biolabs). For the 8.9 kb segment, the primer sequences were: sense, 5′-TCT AAG CCT CCT TAT TCG AGC CGA-3′, antisense, 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′. PCR conditions were: (i) 94°C, 1 min; (ii) 65°C, 12 min—repeat 1 and 2 for 19 cycles; (iii) 65°C, 10 min. For the 117 bp segment, primer sequences were: sense, 5′-CCC CAC AAA CCC CAT TAC TAA ACC CA-3′; antisense, 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′. PCR conditions were: (a) 94°C, 1 min; (b) 58°C, 1 min; (c) 65°C, 1 min—repeat 1–3 for 19 cycles; (d) 65°C, 2 min. PCR products were separated in 1% agarose gel, and visualized with ethidium bromide staining. Quantitation was performed by Fiji software.
Measurement of mtDNA copy number
Total DNA was extracted from cell pellets using the QIAamp DNA Mini kit (51304, QIAGEN). mtDNA copy number analysis was performed by qPCR with the Platinum SYBR Green qPCR SuperMix-UDG reagents(11733038, Thermo Fisher Scientific). mtDNA levels were assessed using primers against the mitochondrial 117bp segment. Primer sequences were: sense, 5′-CCC CAC AAA CCC CAT TAC TAA ACC CA-3′; antisense, 5′-TTT CAT CAT GCG GAG ATG TTG GAT GG-3′; Nuclear actin served as a loading control.
8-oxoG incision assay
Oligonucleotide containing an 8-oxoG lesion at a specific position was annealed to its complementary oligonucleotide. The oligonucleotide sequences are: 5′-GAA CGA CTG T(8-oxoG)A CTT GAC TGC TAC TGA T-3′-TAMRA, and 5′- ATC AGT AGC AGT CAA GTC ACA GTC GTT C-3′. This assay was carried out as previously described with some modifications (52–54). For 8-oxoG incision assay, the DNA substrate (5 pmol) was incubated with the indicated amounts of recombinant human RECQL4 or human OGG1 (ab98249, Abcam) in 20 μl reaction buffer (70 mM HEPES–KOH, 7.4, 5 mM EDTA, 1 mM DTT, 75 mM NaCl, 10% glycerol, 100 μg/ml BSA) for 30 min at 37°C. Reactions were terminated by adding 20 μl of formamide loading buffer (95% formamide, 0.025% w/v bromophenol blue, 0.025% w/v xylene cyanol, 5 mM EDTA pH 8.0). The samples were heated at 95°C for 3 min, and put on ice. For glycosylase and AP lyase activity measurement, the reactions were terminated by adding 20 μl of formamide loading buffer and 1 μl piperidine (99%). The samples were then heated at 95°C for 5 min in order to cleave all AP sites, prior to loading on gels. For 8-oxoG incision assay with whole cell extracts, 30 μg protein extracts were incubated with 5 pmol TAMRA-labeled DNA substrates in reaction buffer containing 20 mM HEPES–KOH pH 7.4, 5 mM EDTA, 5 mM DTT, 150 mM NaCl, 10% glycerol, 100 μg/ml BSA. After incubation for 4 h at 37°C, the reactions were terminated by adding 1 μl 5 mg/ml Proteinase K and 1 μl 10% SDS and incubated at 55°C for 30 min. DNA was precipitated by adding 5 μg glycogen, 4 μl 11 M ammonium acetate, 60 μl ethanol, and overnight incubation at –80°C. Samples were pelleted, dried and suspended in 20 μl of formamide loading buffer. All samples were electrophoresed on 15% polyacrylamide/8 M urea gel in 1× TBE buffer. Fluorescence was detected by ChemiDoc Imaging Systems (Bio-Rad). Quantifications were performed by Fiji software.
In vitro acetylation assay
Recombinant human RECQL4 or human OGG1 (ab98249, Abcam) (1 μg) was incubated with different amounts of recombinant human CBP (0.1 μg, 0.2 μg, 0.5 μg) (catalytic domain) (BML-SE452-0100, Enzo Life Sciences) in 20 μl acetylation buffer containing 50 mM Tris, pH 8.0, 50mM NaCl, 0.1 mM EDTA, 100 μg/ml BSA, 10% glycerol, 1 mM DTT,1 mM PMSF, and 10 mM sodium butyrate in the presence of 2 mM acetyl CoA (A2056-10G, Sigma). Reactions were performed at 30°C for 1 h, stopped by adding 2 × Laemmli buffer (S3401, Sigma-Aldrich), and resolved by SDS-PAGE. The gels were analyzed by Coomassie blue staining and western blot with the indicated antibodies. For detection of the acetylation of RECQL4, an anti-acetylated lysine antibody (9441S, CST) was used. For detection of acetylation of OGG1, an anti-acetylated OGG1 antibody (ab93670, Abcam) was used.
Acetylation of RECQL4 in cells
Cells were co-transfected with the expression constructs for FLAG-tagged RECQL4 and CBP or p300. After 36 h, cells were lysed in lysis buffer (50 mM HEPES–KOH 7.5, 500 mM NaCl, 0.5% NP-40, 10% glycerol, 5 μM TSA, 10 mM nicotinamide, 10 mM sodium butyrate, 1× protease inhibitor cocktail) and sonicated on ice. FLAG-tagged RECQL4 were pulled down with M2 FLAG-magnetic beads at 4°C. After the beads were sequentially washed with wash buffer A (50 mM HEPES–KOH pH7.5, 1 M NaCl, 0.5% NP-40, 10% glycerol, 10 mM nicotinamide), wash buffer B (50 mM HEPES–KOH pH7.5, 500 mM NaCl, 0.5% NP-40, 10% glycerol), and wash buffer C (50 mM HEPES–KOH pH 7.5, 150 mM NaCl, 0.5% NP-40, 10% glycerol), the 3xFLAG-tagged RECQL4 were eluted by incubating the beads in wash buffer C containing 300 μg/ml 3xFLAG peptide (F4799, Sigma-Aldrich). The eluted proteins were analyzed by SDS-PAGE, and acetylation of RECQL4 was probed by western blot with an anti-acetylated lysine antibody (9441S, CST).
In vitro deacetylation assay
This assay was carried out in two steps. First, recombinant human RECQL4 or human OGG1 (ab98249, Abcam) (1 μg) were acetylated by 0.1 μg recombinant human CBP (BML-SE452-0100, Enzo Life Sciences) in 10 μl of acetylation buffer containing 50 mM Tris, pH 8.0, 50 mM NaCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT,1 mM PMSF and 2 mM acetyl CoA. After incubation at 30°C for 1 h, the reaction mixtures were incubated at 30°C for an additional 1 h with the indicated amount of recombinant SIRT1 (1 U = 1 pmol/min at 37°C, 250μM FLUOR DE LYS® substrate (Prod. No. BML-KI104), 500 μM NAD+) (BML-SE239-0100, Enzo Life Sciences) in 20 μl of deacetylation buffer containing 50 mM Tris–HCl, pH 9.0, 50 mM NaCl, 4 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10% glycerol in the presence of 50 μM NAD+ co-factor (20-221, Sigma-Aldrich). The reaction mixtures were resolved by SDS-PAGE and the acetylation status of RECQL4 was analyzed by western blot with a monoclonal anti-acetylated lysine antibody (9441S, CST).
Clonogenic survival assay
Cells were plated into six-well plates at 200 cells per well. After overnight incubation, cells were treated with 50 μM menadione for 1 h. Then the medium were removed, and cells were washed once with DMEM medium and changed to normal culture medium for another 12 days. The colonies were washed with PBS for two times, fixed and stained with staining buffer containing 0.5% crystal violet and 20% ethanol for 1 h. The colonies were then gently washed in water and air-dried. Images were acquired with a CanoScan LiDE 220 scanner and quantification of colonies were performed with Fiji software.
Cell viability assay
Cells (8 × 103 per well) were seeded into 96-well plates. 24 h later, cells were treated as described in the figure legends. Cell viability was measured with CellTiter 96 AQueous One Solution Cell Proliferation Assay MTS kit according to the manufacturer's instructions (G3580, Promega).
EdU incorporation assay
EdU (5-ethynyl-2′-deoxyuridine) incorporation assays were performed using the Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit (Thermo Fisher Scientific, #C10425) according to the manufacturer's instructions. Briefly, the cells were incubated with 10 μM EdU for 1 h, fixed with 4% PFA in DPBS for 15 min at room temperature, permeabilized with 0.4% Triton X-100/1% BSA in PBS for 15 min. After permeabilization, Click-iT reaction was performed. Cells were analyzed using a CytoFLEX flow cytometer (Beckman Coulter, Inc.).
Statistical analyses
Data were analysed using a two-tailed, unpaired Student's t test and are presented as mean ± SD or SEM as indicated in the figure legends. P < 0.05 was considered as statistically significant.
RESULTS
Characterization of RECQL4 knockout cells
Previous studies have greatly increased our knowledge of the molecular function of RECQL4 using RTS patient samples. However, large variations observed in RTS patient cells have hampered understanding of the role of RECQL4 in BER (55–57). To rule out the potential influence of intercellular genetic and epigenetic variations which may result in subtle differences including responses to stress and repair, we generated isogenic RECQL4−/− and control cells using the double nicking CRISPR/Cas9n system. This approach is thought to significantly reduce off-target effects and facilitate gene knockout with high on-target cleavage efficiency (45). The N-terminal 400 amino acids (aa) region of RECQL4 possesses homology to the yeast DNA replication initiation factor Sld2 (7,58), and also contains its nuclear localization signals (59) and a mitochondrial targeting sequence (17), indicating that the N-terminal region of RECQL4 mediates its important functions. Therefore, to completely disrupt the function of RECQL4, we used paired sgRNAs targeting exon 5 of the RECQL4 gene locus (Figure 1A), which encodes most of the amino acids in N-terminal region. Biallelic disruption of RECQL4 was confirmed by genomic PCR and Western blot analysis (Figure 1B and C). A number of studies have demonstrated that RECQL4 plays an important role in DNA replication (7,60–62). Consistent with previous findings, we found that depletion of RECQL4 leads to diminished proliferative capacity, as assessed by colony formation assay (Figure 1D). The number of colonies produced by RECQL4−/− cells was less and the colonies were smaller than those by wild-type cells, indicating that depletion of RECQL4 results in decreased cellular survival and proliferation. Decreased cell proliferation in RECQL4−/− cells was further confirmed by an EdU (5-ethynyl-2′-deoxyuridine) incorporation assay showing that the percentage of EdU-labeled cells in RECQL4−/− cells was significantly less than that in wild type cells (Supplementary Figure S1A). Overexpression of RECQL4 in RECQL4−/− cells rescued these phenotypes, suggesting that depletion of RECQL4 was responsible for decreased cellular survival and proliferation observed in RECQL4−/-cells (Figure 1E and Supplementary Figure S1B). Previously, RECQL4 was shown to be present in mitochondria where it might contribute to mtDNA maintenance (14,15,17,63). Therefore, we assessed the effects of RECQL4 knockout on mitochondrial function. RECQL4 depletion resulted in a significant reduction in mtDNA copy number (Figure 1F), and diminished mtDNA repair capacity as RECQL4−/− cells accumulated more mtDNA damage than wild-type cells (Figure 1G). We generated two RECQL4-deficient cell lines. Analysis of the second RECQL4-deficient cell line (RECQL4−/− #22) showed similar results (Supplementary Figure S1C–E). Thus, we generated an isogenic RECQL4−/− cell line, which exhibits several cellular phenotypes previously observed in RTS patient cells and in RECQL4-deficient cells (7,14–16,64), suggesting that it is suitable for RECQL4 study.
Figure 1.

Characterization of RECQL4 knockout cells. (A) Schematic showing sgRNA sequences targeting the human RECQL4 locus using Cas9 D10A nickases (Cas9n). Red arrows indicate the sites of nicks in DNA. (B) PCR analyses of WT and RECQL4−/− cells using primer pair (P1+P2) to amplify the genomic region ranging from exon 5 to intron 6 of RECQL4. The result shows the RECQL4−/− clone with a homozygous deletion of the RECQL4 locus. (C) Knockout of RECQL4 in U2OS cells was confirmed by immunoblotting. (D) Colony formation assay showing RECQL4−/− cells have clonogenic survival defects. Images show representative colonies stained with crystal violet. Data are shown as mean ± SEM, n = 3. ***P < 0.001 (t-test). (E) Colony formation assay showing that re-introduction of WT-RECQL4 in RECQL4−/− cells rescues the clonogenic survival defects of RECQL4−/-cells. Three independent experiments were performed, each with four replicates. Representative images (left) and quantifications (right) are shown. **P < 0.01, using unpaired two-tailed Student's t test. (F) mtDNA copy number in WT and RECQL4−/− cells. Data are shown as mean ± SEM, n = 3. ***P < 0.001 (t-test). (G) mtDNA damage detection by a PCR-based assay assessing relative quantitative amplification of a small (117 bp) to a large (10 kb) segment of mtDNA. A representative gel (left) and quantifications (right) are shown as mean ± SEM, n = 3. ***P < 0.001.
RECQL4 is required for BER of 8-oxoG
Several studies have shown that RTS patient cells were hypersensitive to oxidative stress (55,56), suggesting that RECQL4 deficiency may impair repair of oxidative DNA lesions. We observed a markedly increased sensitivity of RECQL4−/− cells to menadione, a ROS-generating compound (65), as measured by colony formation assay and MTS assay (Figure 2A, B and Supplementary Figure S2A). Moreover, RECQL4−/− cells showed increased sensitivity to H2O2 treatment (Supplementary Figure S2B). Consistent with some observations in RTS patient cells, we found a significantly higher level of 8-oxoG in RECQL4−/− cells (Figure 2C) (47,48). 8-oxoG:C in the genomic DNA is primarily repaired by the base excision repair (BER) pathway initiated by the removal of 8-oxoG by OGG1 DNA glycosylase (24). Thus, increased sensitivity to oxidative stress and increased endogenous levels of 8-oxoG may be due to reduced 8-oxoG repair capacity in these cells. Therefore, we assessed the 8-oxoG incision ability in RECQL4−/− cells using a TAMRA-labeled DNA substrate containing 8-oxoG (Supplementary Figure S2C). Since RECQL4 and OGG1 are both nuclear and mitochondrial proteins, we used whole cell extracts to better represent the function of these two proteins in both compartments. 8-oxoG incision activity was significantly lower in extracts from RECQL4−/− cells compared with extracts from wild-type control cells (Figure 2D). Addition of purified recombinant RECQL4 protein restored 8-oxoG incision in RECQL4−/− cell extracts to the level of control cells, and significantly increased 8-oxoG incision activity in extracts from control cells (Figure 2E), suggesting that RECQL4 is required for efficient 8-oxoG repair and may be directly involved in 8-oxoG repair. Thus, we conclude that RECQL4 participates in BER of 8-oxoG and as such RECQL4 deficiency leads to defective repair of 8-oxoG.
Figure 2.

RECQL4 is required for BER of 8-oxoG. (A) Colony formation ability of WT and RECQL4−/-cells treated with or without 50 μM menadione. Images show representative colonies stained with crystal violet. Data are shown as mean ± SEM, n = 3. **P < 0.01 (t-test); ***P < 0.001 (t-test). (B) MTS assay showing viability of WT and RECQL4 knockout cells treated with or without menadione. Data are shown as mean ± SEM, n = 3. **P < 0.01 (t-test); ***P < 0.001 (t-test). (C) 8-oxoG base lesion detected in WT and RECQL4−/−cells by immunofluorescence microscopy. Scale bar, 10 μm. The apochromat 63×/1.4 DIC (oil) objective was used. The 8-oxoG level was measured by quantifying the fluorescence intensity with Fiji software. At least 50 cells were counted for each experiment. Data are shown as the mean ± SD of three independent experiments. ***P < 0.001 (t-test). (D) Base excision repair (BER) assay measuring 8-oxoG incision activity using a TAMRA-labeled 8-oxoG containing oligonucleotide duplexes in whole cell extracts of WT and RECQL4−/− cells. Representative gel (left) and quantification (right) of 8-oxoG incision levels are shown. The negative control (NC) was added with the same cell extraction buffer. (E) Quantification of BER analysis of 8-oxoG repair in WT and RECQL4−/-whole cell extracts. WT and RECQL4−/−extracts were supplemented with either recombinant wild-type human RECQL4 protein or BSA. Recombinant human RECQL4 and BSA were used at 50 ng/μl. Data are shown as mean ± SEM from three independent experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001, using unpaired two-tailed Student's t test.
RECQL4 selectively interacts with OGG1 and promotes 8-oxoG repair
We asked whether the stimulation of 8-oxoG repair by RECQL4 in cell extracts involves interaction between RECQL4 and OGG1, the major DNA glycosylase for 8-oxoG repair. To test this, we performed the following experiments. Using immunoprecipitation (IP) with GFP-RECQL4 as bait, we found that RECQL4 directly interacts with OGG1 (Figure 3A), and menadione-induced oxidative stress increased this interaction (Figure 3A), suggesting that RECQL4 may further enhance 8-oxoG repair following oxidative stress. Similarly, IP of endogenous RECQL4 showed direct interaction between RECQL4 and OGG1 in wild-type cells, but not in RECQL4−/-cells (Supplementary Figure S3A). We then mapped the interacting regions between these two proteins. RECQL4 contains a Sld2-like domain at the N-terminus, a highly conserved ATPase and helicase domain in the middle, and a poorly characterized C-terminus (4,66). IP was performed with 3× FLAG-tagged full-length RECQL4 (FL-RECQL4), 3× FLAG-tagged truncated RECQL4 mutants (1−427aa, NT-RECQL4; 427−1208aa, CT-RECQL4) (Figure 3B, upper panel) (12). Only the full-length RECQL4 and NT-RECQL4 interacted with OGG1, but not the CT-RECQL4 (Figure 3B, lower panel). The absence of NEIL1, another major DNA glycosylase for repair of 8-oxoG, in the IP complex indicates the selective RECQL4/OGG1 interaction for repair of 8-oxoG (Figure 3B, lower panel).
Figure 3.

RECQL4 selectively interacts with OGG1 and promotes 8-oxoG repair. (A) Menadione treatment enhances RECQL4/OGG1 interaction. Immunoprecipitated material from cells expressing GFP or GFP-tagged WT-RECQL4 with GFP-TRAP beads analyzed by immunoblotting (IB) with anti-GFP and anti-OGG1 antibodies. (B) N-terminal domain of RECQL4 interacts with OGG1 in cells. Schematic diagram of 3× FLAG-tagged RECQL4 and the truncated fragments (upper panel). FL-RECQL4, NT-RECQL4, and CT-RECQL4 were all tagged with both 3× FLAG and SV40 nuclear-location sequence (NLS). (C) Representative gel (left) and quantification (right) of the stimulation of catalytic activity of OGG1 by RECQL4 in a dose-dependent manner. Data are presented as mean ± SEM from three independent experiments. ****P < 0.0001, using unpaired two-tailed Student's t test. (D) Representative gel (left) and quantification (right) of the stimulation of catalytic activity of OGG1 by RECQL4 in a time-course dependent manner. Data are presented as mean ± SD from two independent experiments. ****P < 0.0001, using unpaired two-tailed Student's t test. (E) Representative gel (left) of the stimulation of catalytic activity of OGG1 by RECQL4 treated with or without piperidine. Quantification (right) of the glycosylase and AP lyase activities of OGG1 stimulated by RECQL4. Data are presented as mean ± SD from two independent experiments. ****P < 0.0001, using unpaired two-tailed Student's t test. (F) BER assay showing that the N-terminal domain of RECQL4 functionally interacts with OGG1. 3 × FLAG-tagged RECQL4 proteins were purified from normal U2OS cell. A representative image (left) and quantifications (right) are shown. **P < 0.01; ns, not significant, using unpaired two-tailed Student's t test. Data are shown as mean ± SEM, n = 4.
To test whether RECQL4 functionally interacts with OGG1, we performed the 8-oxoG incision assay in the presence of RECQL4 and OGG1. RECQL4 significantly stimulated the 8-oxoG incision activity of OGG1 in a concentration-dependent manner (Figure 3C and Supplementary Figure S3B), suggesting a functional interaction. Moreover, as shown in Figure 3D, RECQL4 stimulates the 8-oxoG incision activity of OGG1 in a time-dependent manner. OGG1 is a bifunctional DNA glycosylase, possessing a strong DNA glycosylase activity and a relative weak AP lyase activity (67,68). Next, we tested which activity of OGG1 was most affected by RECQL4. To separate these two activities, after termination, the reactions were followed by treatment with or without hot piperidine. RECQL4 significantly stimulated both the glycosylase and AP lyase activities of OGG1 (Figure 3E). However, the stimulation of the AP lyase activity of OGG1 by RECQL4 was more prominent than that of its glycosylase activity (Figure 3E). These results suggest RECQL4 may coordinate the glycosylase activity and AP lyase activity of OGG1 in BER of 8-oxoG. Moreover, only full-length RECQL4 and NT-RECQL4 stimulated 8-oxoG incision activity of OGG1 (Supplementary Figure S3C and Figure 3F), further supporting that RECQL4 interacted with OGG1 through its N-terminus. Thus, our results strongly support that RECQL4 physically and functionally interacts with OGG1, and that RECQL4 plays a role in 8-oxoG repair through interacting with OGG1.
RECQL4 is an acetylated protein and its acetylation is stimulated by oxidative stress
Increasing number of studies show that the functions and activities of DNA repair proteins are regulated by posttranslational acetylation (69,70). p300, and the closely related protein CBP, are highly conserved acetyltransferases involved in multiple cellular processes including DNA repair (71). Previously, RECQL4 was shown to become acetylated in the N-terminus by p300 (44). We transiently co-transfected cells with 3 × FLAG-WT-RECQL4 together with either p300 or CBP. The level of acetylated RECQL4 was significantly increased in cells co-transfected with RECQL4 together with p300 or CBP (Figure 4A). These results showed that RECQL4 is an acetylated protein, which is consistent with a previous report (44) and suggested that RECQL4 is a potential new target of CBP acetyltransferase. To further confirm that CBP acetylates RECQL4, we performed an in vitro acetylation assay using purified recombinant CBP (catalytic domain) together with recombinant RECQL4. RECQL4 is acetylated by CBP in vitro in the presence of acetyl-coenzyme A (Figure 4B), suggesting that RECQL4 is a possible substrate for CBP. A previous study showed that five lysines (376, 380, 382, 385 and 386) of the total 32 lysines can be acetylated within the N terminus of RECQL4 (44). Therefore, to determine whether CBP may acetylate these residues on RECQL4, we generated two RECQL4 mutants, RECQL4 (KQ) with all five lysine residues mutated to glutamine and RECQL4 (KR) with all five lysine residues mutated to arginine using site-directed mutagenesis to mimic acetylated lysine and non-acetylated lysine, respectively. Compared to the wild type, RECQL4 (WT), both the RECQL4 (KQ) mutant and the RECQL4 (KR) mutant exhibited significantly reduced capacity to become acetylated by CBP in vitro (Figure 4C and Supplementary Figure S4A). This data indicates that at least one of the five lysine residues is the target of acetylation by CBP. Further, it also suggests that CBP may acetylate other lysine residues on RECQL4 in addition to these five lysine residues. Interestingly, the RECQL4 acetylation level was significantly increased following menadione treatment (Figure 4D), suggesting that oxidative stress and likely oxidative DNA damage can trigger RECQL4 acetylation. In addition, we found that under our conditions OGG1 can be acetylated by CBP as well and acetylation significantly increases 8-oxoG incision activity of OGG1 in vitro (Supplementary Figure S4B and Supplementary Figure S4C), which are consistent with previous observations (39,72).
Figure 4.
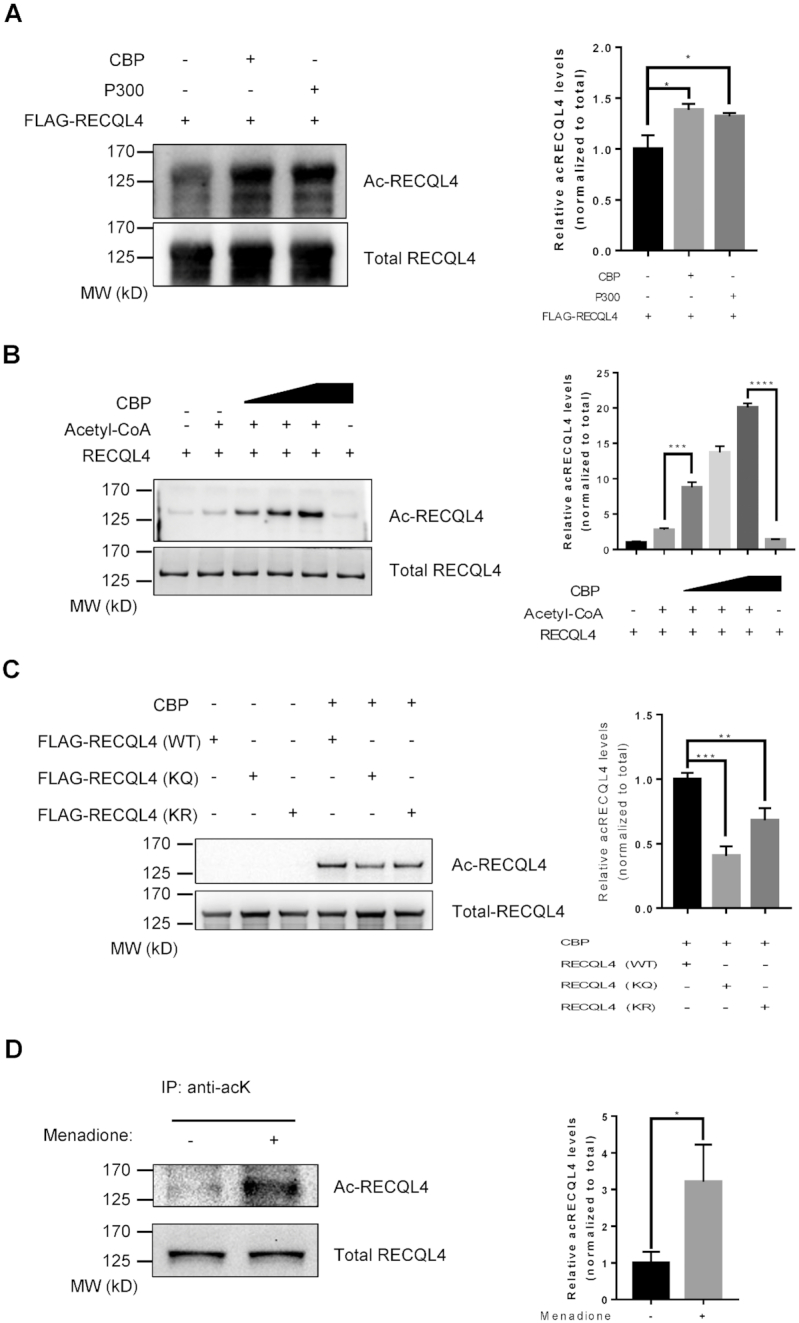
RECQL4 is an acetylated protein and its acetylation is stimulated by oxidative stress. (A) RECQL4 acetylation in transiently transfected U2OS cells with 3xFLAG-tagged RECQL4, p300 or CBP, treated with 2 μM TSA (trichostatin A) and 5 mM NAM (nicotinamide). Immunoprecipitated RECQL4 proteins were then detected by Western blot with anti-acetylated lysine antibody. A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated RECQL4 levels normalized to total RECQL4. Data are presented as mean ± SD from two independent experiments. *P < 0.05, using unpaired two-tailed Student's t test. (B) Acetylation of RECQL4 by CBP in an in vitro acetylation assay. Recombinant RECQL4 (1 μg), Acetyl-CoA (2 mM), and different amounts of recombinant CBP (0.1, 0.2, 0.5 μg) were incubated at 30°C for 1 h. Acetylated RECQL4 proteins were detected with anti-acetylated lysine antibody (top). Total amounts of RECQL4 were assessed with Coomassie blue staining (bottom). The graph on the right shows quantification of relative acetylated RECQL4 levels normalized to total RECQL4. Data are shown as mean ± SD from three independent experiments. ***P < 0.001, ****P < 0.0001, using unpaired two-tailed Student's t test. (C) Acetylation of wild type RECQL4 (WT), RECQL4 (KQ) and RECQL4 (KR) mutants by CBP in an in vitro acetylation assay. 3 × FLAG-tagged RECQL4 proteins were purified from normal U2OS cell. FLAG-tagged RECQL4 proteins (1 μg), Acetyl-CoA (2 mM), and recombinant CBP (0.2 μg) were incubated at 30°C for 1 h. Acetylated FLAG-tagged RECQL4 proteins were detected with anti-acetylated lysine antibody. A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated FLAG-tagged RECQL4 protein levels normalized to total. Data are shown as mean ± SD from two independent experiments. **P < 0.01, ***P < 0.001, using unpaired two-tailed Student's t test. (D) Western blot showing increased endogenous acetylated RECQL4 levels in cells treated with 50 μM menadione for 1 h. Endogenous acetylated proteins were immunoprecipitated with anti-acetyl-lysine antibody-conjugated beads. The presence of RECQL4 in the immunoprecipitated complex and acetylation levels in the extract were measured by Western blot with anti-RECQL4 antibody. Input for immunoprecipitation was used as a loading control for total RECQL4 (bottom). A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated RECQL4 levels normalized to total RECQL4. Data are shown as mean ± SD from two independent experiments. *P < 0.05, using unpaired two-tailed Student's t test.
SIRT1 interacts with and deacetylates RECQL4
Protein acetylation is a highly dynamic post-translational modification that can be reversed by Sirtuin deacetylases. Among the Sirtuins, the nuclear SIRT1 is involved in various cellular processes including DNA repair (73–75). Because RECQL4 is an acetylated protein and the SIRT1 inhibitor, nicotinamide (NAM), affects the acetylation status of RECQL4 (44), we asked whether RECQL4 is a potential target of SIRT1. To test this, we first checked our recently published two separate mass spectrometry data sets of RECQL4 interactome (12,13). SIRT1 was identified in both studies as a potential deacetylase for RECQL4. To further test this, we immunoprecipitated GFP-RECQL4 from U2OS cells and detected a direct interaction between RECQL4 and SIRT1 (Figure 5A). We then mapped the interaction region between RECQL4 and SIRT1 to the N-terminus of RECQL4, which was within the previously reported acetylation region of RECQL4 by p300 (Figure 5B) (44). To determine whether SIRT1 deacetylates RECQL4, we performed a two-step deacetylation assay in vitro. First, we acetylated RECQL4 by incubating recombinant RECQL4 with recombinant CBP acetylase in the presence of acetyl-CoA. Second, we incubated the acetylated form of RECQL4 with recombinant human SIRT1. RECQL4 was deacetylated by SIRT1 in a dose-dependent manner in the presence of NAD+ co-substrate. In addition, the deacetylation was inhibited in the presence of NAM (Figure 5C). Thus, we conclude that RECQL4 is a novel target of SIRT1. To further investigate the five lysine residues as possible targets of SIRT1 deacetylation, we performed the in vitro deacetylation assay using the RECQL4 mutants. Mutations of the five lysines residues did not completely abolish the deacetylation of RECQL4 by SIRT1 (Figure 5D), indicating that deacetylation of lysines other than the five lysine residues by SIRT1 is possible.
Figure 5.

SIRT1 interacts with and deacetylates RECQL4. (A) Immunoprecipitation (IP) in cells expressing GFP or GFP-tagged WT-RECQL4 with GFP-TRAP beads analyzed by immunoblotting (IB) with anti-GFP and anti-SIRT1 antibodies. Representative blot is shown from two independent experiments. (B) Immunoprecipitation (IP) performed in cells expressing vector alone or FLAG-tagged FL-RECQL4, NT-RECQL4, and CT-RECQL4 with FLAG-M2 magnetic beads and analyzed by immunoblotting (IB) with the indicated antibodies shows that the N-terminal domain of RECQL4 interacts with SIRT1 in cells. Representative blot is shown from two independent experiments. (C) Deacetylation of RECQL4 by SIRT1 in an in vitro deacetylation assay. Recombinant RECQL4 (1 μg) was first acetylated by CBP (0.1 μg) for the first 1 h, then acetylated RECQL4 was incubated with NAD+ (50 μM), and different amounts of recombinant SIRT1 (0.5, 1, 2 U) at 30°C for an additional 1 h. Acetylated RECQL4 was probed with anti-acetylated lysine antibody. Total amounts of RECQL4 were assessed with an anti-RECQL4 antibody. NAM served as a SIRT1 inhibitors. A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated RECQL4 levels normalized to total RECQL4. Data are shown as mean ± SD from three independent experiments. **P < 0.01, ***P < 0.001, using unpaired two-tailed Student's t test. (D) Deacetylation of wild type RECQL4 (WT) and RECQL4 (KQ) mutant by SIRT1 in an in vitro deacetylation assay. 3 × FLAG-tagged RECQL4 proteins were purified from normal U2OS cells. FLAG-tagged RECQL4 proteins (1 μg), were first acetylated by CBP (0.1 μg) for the first 1 h, then acetylated proteins were incubated with NAD+ (50 μM), and recombinant SIRT1 (2 U) at 30°C for an additional 1 h. Acetylated FLAG-tagged RECQL4 proteins were probed with anti-acetylated lysine antibody. Total amounts of FLAG-tagged RECQL4 proteins were assessed with an anti-FLAG antibody. A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated RECQL4 protein levels normalized to total. Data are shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, using unpaired two-tailed Student's t test.
It has been reported that SIRT1 negatively regulates the catalytic activity of OGG1 in vivo (39). However, it remains unclear whether SIRT1 deacetylates OGG1 because in a separate study OGG1 activity was not affected by NAM (72). To clarify this, we used an in vitro deacetylation assay using recombinant human OGG1 and recombinant human SIRT1. SIRT1 directly deacetylated OGG1 in the presence of NAD+ co-substrate, and the deacetylation was inhibited in the presence of NAM (Supplementary Figure S5A). Thus, this data provides evidence consistent with OGG1 being a substrate for SIRT1 (39), suggesting that SIRT1 is a potential regulator of BER.
SIRT1 controls the interaction between OGG1 and RECQL4 following oxidative stress and maintains RECQL4 in a hypoacetylated state
Our results showed that SIRT1 deacetylates RECQL4 in vitro, and we next asked whether SIRT1 deacetylates RECQL4 in cells. We assessed RECQL4 acetylation in SIRT1 knockdown cells by immunoprecipitating 3 × FLAG-WT-RECQL4 and then immunoblotting with antibodies against acetylated lysine. Acetylation of RECQL4 was markedly increased in SIRT1 knockdown cells (Figure 6A and Supplementary Figure S6A), indicating that SIRT1 deacetylates RECQL4 in cells.
Figure 6.

SIRT1 controls the interaction between OGG1 and RECQL4 following oxidative stress and maintains RECQL4 in a hypoacetylated state. (A) Western blot analysis of acetylation of RECQL4 in shCtrl or shSIRT1 cells. Knockdown of SIRT1 significantly increased RECQL4 acetylation. shCtrl and shSIRT1 cells were transfected with FLAG-RECQL4. 24 hours after transfection, whole-cell lysates were subjected to immunoprecipitation with FLAG-M2 magnetic beads followed by Western blot analysis of the immunoprecipitated material with anti-acetylated lysine and anti-RECQL4 antibodies. A representative gel is shown on the left. The graph on the right shows quantification of relative acetylated RECQL4 levels normalized to total RECQL4. Data are shown as mean ± SD from two independent experiments. ***P < 0.001, using unpaired two-tailed Student's t test. (B) Immunoprecipitation (IP) was performed in shCtrl and shSIRT1 cells expressing FLAG-tagged RECQL4 with FLAG-M2 magnetic beads and analyzed by western blot with the indicated antibodies. shCtrl and shSIRT1 cells expressing FLAG-tagged RECQL4 were treated with 50μM menadione for 1 h, then the media were replaced with fresh media and the cells were collected at indicated time points. Interaction between RECQL4 and SIRT1 decreased immediately after menadione treatment (0 h recovery) both in shCtrl and shSIRT1 cells. This interaction increased at 6 h recovery, compared with 0 h recovery. Lamin B1 was used as a loading control. A representative gel is shown on the left. The graph on the right shows the relative ratio of immunoprecipitated SIRT1 to FLAG-RECQL4 from shCtrl cells. Data are shown as mean ± SD from two independent experiments. *P < 0.05, **P < 0.01, using unpaired two-tailed Student's t test. (C) Time-course dependent interaction between RECQL4 and SIRT1 following 1 h 50 μM menadione treatment. Cells were transfected with FLAG-RECQL4 and cell lysates were collected at 0, 1, 2, 4, 6, and 12 h after menadione treatment. Western blot analysis of the IP complexes was carried out by anti-FLAG and anti-SIRT1 antibodies. A representative gel is shown on the left. The graph on the right shows the relative ratio of immunoprecipitated SIRT1 to FLAG-RECQL4 from shCtrl cells. Data are shown as mean ± SD from three independent experiments. *P < 0.05, ***P < 0.001, using unpaired two-tailed Student's t test. (D) Immunoprecipitation (IP) was performed in shCtrl and shSIRT1 cells expressing FLAG-tagged RECQL4 with FLAG-M2 magnetic beads and analyzed by Western blot using the indicated antibodies. SIRT1 knockdown significantly affects the interaction between RECQL4 and OGG1. A representative gel is shown on the left. The graph on the right shows the relative ratio of immunoprecipitated OGG1 to FLAG-RECQL4 from shCtrl and shSIRT1 cells. Data are shown as mean ± SD from three independent experiments. ***P < 0.001, using unpaired two-tailed Student's t test. (E) Immunoprecipitation (IP) was performed in cells expressing FLAG-tagged RECQL4 (WT), RECQL4 (KQ) mutant, and RECQL4 (KR) mutant with FLAG-M2 magnetic beads treated with or without menadione, and analyzed by Western blot using the indicated antibodies. Five lysine mutants impaired the interaction between RECQL4 and OGG1. A representative gel is shown on the left. The graph on the right shows relative ratio of immunoprecipitated OGG1 to FLAG-RECQL4. Data are shown as mean ± SD from two independent experiments. *P < 0.05, using unpaired two-tailed Student's t test.
As shown in Figure 4D, we observed that the level of acetylated RECQL4 was significantly increased in cells treated with menadione, suggesting that RECQL4 acetylation is regulated by oxidative stress. To understand how oxidative stress stimulates RECQL4 acetylation, we asked whether oxidative stress affects the interaction between RECQL4 and SIRT1. To test this, we examined the time course of interaction between RECQL4 and SIRT1 after menadione treatment. The interaction between RECQL4 and SIRT1 was significantly disrupted immediately after menadione treatment (Figure 6B), suggesting that the increased acetylation of RECQL4 after menadione treatment is likely due to a reduction in the interaction between RECQL4 and the deacetylase SIRT1. Notably, we observed that the interaction between RECQL4 and SIRT1 was gradually restored after menadione treatment and reached the maximum after 6h recovery (Figure 6C), suggesting that the acetylation of RECQL4 following oxidative stress is tightly controlled by SIRT1. As shown in Figure 3A, the interaction between RECQL4 and OGG1 was enhanced after menadione treatment, which in turn can be counteracted by SIRT1. Indeed, the interaction between RECQL4 and OGG1 was significantly enhanced in SIRT1 knockdown cells (Figure 6D). These results suggest that SIRT1 controls the interaction between RECQL4 and OGG1 following acute oxidative stress, and potentially maintains RECQL4 in a hypoacetylated state. Interestingly, the RECQL4 (KQ) and RECQL4 (KR) mutants almost completely disrupted the interaction between RECQL4 and OGG1 while increasing the interaction between RECQL4 and SIRT1 (Figure 6E). We did not detect any interaction between RECQL4 and APE1 (Figure 6E). Furthermore, the stimulatory effect of RECQL4 on the catalytic activity of OGG1 was enhanced in the presence of CBP and acetyl CoA, and was reduced in the presence of SIRT1 and NAD+ co-substrate (Supplementary Figure S6B).
DISCUSSION
The novel findings in this study are that (i) RECQL4 interacts with OGG1 and stimulates the glycosylase activity and AP lyase activity of OGG1, most prominently the AP lyase activity, (ii) SIRT1 interacts with and deacetylates RECQL4, and (iii) SIRT1 modulates the interaction between RECQL4 and OGG1 in response to oxidative stress. In this study, we provide new evidence that RECQL4 regulates BER of the major oxidative DNA base lesion 8-oxoG. Cellular phenotypes in RECQL4−/-cells suggest defective BER including hypersensitivity to oxidative stress, increased 8-oxoG levels in the genome, and decreasing 8-oxoG incision capacity. We demonstrated that RECQL4 directly interacts with OGG1 DNA glycosylase in cells, and that RECQL4 stimulated OGG1 dependent 8-oxoG repair in vitro. Thus, our results are consistent with previous studies suggesting possible roles of RECQL4 in BER (55,56). However, we show here for the first time that RECQL4 stimulates repair of 8-oxoG through physical and functional interaction with OGG1, and that the possible mechanism for the regulation of this interaction is via acetylation/deacetylation of RECQL4 in response to oxidative stress. Our findings may have important implications for the role RECQL4 in genome stability maintenance and telomere maintenance against aging and cancer.
Many DNA repair proteins have been identified as SIRT1 substrates, including WRN (36), Ku70 (35), XPA (38), NBS1 (37), RPA (76,77). In addition, studies have shown that SIRT1 regulates resistance to oxidative stress through different mechanisms (78–80). Indeed, a number of studies show that the acetylation/deacetylation status of APE1 is regulated by SIRT1, thereby modulating its DNA repair capacity (41,81–83). Here, we report that SIRT1 interacts with RECQL4 and deacetylates RECQL4. Our data shows that oxidative stress readily results in increased acetylation of RECQL4. We found that the interaction between RECQL4 and SIRT1 was disrupted at an early stage of oxidative stress (Figure 6B), and that the interaction between RECQL4 and SIRT1 tends to be restored at a late stage of stress response, possibly to return RECQL4 to a hypoacetylated state when interaction between RECQL4 and OGG1 after acute oxidative stress is no longer urgently needed. This mechanism of regulation of RECQL4 by SIRT1 seems to be different from the regulation of APE1, where the association between APE1 and SIRT1 is increased after oxidative stress and SIRT1 promotes the binding of APE1 to X-ray repair cross-complementing protein 1 (XRCC1) after stress (41). Furthermore, consistent with previous reports that OGG1 may also be a potential target of SIRT1 (39,84), we observed that SIRT1 could deacetylate OGG1 in vitro. Additional experiments are needed to determine whether SIRT1 directly regulates 8-oxoG repair under normal conditions. Finally, the observation that SIRT1 interacts with RECQL4 in the absence of oxidative stress, suggests that SIRT1 may also regulate other functions of RECQL4 in the nucleus, such as DNA replication.
RECQL4 possesses a Sld2-like domain at the N-terminus, a highly conserved ATPase and helicase domain in the middle of the protein, and a poorly characterized C-terminus (2). Our results demonstrate that RECQL4 interacts with OGG1 through its N-terminus domain (1−427 aa) without its ATPase and helicase domain (Figure 3B), and the same N-terminus domain (1−427 aa) of RECQL4 alone is able to stimulate the enzymatic activity of OGG1 (Figure 3F). In addition, RECQL4 stimulates the enzymatic activity of OGG1 in the absence of ATP (Figure 3C−E). These results suggest that the ATPase and helicase of RECQL4 may not be required for the stimulation of OGG1 activity. As we know, RECQL4 possesses a relatively weak helicase activity and a strong strand annealing activity (85–87). Thus, it is reasonable to assume that the RECQL4 strand annealing activity may be involved in the stimulation of OGG1 activities, where double-strand DNA is the substrate for OGG1.
A previous study showed that RECQL4 is acetylated by the p300 acetyltransferase on five lysine residues (376, 380, 382, 385 and 386) (44). In this study, we showed that the CBP acetyltransferase was also able to acetylate RECQL4 in cells and in vitro (Figure 4A and B). Mutations of five lysine residues (376, 380, 382, 385 and 386) out of a total of 18 lysine residues identified within the N-terminus domain of RECQL4 significantly reduced the capacity of RECQL4 to become acetylated by CBP in vitro. However, the RECQL4(KQ) and RECQL4 (KR) mutants can still be acetylated by CBP, suggesting that there are other lysine residues on RECQL4 can be acetylated by CBP in addition to at least one of these five lysine residues. Further, we showed that these five lysine residues may be involved in the regulation of the interaction between RECQL4 and OGG1, because RECQL4 (KQ) and RECQL4 (KR) mutants significantly disrupted the interaction between RECQL4 and OGG1. However, determining which lysine residue/residues are responsible for the interaction disruption still remains to be done. Further, it remains to be determined which lysine/lysines in RECQL4 are deacetylated by SIRT1.
Increasing evidence suggests that OGG1-initiated BER of 8-oxoG in the gene promoter region plays a role in regulating gene expression, especially gene promoters containing the potential G-quadruplex (G4) forming sequence (PQS) (88–91). The N-terminus of RECQL4 has a remarkably high affinity for G4 DNA, 60-fold higher than for other DNA structures (92). In this study, we find that the N-terminus of RECQL4 strongly interacts with OGG1. This raises the possibility that OGG1 together with RECQL4 are involved in the regulation of gene expression in those promoters that contains the potential G-quadruplex (G4) forming sequence (PQS). Indeed, a list of RNA Polymerase II subunits such as POLR2A, POLR2B, POLR2C, POLR2E, POLR2H, POLR3D, POLR2A and POLR2G, were identified as RECQL4 interacting proteins (12,13), which suggests a possible role of RECQL4 in transcription. In addition to gene promoters, G4 structures are also enriched in telomeres (93). Recently, a study reported that targeted and chronic 8-oxoG damage at telomeres contributes to telomere shortening and telomere fragility (94). Previously, our group reported that RTS patient cells had fragile telomeric ends and RECQL4 participates in telomere maintenance through interacting with the shelterin proteins (18). Interestingly, SIRT1 also plays a positive role in telomere maintenance (95,96). Collectively, these observations suggest that RECQL4 may cooperate with OGG1 and SIRT1 in promoters and telomeres.
We propose a model whereby RECQL4 promotes OGG1-initiated BER of 8-oxoG (Figure 7). In response to oxidative stress, RECQL4 becomes hyperacetylated, which enhances its interaction with OGG1 to promote 8-oxoG repair in RECQL4 proficient cells. After repair, SIRT1 outcompetes OGG1 for the interaction with RECQL4 and returns it to a hypoacetylated state, probably to retain RECQL4 in the nucleus (44). We speculate that 8-oxoG repair is impaired in RECQL4-deficient cells because of the loss of stimulatory effect of RECQL4 on OGG1.
Figure 7.
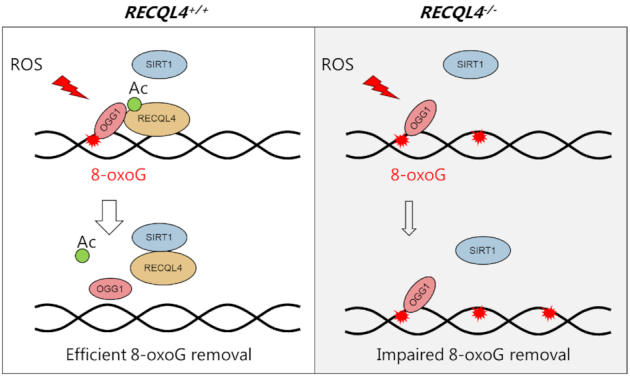
Model for the involvement of RECQL4 in OGG1-mediated removal of 8-oxoG and its regulation by SIRT1 deacetylase. In response to oxidative stress, RECQL4 becomes hyperacetylated, which enhances its interaction with OGG1 to promote 8-oxoG repair. After repair, SIRT1 outcompetes OGG1 from interaction with RECQL4 to return it to a hypoacetylated state. However, 8-oxoG repair is impaired in RECQL4- deficient cells, because of the loss of stimulatory effect of RECQL4 on OGG1, which leads to increased genomic 8-oxoG lesions.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Tomasz Kulikowicz for the help with biochemical experiments. Confocal microscopy experiments were carried out at the Core Facility of Integrated Microscopy (CFIM), University of Copenhagen.
Author contributions: S.D. and V.A.B. designed the study. S.D. and X.H. performed experiments. S.D. wrote the manuscript. X.H., M.A., L.J.R. and V.A.B. helped develop the manuscript. All authors helped to interpret the data and commented on the manuscript.
Contributor Information
Shunlei Duan, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, DK-2200 Copenhagen, Denmark.
Xuerui Han, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, DK-2200 Copenhagen, Denmark.
Mansour Akbari, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, DK-2200 Copenhagen, Denmark.
Deborah L Croteau, Laboratory of Molecular Gerontology, National Institute on Aging, 251 Bayview Blvd, Baltimore, MD, 21224, USA.
Lene Juel Rasmussen, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, DK-2200 Copenhagen, Denmark.
Vilhelm A Bohr, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, DK-2200 Copenhagen, Denmark; Laboratory of Molecular Gerontology, National Institute on Aging, 251 Bayview Blvd, Baltimore, MD, 21224, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NORDEA Foundation, Denmark [02-2013-0220]; EU Joint Programme-Neurodegenerative Disease Research (JPND); Innovation Fund Denmark [5188-00001]; Olav Thon Foundation Norway [531811-710131]; Novo Nordisk Foundation Denmark [NNF17OC0027812]; Intramural Research Program of the NIH, National Institute on Aging [AG000733]; Shunlei Duan received a Travelling Fellowship grant [JCSTF190394] from The Company of Biologists. Funding for open access charge: ICMM, University of Copenhagen.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lu L.C., Jin W.D., Wang L.S.L.. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res. Rev. 2017; 33:30–35. [DOI] [PubMed] [Google Scholar]
- 2. Croteau D.L., Singh D.K., Hoh Ferrarelli L., Lu H., Bohr V.A.. RECQL4 in genomic instability and aging. Trends Genet. 2012; 28:624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brosh R.M., Bohr V.A.. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007; 35:7527–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Croteau D.L., Popuri V., Opresko P.L., Bohr V.A.. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014; 83:519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siitonen H.A., Sotkasiira J., Biervliet M., Benmansour A., Capri Y., Cormier-Daire V., Crandall B., Hannula-Jouppi K., Hennekam R., Herzog D. et al.. The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 2009; 17:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu H., Fang E.F., Sykora P., Kulikowicz T., Zhang Y., Becker K.G., Croteau D.L., Bohr V.A.. Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice. Cell Death. Dis. 2014; 5:e1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sangrithi M.N., Bernal J.A., Madine M., Philpott A., Lee J., Dunphy W.G., Venkitaraman A.R.. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005; 121:887–898. [DOI] [PubMed] [Google Scholar]
- 8. Fang H.B., Niu K.F., Mo D.L., Zhu Y.Q., Tan Q.S., Wei D., Li Y.Y., Chen Z.X., Yang S.C., Balajee A.S. et al.. RecQL4-Aurora B kinase axis is essential for cellular proliferation, cell cycle progression, and mitotic integrity. Oncogenesis. 2018; 7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shin G., Jeong D., Kim H., Im J.S., Lee J.K.. RecQL4 tethering on the pre-replicative complex induces unscheduled origin activation and replication stress in human cells. J. Biol. Chem. 2019; 294:16255–16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Im J.S., Ki S.H., Farina A., Jung S., Hurwitz J., Lee J.K.. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:15628–15632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shamanna R.A., Singh D.K., Lu H.M., Mirey G., Keijzers G., Salles B., Croteau D.L., Bohr V.A.. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis. 2014; 35:2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu H.M., Shamanna R.A., de Freitas J.K., Okur M., Khadka P., Kulikowicz T., Holland P.P., Tian J., Croteau D.L., Davis A.J. et al.. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017; 8:2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu H.M., Shamanna R.A., Keijzers G., Anand R., Rasmussen L.J., Cejka P., Croteau D.L., Bohr V.A.. RECQL4 promotes DNA end resection in repair of DNA Double-Strand breaks. Cell Rep. 2016; 16:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Croteau D.L., Rossi M.L., Canugovi C., Tian J., Sykora P., Ramamoorthy M., Wang Z.M., Singh D.K., Akbari M., Kasiviswanathan R. et al.. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012; 11:456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gupta S., De S., Srivastava V., Hussain M., Kumari J., Muniyappa K., Sengupta S.. RECQL4 and p53 potentiate the activity of polymerase gamma and maintain the integrity of the human mitochondrial genome. Carcinogenesis. 2014; 35:34–45. [DOI] [PubMed] [Google Scholar]
- 16. Kumari J., Hussain M., De S., Chandra S., Modi P., Tikoo S., Singh A., Sagar C., Sepuri N.B., Sengupta S.. Mitochondrial functions of RECQL4 are required for the prevention of aerobic glycolysis-dependent cell invasion. J. Cell Sci. 2016; 129:1312–1318. [DOI] [PubMed] [Google Scholar]
- 17. De S., Kumari J., Mudgal R., Modi P., Gupta S., Futami K., Goto H., Lindor N.M., Furuichi Y., Mohanty D. et al.. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J. Cell Sci. 2012; 125:2509–2522. [DOI] [PubMed] [Google Scholar]
- 18. Ghosh A.K., Rossi M.L., Singh D.K., Dunn C., Ramamoorthy M., Croteau D.L., Liu Y., Bohr V.A.. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol. Chem. 2012; 287:196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J.T., Xu X., Alontaga A.Y., Chen Y., Liu Y.. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014; 7:848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The hallmarks of aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsu G.W., Ober M., Carell T., Beese L.S.. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004; 431:217–221. [DOI] [PubMed] [Google Scholar]
- 23. Shibutani S., Takeshita M., Grollman A.P.. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991; 349:431–434. [DOI] [PubMed] [Google Scholar]
- 24. Krokan H.E., Bjoras M.. Base Excision Repair. Csh. Perspect. Biol. 2013; 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugimura H., Kohno T., Wakai K., Nagura K., Genka K., Igarashi H., Morris B.J., Baba S., Ohno Y., Gao C. et al.. hOGG1 Ser326Cys polymorphism and lung cancer susceptibility. Cancer Epidemiol. Biomarkers Prev. 1999; 8:669–674. [PubMed] [Google Scholar]
- 26. Duan W.X., Hua R.X., Yi W., Shen L.J., Jin Z.X., Zhao Y.H., Yi D.H., Chen W.S., Yu S.Q.. The association between OGG1 Ser326Cys polymorphism and lung cancer Susceptibility: A Meta-Analysis of 27 studies. PLoS One. 2012; 7:e35970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z., Gan L., Nie W., Geng Y.. The OGG1 Ser326Cys polymorphism and the risk of esophageal cancer: a meta-analysis. Genet. Test. Mol. Bioma. 2013; 17:780–785. [DOI] [PubMed] [Google Scholar]
- 28. de Souza-Pinto N.C., Eide L., Hogue B.A., Thybo T., Stevnsner T., Seeberg E., Klungland A., Bohr V.A.. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001; 61:5378–5381. [PubMed] [Google Scholar]
- 29. Xie Y., Yang H., Cunanan C., Okamoto K., Shibata D., Pan J., Barnes D.E., Lindahl T., McIlhatton M., Fishel R. et al.. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004; 64:3096–3102. [DOI] [PubMed] [Google Scholar]
- 30. Haigis M.C., Sinclair D.A.. Mammalian Sirtuins: biological insights and disease relevance. Annu. Rev. Pathol.-Mech. 2010; 5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guarente L. Sirtuins, Aging, and Medicine. N. Engl. J. Med. 2011; 364:2235–2244. [DOI] [PubMed] [Google Scholar]
- 32. Guarente L., Picard F.. Calorie restriction - the SIR2 connection. Cell. 2005; 120:473–482. [DOI] [PubMed] [Google Scholar]
- 33. Longo V.D., Kennedy B.K.. Sirtuins in aging and age-related disease. Cell. 2006; 126:257–268. [DOI] [PubMed] [Google Scholar]
- 34. Vaquero A., Scher M., Lee D.H., Erdjument-Bromage H., Tempst P., Reinberg D.. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell. 2004; 16:93–105. [DOI] [PubMed] [Google Scholar]
- 35. Jeong J., Juhn K., Lee H., Kim S.H., Min B.H., Lee K.M., Cho M.H., Park G.H., Lee K.H.. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007; 39:8–13. [DOI] [PubMed] [Google Scholar]
- 36. Li K., Casta A., Wang R., Lozada E., Fan W., Kane S., Ge Q.Y., Gu W., Orren D., Luo J.Y.. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 2008; 283:7590–7598. [DOI] [PubMed] [Google Scholar]
- 37. Yuan Z., Zhang X., Sengupta N., Lane W.S., Seto E.. SIRT1 regulates the function of the nijmegen breakage syndrome protein. Mol. Cell. 2007; 27:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan W., Luo J.Y.. SIRT1 regulates UV-Induced DNA repair through deacetylating XPA. Mol. Cell. 2010; 39:247–258. [DOI] [PubMed] [Google Scholar]
- 39. Shah A., Gray K., Figg N., Finigan A., Starks L., Bennett M.. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018; 138:1446–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oberdoerffer P., Michan S., McVay M., Mostoslavsky R., Vann J., Park S.K., Hartlerode A., Stegmuller J., Hafner A., Loerch P. et al.. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008; 135:907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamamori T., DeRicco J., Naqvi A., Hoffman T.A., Mattagajasingh I., Kasuno K., Jung S.B., Kim C.S., Irani K.. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010; 38:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Amente S., Di Palo G., Scala G., Castrignano T., Gorini F., Cocozza S., Moresano A., Pucci P., Ma B., Stepanov I. et al.. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019; 47:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rogakou E.P., Boon C., Redon C., Bonner W.M.. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999; 146:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dietschy T., Shevelev I., Pena-Diaz J., Huhn D., Kuenzle S., Mak R., Miah M.F., Hess D., Fey M., Hottiger M.O. et al.. p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. J. Cell Sci. 2009; 122:1258–1267. [DOI] [PubMed] [Google Scholar]
- 45. Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y. et al.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Duan S., Yuan G., Liu X., Ren R., Li J., Zhang W., Wu J., Xu X., Fu L., Li Y. et al.. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015; 6:10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wisnovsky S., Jean S.R., Kelley S.O.. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 2016; 12:567–573. [DOI] [PubMed] [Google Scholar]
- 48. Ohno M., Oka S., Nakabeppu Y.. Quantitative analysis of oxidized guanine, 8-oxoguanine, in mitochondrial DNA by immunofluorescence method. Methods Mol. Biol. 2009; 554:199–212. [DOI] [PubMed] [Google Scholar]
- 49. Furda A., Santos J.H., Meyer J.N., Van Houten B.. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2014; 1105:419–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akbari M., Sykora P., Bohr V.A.. Slow mitochondrial repair of 5′-AMP renders mtDNA susceptible to damage in APTX deficient cells. Sci. Rep. 2015; 5:12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng J., Croteau D.L., Bohr V.A., Akbari M.. Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in Aprataxin-deficient cells. Nucleic Acids Res. 2019; 47:4086–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stuart J.A., Hashiguchi K., Wilson D.M. 3rd, Copeland W.C., Souza-Pinto N.C., Bohr V.A.. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res. 2004; 32:2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stevnsner T., Nyaga S., de Souza-Pinto N.C., van der Horst G.T.J., Gorgels T.G.M.F., Hogue B.A., Thorslund T., Bohr V.A.. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002; 21:8675–8682. [DOI] [PubMed] [Google Scholar]
- 54. Maynard S., de Souza-Pinto N.C., Scheibye-Knudsen M., Bohr V.A.. Mitochondrial base excision repair assays. Methods. 2010; 51:416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schurman S.H., Hedayati M., Wang Z., Singh D.K., Speina E., Zhang Y., Becker K., Macris M., Sung P., Wilson D.M. 3rd et al.. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum. Mol. Genet. 2009; 18:3470–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Werner S.R., Prahalad A.K., Yang J.P., Hock J.M.. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem. Biophys. Res. Commun. 2006; 345:403–409. [DOI] [PubMed] [Google Scholar]
- 57. Jin W., Liu H., Zhang Y., Otta S.K., Plon S.E., Wang L.L.. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum. Genet. 2008; 123:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuno K., Kumano M., Kubota Y., Hashimoto Y., Takisawa H.. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol. Cell. Biol. 2006; 26:4843–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burks L.M., Yin J.H., Plon S.E.. Nuclear import and retention domains in the amino terminus of RECQL4. Gene. 2007; 391:26–38. [DOI] [PubMed] [Google Scholar]
- 60. Kohzaki M., Chiourea M., Versini G., Adachi N., Takeda S., Gagos S., Halazonetis T.D.. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis. 2012; 33:1203–1210. [DOI] [PubMed] [Google Scholar]
- 61. Xu X.H., Rochette P.J., Feyissa E.A., Su T.V., Liu Y.L.. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009; 28:3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thangavel S., Mendoza-Maldonado R., Tissino E., Sidorova J.M., Yin J.H., Wang W.D., Monnat R.J., Falaschi A., Vindigni A.. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol. Cell. Biol. 2010; 30:1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chi Z.F., Nie L.H., Peng Z., Yang Q., Yang K., Tao J.H., Mi Y., Fang X.D., Balajee A.S., Zhao Y.L.. RecQL4 cytoplasmic localization: implications in mitochondrial DNA oxidative damage repair. Int. J. Biochem. Cell B. 2012; 44:1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Im J.S., Park S.Y., Cho W.H., Bae S.H., Hurwitz J., Lee J.K.. RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle. 2015; 14:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thor H., Smith M.T., Hartzell P., Bellomo G., Jewell S.A., Orrenius S.. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes - a study of the implications of oxidative stress in intact-cells. J. Biol. Chem. 1982; 257:2419–2425. [PubMed] [Google Scholar]
- 66. Kaiser S., Sauer F., Kisker C.. The structural and functional characterization of human RecQ4 reveals insights into its helicase mechanism. Nat. Commun. 2017; 8:15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kuznetsov N.A., Koval V.V., Zharkov D.O., Nevinsky G.A., Douglas K.T., Fedorova O.S.. Kinetics of substrate recognition and cleavage by human 8-oxoguanine-DNA glycosylase. Nucleic Acids Res. 2005; 33:3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vidal A.E., Hickson I.D., Boiteux S., Radicella J.P.. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Res. 2001; 29:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Narita T., Weinert B.T., Choudhary C.. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019; 20:156–174. [DOI] [PubMed] [Google Scholar]
- 70. Glozak M.A., Sengupta N., Zhang X.H., Seto E.. Acetylation and deacetylation of non-histone proteins. Gene. 2005; 363:15–23. [DOI] [PubMed] [Google Scholar]
- 71. Dancy B.M., Cole P.A.. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015; 115:2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bhakat K.K., Mokkapati S.K., Boldogh I., Hazra T.K., Mitra S.. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006; 26:1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schiedel M., Robaa D., Rumpf T., Sippl W., Jung M.. The current state of NAD(+)-dependent histone deacetylases (Sirtuins) as novel therapeutic targets. Med. Res. Rev. 2018; 38:147–200. [DOI] [PubMed] [Google Scholar]
- 74. Wang Y.J., He J., Liao M.Y., Hu M.X., Li W.Z., Ouyang H.L., Wang X., Ye T.H., Zhang Y.W., Ouyang L.. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019; 161:48–77. [DOI] [PubMed] [Google Scholar]
- 75. Chalkiadaki A., Guarente L.. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer. 2015; 15:608–624. [DOI] [PubMed] [Google Scholar]
- 76. He H.Q., Wang J.J., Liu T.. UV-Induced RPA1 acetylation promotes nucleotide excision repair. Cell Rep. 2017; 20:2010–2025. [DOI] [PubMed] [Google Scholar]
- 77. Zhao M.M., Geng R., Guo X., Yuan R.S., Zhou X., Zhong Y.Y., Huo Y.F., Zhou M., Shen Q.J., Li Y.G. et al.. PCAF/GCN5-mediated acetylation of RPA1 promotes nucleotide excision repair. Cell Rep. 2017; 20:1997–2009. [DOI] [PubMed] [Google Scholar]
- 78. Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y.X., Tran H., Ross S.E., Mostoslavsky R., Cohen H.Y. et al.. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004; 303:2011–2015. [DOI] [PubMed] [Google Scholar]
- 79. Alcendor R.R., Gao S.M., Zhai P.Y., Zablocki D., Holle E., Yu X.Z., Tian B., Wagner T., Vatner S.F., Sadoshima J.. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007; 100:1512–1521. [DOI] [PubMed] [Google Scholar]
- 80. Braidy N., Guillemin G.J., Mansour H., Chan-Ling T., Poljak A., Grant R.. Age related changes in NAD plus metabolism oxidative stress and sirt1 activity in wistar rats. PLoS One. 2011; 6:e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81. Sengupta S., Mantha A.K., Song H., Roychoudhury S., Nath S., Ray S., Bhakat K.K.. Elevated level of acetylation of APE1 in tumor cells modulates DNA damage repair. Oncotarget. 2016; 7:75197–75209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Roychoudhury S., Nath S., Song H., Hegde M.L., Bellot L.J., Mantha A.K., Sengupta S., Ray S., Natarajan A., Bhakat K.K.. Human Apurinic/Apyrimidinic Endonuclease (APE1) is acetylated at DNA damage sites in chromatin, and acetylation modulates its DNA repair activity. Mol. Cell. Biol. 2017; 37:e00401-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lirussi L., Antoniali G., Vascotto C., D’Ambrosio C., Poletto M., Romanello M., Marasco D., Leone M., Quadrifoglio F., Bhakat K.K. et al.. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell. 2012; 23:4079–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. O’Hagan H.M., Wang W., Sen S., Destefano Shields C., Lee S.S., Zhang Y.W., Clements E.G., Cai Y., Van Neste L., Easwaran H. et al.. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011; 20:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Macris M.A., Krejci L., Bussen W., Shimamoto A., Sung P.. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair (Amst.). 2006; 5:172–180. [DOI] [PubMed] [Google Scholar]
- 86. Xu X.H., Liu Y.L.. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009; 28:568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Suzuki T., Kohno T., Ishimi Y.. DNA helicase activity in purified human RECQL4 protein. J. Biochem. 2009; 146:327–335. [DOI] [PubMed] [Google Scholar]
- 88. Fleming A.M., Ding Y., Burrows C.J.. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cogoi S., Ferino A., Miglietta G., Pedersen E.B., Xodo L.E.. The regulatory G4 motif of the Kirsten ras (KRAS) gene is sensitive to guanine oxidation: implications on transcription. Nucleic Acids Res. 2018; 46:661–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pastukh V., Roberts J.T., Clark D.W., Bardwell G.C., Patel M., Al-Mehdi A.B., Borchert G.M., Gillespie M.N.. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am. J. Physiol.-Lung C. 2015; 309:L1367–L1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wang R.X., Hao W.J., Pan L., Boldogh I., Ba X.Q.. The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 2018; 75:3741–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Keller H., Kiosze K., Sachsenweger J., Haumann S., Ohlenschlager O., Nuutinen T., Syvaoja J.E., Gorlach M., Grosse F., Pospiech H.. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res. 2014; 42:12614–12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hansel-Hertsch R., Di Antonio M., Balasubramanian S.. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 94. Fouquerel E., Barnes R.P., Uttam S., Watkins S.C., Bruchez M.P., Opresko P.L.. Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell. 2019; 75:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Palacios J.A., Herranz D., De Bonis M.L., Velasco S., Serrano M., Blasco M.A.. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 2010; 191:1299–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen J., Zhang B., Wong N., Lo A.W.I., To K.F., Chan A.W.H., Ng M.H.L., Ho C.Y.S., Cheng S.H., Lai P.B.S. et al.. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 2011; 71:4138–4149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.