

Abstract
Accurate, traceable quantification of ribonucleotide or deoxyribonucleotide oligomers is achievable using acid hydrolysis and isotope dilution mass spectrometry (ID-MS). In this work, formic acid hydrolysis is demonstrated to generate stoichiometric release of nucleobases from intact oligonucleotides, which can be then measured by ID-MS, facilitating true and precise absolute quantification of RNA, short linearized DNA, or genomic DNA. Surrogate nucleobases are quantified with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) workflow, using multiple reaction monitoring (MRM). Nucleobases were chromatographically resolved using a novel cation exchange separation, incorporating a pH gradient. Trueness of this quantitative assay is estimated from agreement among the surrogate nucleobases, and by comparison to concentrations provided for commercial materials, or Standard Reference Materials (SRMs) from the National Institute of Standards and Technology (NIST). Comparable concentration estimates using NanoDrop Spectrophotometry or established from droplet-digital PCR (ddPCR) techniques agree well to the results. Acid hydrolysis-ID-LC-MS/MS provides excellent quantitative selectivity and accuracy while enabling traceability to mass unit. Additionally, this approach can be uniquely useful for quantifying modified nucleobases, or mixtures.
Graphical Abstract
Introduction
Oligonucleotide quantification supports a diverse list of clinical and commercial fields, including microbial analysis, food safety (GMOs), medicine, biotherapeutics development, forensics, and more. Accurate and traceable quantitative assays to underpin these disciplines will be essential as those communities moves towards standardized measurement systems1–4.
Conventionally, UV and fluorescence spectroscopy techniques have been broadly applied towards quantification of oligonucleotides2, 5, 6. But although spectroscopic approaches may be fast, inexpensive, and simple, these relative quantification methods lack specificity, traceability, and calibration potentially resulting in significant quantitative bias. Still, fluorescence approaches can be highly sensitive, and UV can be fit-for-purpose when absolute accuracy is not essential. PCR-based techniques such as droplet digital polymerase chain reaction (ddPCR) can be robust, precise, and highly sensitive means toward oligonucleotide quantification, and have broad application in this area. Caution must be exercised however, as ddPCR assumes stoichiometric amplification of nucleic acid targets, which is not always true. Incomplete amplification is especially prevalent in reverse transcription assays (RT-dPCR) for RNA measurements7, 8. Furthermore, ddPCR quantitative trueness is directly correlated to droplet size, which has been demonstrated to vary among different master stocks, between instrumentation, and over time6, 9, 10; and ddPCR faces analytical limitations including for very large, truncated, high G/C, or non-linearized sequences.
Isotope dilution (ID) techniques are considered the gold-standard for biomolecule quantification. ID employs pre-spiked stable-isotope labeled (SIL) analogs (i.e., 13C or 15N) to account for quantitative biases assumed during sample preparation or subsequent detection by mass spectrometry (MS). In order to overcome the analytical limitations of detecting large oligos by MS, or the costs/challenges associated with synthesizing SIL oligos, bottom-up approaches have been previously described where RNA or DNA could be enzymatically digested into constituent nucleosides11–19 or nucleotides3, 20, 21 prior to ID-MS analysis with nucleosides or nucleotides serving as quantitative surrogates of oligo concentration. Previously, this was achieved by other groups using endonucleases (RNAse A, nuclease P1, S1, DNAse I…) or exonuclease cocktails (phosphodiesterase I, alkaline phosphatase…). Unfortunately, enzymatic approaches have limitations – complete digestion may be inhibited by oligomeric size, by higher order structure, enzyme specificity, or covalent modifications22. In this manuscript, chemical hydrolysis rather than enzymatic cleavage is proposed as an alternative method to exploit bottom-up quantification of oligonucleotides.
Chemical approaches for RNA or DNA hydrolysis have been around for decades23,24, and have evolved continuously with improvements in accuracy and specificity. Predominantly though, acid hydrolysis has been applied for atypical or modified nucleobase detection (DNA damage or carcinogenic adduction25–32), rather than with the intent to quantify the intact oligonucleotide. Recently chemical hydrolysis was demonstrated using double-stranded genomic DNA from bacteriophage lambda (≈48.5 kbp) showing that DNA can be accurately quantified using just such an approach33. In this work, we demonstrate a modified chemical hydrolysis workflow for any sized DNA using formic acid hydrolysis and nucleobase calibrators applied to small dsDNA (7.9 kbp linearized plasmid containing 5.2 kbp BK virus), and for the first time, to human genomic DNA (3.2×109 base-pairs). But most importantly, we demonstrate for the first time that this approach can be applied to ribonucleotide oligomers (RNA).
Purine and pyrimidine bases are known to be disproportionately or incompletely hydrolyzed under acidic conditions24. A previous report34 estimated nucleic acid concentrations in cellular material using 6 mol/L hydrochloric acid (HCl) hydrolysis with diode array detection (DAD), noting significant deamination or other losses for all nucleobases in 6 mol/L HCl hydrolysis, while separately hydrolyzing purines and pyrimidines. Another study35 using dilute HCl (0.2 mol/L) observed complete purine hydrolysis from DNA, but with diminutive release of pyrimidines. Quantitative analysis of purine base adducts in genomic DNA has been reported previously using mild acidic treatment for short hydrolysis times26, 36. Most recently, the National Metrology Institute of Japan (NMIJ) demonstrated formic acid’s utility for hydrolyzing purines and pyrimidines from intermediate-sized bacterial dsDNA33. Here, we expand on that effort by demonstrating hydrolysis at extreme sizes (for human genomic dsDNA) and for quantification of ribo-oligos (RNA). Neat formic acid (≈26 mol/L) can be used to hydrolyze samples in either the gas phase or liquid phase. Purine and pyrimidine stability is demonstrated for the nucleobases A, G, T, and U under the optimized hydrolysis conditions, while some deamination of cytosine to uracil was observed ≈ 3 % (m/m), as reported previously37. Here, we demonstrate that acid hydrolysis is well-suited to RNA hydrolysis and DNA hydrolysis, and in conjunction with ID-MS can achieve traceable, accurate absolute quantification.
A ‘double isotope dilution’ scheme is designed for this work, involving the use of SIL nucleobase analogs (13C and/or 15N-adenine, -cytosine, -guanine, -thymine, -uracil) pre-spiked as internal standards into samples and nucleobase calibrators, enabling true and precise quantification, traceable through reference standards to the US Pharmacopeia (USP), European Pharmacopeia (EP), and British Pharmacopeia (BP) primary standards. (Certified Reference Materials (CRMs) necessary to establish SI-traceability do not yet exist for pure nucleobases or nucleotides.) This unique approach establishes a process for using a single set of SIL nucleobases along with a single set of well-characterized nucleobase standards to be broadly applied to calibrate quantitative measurement of multiple, unrelated RNA or DNA oligomers.
Experimental
Materials
Nucleobase calibrators were purchased from Millipore Sigma. Calibrators are categorized as “Pharmaceutical Secondary Standard Certified Reference Materials” with traceability to the USP, EP (PhEur) and BP primary standards. No further chemical purity or water analysis was performed in-house. [Adenine (PHR1383, traceable to USP 1012101, PhEur A0230000); Cytosine (PHR1350, traceable to USP 1162148); Guanine (PHR1243, traceable to BP 879 and USP 1302156); Thymine (PHR1345, traceable to USP 1754532); Uracil (PHR1581, traceable to USP 1705753 and PhEur Y0000764); 5-methylcytosine hydrochloride (Sigma M6751, ≥99% pure)].
Stable isotope-labeled nucleobase standards were purchased through Cambridge Isotope Laboratories, Inc. (Andover, MA) for use as internal standards, as described below (Table S1):
Deoxynucleoside monophosphate standards (dNMPs) were purchased from commercial sources.
2’-deoxyadenosine 5’-monophosphate disodium salt hydrate (MP Biomedicals 02150795; MW 331.225)
2’-deoxycytidine 5’-monophosphate sodium salt (Sigma D7625; MW 351.16)
2’-deoxyguanosine 5’-monophosphate disodium salt hydrate (Sigma 852228; MW 391.18)
thymidine 5’-monophosphate disodium salt hydrate (Sigma T7004; MW 366.17)
2’-deoxyuridine 5’-monophosphate disodium salt (Sigma D3876; MW 352.15)
An RNA standard was acquired from Ambion (Invitrogen) through ThermoFisher (AM7155, RNA Control 250). DNA standards were acquired in-house. BK virus linearized plasmid DNA was acquired from a stock solution used in the preparation of NIST SRM 2365. Human genomic DNA was from NIST SRM 2372a (Component A)38. 5-methylcytosine powder was purchased through Sigma-Aldrich (M6751, 5mC•HCl). Formic acid was LC-MS grade from Honeywell (56302).
Acid hydrolysis
RNA or DNA samples were gravimetrically added to 400 μL glass flat-bottomed autosampler inserts (Agilent, 5181–3377), and dried to dryness in a speed-vac without heat. A solution of a mixture of SIL-nucleobases was pre-spiked into samples and calibrants and used as an internal standard to normalize MS signal. Glass inserts were placed into acid-resistant, temperature-safe Teflon vessels within a steel compression pressure-bomb. Approximately two (2) mLs of neat formic acid was pipetted into the vessels, external to the glass inserts. Vessels were tightened to ensure vapor pressure retention, and then placed into an oven at 140 °C for gas-phase hydrolysis. Hydrolysis was performed for 24–48 hours depending on the sample type. After hydrolysis, glass inserts were dried to dryness in a speed-vac and samples were reconstituted in water for subsequent LC-MS analysis.
LC-MS/MS analysis
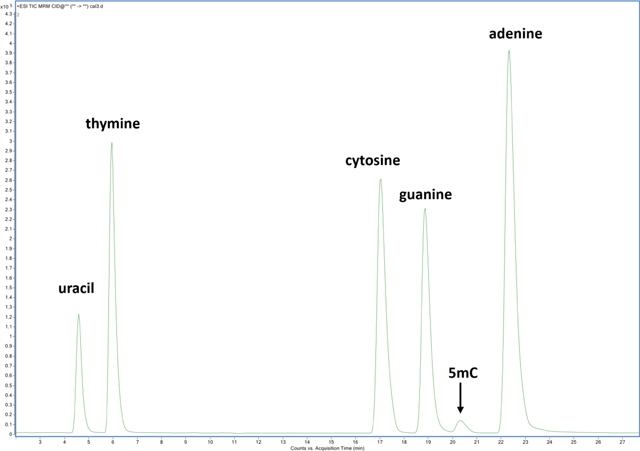
For the chromatographic separation of nucleobases, an ion-exchange mechanism with a decreasing pH gradient on a mixed-mode column enabled complete resolution of nucleobases (Figure 1). Gradient elution began at 99 % mobile phase A (0.5 mL/L TFA) and increased linearly with additional mobile phase B (4.5 mL/L TFA in 0.2 L/L aqueous acetonitrile (ACN)) to 10 % over 10 min., and to 40 % over the next 20 min. followed by a column wash at 95 % B and re-equilibration. The column was regularly washed with 95 % ACN in-between batches. A constant mobile phase flow rate of 200 μL/min was used. A mixed-mode ion exchange / reversed-phase Primesep 100 column (SIELC Technologies, Wheeling, IL), 2.1 × 250 mm, 3 μm particles, was maintained at 50 °C in a thermostatted column compartment. Samples were maintained at 5 °C in the autosampler. Fresh mobile phases were prepared weekly.
Figure 1:

Liquid chromatographic separation of nucleobase standards using a mixed-mode ion exchange / reversed-phase stationary phase with a decreasing pH gradient. Fragmentation transitions were detected using multiple-reaction monitoring on a triple quadrupole mass spectrometer.
For quantification, a multiple-reaction monitoring (MRM) assay was developed targeting two specific fragmentation transitions of each nucleobase and two fragmentation transitions of a stable-isotope-labeled analog of each nucleobase (Figure S1). Precursor-to-product ion fragmentation transitions and ionization conditions were optimized using purified standards of nucleobases. An Agilent 1290 Infinity liquid chromatography system (Agilent Technologies, Santa Clara, CA) was used in-line with an Agilent 6460A triple quadrupole (QQQ) mass spectrometer. The LC separation was optimized for complete chromatographic resolution of all nucleobases in order to ensure specificity of the quantitative assay. Electrospray ionization (ESI) was achieved in positive ion polarity using one continuous MRM scan segment. All analyses were performed with the following Agilent 6460A MS source parameters: source gas temperature = 300 °C, source gas flow = 13 L/min, nebulizer = 345 kPa (50 psi), sheath gas temperature = 250 °C, sheath gas flow = 12 L/min, capillary voltage = 3500 V, nozzle voltage = 1500 V. Each MRM fragmentation scan was acquired in unit resolution for MS1 and MS2, over a 100 ms dwell time, and with a cell accelerator voltage of 7 V. All other instrument parameters for MRM scans used in the measurement of each set of labeled and unlabeled nucleobases are listed in Table S2.
Agilent MassHunter Workstation software (version B.09.01) was used for peak selection and integration. Peak retention times and integrated peak areas were automatically determined by MassHunter. All peak integrations were visually inspected, and in some cases, manual integration was necessary. Peak area ratios were exported into Microsoft Excel for quantitative analysis. Unlabeled/labeled integrated peak area ratios were calculated from calibrant data and plotted against gravimetric mass ratios into calibration curves. For quantitative analysis of sample, molar mass ratios were extrapolated from the calibration curves according to the measured peak area ratios. From this data, nucleobase concentrations were calculated and converted to intact oligonucleotide concentration.
For high-resolution MS analysis of modified nucleobases (5mC) on an Orbitrap Elite MS system (Thermo), a data-dependent analysis with dynamic exclusion was used with FTMS resolution of 30,000 for MS1 acquisition in positive polarity within the scan range of m/z 50.0–300.0. Standard source and fragmentation parameters were used with a CID normalized collision energy of 35, and MS2 data acquisition in the ion-trap.
Isotope dilution and sample preparation
A double exact-matching isotope dilution (ID) workflow was designed for the quantitative assay. This design requires the use of well-characterized SIL internal standard nucleobases pre-spiked into samples and external calibrants to normalize for instrument and sample preparation variability39. Calibration is achieved with a five-point bracketing calibration curve using traceable, pure standards of nucleobases with known concentrations. For hydrolysis assays of intact RNA and DNA, six replicates were prepared gravimetrically and were pre-spiked with a mixture of SIL internal standard nucleobases prior to hydrolysis. Samples were incubated at 140 °C for 24 h −48 h using gas phase formic acid hydrolysis, dried, and reconstituted in mobile phase A prior to LC-MS/MS analysis. Calibrants consisted of nucleobase standards in a mixture with SIL nucleobases, prepared at unlabeled/labeled mass ratios of approximately 0.5, 0.75, 1.0, 1.25, and 1.5 (m/m %), where the midpoint of the calibration range is estimated as the expected concentration of nucleobase content of the RNA or DNA sample. SIL internal standard concentrations varied among RNA or DNA samples and were targeted at a ≈1:1 molar equivalence to the expected sample concentration, while remaining constant among calibrators for a given sample. All calibration plots were reported with coefficients of determination (R2) for regression lines > 0.99 for both MRM transitions of all nucleobases.
Results and Discussion
Chromatography
The most commonly used liquid chromatographic approach for nucleic acids is reversed-phase separation40, 41. Although C18 reversed-phase is both robust and MS-friendly, nucleobase retention on this phase is relatively weak. Here, we demonstrate better retention and complete resolution of nucleobases using a mixed-mode column consisting of a C18 stationary phase embedded with acidic ion-pairing groups. This phase (SiELC, Primesep 100) improves retention of basic compounds (nucleobases) by cation-exchange mechanisms while using gradient elution of decreasing pH and increasing organic mobile phase composition. Figure 2 provides representative MRM extracted ion chromatograms of canonical nucleobase standards on a) C18 analytical column using common mobile phases (A:B = acidified H2O: acidified ACN), b) on a porous-graphitized carbon column (HyperCarb, Thermo) using the same mobile phases, and c) using the mixed-mode Primesep column as described in Experimental. Retention under several gradient conditions on the C18 column (shown in Fig. 2) proved inadequate for complete peak resolution resulting in less precise quantification. However, it should be noted that not all C18 columns are alike, and many chromatographic options are available. PGC is a unique phase with distinct separating mechanisms for polar or non-polar analytes. Under MS-friendly conditions PGC retains nucleobases well with near baseline resolution, however, purines tend to tail under these LC conditions leading to integration bias. Ion exchange/ reversed-phase separations using the Primesep column were observed with moderate retention using MS-amenable mobile phases and with baseline resolution of A, C, T, G, and U (Figure 1). The separation was found to be robust and repeatable. In addition to facilitating chromatographic separation of canonical nucleobases, this column was demonstrated capable of retaining other modified nucleobases including 5-methylcytosine (5mC).
Figure 2:

MRM EICs for nucleobase standards using a) C18 column with reversed-phase chromatography, b) PGC column, or c) mixed-mode cation-exchange with C18 column using a pH and organic gradient elution.
Acid hydrolysis optimization
Time-course assays of calibration standards were designed to test nucleobase stability under hydrolysis conditions (Figure S2). Hydrolysis was ultimately achieved in this work using formic acid, however, initially three acids were tested: 1) trifluoroacetic acid (TFA; CF3CO2H) rapidly degraded nucleobases in less than one hour at each temperature tested (60 °C, 120 °C, and 140 °C); 2) hydrochloric acid (HCl) was tested at 8 mol/L and ≈2 mol/L (120 °C and 140 °C) with only slight differences observed – nucleobase pyrimidines degraded rapidly (less than one hour), while purines were more tolerant but degraded partially or entirely within 2–4 hours. Hydrolysis using ≈0.2 mol/L HCl yielded incomplete pyrimidine release and significant deamination of cytosine to uracil at longer timepoints; 3) hydrolysis using neat formic acid (HCOOH) was demonstrated to preserve purines over the full time-course at 140 °C. For pyrimidines, both uracil and thymine were shown stable when subjected to these hydrolysis conditions, while cytosine was observed to be slightly deaminated into uracil at longer time-points. Semi-quantitative assays estimated cytosine deamination rates of ≈3 % (relative peak area) at 24 hours of hydrolysis (Figure 3). A 5-methylcytosine (5mC) standard was tested using only formic acid and although some deamination into thymine was expected, little to no thymine degradation product was detectable.
Figure 3:

Cytosine stability timecourse under hydrolysis conditions (gas-phase formic acid, 140°C). Uracil is quantified by LC-MS/MS (MRM) analysis following the deamination of cytosine into uracil.
Time-course assays were performed on dNMP standards to determine a target hydrolysis time which ensures reaction completion. dNMPs were hydrolyzed with formic acid, as above for nucleobase standards, with liberated nucleobases being detected by LC-MS/MS. Saturation of signal was monitored for reaction completion (Figure S3). Several temperatures were tested over the 24 h time-course. It was observed that at the highest temperature tested (160 °C), deamination of cytosine was accelerated. At lower temperatures (60 °C and 120 °C), complete hydrolysis of pyrimidine-based dNMPs was not guaranteed (dUMP and dTMP specifically). The optimal temperature was determined to be 140 °C, with complete hydrolysis being observed for all dNMPs by ≈24 h. It was observed that for the RNA material full release of the pyrimidines (T and U) was not complete until >24 h, as detailed previously34. A hydrolysis time of 24 h (or 48 h) was possible for analysis of the intact DNA samples described in this work, and 48 h of hydrolysis was used for RNA samples, to ensure complete hydrolysis. For any given target oligonucleotide, whether DNA or RNA, it is important to test for hydrolysis completion under the given conditions and ensure that the optimized conditions are appropriate.
Absolute Quantification of RNA
A single-strand RNA control solution used routinely for verifying the accuracy of NanoDrop Spectrophotometer measurements was acquired from commercial sources (Invitrogen, part AM7155). The RNA Control 250 material is provided as an ultra-pure RNA transcript of a precisely defined sequence, molecular weight, and concentration (255 ± 13 ng/μL), as determined by the manufacturer using absorbance @ 260nm. Individual nucleobase concentrations ([A], [C], [G], [U]) were inferred from the provided RNA sequence and its stated concentration, assuming stoichiometric release following hydrolysis, and expressed as nmol/L (nM). The manufacturer-provided concentrations of A, C, G, and U, respectively, are 176.0 ± 8.8, 224.3 ± 11, 194.3 ± 9.7, and 185.0 ± 9.3 nmol/L. RNA samples were thawed only once from −20 °C, and replicates were gravimetrically prepared with SIL-nucleobase internal standard. Approximately three (3) μLs of the RNA 250 control solution was hydrolyzed per replicate, providing abundant signal for robust quantification (typical MRM peak height 105–106). Hydrolysis was completed after 48 hours at 140 °C using gas-phase hydrolysis with neat formic acid. ID-MS analysis was performed on three (3) separate days using six sample replicates per day and freshly prepared calibration curves.
The average (μ) results from ID-LC-MS/MS analyses of the RNA control 250 solution are provided in Table 1, reporting standard deviation (σ) and relative standard deviation (RSD) of each nucleobase (throughout this text, statistical analysis is represented as μ ± σ, with RSD expressed as appropriate). Expected values and measured values of nucleobase concentrations agree within ±1σ (Figure S4). The measurement precision for nucleobases ranged from 3.4 % – 4.5 % RSD. Both purines trended slightly high but fell within uncertainty estimates. Purine nucleobases were observed to be completely released from the intact oligomer quickly under the hydrolysis conditions described here. Both adenine and guanine reached steady-state within one hour of hydrolysis (Figure 4). Due to practical limitations on rapid heating and cooling of the steel hydrolysis bomb, it is difficult to make estimates on shorter time-points. Regardless, it is possible to quantify the purines at quite early timepoints using this ID-MS approach as compared with pyrimidine nucleobases. Pyrimidine bases were released slowly under formic acid hydrolysis, requiring greater than 24 h to reach steady-state for this intact RNA. Fortuitously, due to the robust stability of nucleobases under these hydrolysis conditions, it is possible to measure the purines and pyrimidines simultaneously using a single assay. Pre-spiked SIL-nucleobase internal standards further guarantee any potential quantitative biases associated with losses or degradation during the sample preparation are accounted for.
Table 1:
Quantitative results for RNA 250 control solution from Ambion (Invitrogen)
| adenine | cytosine | guanine | uracil | |
|---|---|---|---|---|
| [average], nM | 189.4 | 225.5 | 206.6 | 183.3 |
| stdev, nM | 7.9 | 10.2 | 7 | 7.3 |
| % CV | 4.2 | 4.5 | 3.4 | 4.0 |
| [expected], nM | 176.0 | 224.3 | 194.3 | 185.0 |
| stdev, nM | 8.8 | 11 | 9.7 | 9.3 |
| % of theoretical | 107.6 | 100.5 | 106.3 | 99.1 |
Figure 4:

Purines and pyrimidines release from RNA – hydrolysis timecourse using gas-phase formic acid at 140°C. Purines (adenine and guanine) are rapidly released from RNA (or DNA) and can be quantified within 2 hours of hydrolysis, while pyrimidines (cytosine and uracil) do not reach complete hydrolysis until more than 24 hours after hydrolysis begins.
It is useful to provide an estimated value or a ‘predicted’ value for the concentration of the intact RNA sequence based on the concentration of detected nucleobases. Although this work cannot establish the origin of the detected nucleobases after they are liberated from the oligomer, it is possible to provide a predicted concentration of RNA with the reasonable assumption that all nucleobases originated from an intact, homogenous, pure oligomer of known structure. (Techniques like ddPCR or UV/Vis make similar assumptions by amplifying and detecting oligomer fragments, or detecting excited light from non-specific electronic transitions.) Predicted RNA concentration was calculated using the mean of the percent deviation of each nucleobase from its expected nucleobase concentration and multiplying this value against the RNA concentration provided by the manufacturer. This assumes that the mean measurement deviation of the nucleobases from theoretical is proportional to the measurement deviation of the intact RNA oligomer from the theoretical value. The predicted value from ID-MS analysis of the RNA Control 250 material of 263.6 ng/μL agrees statistically to the manufacturer-provided value of 255 ± 13 ng/μL.
Absolute Quantification of DNA
NIST SRM 2365 (BK virus DNA Quantitative Standard42) is a 7934 bp, linearized plasmid DNA material containing BK virus DNA that is provided within a solution of yeast tRNA, for material stability. BK virus DNA copy number was certified at NIST for SRM 2365 using a robust ddPCR assay. In this work, a concentrated (5.58×109 copies/μL ± 1.2×108) tRNA-free stock solution of SRM 2365 was used, as acid hydrolysis does not discriminate between nucleobases originating from DNA or from the tRNA. The stock solution used to prepare NIST SRM 2365 was subjected to acid hydrolysis using formic acid at 140 °C. Time-course analysis of the BK plasmid demonstrates full hydrolysis of its nucleobases by 24 h (Figure S5). As described above for the RNA material, the BK concentration can be expressed in terms of nucleobase concentration and for calculated in nmol/L: A, C, G, and T concentrations are 41.3 ± 0.91, 32.0 ± 0.70, 32.0 ± 0.70, and 41.8 ± 0.92 nmol/L, respectively. The BK DNA is stable at 4 °C, and replicate samples were gravimetrically prepared with mixed SIL-nucleobase internal standard. Approximately fifteen (15) μLs of the BK solution was lyophilized and hydrolyzed per replicate. Hydrolysis was completed after 24 h at 140 °C using gas-phase hydrolysis with neat formic acid. ID-MS analysis was performed on three (3) separate days using six sample replicates per day and freshly prepared calibration curves.
The average (μ) results from ID-LC-MS/MS analyses of the BK solution are provided in Table 2, with standard deviation (σ) and relative standard deviation (RSD) of each nucleobase specified. Expected values and measured values of nucleobase concentrations agree within ±1σ (Figure S6). The total precision of the assay was excellent, with RSDs for nucleobases A, C, G, and T equal to 2.8, 3.1, 3.9, and 3.0 %, respectively. As mentioned earlier, complete hydrolysis of purines was observed within one (1) hour, significantly faster than hydrolysis of pyrimidines. In addition to the canonical nucleobases expected from DNA, a non-negligible concentration of uracil was measured in this material. Although the precision of the uracil quantification was poor (≈17% RSD), the mean of the measurements suggested at least some contribution from sources other than deamination of cytosine to uracil. Based on the hydrolysis stability assays for a cytosine standard material (Figure 3), deamination of cytosine should contribute to roughly 1.1 nmol/L of uracil following hydrolysis of the BK material. RNA contamination of the stock solution is one potential source of the unexpected uracil signal. Another possible contribution is from a measurement calibration system bias at exceedingly low analyte concentrations.
Table 2:
Quantitative results from ID-LC-MS/MS analysis of BK virus solution
| Adenine | Cytosine | Guanine | Thymine | Uracil | |
|---|---|---|---|---|---|
| [average], nM | 40.5 | 31.2 | 30.9 | 40.1 | 2.8 |
| stdev, nM | 1.1 | 1.0 | 1.2 | 1.2 | 0.5 |
| %CV | 2.8 | 3.1 | 3.9 | 3.0 | 16.7 |
| [expected], nM | 41.3 | 32.0 | 32.0 | 41.8 | - |
| stdev, nM | 0.91 | 0.70 | 0.70 | 0.92 | - |
| % of expected | 98.2 | 97.5 | 96.7 | 96.0 | - |
A predicted value for the intact DNA oligomer can be estimated in a similar fashion to the calculation above of RNA concentration from its constituent nucleobases. For this ID-MS assay, the predicted value of the BK virus DNA oligomer of 5.42×109 ± 1.7×108 copies/μL is statistically equivalent to the value predicted from ddPCR of 5.58×109 ± 1.2×108 copies/μL.
Quantification of human genomic DNA
The acid hydrolysis procedure was next applied to a human genomic DNA material – NIST SRM 2372a (Component A) – for a proof-of-principle experiment aiming to demonstrate that even the largest biological genomic materials are amenable to complete hydrolysis using formic acid and ID-MS quantification. This DNA material was prepared from human buffy coat genomic DNA from a single male donor (the buffy coat contains leukocytes and platelets). The certified value for DNA copy number was based on ddPCR quantification assays of ten targets on eight (8) chromosomes. The certified DNA mass concentration of 49.8 ± 5.0 ng/μL is calculated based on a reference human haploid genome equivalent and the mean MW of sodium salts of nucleotide monomers. In order to calculate nucleobase concentrations from the certified values provided in the NIST CoA it is necessary to make the assumption that the AT:GC for human genomic DNA ≈ 60:40 and therefore the average MW of a nucleoside in genomic DNA is 330 g/mol43. A second assumption was made that the density of the buffy coat sample ≈ 1 g/mL.
SRM 2373a(A) was subjected to 24 hours of formic acid hydrolysis as described previously. The analysis was performed in triplicate subjecting ≈15 μL of the SRM material per replicate to hydrolysis. Calibration was achieved using a similar scheme as described above. The absolute concentrations for nucleobases in SRM 2372a(A) were determined for A, C, G, and T as 43.3, 30.8, 32.9, and 43.3 nmol/L, respectively. RSDs for nucleobase measurements ranged from 2.8 – 8.4 % with standard deviations for A, C, T, and G equal to 1.80, 0.848, 1.98, and 3.62 nmol/L, respectively. It follows that the product of the average MW of nucleosides in genomic DNA (330 g/mol) with the summation of the molar concentrations (from ID-MS) of A, C, G, and T (≈150.2 nmol/L) is approximately equal to 49.6 ng/μL of DNA in SRM 2372a(A). This value agrees well to the certified DNA mass concentration on the CoA (49.8 ± 5.0 ng/μL) determined by ddPCR, and suggests that acid hydrolysis with ID-MS is well-suited to quantitatively measure complete, human genomic DNA. Other considerations beyond the scope of this manuscript that might affect the ID-MS work include the potential contribution of nucleobase signal from mitochondrial DNA in the buffy coat, which is expected to be negligible in this case when assuming the mass of mtDNA < 2 % (m/m) that of nDNA. Also, the detection of uracil from deamination of cytosine, from RNA, or mtRNA was not addressed.
(It should be clearly noted that the work presented here did not in any way contribute to the certification of NIST SRM 2372a.)
Detection of covalently-modified nucleobases
Formic acid hydrolysis with LC-MS/MS analysis is capable of detecting or quantifying modified nucleobases – among others, 5-methyl cytosine (5mC). During LC-MS/MS analysis of the BK sample described above, relative quantification was performed for the modified nucleobase 5mC using LC-MRM-MS, and later we confirmed the identity of 5mC by high-resolution LC-MS/MS analysis on an Orbitrap MS system. The 5mC standard was used to optimize chromatographic retention time, and the ionization and fragmentation conditions for LC-MS/MS assays. However, as there are no CRMs or primary quantitative standards available for 5mC analysis, this work demonstrates only a semi-quantification of 5mC in the BK solution. Figure S7 shows an extracted ion chromatogram (XIC) from the MRM analysis of a calibration solution and for a BK plasmid sample, as performed on a QQQ instrument. 5mC is chromatographically resolved from other nucleobases, enabling use of low-resolution techniques such as MRM analysis, even considering a mass difference (Δ) from thymine of only −1 Da. 5mC was monitored during method development using two MRM transitions (126.1→ 82.4, 110.3), however, due to its low biological abundance, only the 82.4 m/z fragment ion was observed with good S/N in samples. A calibration curve was generated without SIL-internal standard for 5mC. Semi-quantification indicates ≈92 pmol/L of 5mC in the BK virus sample. This is equivalent to ≈0.3% relative abundance to cytosine – a value that tends towards the low end of agreement with other semi-quantitative work44. To verify the presence of 5mC in the BK sample, high-resolution LC-MS analysis was performed using an Orbitrap MS and fragmentation data acquired in the ion trap. Figure S8 provides XICs and MS2 spectra of 5mC comparing standards to the BK sample.
Analytical approach
The results of this work establish a robust and accurate, mass-traceable method for multiplexed nucleic acid measurement, suitable for absolute quantification of homogeneous RNA or DNA samples. The approach is broadly applicable to many classes of nucleic acid polymers, whether single or double stranded or consisting of ribo- or deoxyribo-nucleic acids, and this approach is demonstrated to be responsive to covalently-modified (stable) nucleobases. Measurement precision for this acid hydrolysis with ID-MS assay typically ranges between 2–5 % RSD for quantification of nucleobase concentration – equal to or better than many other measurement techniques. The principal benefit of using an MS-based approach for oligonucleotide quantification as compared to prevailing techniques is the capability to apply an isotope dilution (ID) framework. ID techniques make it feasible to use external and internal calibration of the measurement assay, simultaneously linking the quantitative traceability chain to a higher-order reference standard – a Standard or Certified Reference Material – while also accounting for experimental biases associated with sample preparation or chromatographic or mass spectrometric variability. Conventional techniques such as UV absorbance or fluorescence detection lack specificity and traceability and can be susceptible to interferences; copy number measurements by ddPCR require an accurate determination of droplet size to achieve absolute quantification and can measure only accessible, intact targets – while all three approaches require nucleic acid oligomer standards, or unique primers, that exclusively match only the sample of interest. In this work, calibration solutions and SIL-internal standards can be developed from nucleobase monomers. The unvarying result of hydrolyzing any RNA or DNA sequence is the same subset of five canonical nucleobases (ignoring covalently-modified nucleobases detectable at considerably lower levels). This congruence means that any pure RNA or DNA sample can be quantified by ID-MS using the same set of calibrators where all nucleobases are useful to determine a ‘consensus’ quantity of the intact oligomer. Alternatively, is it possible to differentiate DNA from RNA in a mixture through quantification of unique surrogate bases – cytosine or uracil, respectively. In this approach, the deamination rate will necessarily need to be considered for mixtures where [DNA] >> [RNA].
There are clear limitations to this technology, as well. All nucleic acid-containing compounds contribute to the signal, so it is not possible to distinguish between, say, adenine from different sources. However, it is conceivable to differentiate a mixture of DNA and RNA based on differences in thymine or uracil signal. The other major limitation is analytical sensitivity when compared to amplification-based approaches like PCR, or fluorescence detection which might be >100 fold more sensitive than mass spectrometry. Nevertheless, acid hydrolysis with mass spectrometry detection should find its niche, being exactly well-suited to provide traceable quantification necessary for reference material development, and for applications such as “total” nucleobase quantification of mixtures, for quality control of droplet size for ddPCR, or for relative quantification or optimization of cell-based sample preparation procedures.
Supplementary Material
Acknowledgements:
The authors would like to thank Dr. Megan Cleveland (NIST) for providing stock solutions of NIST SRM 2365, and Dr. David Duewer and Margaret Kline (NIST) for providing aliquots of NIST SRM 2372a(A).
Footnotes
Disclaimer:
Certain commercial equipment, instruments, and materials are identified in this paper to specify the experimental procedures and analytical methods adequately. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment are necessarily the best available for the purpose.
Supporting Information Available:
Figures and tables are provided in Supporting Information: MRM mass chromatograms of standard nucleobases; quantitative results from hydrolysis of DNA and RNA samples; nucleobase stability timecourses; dNMP stability timecourses; mass spectrometry analysis of 5mC; hydrolysis timecourse of DNA samples; nucleobase internal standards; MRM fragmentation transitions for MS analysis of nucleobases.
References
- 1.Corbisier P; Broothaerts W; Gioria S; Schimmel H; Burns M; Baoutina A; Emslie KR; Furui S; Kurosawa Y; Holden MJ; Kim HH; Lee YM; Kawaharasaki M; Sin D; Wang J, Toward metrological traceability for DNA fragment ratios in GM quantification. 1. Effect of DNA extraction methods on the quantitative determination of Bt176 corn by real-time PCR. Journal of agricultural and food chemistry 2007, 55 (9), 3249–57. [DOI] [PubMed] [Google Scholar]
- 2.Bhat S; Curach N; Mostyn T; Bains GS; Griffiths KR; Emslie KR, Comparison of methods for accurate quantification of DNA mass concentration with traceability to the international system of units. Analytical chemistry 2010, 82 (17), 7185–92. [DOI] [PubMed] [Google Scholar]
- 3.Burke DG; Griffiths K; Kassir Z; Emslie K, Accurate measurement of DNA methylation that is traceable to the international system of units. Analytical chemistry 2009, 81 (17), 7294–301. [DOI] [PubMed] [Google Scholar]
- 4.Pavsic J; Devonshire AS; Parkes H; Schimmel H; Foy CA; Karczmarczyk M; Gutierrez-Aguirre I; Honeyborne I; Huggett JF; McHugh TD; Milavec M; Zeichhardt H; Zel J, Standardization of Nucleic Acid Tests for Clinical Measurements of Bacteria and Viruses. Journal of clinical microbiology 2015, 53 (7), 2008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen K; Mogensen HS; Hedman J; Niederstatter H; Parson W; Morling N, Comparison of five DNA quantification methods. Forensic science international. Genetics 2008, 2 (3), 226–30. [DOI] [PubMed] [Google Scholar]
- 6.He HJ; Stein EV; DeRose P; Cole KD, Limitations of methods for measuring the concentration of human genomic DNA and oligonucleotide samples. BioTechniques 2018, 64 (2), 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders R; Mason DJ; Foy CA; Huggett JF, Evaluation of digital PCR for absolute RNA quantification. PloS one 2013, 8 (9), e75296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stein EV; Duewer DL; Farkas N; Romsos EL; Wang L; Cole KD, Steps to achieve quantitative measurements of microRNA using two step droplet digital PCR. PloS one 2017, 12 (11), e0188085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emslie KR; JL HM; Griffiths K; Forbes-Smith M; Pinheiro LB; Burke DG, Droplet Volume Variability and Impact on Digital PCR Copy Number Concentration Measurements. Analytical chemistry 2019, 91 (6), 4124–4131. [DOI] [PubMed] [Google Scholar]
- 10.Kosir AB; Divieto C; Pavsic J; Pavarelli S; Dobnik D; Dreo T; Bellotti R; Sassi MP; Zel J, Droplet volume variability as a critical factor for accuracy of absolute quantification using droplet digital PCR. Analytical and bioanalytical chemistry 2017, 409 (28), 6689–6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarin LP; Kienast SD; Leufken J; Ross RL; Dziergowska A; Debiec K; Sochacka E; Limbach PA; Fufezan C; Drexler HCA; Leidel SA, Nano LC-MS using capillary columns enables accurate quantification of modified ribonucleosides at low femtomol levels. RNA (New York, N.Y.) 2018, 24 (10), 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu L; Amato NJ; Wang P; McGowan SJ; Niedernhofer LJ; Wang Y, Simultaneous Quantification of Methylated Cytidine and Adenosine in Cellular and Tissue RNA by Nano-Flow Liquid Chromatography-Tandem Mass Spectrometry Coupled with the Stable Isotope-Dilution Method. Analytical chemistry 2015, 87 (15), 7653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heiss M; Reichle VF; Kellner S, Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS. RNA biology 2017, 14 (9), 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song L; James SR; Kazim L; Karpf AR, Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Analytical chemistry 2005, 77 (2), 504–10. [DOI] [PubMed] [Google Scholar]
- 15.Addepalli B; Limbach PA, Mass spectrometry-based quantification of pseudouridine in RNA. Journal of the American Society for Mass Spectrometry 2011, 22 (8), 1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaston KW; Limbach PA, The identification and characterization of non-coding and coding RNAs and their modified nucleosides by mass spectrometry. RNA biology 2014, 11 (12), 1568–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thuring K; Schmid K; Keller P; Helm M, LC-MS Analysis of Methylated RNA. Methods in molecular biology (Clifton, N.J.) 2017, 1562, 3–18. [DOI] [PubMed] [Google Scholar]
- 18.Richardson FC; Zhang C; Lehrman SR; Koc H; Swenberg JA; Richardson KA; Bendele RA, Quantification of 2’-fluoro-2’-deoxyuridine and 2’-fluoro-2’-deoxycytidine in DNA and RNA isolated from rats and woodchucks using LC/MS/MS. Chemical research in toxicology 2002, 15 (7), 922–6. [DOI] [PubMed] [Google Scholar]
- 19.Dizdaroglu M; Jaruga P; Rodriguez H, Measurement of 8-hydroxy-2’-deoxyguanosine in DNA by high-performance liquid chromatography-mass spectrometry: comparison with measurement by gas chromatography-mass spectrometry. Nucleic acids research 2001, 29 (3), E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang HP; Liu T; Guo N; Yu L; Yuan BF; Feng YQ, Determination of formylated DNA and RNA by chemical labeling combined with mass spectrometry analysis. Analytica chimica acta 2017, 981, 1–10. [DOI] [PubMed] [Google Scholar]
- 21.Li QY; Xie NB; Xiong J; Yuan BF; Feng YQ, Single-Nucleotide Resolution Analysis of 5-Hydroxymethylcytosine in DNA by Enzyme-Mediated Deamination in Combination with Sequencing. Analytical chemistry 2018, 90 (24), 14622–14628. [DOI] [PubMed] [Google Scholar]
- 22.Dong L; Zang C; Wang J; Li L; Gao Y; Wu L; Li P, Lambda genomic DNA quantification using ultrasonic treatment followed by liquid chromatography-isotope dilution mass spectrometry. Analytical and bioanalytical chemistry 2012, 402 (6), 2079–88. [DOI] [PubMed] [Google Scholar]
- 23.Smith JD; Markham R, Chromatographic studies on nucleic acids; the quantitative analysis of ribonucleic acids. The Biochemical journal 1950, 46 (5), 509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyatt GR, The purine and pyrimidine composition of deoxypentose nucleic acids. The Biochemical journal 1951, 48 (5), 584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villalta PW; Hochalter JB; Hecht SS, Ultrasensitive High-Resolution Mass Spectrometric Analysis of a DNA Adduct of the Carcinogen Benzo[a]pyrene in Human Lung. Analytical chemistry 2017, 89 (23), 12735–12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vanden Bussche J; Moore SA; Pasmans F; Kuhnle GG; Vanhaecke L, An approach based on ultra-high pressure liquid chromatography-tandem mass spectrometry to quantify O6-methyl and O6-carboxymethylguanine DNA adducts in intestinal cell lines. Journal of chromatography. A 2012, 1257, 25–33. [DOI] [PubMed] [Google Scholar]
- 27.Ham AJ; Ranasinghe A; Morinello EJ; Nakamura J; Upton PB; Johnson F; Swenberg JA, Immunoaffinity/gas chromatography/high-resolution mass spectrometry method for the detection of N(2),3-ethenoguanine. Chemical research in toxicology 1999, 12 (12), 1240–6. [DOI] [PubMed] [Google Scholar]
- 28.Yen TY; Christova-Gueoguieva NI; Scheller N; Holt S; Swenberg JA; Charles MJ, Quantitative analysis of the DNA adduct N2,3-ethenoguanine using liquid chromatography/electrospray ionization mass spectrometry. Journal of mass spectrometry : JMS 1996, 31 (11), 1271–6. [DOI] [PubMed] [Google Scholar]
- 29.Swarts SG; Smith GS; Miao L; Wheeler KT, Effects of formic acid hydrolysis on the quantitative analysis of radiation-induced DNA base damage products assayed by gas chromatography/mass spectrometry. Radiation and environmental biophysics 1996, 35 (1), 41–53. [DOI] [PubMed] [Google Scholar]
- 30.Djuric Z; Luongo DA; Harper DA, Quantitation of 5-(hydroxymethyl)uracil in DNA by gas chromatography with mass spectral detection. Chemical research in toxicology 1991, 4 (6), 687–91. [DOI] [PubMed] [Google Scholar]
- 31.Catania J; Keenan BC; Margison GP; Fairweather DS, Determination of 5-methylcytosine by acid hydrolysis of DNA with hydrofluoric acid. Analytical biochemistry 1987, 167 (2), 347–51. [DOI] [PubMed] [Google Scholar]
- 32.Chen HC; Chang YL; Teng YC; Hsiao CF; Lin TS, A Stable Isotope Dilution Nanoflow Liquid Chromatography Tandem Mass Spectrometry Assay for the Simultaneous Detection and Quantification of Glyoxal-Induced DNA Cross-Linked Adducts in Leukocytes from Diabetic Patients. Analytical chemistry 2017, 89 (24), 13082–13088. [DOI] [PubMed] [Google Scholar]
- 33.Shibayama S; Fujii SI; Inagaki K; Yamazaki T; Takatsu A, Formic acid hydrolysis/liquid chromatography isotope dilution mass spectrometry: An accurate method for large DNA quantification. Journal of chromatography. A 2016, 1468, 109–115. [DOI] [PubMed] [Google Scholar]
- 34.Qinghui Huang KK, Ronald Benner A simple high performance liquid chromatography method for the measurement of nucleobases and the RNA and DNA content of cellular material. Limnology and Oceanography: Methods 2012, 10 (10), 608–616. [Google Scholar]
- 35.de Bruin OM; Birnboim HC, A method for assessing efficiency of bacterial cell disruption and DNA release. BMC microbiology 2016, 16 (1), 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yates SA; Dempster NM; Murphy MF; Moore SA, Quantitative analysis of malondialdehyde-guanine adducts in genomic DNA samples by liquid chromatography/tandem mass spectrometry. Rapid communications in mass spectrometry : RCM 2017, 31 (9), 762–770. [DOI] [PubMed] [Google Scholar]
- 37.Bonilla J SG, Handbook of Analysis of Oligonucleotides and Related Products 1st ed; CRC Press: 2016; p 511. [Google Scholar]
- 38.Romsos EL KM, Duewer DL, Toman B, Farkas N., Certification of Standard Reference Material® 2372a Human DNA Quantitation Standard. . NIST Special Publication; 2018, 260–189, 10.6028/NIST.SP.260-189. [DOI] [Google Scholar]
- 39.Bunk DM; Lowenthal MS, Isotope Dilution Liquid Chromatography-Tandem Mass Spectrometry for Quantitative Amino Acid Analysis. Methods in molecular biology (Clifton, N.J.) 2019, 2030, 143–151. [DOI] [PubMed] [Google Scholar]
- 40.Apffel A; Chakel JA; Fischer S; Lichtenwalter K; Hancock WS, New procedure for the use of high-performance liquid chromatography–electrospray ionization mass spectrometry for the analysis of nucleotides and oligonucleotides. Journal of Chromatography A 1997, 777 (1), 3–21. [Google Scholar]
- 41.Apffel A; Chakel JA; Fischer S; Lichtenwalter K; Hancock WS, Analysis of Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Analytical chemistry 1997, 69 (7), 1320–5. [DOI] [PubMed] [Google Scholar]
- 42.Cleveland MHFN, Kiesler KM; Toman B; Vallone PM; Certification of Standard Reference Material® 2365 BK Virus DNA Quantitative Standard. NIST Special Publication 260–191, available at: 10.6028/NIST.SP.260-191 (accessed Sep 2018). [DOI] [Google Scholar]
- 43.Duewer DL; Kline MC; Romsos EL; Toman B, Evaluating droplet digital PCR for the quantification of human genomic DNA: converting copies per nanoliter to nanograms nuclear DNA per microliter. Analytical and bioanalytical chemistry 2018, 410 (12), 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le T; Kim KP; Fan G; Faull KF, A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Analytical biochemistry 2011, 412 (2), 203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.