

Abstract
Proper brain function relies on the precise arrangement and flow of information between diverse neural subtypes. Developing improved human cell-based models which faithfully mimic biologically relevant connectivity patterns may improve drug screening efforts given the limited success of animal models to predict safety and efficacy of therapeutics in human clinical trials. To address this need, we have developed experimental models of defined neural circuitries through the compartmentalization of neuronal cell subtypes in a 96 well plate-based platform where each microwell is divided into two compartments connected by microchannels allowing high-throughput screening (HTS) of small molecules. We demonstrate that we can generate subtype-specific excitatory and inhibitory induced neuronal cells (iNs) from human stem cell lines and that these neurons form robust functional circuits with defined connectivity. Through the use of the genetically encoded calcium indicator GCaMP6f, we monitor calcium ion transients generated during neuronal firing between and within compartments. We further demonstrate functionality of the circuit by perturbing network activity through the addition of glutamate receptor blockers using automated liquid handling. Lastly, we show that we can stimulate network activity in defined neuronal subtypes through the expression of the designer receptor exclusively activated by designer drugs (DREADD) hM3Dq and application of the ligand clozapine-N-oxide (CNO). Our results demonstrate the formation of functional neural circuits in a high-throughput platform that is compatible with compound screening, representing an important step towards developing new screening platforms for studying and ultimately treating psychiatric brain disorders that arise from disordered neural circuit function.
Introduction
The use of human neurons for modeling neurodevelopmental and neuropsychiatric disorders has grown in popularity due to advances in stem cell biology, such as the development of induced pluripotent stem (iPS) cells and induced neuronal cells (iNs) that preserve human genetic information relevant to disease. Much of the focus of human neurons has been on mechanistic studies: uncovering disease etiology and pathophysiology, such as revealing the cellular and molecular phenotypes produced by disease-associated genetic variants in vitro. These mechanistic studies will lead to improved therapeutic development. However, in the brain, neurons do not exist as single cell populations alone, but rather as complex interconnected networks formed between distinct brain regions, or nuclei, that relay information via synapses. To further the goal of improving new therapy development for human neuropsychiatric disorders, it is important to consider disease mechanisms within the appropriate neurocircuitry context and the complex crosstalk between distinct neuronal subpopulations. For neuropsychiatric disorders, this is especially important since effects in one brain region can impact circuit-level functions in complex and poorly understood ways. While preservation of circuitry and anatomical arrangement is an advantage of animal models, the data obtained may not always directly apply to humans. Furthermore, given the limited translation of many candidate drugs through human clinical trials despite passing early research and development stages using animal models,1 there is clearly a need to develop better initial methods for identifying effective drug candidates. We propose that this would in part be achieved by the creation of novel in vitro human cell models that can extend mechanistic work at the single cell population level by incorporating biologically-relevant connectivity patterns.
Recently, microfabrication has presented itself as a major tool for generating compartmentalized neural circuit models, many of which utilize a design and fabrication approach similar to that originally developed by the Jeon group2–6 in which connecting microchannels between compartments facilitate axonal extension while preventing cell body migration between adjacent chambers. While these compartmentalized devices were initially designed for modeling axonal injury, they demonstrated the utility of microfabrication to create similar devices for neuronal communication studies. Since then, a variety of devices have been developed based on this approach for the investigation of neuronal injury and circuit dynamics,7–11 some of which have recently translated measured circuit function into drug screening models producing disease-specific results.12,13 While the utility of compartmentalized devices in the study of neurocircuits has been demonstrated, the majority of reported studies have been limited to the use of primary cultures, which do not preserve the genetic information relevant to disease inherent to patient-specific iPS cell-derived neurons. Reported compartmentalized systems have been shown to capture certain pathological hallmarks of neuropsychiatric disorders, but are often not conducive to parallelization for high-throughput screening (HTS). Furthermore, human neurons have been utilized in HTS platforms,14–18 but primarily with single cell populations, and not in contexts where distinct neuronal groups interact with one another in a defined connectivity.
To address some of these limitations, we have created a parallelized microfluidic plate-based system designed to be coupled with a liquid handling microscopy platform, the GE IN Cell Analyzer 6000, and demonstrate its ability to assay mini-neurocircuits for functional and morphological properties using human neurons derived from iPS cells. We employed soft lithography techniques to design a 96 well plate bottom which introduces two distinct compartments connected by a series of microchannels into a single well of a 96 well plate. We demonstrate that distinct human neuronal populations can be seeded in each compartment and that these cells can be used for functional and morphological screening assays. As a readout of circuit-level connectivity, we employed lentiviral-mediated expression of the genetically-encoded calcium indicator GCaMP6f to measure neural activity-induced depolarization between connected subpopulations of human neurons.19 To demonstrate HTS capabilities and to confirm synaptic communication between isolated neuronal populations within our circuit models, we used two experimental configurations: 1) glutamate receptor antagonists to silence glutamate signaling from excitatory neurons, as well as 2) excitatory neurons expressing designer receptors exclusively activated by designer drugs (DREADDs). For DREADDs, we used a mutated human muscarinic receptor type 3 (hM3Dq) which is a Gq-coupled 7-transmembrane receptor that signals calcium mobilization upon activation by its designer ligand clozapine-N-oxide (CNO).20–22 We demonstrate that we can achieve a circuit-level readout of calcium activity in excitatory (glutamate)-inhibitory (GABA) two-way circuits. We also replicate the synaptogenetic property of neuroligin-3 in our device to demonstrate its use in high-throughput morphometric and synapse formation assays on increased synaptogenesis. We optimized these experiments for automated confocal microscopy and reagent dispensing using a commercially available GE IN Cell Analyzer 6000, the industry standard for laser-based confocal imaging. This system enables high-throughput image acquisition for high-content assays and provides a quantitative platform for cell viability and morphological analyses. Incorporation of this plate within the IN Cell system demonstrates the utility of our approach to serve as an HTS platform for neural circuit studies and its potential as a therapeutic development tool.
Materials and methods
Device fabrication
Device fabrication and plate assembly are summarized in Fig. 1. Device photomasks were designed using DraftSight™ software (Dassault Systèmes). Half of one well plate (48 wells) was designed to fit on one 4-inch wafer, due to wafer size constraints. Each individual well was designed to have the same footprint as in a 96 well culture dish: each well had a diameter of 6.4 mm in which two distinct compartments were separated by a series of 10 μm wide × 3 μm tall × 500 μm long microchannels, with a 9 mm center-to-center spacing between wells. The lithographic process was performed in two distinct layers, as described previously.11 Briefly, a thin layer of SU-8 2002 photoresist (Microchem) was spun onto a clean wafer at 1000 rpm to produce a 3 μm resist layer. The second layer for the chamber structures involved two stacked coats of SU-8 as described previously. This results in a total thickness of approximately 320 μm. Following completion of photolithography, the wafer was coated in trichloro(1H,1H,2H,2H perfluoro-octyl)silane for 30 minutes. Once completed, a 1 : 10 mixture of PDMS curing agent to PDMS monomer (Sylgard® 184) was dispensed onto the wafer. Due to the microplate design, the height of the wall separation was restricted to the skirt height of the microplate (approximately 2.5 mm). The PDMS mixture was degassed for approximately 2 hours. Entrained bubbles were removed using a pipettor. Once devoid of bubbles, the wafer with PDMS was cured at room temperature overnight. The plate was placed such that it remained level during curing to ensure that the PDMS thickness was uniform across the wafer, which improves the focusing on each individual well within the IN Cell analyzer. The following day, the dish was placed in a 65 °C oven to complete the curing process.
Fig. 1.
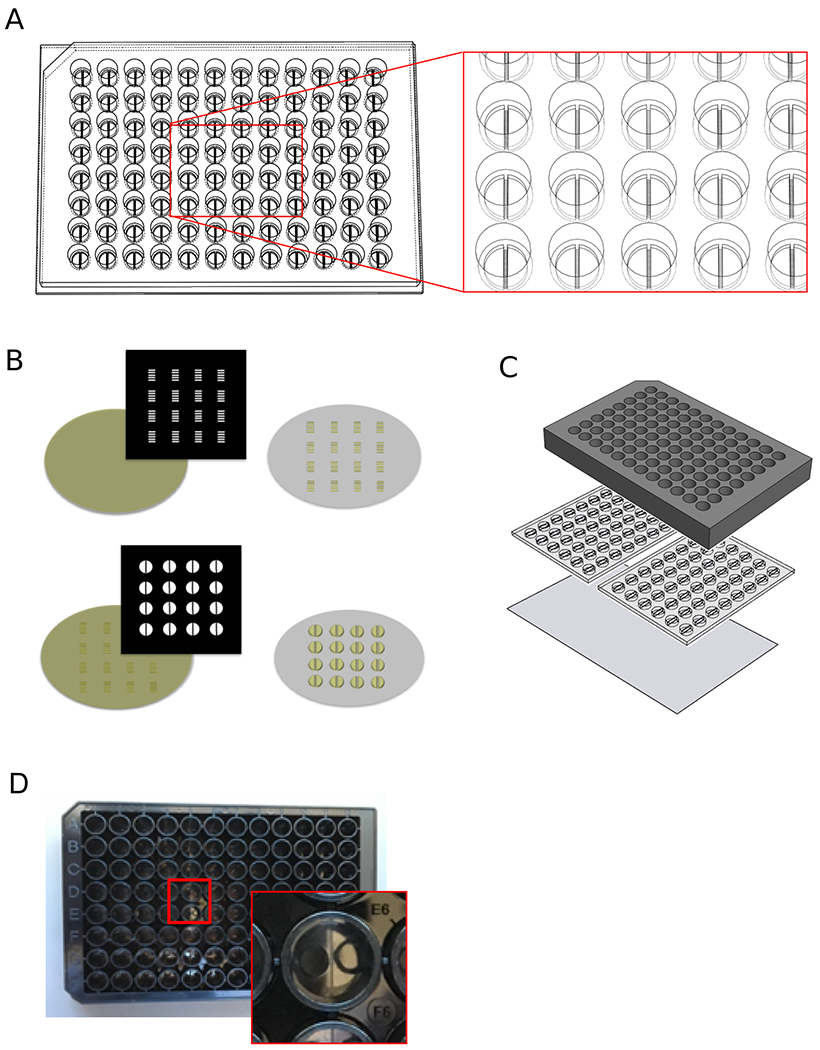
Device fabrication and assembly. A) SolidWorks rendering of the device design concept. Red box indicates a zoomed in area for visualization. Each well is divided into two compartments separated by a wall containing microchannels. B) Fabrication process involving two rounds of photolithography. The first round creates 3 μm tall microchannels for axonal access (top). The second round (bottom) defines the microwell reservoirs for cell culture. C) Assembly of device, which for a full plate requires two PDMS slabs bonded to a glass plate. The PDMS slabs are then attached to the bottom of a 96 well plate. D) Photograph of the assembled plate with a two-compartment well shown in the inset.
Device assembly
PDMS slabs were removed using a razor blade. Three-millimeter biopsy punches (Tru-punch, Skylar Instruments) were compressed using pliers to create a slight oval shape. Two holes were punched into the PDMS to form each of the two compartments within a well (4 punches per well, 2 on each side to allow air escape during filling). The PDMS slabs were then washed with isopropanol followed by water and allowed to fully dry. The PDMS was bonded to a 74 mm × 108 mm × 0.17 mm thick glass plate (0CON-160, Logitech, Ltd., Glasgow, UK) using oxygen plasma (March PX-250, 100 W, 60 s, 60% O2), bonded, and placed on a hotplate at 95 °C under light weight for 30 minutes. The PDMS/glass substrate was next bonded to the bottom of a μClear 96 well plate (Greiner Bio-One), which was used as a structure on which to attach the microfluidic PDMS slab. Prior to bonding, the well bottoms of the microplate were punched out using a 10 μl pipette tip (the tip head was pressed into the well bottom to punch out the plastic) to provide fluid access to the PDMS slab. To facilitate bonding of the PDMS to the polystyrene plate, the plate was treated with aminopropyltriethoxysilane (APTES), in which the polystyrene plate was first exposed to oxygen plasma (100 W, 60 s, 60% O2). It was then placed in a bath of 5% APTES in distilled water at 80 °C for 20 minutes.23 The polystyrene plate was then washed with distilled water and allowed to fully dry. Lastly, the plate and PDMS were exposed to oxygen plasma at 100 W, 60% O2, for 20 seconds, and then bonded overnight in a 65 °C oven under light weight. To re-oxidize the surface, the completed plate was placed in oxygen plasma at 100 W, 60% O2, for 60 seconds. Immediately following this treatment, sterile distilled water was added to the wells to maintain hydrophilicity. The plate with its lid were then wrapped in Parafilm until sterilization and seeding.
Cloning hM3Dq-mCherry into lentiviral construct
pAAV-hSyn-hM3Dq(Gq)-mCherry was a gift from Bryan Roth (Addgene viral prep # 50474-AAV8) and pGP-CMV-GCaMP6f was a gift from Dr. Kelvin Kwan. To clone the hSyn-hM3Dq-mCherry and hSyn-GCaMP6f lentiviral constructs used in this study, the coding sequences were amplified from these templates by PCR (Primestar Takara) and incorporated into a lentiviral backbone vector under the human synapsin promoter (hSyn) using isothermal assembly.24 The primers used were as follows:
hSyn-hM3Dq-mCherry Forward: 5′-CGTGTCGTGCCTGAGAGCGCAGTCTAGAATGACCTTGCACAATAACAGTACAAC-3′
hSyn-hM3Dq-mCherry Reverse: 5′-AGAGGTTGATTATCGATAAGCTTGATATCGAATTCTTACTTGTACAGCTCGTCCATGC-3′
hSyn-GCaMP6f Forward: 5′-TCGTGTCGTGCCTGAGAGCGCAGTCTAGAGAATTCATGGCTAGCATGACTGGTGGG-3′
hSyn-GCaMP6f Reverse: 5′-ATCGATAAGCTTGATATCGAATTGTTAACGGATCCTCACTTCGCTGTCATCATTTGTA-3′.
Lentiviral production
All lentiviruses were produced through transfection of human embryonic kidney cells expressing the SV40 large T-antigen (HEK293T).25 HEK293T cells were grown to approximately 50% confluence for calcium phosphate transfection in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Prior to preparation, the media was refreshed. Lentiviral envelope and capsid plasmids (VsVg, RRE, and Rev) were mixed with water and calcium (2.5 M) for a final volume of 0.5 ml. This solution was added dropwise to a 0.5 ml solution of 2× HEPES-buffered phosphate solution. The solution was incubated at room temperature in the dark for 30 minutes, and then added to the culture media. Using this procedure, the following lentiviruses were produced: reverse transcription transactivator (rtTa), transcription factors Ngn2 (with puromycin resistance), Dlx2 (with hygromycin resistance), Ascl1 (with puromycin resistance), hM3Dq-mCherry, tdTomato, and wild-type neuroligin-3 (NL3). Medium containing the packaged lentiviruses was collected on day 2 and 3 after transfection, aliquoted, and stored at −80 °C.
Induced pluripotent stem cell sources and maintenance
Induced pluripotent stem (iPS) cells were prepared from human primary lymphocytes using Sendai viral vectors (CytoTune™, Life Technologies, Grand Island, NY).26,27 Source biomaterials were obtained from a de-identified repository and were therefore exempt from human subject regulations. Stem cells were maintained using mTeSR1 medium (STEMCELL Technologies).
iPS cell infection to generate induced neurons (iNs)
The iPS cell differentiation and iN seeding and analysis procedures are summarized in Fig. 2. iPS cells were passaged into a 6 well plate at a density of 500 000–750 000 cells per well on day 1 using mTeSR™1 medium (STEMCELL Technologies) with 5 μM Y-compound (Y-27632, a ROCK1 and ROCK2 inhibitor which promotes the survival of stem cells).28 On day 0, iPS cell medium was changed to mTESR containing 5 μM Y-compound and lentivirus specific to the desired neuronal phenotype. Excitatory neurons were generated by the addition of Ngn2 lentivirus and inhibitory neurons were generated by the addition of Dlx2 and Ascl1 lentiviruses.26 Since Ngn2, Dlx2, and Ascl1 virus use the Tet-on operator, all cells were also infected with rtTa lentivirus at a ratio of 1.5× the volume of the other viruses.
Fig. 2.
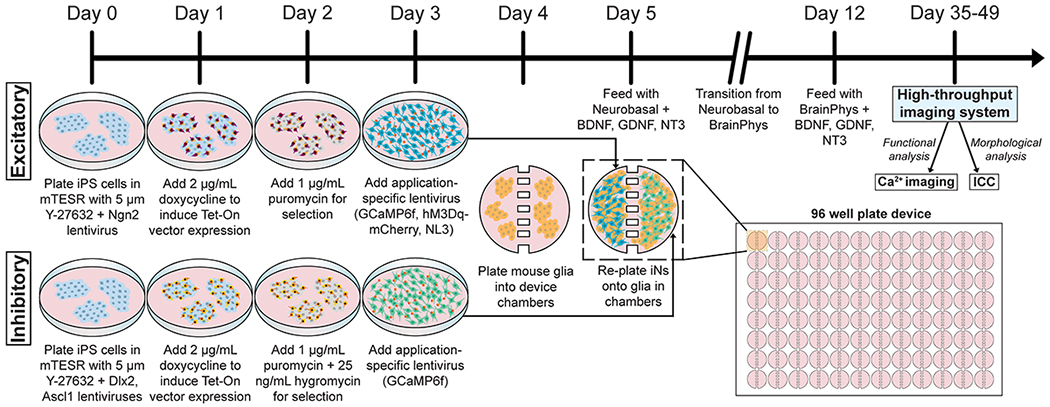
Overview schematic for induction of human iNs, seeding iNs in 96 compartmental devices, and analysis. Human iPS cells are cultured and differentiated toward an excitatory or inhibitory phenotype from day 0 using subtype-specific lentiviruses. Following the addition of doxycycline to induce gene expression on day 1 and antibiotics for further selection on day 2, lentiviruses specific to the desired functional application are introduced to the culture on day 3 – specifically, GCaMP6f vectors for calcium (Ca2+) imaging analysis, hM3Dq-mCherry for subsequent manipulation of neuronal activity, or neuroligin-3 (NL3) for facilitating synapse formation. After device seeding with iNs, a slow transition from Neurobasal to BrainPhys medium begins, each supplemented with growth factors including BDNF, GDNF, and NT3. After 5–7 weeks, human iNs are analyzed with a high-content imaging system, in this case the GE IN Cell Analyzer 6000. For neuronal morphometric analysis, immunocytochemistry (ICC) with specific antibodies is performed after fixation of human iNs prior to IN Cell analysis and quantification.
Doxycycline, which is used to induce expression of Tet-On vectors, was added to the Neurobasal media with Y-compound on day 1 at a concentration of 2 μg ml−1 and maintained in media throughout all experiments. On day 2, puromycin (1 μg ml−1) was added for selection. Inhibitory neurons were also selected with hygromycin (25 ng ml−1), since a hygromycin resistance was built into the Dlx2 vector. Concentrations of puromycin and hygromycin were occasionally increased, as needed, to more aggressively select for iNs against surviving iPS cells. Additional lentivirus was added on day 3, as applicable: vectors carrying DNA for GCaMP6f (200 μl ml−1 in Neurobasal media) were added, as needed, for calcium imaging studies on day 3, lentiviral vectors for hM3Dq-mCherry for DREADDs stimulation of excitatory neurons, and neuroligin-3 were added to the excitatory neuron cultures at 100 μl ml−1 in Neurobasal media on day 3. Induced neurons were maintained in Neurobasal media with puromycin and doxycycline until plating.
Device preparation, cell seeding, and maintenance
To prepare microplates for cellular seeding, the majority of the distilled water in all wells was removed, with a small amount (~10 μl per compartment) remaining in each well to preserve hydrophilicity. Microplates were angled toward the UV bulb and sterilized under UV radiation for 1 hour. Afterward, Matrigel™ (Corning) was added to cover the cell culture surface in each well. The devices were placed in the incubator for approximately one hour. Glia isolated from P0 pups were seeded into the wells one day prior (day 4–5) to iN seeding (day 5–6) with a glial cell concentration of approximately 300 cells per mm2. Glial cells were removed from the flask using Trypsin/EDTA and re-seeded into the microdevice in DMEM with 10% FBS and 1% P/S. In the microplate, remaining water was removed from the well and a 15 μl bolus of cells was added to the bottom of each compartment (3200 cells per 15 μl). Approximately one hour was given for the cells to adhere to the surface, after which approximately 200 μl of media was added to the well. This additional step was necessary to ensure cell restriction in their respective compartments.
Induced neurons were removed from the 6 well plate surface using Accutase and plated in a 10 μl bolus of 20 000 iNs per compartment. Prior to adding the 10 μl bolus, all DMEM for glial cell seeding was removed via a pipette. Cells were allowed to attach to the surface for five hours, after which approximately 200 μl of Neurobasal medium with 5% FBS, 1% P/S, 10 ng ml−1 BDNF, 10 ng ml−1 GDNF, 10 ng ml−1 NT3, and 2 μg ml−1 doxycycline was added; care must be taken to avoid cellular crossing over the microchannel wall during seeding. After plating the iNs, 96 well plate cultures began a slow transition from Neurobasal medium to BrainPhys™ medium (Stem Cell Technologies). On day 4 after iN seeding, 50 μl were removed from each well and replaced with 50 μl of BrainPhys™ with L-glutamine and NeuroCult™ SM1 supplements with 1% P/S, 10 ng ml−1 BDNF, 10 ng ml−1 GDNF, 10 ng ml−1 NT3, and 2 μg ml−1 doxycycline. At day 8, 80 μl of media were removed from each well, followed by the addition of 100 μl of BrainPhys™ with supplements. On day 12, 100 μl were removed from each well and replaced with 100 μl of BrainPhys™ media with supplements. After day 12,100 μl were removed from each well and replaced with approximately 250 μl of fresh media. Some wells required the addition of extra media during each feeding due to their plate location resulting in greater media evaporation. Devices were cultured for 5–7 weeks prior to analysis. Mature cultures expressing GCaMP6f projected axonal bundles through the microchannels (Fig. 3).
Fig. 3.
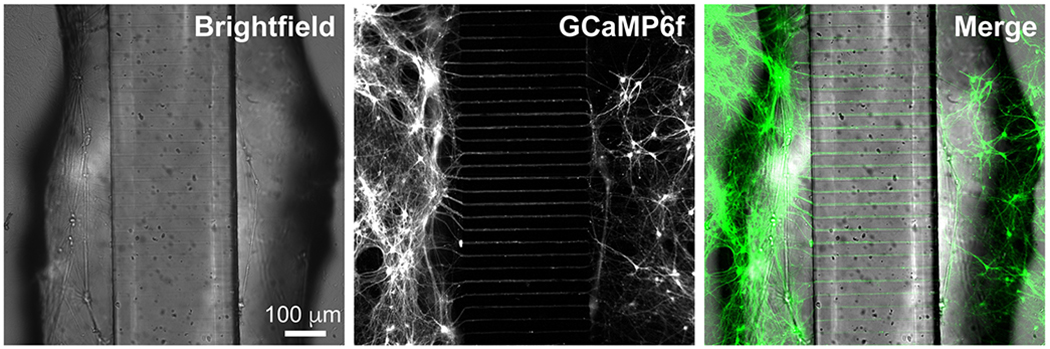
Axons project through microchannels. Brightfield image shows device structure. Shadows are produced from holes punched in PDMS. GCaMP6f-positive neurons project axons through the microchannels between the two compartments.
Immunocytochemistry
Each neuronal subtype was verified via immunocytochemistry. Cells were fixed in 4% paraformaldehyde for 15 minutes, and then permeabilized with 0.2% Triton X-100 for 10 minutes, followed by blocking buffer (2% normal goat serum, 4% bovine serum albumin in PBS) for 1.5 hours. Primary antibodies used were anti-synapsin (E028, a gift from the Sudhof lab, 1:3000), mouse anti-vGlut2 (Synaptic Systems,1: 250), anti-MAP2 (Sigma M1406, 1:1000, Millipore 3418, 1:200, AB5543 1:500), and rabbit anti-vGAT (Millipore, AB5062P, 1: 500). Primary antibodies were incubated overnight at 4 °C. AlexaFluor conjugated species matched secondary antibodies were incubated at room temperature for 2 hours.
Calcium imaging
Calcium imaging experiments were performed using the IN Cell Analyzer 6000 (GE Healthcare Life Sciences). Media was removed from each well in the 96 well plate with a volume of 10 μl remaining. To achieve a final volume of 190 μl, 180 μl of HEPES buffer was added to the culture. For liquid handling operations, 10 μ! was removed from the matching well of the reagent plate and added to the 190 μl in the analysis plate. For the addition of KCl, 80 μl of “High KCl” HEPES buffer (i.e., HEPES buffer where Nad was exchanged for Kd for a final Kd concentration of 145 mM) was added to the 200 μl buffer in the analysis plate to yield a final concentration of 41.4 mM. CNO or clozapine were added to the cultures at varying concentrations for comparison. Expression of glutamate receptor AMPA was investigated and CNQX, a commonly used competitive AMPA receptor antagonist, was used to silence activity due to glutamatergic inputs from excitatory neurons in the culture. CNQX was added to the cultures during experiments at a final concentration of 20 μM. Images were collected at approximately 9 Hz. Reagents were dispensed at frame 900 for CNO or clozapine treatment or at frame 1200 for CNQX addition. After an additional 1500–1800 frames were collected, the analysis was complete, and the reagent plate was switched out for a plate containing High KCl buffer. An additional 600 or 900 frames were collected after the plates were exchanged, with a transfer of 80 μl KCl occurring at frame 300 of the additional set. The KCl addition depolarized all neuronal cells and verified cell viability at the end of each experiment.
For both CNQX and CNO experiments, a blank of HEPES buffer was added as a control, and the orientation of excitatory and inhibitory neurons in the well was altered to control for the position of each cell type within the device.
Clozapine-N-oxide dose response for chemogenetic interventions of neuronal network
Clozapine-N-oxide (CNO) and clozapine were added to microwells containing DREADD-infected excitatory neurons and DREADD-negative excitatory neurons, as a control. CNO doses ranged from 500 nM to a final concentration of 0.8 nM, consistent with previous studies.29,30 Varying concentrations of CNO were added to different plate locations to avoid plate bias. The reagent plate contained CNO concentrations that were 20× the final, due to the 20× dilution in the media after addition (Table 1).
Table 1.
CNO dose response experimental concentrations and setup
| Reagent plate [CNO] (nM) | Final [CNO] (nM) |
|---|---|
| 10 000 | 500 |
| 2000 | 100 |
| 400 | 20 |
| 80 | 4 |
| 16 | 0.8 |
| 0 | 0 |
In addition to CNO, we also investigated the effects of 500 nM clozapine on the circuit in both hM3Dq-infected cultures and non-infected cultures as a control for CNO stimulation.
Image processing
To quantify network activity, raw TIFF images of GCaMP6f fluorescence were collected at 9 Hz (frames per second) using the IN Cell Analyzer with a GFP bandpass filter. Using image processing software FIJI, images were stacked, concatenated, and uniformly contrast adjusted (to 85 and 535 intensity values). AVI videos were saved at 70 Hz. For region of interest (ROI) analysis, ROIs were drawn and stored in ImageJ’s ROI manager. The Multi Measure function in ImageJ was used to collect signal intensity information. To process the data as a ratio of signal over baseline, a custom MATLAB script which collects the lowest fluorescence intensity value from the first 200 frames, represented as F0, was used. Fluorescence signals were then normalized using the ratio calculation (F – F0)/(Fsat – F0), where F is the measured fluorescence signal within an ROI and Fsat is the highest fluorescence intensity value following KCl addition. To investigate signal change after CNO/clozapine addition, two 90 second windows, equivalent to 818 frames, on either side of the addition were considered. The first 90 second window lasted from frame 80 to 898. The second window after the addition of CNO/clozapine lasted from frame 1580 to 2398, which are the latest time points of the pre- and post- CNO addition timelines. Ten cells from both the excitatory and inhibitory sides were then individually analyzed.
Synapse formation assay in neurons with overexpression of wildtype neuroligin-3
To validate whether the device can be used for morphometric imaging analyses, we investigated whether we could recapitulate the synaptogenic function of neuroligin-3.31 In the left well compartment, wild-type excitatory neurons were seeded. In the right well compartment, two different excitatory neuronal populations were seeded: wild-type excitatory neurons (no over-expression of NL3) as a control, and neurons with lentiviral infection for wild-type NL3 (overexpression). Images were acquired using the IN Cell Analyzer 6000 (GE Healthcare Life Sciences) with a 40× objective and numerical aperture of 0.6. Three to four wells were used for two batches of culture. Between two and four images were captured as Z-stacks from each side of the well by the IN Cell Analyzer 6000. Images were merged and formatted as maximum intensity projections. Synapse quantification was performed using our previously developed quantification platform Intellicount32 to identify synapsin-positive puncta co-localized with MAP2. Data were expressed as a ratio of right-compartment synapses over left compartment synapses. The left compartment functions as an internal control for the synaptogenic potential of each well (which could vary with cell density, neuronal maturity, etc.) since NL3 is a post-synaptic protein and dendrites do not cross the microchannels, NL3 is not over-expressed in the left side of the chamber.
Statistics
Multi-group comparisons were performed using two-way ANOVA followed by a post hoc Tukey–Kramer test in MATLAB. For synapse formation quantification with Neuroligin-3, a paired student’s t-test was performed between the wildtype and NL3 overexpression groups on puncta density.
Results
Compartmentalized culturing of human iN cells
We have developed a two-chamber multi-well device capable of housing distinct neuronal subtypes in different compartments. The induced neurons were derived from human iPS cell lines, where subtype identities have been extensively characterized elsewhere.11,33,34 First, we wanted to verify that the seeding technique preserved distinct cell compartmentation, since shortly after seeding, neurons share the same medium within the well.
To test that neurons remain in their distinct compartments, one excitatory neuronal population was seeded against another. Neurons in the left chamber were infected with lentivirus encoding tdTomato prior to seeding, to aid in visualization of their position within the two-compartment device. Cells were given five hours to attach before filling the well with fresh media. After approximately four weeks in culture, tdTomato-labeled axonal projections were seen traversing the microchannels. Importantly, tdTomato-labeled neuronal cell bodies were retained on the left side of the device (Fig. S1a and b†). Examination of an entire well revealed that the seeded neurons grow uniformly within the entire chamber, but do not migrate into the adjacent chamber (Fig. S1c†).
Neuroligin-3 overexpression used for morphological assay
We conducted a morphological assay on human neuronal cells in our platform using the IN CeII Analyzer. Previous studies have found that neuroligin-3 (NL3), a protein encoded by the NLGN3 gene in humans, plays an important role in facilitating synapse formation and that overexpression of NL3 can increase synapse number in mouse neurons.31 Due to the fact that mutations in human neuroligin genes are implicated in neurodevelopmental disorders such as autism, we sought to use the role of NL3 on synapse formation as a paradigm to demonstrate the utility of the 96 well plate microdevice in performing a synaptogenesis/morphological assay. We used lentiviruses to overexpress NL3-WT in the excitatory neurons while control iPS cell lines were infected to produce excitatory neurons. Devices were cultured for at least 5 weeks prior to staining for MAP2 and synapsin. We used Intellicount to identify synapsin-positive puncta co-localized within a defined region of MAP2 signal (1.5 μm away from dendrite), used as the metric for synapse number. The quantification reproduced a trend consistent with previous literature, showing an increase in puncta density with NL3 overexpression (Fig. 4). This experiment demonstrates the utility of the 96 well device in performing morphological assays within the IN Cell Analyzer 6000 for high-throughput investigation of specific neuropathological mechanisms.
Fig. 4.
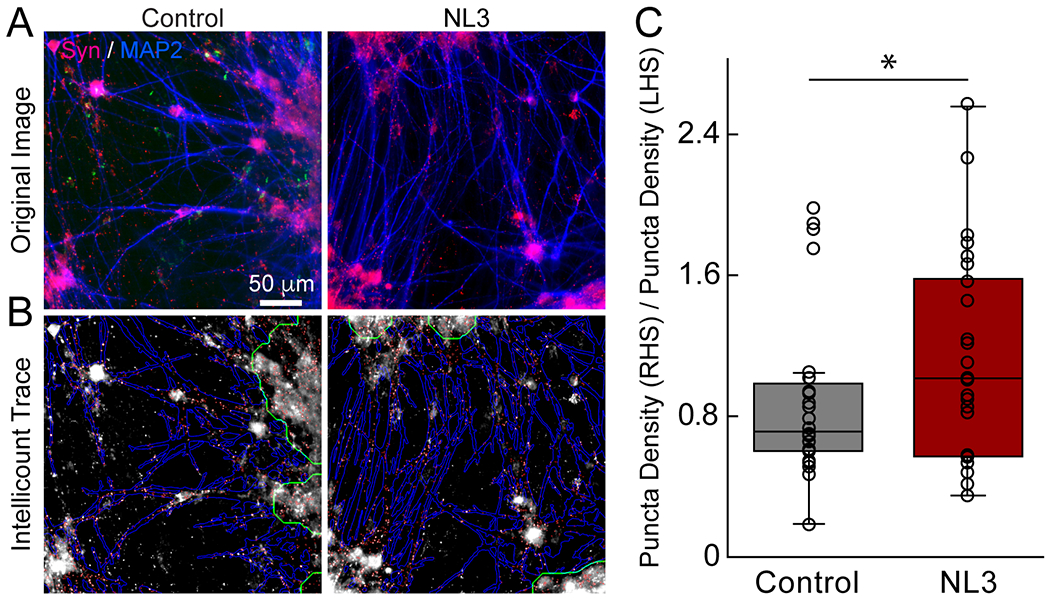
Comparison of synaptic puncta number between different neuroligin-3 conditions. A) Images taken from right compartment of microwell. Wells were fixed and stained for synapsin (red) and MAP2 (blue). B) Puncta and neurite traces obtained by Intellicount synapse quantification software. C) Ratio of synaptic puncta in right-hand side (RHS) compartment to left-hand side (LHS) compartment. A total of seven wells in two different plates was used for each condition (7/2). A one-tailed t-test was used to test for significance between groups (p < 0.04).
Induced neurons display synchronized circuit activity between compartments
Next, we sought to determine the functionality of the circuit formed between the two chambers. To do so, we paired excitatory neurons with inhibitory neurons in each well. Both neuronal populations were infected with GCaMP6f, which enables visualization of changes in intracellular free calcium levels via fluorescence intensity as a metric for cellular activity/neuronal firing. To use calcium imaging as a high-throughput readout, it is convenient to have access to a semi-/fully-automated system such as the IN Cell Analyzer for data collection. To capture both chambers in the excitatory-inhibitory circuit, we used 2048 × 2048 images to capture the 500 μm width of the microchannels and approximately an additional 200 μm on either side of the wall. This provided a large enough number of cells within the field of view to simultaneously record network activity populations of human neurons. Baseline activity was captured for 2:12 minutes (1 frame captured every 0.11 seconds), after which the AMPA receptor blocker CNQX was added to the chamber at a final concentration of 20 μM to suppress excitatory synaptic currents and an additional 3:51 minutes were recorded. During the recording of basal activity, neurons in both chambers demonstrated isolated spontaneous activity, as well as coordinated network activity between and within compartments (Fig. 5 and Video S1†). Importantly, this circuit activity was often coordinated between chambers, indicating that axons traversed the microchannels and formed synaptic contacts with neurons within the opposing chamber (Fig. 5A and B). Excitatory neurons demonstrated low frequency circuit bursts that influenced inhibitory neuron firing. Inhibitory neurons displayed frequent network bursts, with many correlated to the activity of the excitatory neurons. After CNQX application, excitatory neuronal firing was no longer able to coordinate large circuit bursting activity (Fig. 5C), with only one exception, and only after 3 minutes of no bursting activity. To confirm that the effect was due to CNQX and not mechanical disruption of the cells from reagent dispensing, we added 10 μl HEPES (vehicle) as a negative control. When vehicle-only was added, the burst calcium spikes likely driven by the excitatory neurons were largely unaffected (Fig. 5D). This experiment indicates that synapses were formed between the excitatory neurons in one compartment and inhibitory neurons in the connected compartment, and that calcium signals can be recorded as an indicator of functional network activity.
Fig. 5.
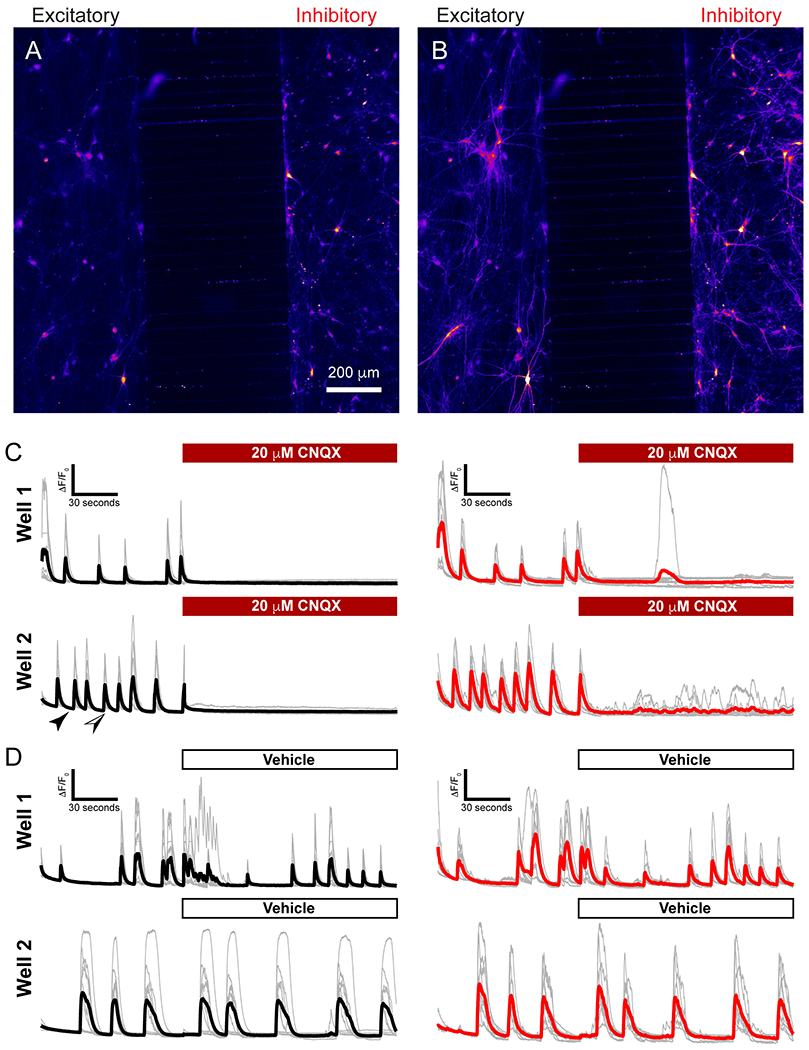
Spontaneous circuit activity and disruption by CNQX. A and B) Frames taken from calcium imaging of device well. Excitatory neurons cultured on left side, inhibitory neurons on right side. Frames correspond to specific times indicated in C): Black arrow corresponds to time at frame in A, and black/white arrow corresponds to frame B. C and D) Traces acquired from representative cells in two different wells. Average trace for excitatory neurons shown in black, with gray traces indicating the cells averaged. Inhibitory neurons shown in gray, with a red average trace. CNQX or vehicle added after 222 seconds. Traces are represented as ΔF/F0.
Utilization of chemogenetics and synthetic compounds to demonstrate drug screening capability
Next, we aimed to further demonstrate the capacity for perturbing the in vitro neural circuitry function with different concentrations of small molecules or drugs. We applied a chemogenetic approach by expressing hM3Dq DREADDs in human neurons and applying the synthetic molecule clozapine-N-oxide (CNO), a synthetic agonist for hM3Dq.29 We infected excitatory neurons with hM3Dq and paired them within the circuit model with inhibitory neurons expressing GCaMP6, which were not infected with the hM3Dq lentivirus. Excitatory and inhibitory neurons were also cultured on coverslips for morphological analysis of their subtype identity (Fig. S2†). CNO (at varying concentrations), clozapine (at 500 nM), and vehicle (HEPES) were added to the system to measure network stimulation (Fig. 6). Calcium imaging was performed in the same manner as described above. Basal activity was recorded for 100 seconds, after which the CNO drug was added. An additional 2:45 minutes were then recorded to capture the effect of the drug. Excitatory neurons which were mCherry (and therefore hM3Dq) positive showed a strong response to 500 nM CNO addition (Fig. 6A and B) while mCherry-hM3Dq-negative excitatory neurons did not show any signs of signal change (Fig. 6C and D).
Fig. 6.
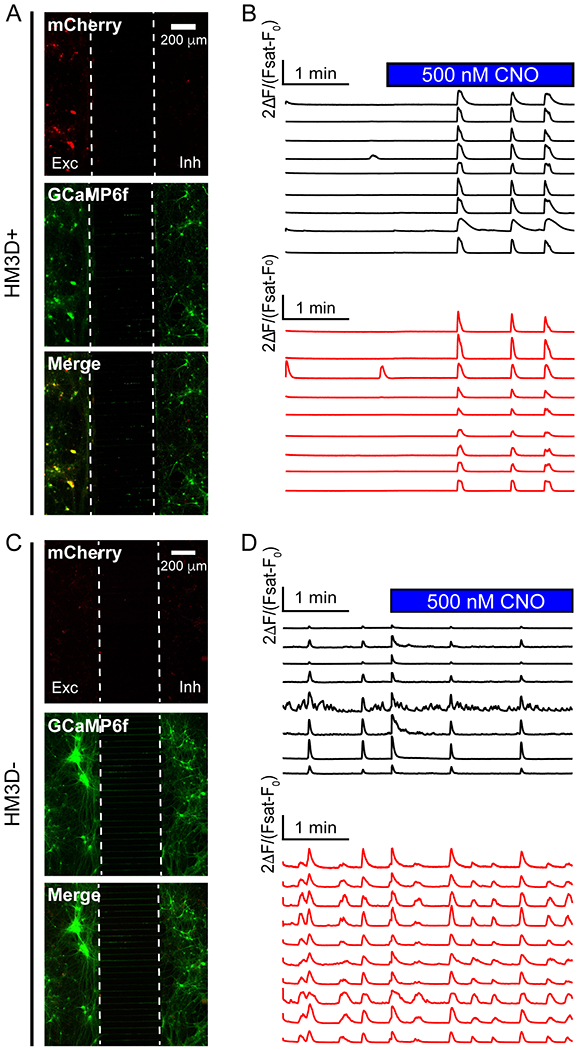
Use of DREADDs to stimulate compartmentalized neural circuits. A) One microwell containing excitatory neurons infected with hM3Dq-mCherry in the left compartment. Co-localization of mCherry and GCaMP6f highlight GCaMP6f-positive neurons containing the hM3Dq-mCherry vector. B) Traces obtained from the well in A). The top black lines represent traces from the excitatory (left side) neurons. The bottom red lines are traces of the inhibitory (right side) neurons. 500 nM CNO was added at frame 900 (99 seconds). Time frame of CNO dosing shown by blue bar. CNO was applied to the entire well. C) A well containing excitatory neurons (left side) and inhibitory neurons (right side) without hM3Dq-mCherry. D) Calcium signals obtained from neurons in well C). The top black lines represent traces from the excitatory (left side) neurons. The bottom red lines are traces of the inhibitory (right side) neurons. All calcium traces are expressed as ΔF/(Fsat – F0) where Fsat is the maximum intensity after KCl addition (data not shown).
For calcium signal quantification, the number of individual peaks were difficult to quantify, since there were either large bursts of activity spanning tens of seconds (likely the result of trains of action potentials) or no network bursts seen prior to CNO addition. When there are no peaks prior to CNO addition, it is impossible to establish a ratio of firing based on peak number. Therefore, we used the area under curves (AUC) to quantify the degree of total network activity. To normalize all signals to the baseline, a custom-made MATLAB script containing both baseline subtraction by least-squares fitting was performed followed by intensity curve area calculation using the trapezoid rule. The MATLAB script prints an output of the intensity curve areas before and following CNO addition (highlighted in red and blue, respectively, Fig. 7A and B). Results were processed as (fluorescence curve area of post-CNO)/(area of pre-CNO) ratio, determined in the analysis time windows over all conditions (Fig. 7C). While there was variation in the area ratio between wells of the same condition, the CNO concentrations of 500 nM and 100 nM showed significantly (p < 0.001) higher total bursting activity following CNO addition compared to hM3Dq negative and vehicle controls in both the excitatory and inhibitory neuronal chambers (Videos S2–S4†). Other CNO concentrations show a trend of effect that did not reach statistical significance. Clozapine at 500 nM did show an increase in area but was not significantly different from the vehicle control (HEPES only). Importantly, high concentrations of CNO showed significant differences from both vehicle-treated hM3Dq-positive cells and 500 nM CNO-treated hM3Dq-negative cells (Fig. 7C). We generated a dose response curve for both excitatory and inhibitory neurons within the device (Fig. 7D). hM3Dq-negative inhibitory neurons showed a similar increase in activity with increasing CNO concentration, indicating the presence of excitatory input via synaptic connections across the microchannels. Higher CNO concentrations also showed significant differences compared to vehicle and hM3Dq-negative cells in inhibitory neurons (Fig. 7C and D). This experiment indicates that functionality of neurons within a defined neurocircuitry can be assayed in a high-throughput manner using our platform.
Fig. 7.
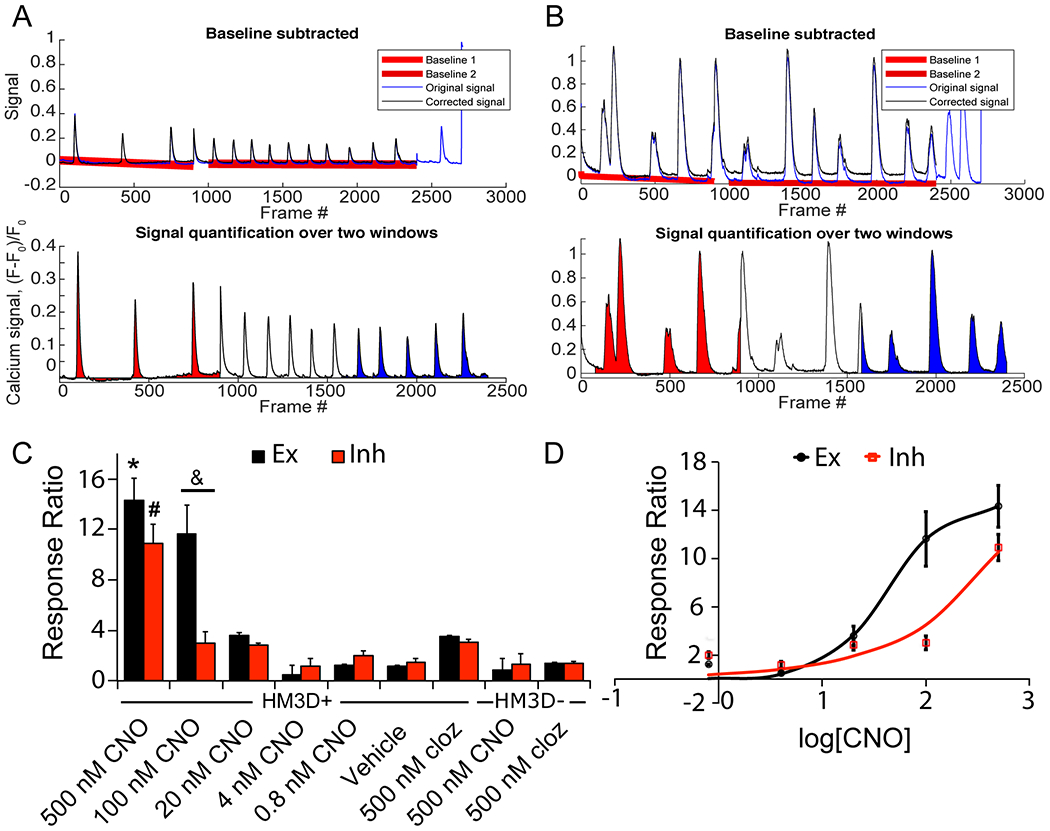
Quantification of hM3Dq/CNO dose response within excitatory and inhibitory neurons. A) A custom MATLAB script was used to perform signal scaling and baseline subtraction using a least-squares fit. Original signal displayed in blue, the baseline-corrected signal shown in black. The two baselines are shown in red. Bottom graph shows the quantification of area under the curve (AUC) of pre-drug addition (red) and post-drug addition (blue) of a neuron from an hM3Dq-positive well. B) The same process applied to an inhibitory neuron in an hM3Dq-negative well. C) Area ratio quantification for excitatory neurons (black bars) and inhibitory neurons (red bars). Ten cells were processed from 3 to 4 wells over 2 plates. * indicates p < 0.001 for comparisons between excitatory concentrations, # for p < 0.001 for inhibitory concentrations, and & for p < 0.001 for comparisons between excitatory and inhibitory neurons at the same concentration. D) Dose–response curve generated from area data. Black line corresponds to excitatory neuron area ratios and red line to inhibitory neurons.
Discussion
The advent of technology for inducing defined neuronal subtypes from human cells has provided an important step toward understanding and modeling neuropsychiatric diseases. Progress in this understanding is still greatly limited and heavily based on non-primate animal models or mixed two-dimensional human neuronal cultures lacking defined connectivity. Animal models have provided clues regarding possible circuitry and mechanisms involved in human disease; however, they have not delivered a high level of successful translation of scientific findings into new therapeutics. To bridge this gap, better modeling systems are greatly needed. We have developed a high-throughput compartmentalized system for human neurocircuitry studies, which builds upon previous approaches using microchannels to segregate cell bodies and enable axons to relay cell to cell communication between chambers.2–4,7,8,11 Our device has two compartments built into each well within a 96 well plate to allow HTS of therapeutic candidates within a well-defined, simplified neurocircuitry. Neurons seeded into the device remain in distinct compartments up to 7 weeks in culture after plating (Fig. 3) and could enable morphological analysis of cultures (Fig. 4). Importantly, these neurons establish functional synaptic contacts and coordinated network activity between compartments (Fig. 5–7).
Since we designed the device to function as a screening system for therapeutic development, we tested a specific receptor-ligand interaction. We purposefully chose a receptor that would be present in neurons on only one side of the device in order to measure a purely post-synaptic response. To control the cells we were stimulating, we utilized DREADDs, specifically the combination of hM3Dq/CNO. Only excitatory neurons infected with mCherry-hM3Dq showed the presence of mCherry throughout the compartment, indicating successful cloning of the hM3Dq-mCherry sequence, as well as the ability to co-culture infected excitatory iNs with non-infected inhibitory iNs in the same microwell. DREADD-positive neurons demonstrate a robust response to high concentrations of CNO, but little to no reaction to doses lower than 100 nM (Fig. 7). These results reveal the utility of the system as a screening platform for neuroactive compound screening. Neuronal circuits can be parallelized in the microplate and several different conditions (in this case, drug concentrations) can be tested in distinct wells. Expansion of this approach to multiple plates can further extend the opportunity to screen different drugs and different cell lines in a micro-circuit context.
In addition to testing various CNO concentrations, we also screened the circuit response to clozapine, the metabolite of CNO. Gomez et al. recently demonstrated the differences between clozapine-N-oxide (CNO) and clozapine, a metabolite of CNO.30 They demonstrated, using both human embryonic kidney cells (HEK293) in vitro and mouse in vivo experiments, that clozapine has much greater reactivity and affinity for the hM3Dq receptor than CNO. Gomez et al. argue that CNO likely only reacts with hM3Dq at high concentrations due to its ability to convert to significant clozapine levels for binding hM3Dq. However, in our system, we only observed a minimal, non-significant, increase in activity when a high concentration (500 nM) of clozapine was added to our cultures compared to a large stimulation following an addition of 500 nM CNO. For the efficacy of CNO on activation of neuronal activity in neurons expressing hM3Dq, we were able to estimate a half maximal effective concentration (EC50) of approximately 30 nM based on our dose-response curve. This is within the same order of magnitude as the EC50 of 85 ± 17 nM established in HEK293 cells, reported by Gomez et al.30 These experiments demonstrated the formation of a neural circuit model within a microdevice that can be subjected to HTS assays conducted within an automated instrument.
Further development of this device involves creating a taller vertical barrier between the adjacent compartments within each microwell to allow for distinct media reservoirs. One advantage of compartmentalized devices is to unbalance the liquid heights between compartments so that individual chambers can be treated with distinct chemicals without diffusion between chambers.5,6 Creation of a thicker PDMS insert into the microplate would extend the wall between compartments, allowing these individual treatments. To facilitate automation in the image processing of neural activity data, the incorporation of an automated ROI tracer should be explored in future work.35 Although the proposed platform provides a powerful means for automated HTS of therapeutic reagents in established neurocircuits, introducing an automated method for subsequent analysis would further enhance its usability in handling large datasets beyond manual and semiautomated methods.
We have demonstrated the development and application of a novel HTS platform for the investigation of human neuronal circuitry function with defined connectivity patterns in vitro. We have shown that defined neuronal subtypes can form functional synaptic connections and actively communicating neural networks, and that manipulation of neuronal activity in upstream neurons can be functionally correlated with the activity of downstream neurons receiving synaptic inputs. This synaptic relay of information and coordination of neural circuit activity can be pharmacologically modulated and has been validated with the use of human neurons derived from stem cells in our high-throughput platform. Thus, this device is well suited for drug screening applications. Future work extending this approach would involve the use of neurons derived from individuals with different genetic backgrounds and their perturbation with pharmacologic agents aimed to target disease-relevant mechanisms in order to provide testing of “personalized” drug therapies and drug efficacy improvements. Thus, future work using this platform will capitalize on the combined benefits of novel human stem cell technology and microfluidic device engineering to facilitate enhanced drug screening by placing cells within the appropriate circuit context and retaining the high-throughput capacity needed for pharmacological discovery.
Conclusion
Using microfluidics and human iN cell technology, we have created a high-throughput screening platform and culturing device for simplified two-way neuronal circuit studies using human stem cell derived neurons. Within the platform, human iN cells form functional circuits which can be studied using calcium imaging and morphological analyses in high-throughput imaging systems such as the IN Cell Analyzer 6000. The impact of small molecules on neurocircuitry function can be efficiently probed in a parallelized manner using automated liquid handling and confocal microscopy. Thus, we have provided a proof-of-concept device and approach that can be further developed into circuitry-based models for studying human neuropsychiatric disorders. We hope that this platform will advance therapeutic developments targeting neuronal circuit function and provide a conceptual and technical framework for furthering novel cell-based assays and platforms in the treatment of neuropsychiatric disorders.
Supplementary Material
Acknowledgements
We thank our funding sources: the National Institute on Drug Abuse (NIDA) awards 5R21DA039686 and 5R21DA035594. VRM is supported by the Ruth L. Kirschstein Institutional National Research Service Award from National Institute of Mental Health (NIMH) (F30 MH108321). DAR is supported by the Rutgers University Biotechnology Training Program (T32 GM008339). The Child Health Institute of New Jersey is funded by the Robert Wood Johnson Foundation Grant #74260. We also thank Drs. Kelvin Kwan and Noriko Kane-Goldsmith for their assistance with the IN Cell Analyzer 6000 and the CBN Imaging Facility.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc01179j
Conflicts of interest
There are no conflicts to declare.
References
- 1.Kesselheim AS, Hwang TJ and Franklin JM, Two decades of new drug development for central nervous system disorders, Nat. Rev. Drug Discovery, 2015, 14, 815–816. [DOI] [PubMed] [Google Scholar]
- 2.Park JW, Vahidi B, Taylor AM, Rhee SW and Jeon NL, Microfluidic culture platform for neuroscience research, Nat. Protoc, 2006, 1, 2128–2136. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AM and Jeon NL, Microfluidic and Compartmentalized Platforms for Neurobiological Research, Crit. Rev. Biomed. Eng, 2011, 39, 185–200. [DOI] [PubMed] [Google Scholar]
- 4.Taylor AM, et al. , Microfluidic Multicompartment Device for Neuroscience Research, Langmuir, 2003, 19, 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fantuzzo JA, Hart RP, Zahn JD and Pang ZP, Compartmentalized devices as tools for investigation of human brain network dynamics, Dev. Dyn, 2019, 248(1), 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor AM, et al. , A microfluidic culture platform for CNS axonal injury, regeneration and transport, Nat. Methods, 2005, 2, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renault R, Durand JB, Viovy JL and Villard C, Asymmetric axonal edge guidance: a new paradigm for building oriented neuronal networks, Lab Chip, 2016, 16, 2188–2191. [DOI] [PubMed] [Google Scholar]
- 8.Renault R, et al. , Combining microfluidics, optogenetics and calcium imaging to study neuronal communication in vitro, PLoS One, 2015, 10, e0120680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deleglise B, et al. , Synapto-protective drugs evaluation in reconstructed neuronal network, PLoS One, 2013, 8, e71103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyrin JM, et al. , Axon diodes for the reconstruction of oriented neuronal networks in microfluidic chambers, Lab Chip, 2011, 11, 3663–3673. [DOI] [PubMed] [Google Scholar]
- 11.Fantuzzo JA, et al. , 4Neurocircuitry: Establishing in vitro models of neurocircuits with human neurons, Technology, 2017, 5, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, et al. , TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, E5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virlogeux A, et al. , Reconstituting Corticostriatal Network on-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease, Cell Rep, 2018, 22, 110–122. [DOI] [PubMed] [Google Scholar]
- 14.Darville H, et al. , Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome, EBioMedicine, 2016, 9, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desbordes SC and Studer L, Adapting human pluripotent stem cells to high-throughput and high-content screening, Nat. Protoc, 2013, 8, 111–130. [DOI] [PubMed] [Google Scholar]
- 16.McNeish J, et al. , High-throughput screening in embryonic stem cell-derived neurons identifies potentiators of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate-type glutamate receptors, J. Biol. Chem, 2010, 285, 17209–17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haggarty SJ and Perlis RH, Translation: screening for novel therapeutics with disease-relevant cell types derived from human stem cell models, Biol. Psychiatry, 2014, 75, 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theodorou E, et al. , A high throughput embryonic stem cell screen identifies Oct-2 as a bifunctional regulator of neuronal differentiation, Genes Dev, 2009, 23, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen T-W, et al. , Ultra-sensitive fluorescent proteins for imaging neuronal activity, Nature, 2013, 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith KS, Bucci DJ, Luikart BW and Mahler SV, DREADDS: Use and application in behavioral neuroscience, Behav. Neurosci, 2016, 130, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H-M, Giguere PM and Roth BL, DREADDs: novel tools for drug discovery and development, Drug Discovery Today, 2014, 19, 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferguson SM and Neumaier JF, Grateful DREADDs: engineered receptors reveal how neural circuits regulate behavior, Neuropsychopharmacology, 2012, 37, 296–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aran K, et al. , Microfiltration platform for continuous blood plasma protein extraction from whole blood during cardiac surgery, Lab Chip, 2011, 11, 2858–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson DG, et al. , Enzymatic assembly of DNA molecules up to several hundred kilobases, Nat. Methods, 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 25.Tiscornia G, Singer O and Verma IM, Production and purification of lentiviral vectors, Nat. Protoc, 2006, 1, 241–245. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. , Rapid single-step induction of functional neurons from human pluripotent stem cells, Neuron, 2013, 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore JC, Sheldon MH and Hart RP, Biobanking in the era of the stem cell: a technical and operational guide: The Use of iPSC to Study Genetic Diseases in Mental Health Disorders, Morgan-Claypool Publishers, 2012. [Google Scholar]
- 28.Davies SP, Reddy H, Caivano M and Cohen P, Specificity and mechanism of action of some commonly used protein kinase inhibitors, Biochem. J, 2000, 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guettier JM, et al. , A chemical-genetic approach to study G protein regulation of beta cell function in vivo, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 19197–19202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez J, et al. , Chemogenetics revealed: DREADD occupancy and activation via converted clozapine, Science, 2017, 357, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chih B, Afridi SK, Clark L and Scheiffele P, Disorder-associated mutations lead to functional inactivation of neuroligins, Hum. Mol. Genet, 2004, 13, 1471–1477. [DOI] [PubMed] [Google Scholar]
- 32.Fantuzzo JA, et al. , Intellicount: High-Throughput Quantification of Fluorescent Synaptic Protein Puncta by Machine Learning, eNeuro, 2017, 4, ENEURO.0219-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang N, et al. , Generation of pure GABAergic neurons by transcription factor programming, Nat. Methods, 2017, 14, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oni EN, et al. , Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele, Sci. Rep, 2016, 6, 34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukamel EA, Nimmerjahn A and Schnitzer MJ, Automated analysis of cellular signals from large-scale calcium imaging data, Neuron, 2009, 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.