

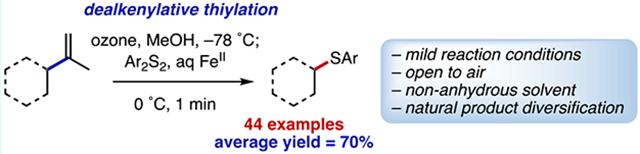
Abstract
Carbon–carbon bond fragmentations are useful methods for the functionalization of molecules. The value of such cleavage events is maximized when paired with subsequent bond formation. Herein we report a protocol for the cleavage of an alkene C(sp3)–C(sp2) bond, followed by the formation of a new C(sp3)–S bond. This reaction is performed in nonanhydrous solvent and open to the air, employs common starting materials, and can be used to rapidly diversify natural products.
Graphical Abstract
The formation of carbon–heteroatom bonds in organic compounds is an important step in the synthesis of many drug molecules. After oxygen and nitrogen, sulfur is the heteroatom found most frequently in FDA-approved drugs (Figure 1A).1 Thioethers and their higher oxidation state derivatives are also versatile and widely used synthetic intermediates.2 Some of the most prevalent means of alkyl aryl thioether synthesis are alkylation (e.g., SN2, Mitsunobu reaction), addition to unsaturated bonds (e.g., Michael addition, hydrothiylation), and cross-coupling.2 Less frequent are reports of trapping of a carbon radical with an aryl disulfide species (Figure 1B), a transformation that typically requires the use of radical precursors (e.g., organometallic species, Barton esters, trialkyl boranes), carboxylic acids, simple alkanes, or a peroxide species.3–11 This approach is sometimes limited in its applicability because of the syntheses of the requisite coupling partners, the harsh reaction conditions (high temperatures, long reaction times), or the low selectivity for C–H abstraction (in the case of alkane starting materials). Consequently, a method for the generation of carbon radicals under mild reaction conditions from readily available starting materials, including natural products, would be highly useful.
Figure 1.
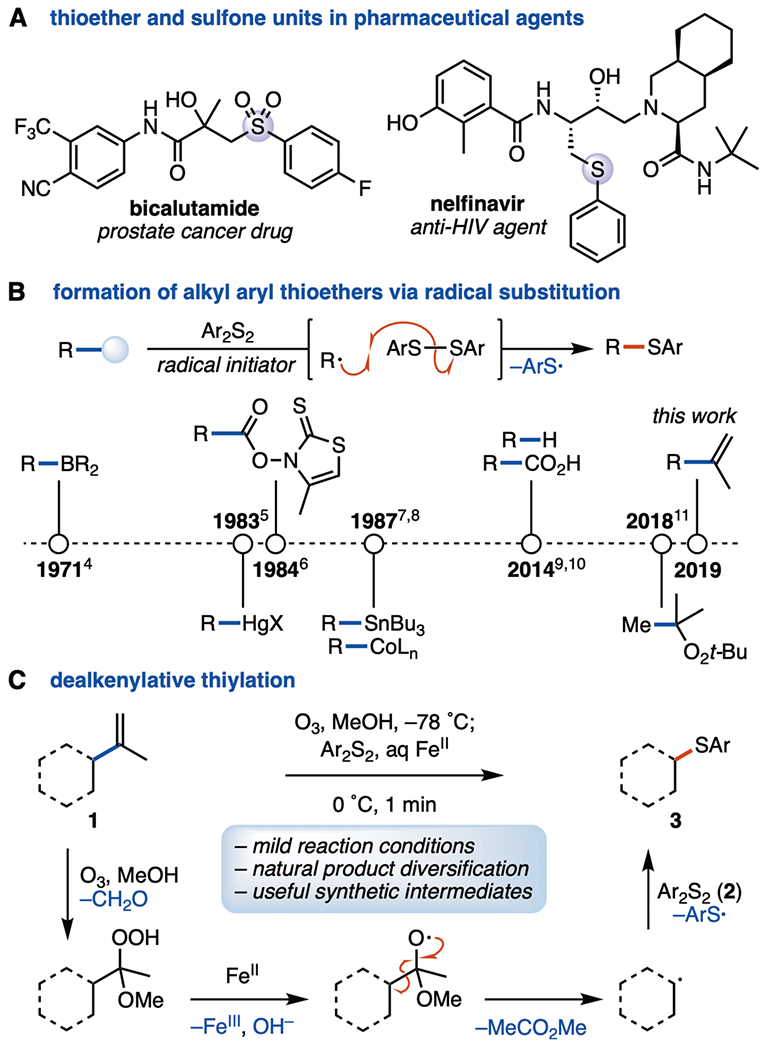
(A) Pharmaceuticals featuring thioether and sulfone units. (B) Timeline of aryl alkyl thiother formation via radical substitution. (C) Mechanism of dealkenylative thiylation.
Recent methodological advancements in the area of C–C bond activation have enabled a number of powerful transformations.12 Specifically, C(sp3)–C(sp3) and C(sp3)–C(sp2) bond fragmentations have proven to be effective methods for functionalizing molecules with regard to both the total synthesis and the preparation of pharmaceutically relevant compounds.12,13 The value of such transformations can be increased when paired with a subsequent bond-forming event. Nevertheless, reports of alkene C(sp3)–C(sp2) bond cleavage, followed by C(sp3)–heteroatom bond formation remain uncommon13a,e,14 despite the ubiquity of alkenes in organic molecules, especially within natural products.15 In this Letter, we report a simple method for the dealkenylative thiylation of alkenes (1, 4, and 6) to give alkyl aryl sulfides (3, 5, and 7) under mild conditions (Figure 1C). We have found that alkyl radicals generated through the single electron transfer (SET)-based reductive cleavage of α-alkoxy hydroperoxides13,14,16 (generated during the ozonolysis of alkenes 1)17 can be trapped with an aryl disulfide (2) to form a new C(sp3)–S bond in 3. In contrast with previously reported carbon-radical-based methods for C(sp3)–S bond formation, this transformation is performed under mild reaction conditions (below room temperature, within 1 min, open to air, nonanhydrous solvent), employs common olefins as starting materials, and is stereoselective when the starting materials contain stereocenters.
A survey of the reaction parameters revealed the optimal conditions for the dealkenylative thiylation to be ozonolysis of the alkene at −78 °C in methanol, followed by treatment with 3.0 equiv of the aryl disulfide and 1.2 equiv of ferrous sulfate heptahydrate (added as a 5% w/v aqueous solution16f) at 0 °C. (See Table S1 in the Supporting Information for details.) Under these optimized conditions and using 4-isopropenylte-trahydropyran (1a) as our alkene substrate, we examined the scope of the aryl disulfide reaction partner (Figure 2). Phenyl disulfide (2a) gave the product 3aa in 80% yield, whereas various tolyl and xylyl disulfides also produced their thiylation products 3ab–af in yields of 62–77%. Electron-rich and -poor aryl disulfides were competent partners, generating the expected products 3ag–aj in yields of 50–75%. 1-Phenyl-tetrazol-5-yl and 2-pyridyl disulfides, notable for the use of their thioether and sulfone derivatives in cross-coupling reactions,18 were also compatible, providing the thioethers 3ak and 3al in yields of 51 and 75%, respectively. Attempts to employ dialkyl disulfides as coupling partners were unfruitful, presumably because of the side reactions resulting from the generation of reactive alkylthiyl radical intermediates.3
Figure 2.
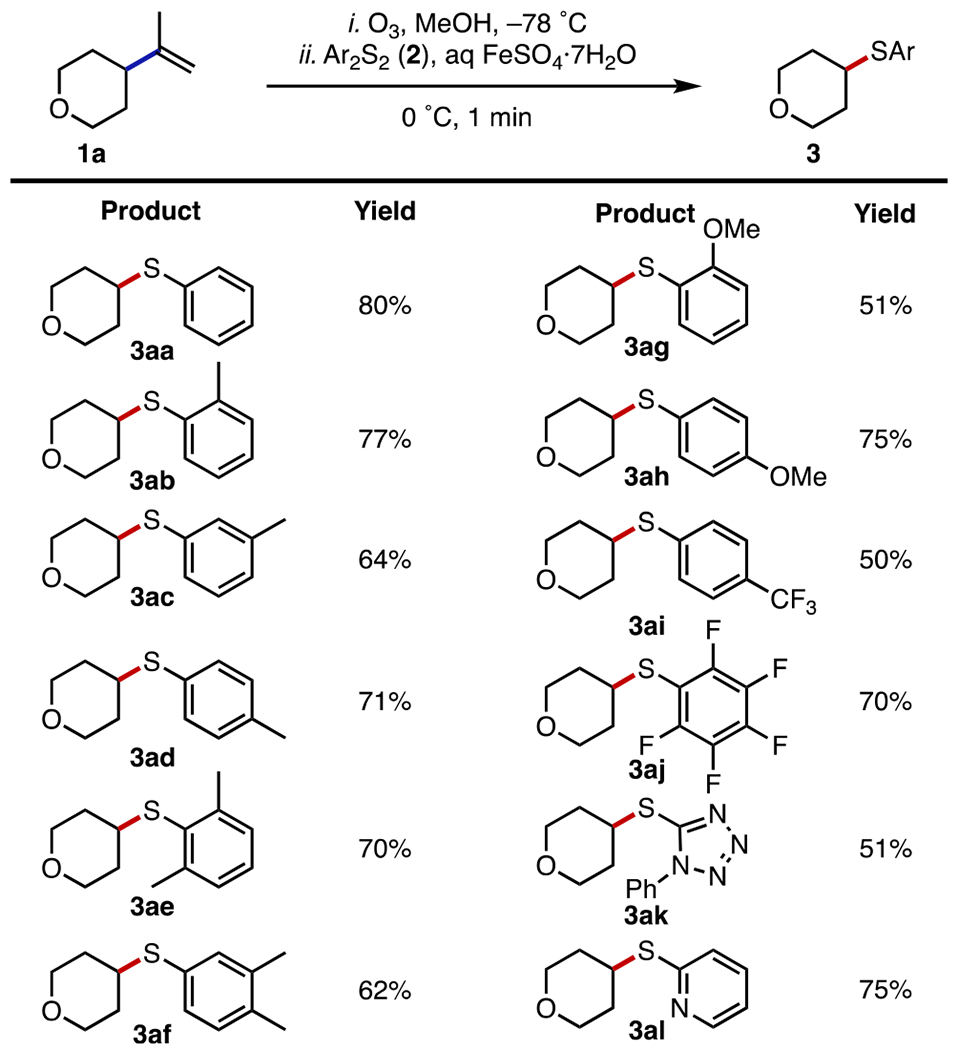
Substrate scope of the aryl disulfide coupling partner in the dealkenylative thiylation of 1a. Experiments were performed on a 1.0 mmol scale. Isolated yields are after SiO2 chromatography. See the SI for experimental details.
Subsequently, we investigated the substrate scope of the alkene coupling partner (Figure 3). The primary radical precursors 1b–d supplied the thioethers 3ba–da in yields of 58–80%. Secondary (1e, 1f) and tertiary (1g) radical precursors were also compatible, furnishing the desired products 3ea–ga in yields of 74–82%. The reaction was tolerant to a range of functionalities, including alcohol, imide, amide, carboxylic ester, and carbamate groups. A powerful feature of the reaction is the ability to introduce heteroatom functionality in terpenoid (e.g., (+)-nootkatone, 1i) and terpenoid-derived starting materials. The bicyclic ketones 1h–k gave their corresponding products in yields of 67–77% (5.9:1 to 7.5:1 d.r.). Notably, dealkenylative thiylation of the diastereoisomeric enones 1j and 1k resulted in the same distribution of product isomers. This observation is consistent with stereoselectivity trends commonly observed in reactions with cyclic radicals, in which the stereoselectivity of the addition is dictated by a combination of torsional and steric effects.19 The commonly available terpenoids (−)-isopulegol (1l), trans-(+)-dihydrocarvone (1m), cis-(−)-limonene oxide (1n), (−)-dihydrocarveol (1o), and (−)-limonene-1,2-diol (1p) were also viable substrates, producing their products 31a–pa, respectively, in yields of 60–84%. When run using 10 mmol of trans-(+)-dihydrocarvone (1m), the reaction produced the thioethers 3ma/3ma′ in 66% yield. With the exception of 3na (12:1 d.r.), the diastereoisomeric ratios of the products from the monoterpenoids were lower (in the range from 1.5:1 to 4:1) than those of the decalinone substrates 1h–k.
Figure 3.
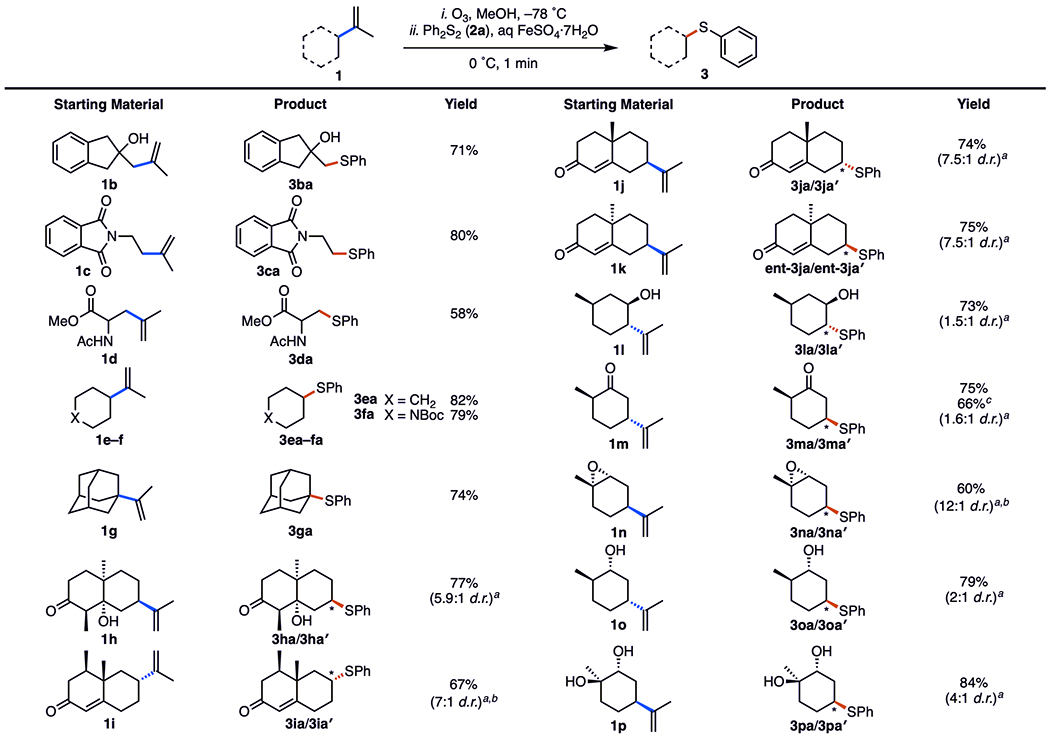
Substrate scope of alkene coupling partner. Experiments were performed on 1.0 mmol scale. Isolated yields are after SiO2 chromatography. See the SI for experimental details. aMajor diastereoisomer displayed. bInseparable mixture of diastereoisomers. c10.0 mmol scale.
Next, we found that alkenes containing exo-methylene groups (4) were converted into the corresponding phenyl-thiyl-containing methyl carboxylates (5) (Figure 4). The simple cycloalkenes 4a and 4b generated their products 5aa and 5ba cleanly in yields of 73 and 71%, respectively. 1-Methylene-2-methylcyclohexane (4c) provided its product 5ca in 75% yield, whereas the Boc-protected piperidine 4d also cleanly supplied the thioether 5da in 80% yield. 1-Methyleneindane (4e) furnished its product 5ea in 51% yield, whereas 2-methylenetetralin (4f) displayed the poorest efficiency, providing 5fa/5fa′ (1.6:1 r.r.) in only 35% yield. The naturally occurring terpene (±)-sabinene (4g) produced a single diastereoisomer of the trisubstituted cyclopropane 5ga in 74% yield. The fragmentative coupling of methyleneadamantane (4h) also generated the exo-phenylthio ester 5ha exclusively in 51% yield.
Figure 4.
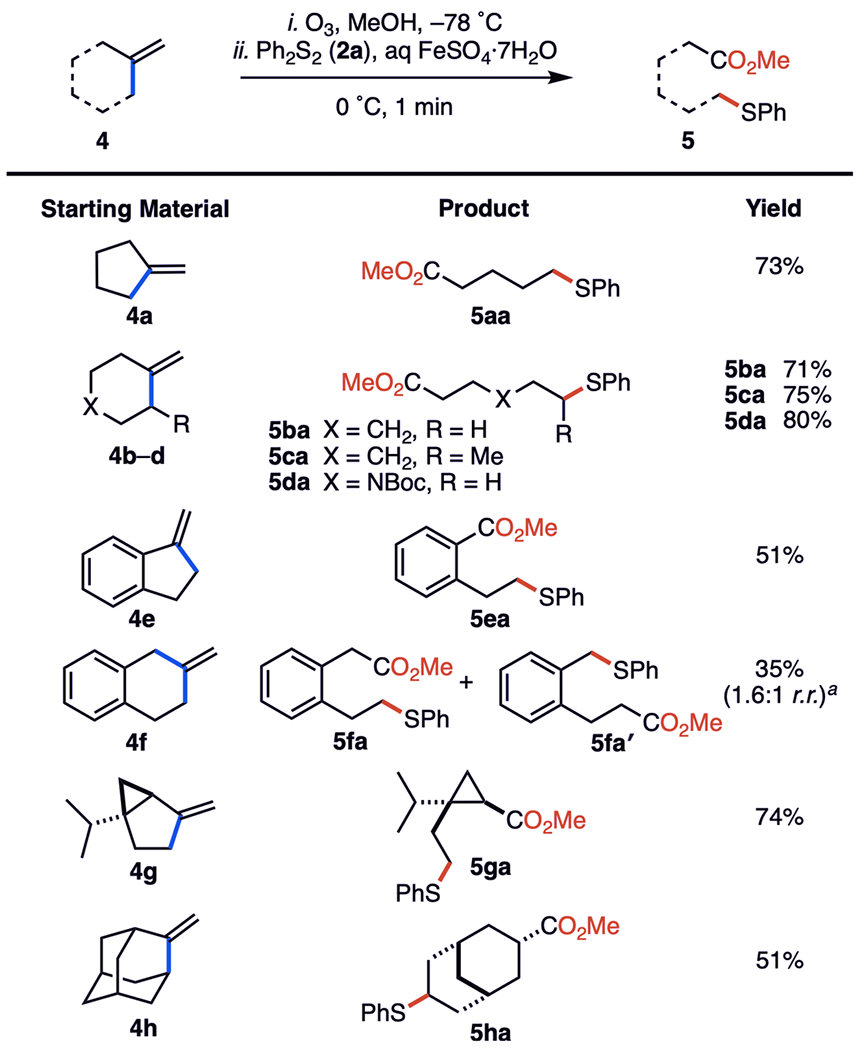
Dealkenylative thiylation of exo-methylene cycloalkanes. Experiments were performed on 1.0 mmol scale. Isolated yields are after SiO2 chromatography. See the SI for experimental details. aInseparable mixture of regioisomers.
Cycloalkenes bearing endocyclic olefins (6) were also competent substrates, providing phenylthio-aldehydes (7) as products (Figure 5). Simple hydrocarbon substrates (6a–c) supplied the expected products 7aa–ca, respectively, in yields from 63 to 75%. (+)-2-Carene (6d) furnished a single diastereoisomer of the tetrasubstituted cyclopropane 7da in 65% yield. α-Terpineol (6e) gave the cyclic ketals 7ea/7ea′ in 42% yield (1.2:1 d.r.), the result of intramolecular trapping of the aldehyde; in contrast, the corresponding acetate 6f produced the uncyclized product 7fa in 61% yield. Upon fragmentation, norbornylene (6g) generated the products 7ga and 7ga′ (1.2:1 d.r.) in 50% yield. Intriguingly, attempts to extend this transformation to generate benzaldehydes resulted in the cyclization of the intermediate radical rather than trapping with the disulfide species. 6-Methyl-5,6-didehydrobenzocycloheptane (6h) provided the desired product 7ha in only 5% yield while generating α-tetralone (7ha′) in 90% yield.20
Figure 5.
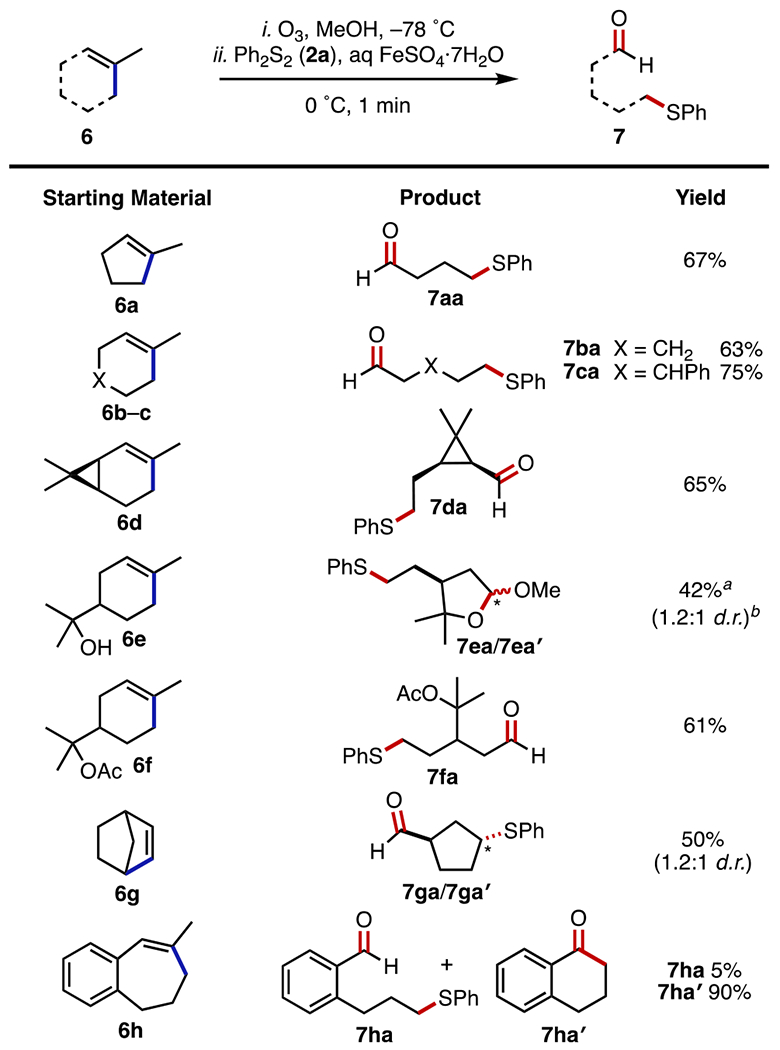
Dealkenylative thiylation of cyclic alkenes. Experiments were performed on a 1.0 mmol scale. Isolated yields are after SiO2 chromatography. See the SI for experimental details. aSolid FeSO4.7H2O was added at room temperature. bInseparable mixture of diastereoisomers.
Figure 6 presents several examples of synthetic applications in which we have employed this transformation. With regard to medicinal agents, this process can be a tool for the functionalization of biologically active compounds. We examined the reaction of betulin (1q), which has wide biological activities (in particular, anticancer and anti-HIV properties)21 yet few functional group handles on the skeleton. Notably, the cleavage of the C-19 C(sp3)–C(sp2) bond, followed by the introduction of a heteroatom at this position is rare,22 with most modifications occurring at the C-3 or C-28 positions.23 Dealkenylative thiylation of betulin readily furnished the thioethers 3ql and 3ql′ in 78% yield (1.2:1 d.r.). The subsequent oxidation of the major diastereoisomer (3ql) gave the sulfone 8 (see ORTEP of the solid-state structure in Figure 6) in 43% overall yield from betulin, providing facile access to previously inaccessible derivatives of this natural product. Sulfones and sulfides are also useful functional group handles that are widely used in organic synthesis. When dealkenylative thiylation is combined with terpenoid precursors, enantiopure synthetic intermediates are readily generated. Oxidation, mesylation, and intramolecular alkylation of the dihydrocarveol-derived thioether 3oa produced the enantiomerically pure bicyclo[3.1.0]hexane 9 in 85% overall yield. Vinyl sulfides and sulfones are also useful synthetic precursors.24 A sequence of oxidation, mesylation, and elimination generated a single enantiomer of the vinyl sulfone 10 in 96% yield from the thioether 3la. When employing the dihydrocarvone-derived thioether 3ma′, we found that the protected sulfone underwent iron-catalyzed cross-coupling18a to provide the arylated products 11/11′ in 47% overall yield (4.3:1 d.r.). Finally, the oxidation of the thioether 3ma to the sulfone, followed by Baeyer–Villiger oxidation, gave the lactone 12 in 70% yield. Caprolactones are employed widely as monomers for polymer synthesis,25 and this approach establishes a route toward biorenewable terpenoid-based caprolactones that are both diastereomerically and enantiomerically pure.
Figure 6.
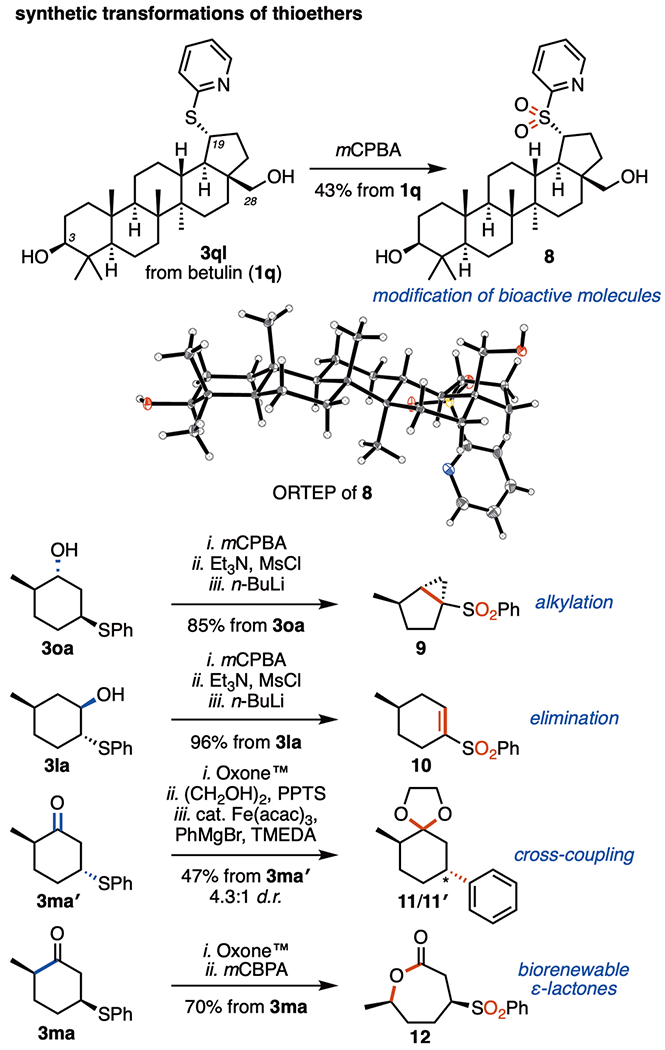
Synthetic transformations of the thioether products. Experiments were performed on ≥0.5 mmol scale. Isolated yields after SiO2 chromatography. See the SI for experimental details.
In summary, we have demonstrated that alkene C(sp3)–C(sp2) bond fragmentation and C(sp3)–S bond formation can be combined under mild operating conditions when employing abundantly available starting materials.26 This transformation, which occurs upon ozonolysis and the subsequent FeII-mediated SET-based reduction, is a facile means for the diversification of natural products. We have also demonstrated that the thiylated adducts could be elaborated for use in organic synthesis and in the preparation of biologically relevant materials. On a fundamental level, we have established a deconstructive strategy of using olefins for the introduction of heteroatoms in organic compounds. Further efforts directed toward the application of this technology in the preparation of pharmaceutically and synthetically useful adducts are underway.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this study was provided by the NIH (R01GM071779). A.J.S. thanks the Majeti–Alapati fellowship for funding. We thank the UCLA Molecular Instrumentation Center for the NMR spectroscopy and mass spectrometry instrumentation and for the X-ray diffraction studies (S. I. Khan).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b03186.
Experimental procedures, detailed discussions, compound characterization, and NMR spectra (PDF)
Accession Codes
CCDC 1913869 and 1917888 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Feng M; Tang B; Liang SH; Jiang X Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem 2016, 16, 1200–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scott KA; Njardarson JT Analysis of US FDA-approved drugs containing sulfur atoms. Top. Curr. Chem 2019, 376, 1–34. [DOI] [PubMed] [Google Scholar]
- (2).(a) Block E Organic sulfur compounds in organic synthesis. J. Chem. Educ 1971, 48, 814–824 [Google Scholar]; (b) Patai S The Chemistry of Functional Groups–The Chemistry of the Thiol Group; Wiley: London, 1974 [Google Scholar]; (c) Block E Reactions of Organosulfur Compounds; Blomquist AT, Wasserman HH, Eds.; Academic Press: New York, 1978 [Google Scholar]; (d) Jones DN Comprehensive Organic Chemistry; Barton DJ, Ollis DW, Eds.; Pergamon: New York, 1979; Vol. 3 [Google Scholar]; (e) Metzner P; Thuillier A Sulfur Reagents in Organic Synthesis; Katritzky AR, Meth-Cohn O, Rees CW, Eds.; Academic Press: San Diego, CA, 1994 [Google Scholar]; (f) Cremlyn RJ An Introduction to Organosulfur Chemistry; Wiley: New York, 1996 [Google Scholar]; (g) Clayden J; MacLellan P Asymmetric synthesis of tertiary thiols and thioethers. Beilstein J. Org. Chem 2011, 7, 582–595 [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Eichman CC; Stambuli JP Transition metal catalyzed synthesis of aryl sulfides. Molecules 2011, 16, 590–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Dénès F; Schiesser CH; Renaud P Thiols, thioethers, and related compounds as sources of C-centered radicals. Chem. Soc. Rev 2013, 42, 7900–7942 [DOI] [PubMed] [Google Scholar]; (b) Denes F; Pichowicz M; Povie G; Renaud P Thiyl radicals in organic synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (4).Brown HC; Midland MM Facile photochemical or oxygen initiated free-radical chain reactions of trialkylboranes with organic disulfides. Convenient new synthesis of organic sulfides via hydroboration. J. Am. Chem. Soc 1971, 93, 3291–3293. [Google Scholar]
- (5).(a) Russell GA; Tashtoush H Free-radical chain-substitution reactions of alkylmercury halides. J. Am. Chem. Soc 1983, 105, 1398–1399 [Google Scholar]; (b) Russell GA; Ngoviwatchai P; Tashtoush HI; Pla-Dalmau A; Khanna RK Reactions of alkylmercurials with heteratom-centered acceptor radicals. J. Am. Chem. Soc 1988, 110, 3530–3538. [Google Scholar]
- (6).Barton DHR; Bridon D; Zard SZ New decarboxylative chalcogenation of aliphatic and alicyclic carboxylic acids. Tetrahedron Lett 1984, 25, 5777–5780. [Google Scholar]
- (7).(a) Patel VF; Pattenden G Radical reactions in synthesis. Homolysis of alkyl cobalt salophens in the presence of radical trapping agents. Tetrahedron Lett 1987, 28, 1451–1454 [Google Scholar]; (b) Branchaud BP; Meier MS; Malekzadeh MN New synthetic methods via free radicals. Free-radical generation via photolytic hemolysis of alkylcobaloxime carbon-cobalt bonds. Efficient radical trapping with useful functional groups. J. Org. Chem 1987, 52, 212–217. [Google Scholar]
- (8).Russell GA; Ngoviwatchai P; Tashtoush H; Hershberger J Reaction of 1-alkenyl and 1-alkynl derivatives of tin and mercury with hetero-centered radicals. Organometallics 1987, 6, 1414–1419. [Google Scholar]
- (9).Wang P-F; Wang X-Q; Dai J-J; Feng Y-S; Xu H-J Silver-mediated decarboxylative C-S cross-coupling of aliphatic carboxylic acids under mild conditions. Org. Lett 2014, 16, 4586–4589. [DOI] [PubMed] [Google Scholar]
- (10).(a) Zhao J; Fang H; Han J; Pan Y; Li G Metal-free preparation of cycloalkyl aryl sulfides via di-tert-butyl peroxide-promoted oxidative C(sp3)–H bond thiolation of cycloalkanes. Adv. Synth. Catal 2014, 356, 2719–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Du B; Jin B; Sun P Syntheses of sulfides and selenides through direct oxidative functionalization of C(sp3)–H bond. Org. Lett 2014, 16, 3032–3035 [DOI] [PubMed] [Google Scholar]; (c) Sahoo SK A comparative study of Cu(II)-assisted vs Cu(II)-free chalcogenation on benzyl and 2/3-cycloalkyl moieties. J. Chem. Sci 2015, 127, 2151–2157. [Google Scholar]
- (11).Wu X; Wang Y Metal-free S-methylation of diaryl disulfides with di-tert-butyl peroxide. Tetrahedron Lett. 2018, 59, 1240–1243. [Google Scholar]
- (12).(a) C–C Bond Activation; Dong G, Ed.; Springer Verlag: Berlin/Heidelberg, 2014 [Google Scholar]; (b) Nairoukh Z; Cormier M; Marek I Merging C–H and C–C bond cleavage in organic synthesis. Nat. Rev. Chem 2017, 1, 1–16 [Google Scholar]; (c) Morcillo SP Radical-promoted C–C bond cleavage: A deconstructive approach for selective functionalize-tion. Angew. Chem 2019, 58, 2–13. [DOI] [PubMed] [Google Scholar]
- (13).(a) Gui J; Wang D; Tian W Biomimetic synthesis of 5,6-dihydro-glaucogenin C: Construction of the disecopregnane skeleton by iron(II)-promoted fragmentation of an α-alkoxy hydroperoxide. Angew. Chem., Int. Ed 2011, 50, 7093–7096 [DOI] [PubMed] [Google Scholar]; (b) Schuppe AW; Newhouse TR Assembly of the limonoid architecture by a divergent approach: Total synthesis of (±)-andirolide N via (±)-8α-hydroxycarapin. J. Am. Chem. Soc 2017, 139, 631–634 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Manoni F; Rumo C; Li L; Harran PG Unconventional fragment usage enables a concise total synthesis of (−)-callyspongiolide. J. Am. Chem. Soc 2018, 140, 1280–1284 [DOI] [PubMed] [Google Scholar]; (d) Roque JB; Kuroda Y; Gottemann LT; Sarpong R Deconstructive diversification of cyclic amines. Nature 2018, 564, 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smaligo AJ; Swain M; Quintana JC; Tan MF; Kim DA; Kwon O Hydrodealkenylative C(sp3)–C(sp2) bond fragmentation. Science 2019, 364, 681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Schreiber SL; Liew W-F Criegee rearrangement of α-alkoxy hydroperoxides. A synthesis of esters and lactones that complements the Baeyer–Villiger oxidation of ketones. Tetrahedron Lett 1983, 24, 2363–2366 [Google Scholar]; (b) Sato T; Oikawa T; Kobayashi K Metal-catalyzed organic photoreactions. Iron(III) chloride catalyzed photooxidation of cyclic olefins and its application to the synthesis of exo-brevicomin. J. Org. Chem 1985, 50, 1646–1651 [Google Scholar]; (c) Bao J; Tian H; Yang P; Deng J; Gui J Modular Synthesis of Functionalized Butenolides by Oxidative Furan Fragmentation. 2019, chemrxiv.8874455.v1. ChemRxiv Preprint Server. 10.26434/chemrxiv.8874455.v1. [DOI] [Google Scholar]
- (15).Ertl P; Schuhmann T A systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod 2019, 82, 1258–1263. [DOI] [PubMed] [Google Scholar]
- (16).(a) Hawkins EGE Reactions of organic peroxides. Part VII. Reaction of 1-hydroxycycloalkyl hydroperoxides with ferrous compounds. J. Chem. Soc 1955, 3463–3467 [Google Scholar]; (b) Kumamoto J; De La Mare HE; Rust FF The use of cupric and ferric chlorides in the trapping of radical intermediates and the synthesis of alkyl chlorides. J. Am. Chem. Soc 1960, 82, 1935–1939 [Google Scholar]; (c) De La Mare HE; Kochi JK; Rust FF The oxidation of free radicals by metal ions. J. Am. Chem. Soc 1961, 83, 2013 [Google Scholar]; (d) Murai S; Sonoda N; Tsutsumi S The redox reaction of 1-ethoxy-n-heptyl hydroperoxide. Bull. Chem. Soc. Jpn 1964, 37, 1187–1190 [Google Scholar]; (e) Schreiber SL Fragmentation reactions of α-alkoxy hydroperoxides and application to the synthesis of the macrolide (±)-recifeiolide. J. Am. Chem. Soc 1980, 102, 6163–6165 [Google Scholar]; (f) Huang D; Schuppe AW; Liang MZ; Newhouse TR Scalable procedure for the fragmentation of hydroperoxides mediated by copper and iron tetrafluoroborate salts. Org. Biomol Chem 2016, 14, 6197–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Fisher TJ; Dussault PH Alkene ozonolysis. Tetrahedron. Tetrahedron 2017, 73, 4233–4258. [Google Scholar]
- (17).Criegee R; Wenner G Die ozonisierung des 9,10-oktalins. Justus Liebig Ann. Chem 1949, 564, 9–15. [Google Scholar]
- (18).(a) Denmark SE; Cresswell AJ Iron-catalyzed cross-coupling of unactivated secondary alkyl thio ethers and sulfones with aryl Grignard reagents. J. Org. Chem 2013, 78, 12593–12628 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Merchant RR; Edwards JT; Qin T; Kruszyk MM; Bi C; Che G; Bao D-H; Qiao W; Sun L; Collins MR; Fadeyi OO; Gallego GM; Mousseau JJ; Nuhant P; Baran PS Modular radical cross-coupling with sulfones enables access to sp3-rich (fluoro)alkylated scaffolds. Science 2018, 360, 75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hughes JME; Fier PS Desulfonylative arylation of redox-active alkyl sulfones with aryl bromides. Org. Lett 2019, 21, 5650–5654. [DOI] [PubMed] [Google Scholar]
- (19).(a) Giese B The stereoselectivity of intermolecular free radical reactions. Angew. Chem., Int. Ed. Engl 1989, 28, 969–1146 [Google Scholar]; (b) Bar G; Parsons AF Stereoselective radical reactions. Chem. Soc. Rev 2003, 32, 251–263. [DOI] [PubMed] [Google Scholar]
- (20).See the Supporting Information for a proposed mechanism for the formation of 7ha′ from 6h.
- (21).(a) Alakurtti S; Makela T; Koskimies S; Yli-Kauhaluoma J Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci 2006, 29, 1–13 [DOI] [PubMed] [Google Scholar]; (b) Genet C; Strehle A; Schmidt C; Boudjelal G; Lobstein A; Schoonjans K; Souchet M; Auwerx J; Saladin R; Wagner A Structure-activity relationship study of betulinic acid, a novel and selective TGR5 agonist, and its synthetic derivatives: Potential impact in diabetes. J. Med. Chem 2010, 53, 178–190. [DOI] [PubMed] [Google Scholar]
- (22).Baddeley GV; Eade RA; Ellis J; Harper P; Simes JJH Oxidation by mercuric acetate in the lup-20(29)-ene and related series. Tetrahedron 1969, 25, 1643–1650. [Google Scholar]
- (23).(a) Aplin RT; Chan RPK; Halsall TG The chemistry of triterpenes and related compounds. Part XLVI. Some novel products from the ozonolysis of methyl acetylbetulinate. J. Chem. Soc. C 1969, 2322–2327 [Google Scholar]; (b) Dutta G; Bose SN Preparation and circular dichroism studies of triterpene lactones of lupane series. Tetrahedron Lett 1988, 29, 5807–5810 [Google Scholar]; (c) Heller L; Kahnt M; Loesche A; Grabandt P; Schwarz S; Brandt W; Csuk R Amino derivatives of platanic acid act as selective and potent inhibitors of butyrylcholinesterase. Eur. J. Med. Chem 2017, 126, 652–668 [DOI] [PubMed] [Google Scholar]; (d) Kahnt M; Heller L; Grabandt P; Al-Harrasi A; Csuk R Platanic acid: A new scaffold for the synthesis of cytotoxic agents. Eur. J. Med. Chem 2018, 143, 259–265. [DOI] [PubMed] [Google Scholar]
- (24).(a) Creech GS; Kwon O Synthesis of nitrodienes, nitrostyrenes, and nitrobiaryls through palladium-catalyzed couplings of β-nitrovinyl and o-nitroaryl thioethers. Chem. Sci 2013, 4, 2670–2674 [Google Scholar]; (b) Fang Y; Luo Z; Xu X Recent advances in the synthesis of vinyl sulfones. RSC Adv 2016, 6, 59661–59676. [Google Scholar]
- (25).(a) Lowe JR; Martello MT; Tolman WB; Hillmyer MA Functional biorenewable polyesters from carvone-derived lactones. Polym. Chem 2011, 2, 702–708 [Google Scholar]; (b) Tschan MJ-L; Brule E; Haquette P; Thomas CM Synthesis of biodegradable polymers from renewable resources. Polym. Chem 2012, 3, 836–851. [Google Scholar]
- (26).This paper first appeared on chemRxiv:; Smaligo AJ; Kwon O Dealkenylative Thiylation of C(sp3)–C(sp2) Bonds. 2019, chemrxiv.8847962.v1. ChemRxiv Preprint Server. 10.26434/chemrxiv.8847962.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.