

Abstract
The ESCRT complexes drive membrane scission in HIV-1 release, autophagosome closure, MVB biogenesis, cytokinesis, and other cell processes. ESCRT-I is the most upstream complex and bridges the system to HIV-1 Gag in virus release. The crystal structure of the headpiece of human ESCRT-I comprising TSG101–VPS28–VPS37B–MVB12A was determined, revealing an ESCRT-I helical assembly with a 12 molecule repeat. Electron microscopy confirmed that ESCRT-I subcomplexes form helical filaments in solution. Mutation of VPS28 helical interface residues blocks filament formation in vitro and autophagosome closure and HIV-1 release in human cells. Coarse grained simulations of ESCRT assembly at HIV-1 budding sites suggest that formation of a 12-membered ring of ESCRT-I molecules is a geometry-dependent checkpoint during late stages of Gag assembly and HIV-1 budding, and templates ESCRT-III assembly for membrane scission. These data show that ESCRT-I is not merely a bridging adaptor, but has an essential scaffolding and mechanical role in its own right.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
The Endosomal Sorting Complexes Required for Transport (ESCRTs) catalyze so-called “reverse topology” membrane scission events, defined as those in which budding is directed away from the cytosol1,2. Such events include, but are not limited to, multivesicular body (MVB) biogenesis, HIV-1 particle egress3,4; cytokinesis5; autophagosome closure6–8; and nuclear envelope (NE) reformation9,10. The core ESCRT machinery consists of the ESCRT-I, ESCRT-II and ESCRT-III complexes, ALIX, and the AAA+ ATPase VPS4. ESCRT-III and VPS4 are considered to be the minimal machinery for membrane scission, in that they can carry this reaction out in vitro in the absence of the upstream components11. The ESCRT-IIIs are a family of small alpha helical proteins that can self-assemble into membrane-associated, homo- and hetero-polymeric structures. These polymeric assemblies can adopt many different morphologies, including cones12, coils13, flat spirals13–17, and helical tubes12,14,18,19. The subunits within ESCRT-III polymers are exchanged with cytosolic monomers by VPS420, leading to scission11.
Viral proteins and host cargoes are recognized by the upstream ESCRT-I complex21–24 or ALIX25,26. ESCRT-I in turn recruits ESCRT-II and then CHMP6, the most upstream of the ESCRT-III proteins. ALIX, and its paralog HD-PTP, provide a parallel route for ESCRT-III recruitment via CHMP427–30. It is still unclear how ESCRT-I/-II and ALIX orchestrate ESCRT-III polymerization for scission. Each of these complexes are rigid and elongated, and their physical dimensions approximate those of the membrane necks with which they function. A central question is whether ESCRT-I (and other upstream ESCRTs) merely bridges cargo and the downstream ESCRTs, or whether it has a more specific and active mechanical role in scaffolding membrane remodeling and scission.
The critical Pro-Thr/Ser-Ala-Pro (PTAP) motif of HIV-1 Gag is recognized by the ubiquitin E2 variant (UEV) domain of TSG10131–34. Human ESCRT-I is a heterotetrameric complex consisting of a 1:1:1:1 complex of (a) TSG101, (b) VPS28, (c) one of VPS37A, B, C, or D, and (d) one of the MVB12A, MVB12B, UBAP1, UBA1L, or UMAD135–40. The structure of the human core has not been determined, but TSG101, VPS28, and VPS37A-D have enough sequence homology to yeast Vps23, Vps28, and Vps37, respectively, to infer that similar general principles apply.
The crystal structure of the yeast ESCRT-I complex consisting of Vps23, Vps28, Vps37, and Mvb12 has an 18 nm rod-like core35. With the inclusion of linkers and non-core domains, the yeast assembly spans up to 22 nm41. The UEV domain is connected by a ~90 residue Pro-rich linker to the stalk, which is followed by the fan shaped headpiece35,42,43. The headpiece and stalk are collectively referred to as the core. The headpiece is capped by VPS28, which has a C-terminal alpha-helical bundle (VPS-CTD) that binds directly to the ESCRT-II complex and is distal to the UEV cargo recognition end35,42,44–46. Yet, despite the similar function and nomenclature, human MVB12A/MVB12B and the other UBAP1-MVB12A associated (UMA) domain proteins37 have no sequence homology to yeast Mvb12. They are integrated into ESCRT-I via their UMA domain39,47. Thus, with no reported structures including UMA sequences, nothing is known at the structural level about how the UMA-containing subunits are incorporated into ESCRT-I. This has left a major gap in understanding how human ESCRT-I complexes are organized.
Here we describe the structure of the human ESCRT-I headpiece including VPS37B and MVB12A. The structure reveals that human MVB12A and yeast Mvb12 bind to equivalent regions of the ESCRT-I head, albeit with altered conformations, and that MVB12A occludes the TSG101 PTAP motif. Upon determining this structure, we made the unexpected observation that the ESCRT-I head self-assembles into helical tubes within the crystal. This has led us to propose a model wherein cargo-dependent clustering of ESCRT-I triggers helix formation providing a nucleating platform upon which the downstream ESCRTs can assemble into membrane-deforming polymers. By mutating filament contact residues, we were able to test this model in HIV-1 release and autophagosome closure. These observations show that ESCRT-I is not merely an adaptor, but forms highly organized assemblies that template the initiation of membrane remodeling.
Results
Structure of the MVB12A-containing human ESCRT-I headpiece
Sequence alignments of metazoan MVB12 homologs showed that their UMA contains two distinct segments of conservation in its N- and C-terminal regions, respectively (Fig. 1a). We found that a 22-amino acid fragment of MVB12A (206–228; Fig. 1b) was sufficient to pull down the entire human ESCRT-I headpiece (Fig. 1c). This region is the most N-terminal of the two conserved segments and will subsequently be referred to as ‘UMA-N’. On the basis of this information, we co-expressed MVB12A UMA-N in complex with the trimeric human ESCRT-I (TSG101–VPS28 –VPS37B) headpiece. We determined the crystal structure of the resulting ESCRT-I tetramer at 2.2 Å resolution using a search model derived from the Vps23–Vps28 –Vps37 headpiece portion of the yeast ESCRT-I core (Fig. 1d). As expected, these three subunits (TSG101, VPS37B, VPS28) are similar to the yeast structure. The conformation of the novel component, MVB12A, resembles an ‘S’ shape comprised of two short 310 helices followed by an anti-parallel beta-sheet formed with TSG101 (Fig. 1d–g). While yeast Mvb12 binds to the same region, it adopts an α-helical conformation (Fig. 1h)35. Like human MVB12A, this is followed by an antiparallel beta-sheet formed with Vps23, the yeast TSG101 ortholog. Thus the mammalian UMA-N domain allows UMA-containing subunits to incorporate into the same site on the ESCRT-I headpiece as yeast Mvb12 despite the lack of any sequence homology and the differences in conformation.
Fig. 1: Structure of the heterotetrameric human ESCRT-I head.
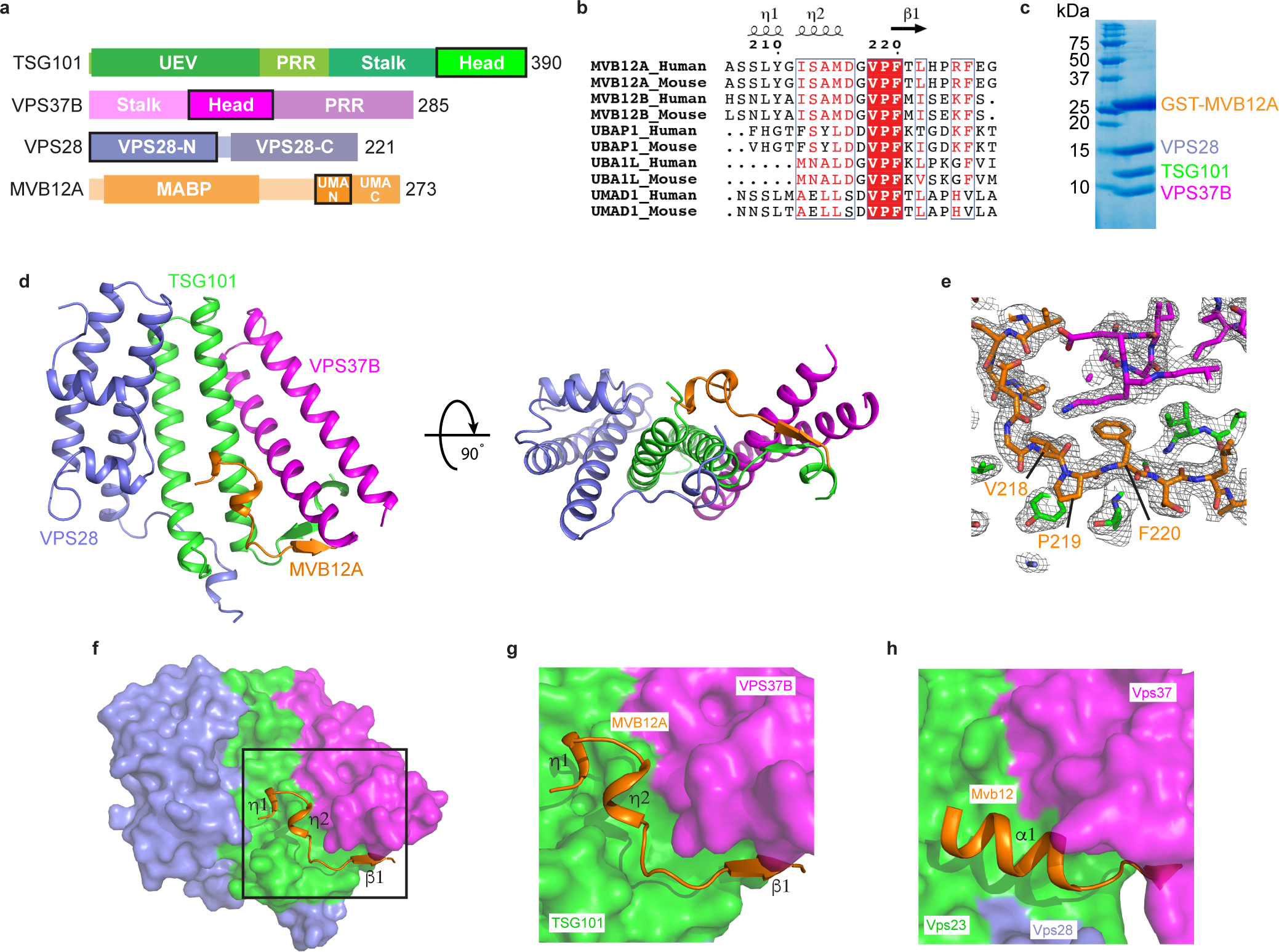
a, Domain architecture of human ESCRT-I subunits. Constructs used for structural analysis are highlighted. b, Sequence alignment of MVB12A paralogs and orthologs. Secondary structure displayed above the alignment is derived from the human ESCRT-I head structure. Alignment was generated using ClustalW and ESPript. c, GST-tagged MVB12A 206–228 was co-expressed with trimeric ESCRT-I head (TSG101 308–388, VPS37B 97–167, VPS28 1–122) in E.coli and analyzed by SDS-PAGE. Lysate was incubated with glutathione beads and complex integrity analyzed by SDS-PAGE following multiple washes. d, Ribbon diagram of the human ESCRT-I head crystal structure. e, MVB12A 2Fo-Fc omit map. MVB12A chain was removed and model re-refined. Resulting 2Fo-Fc map is contoured at 1.0 σ and represented as a grey mesh. MVB12A chain replaced for clarity. f, Surface representation of the human ESCRT-I head with MVB12A shown as an orange ribbon. g, Enlargement of boxed region shown in (f). h, Mvb12 binding site from the previously determined heterotetrameric yeast ESCRT-I head (PDB 2P22). Mvb12, Vps37, Vps23 and Vps28 are colored orange, magenta, green and purple respectively. Uncropped image for panel (c) is in Supplementary Figure 1.
MVB12A interaction network in the headpiece
Within the UMA-N region, the residues VPF comprise a completely conserved bloc (Fig. 1b). The side chains of Val218 and Phe220 of the VPF motif protrude into a deep hydrophobic pocket formed by both TSG101 and VPS37B (Fig. 2a–c). The phenyl group of MVB12A Phe220 is wedged between Pro320 of TSG101 and the aliphatic chains of Lys155 and Lys158 and the side-chain of Val154 from VPS37B. MVB12A Val218 is buried against TSG101 Tyr325, Ile328, and Leu386. Pro219 is held in place by a hydrophobic interaction formed with Tyr325 of TSG101. The side-chain of Lys158 donates multiple hydrogen bonds with the carbonyl groups of residues Met215 and Val218 of the MVB12A main-chain, helping to locking this region of main-chain in place. The contributions of these key residues to MVB12A integration into the headpiece were validated by in vitro pull-downs. Mutation of both TSG101 Tyr325 and VPS37B Lys158 to Asp essentially abolished the interaction with GST-MVB12A as judged by pull down experiments (Fig. 2d and Extended Data Fig. 1). Likewise, mutation of the VPF sequence to AAA also blocked incorporation (Fig. 2d and Extended Data Fig. 1).
Fig. 2: MVB12A VPF motif is required for the interaction with the ESCRT-I head and occludes the TSG101 PTAP motif.
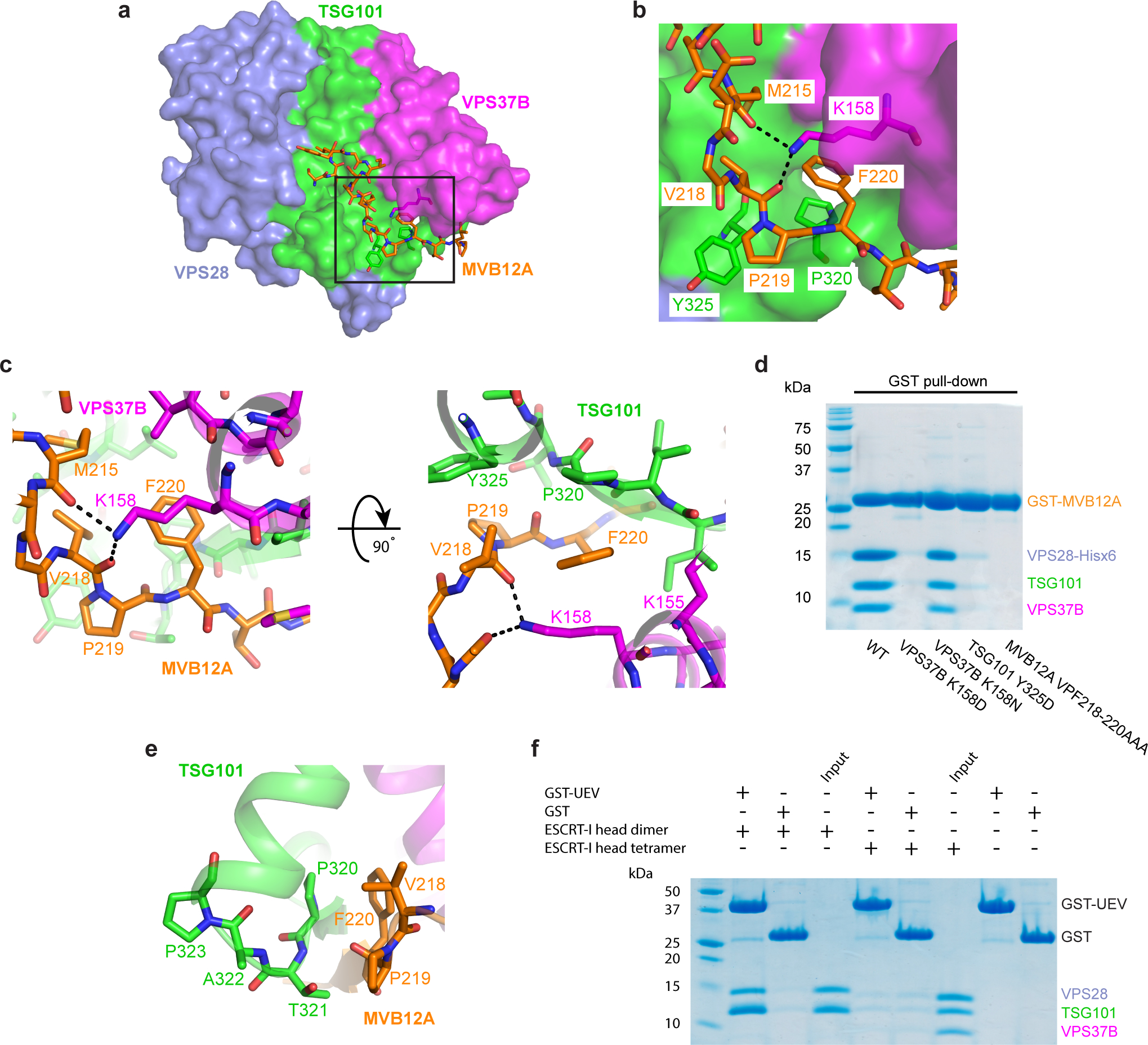
a, Surface representation of the human ESCRT-I head with MVB12A and other key residues shown as sticks. b, Enlargement of boxed region in (a) showing the conserved MVB12A VPF motif binding pocket. TSG101, VPS37B, MVB12A colored green, magenta and orange respectively. Selected hydrogen bonds are shown as dashed lines. c, Stick representation of the MVB12A VPF motif binding pocket. d, Mutant versions of the GST-tagged ESCRT-I head complex were expressed in E. coli. Lysate was incubated with glutathione beads and complex integrity analyzed by SDS-PAGE following multiple washes. e, Interface between TSG101 PTAP motif and MVB12A VPF motif. f, GSH beads were coated with GST-TSG101 UEV or GST bait and incubated with binary (TSG101–VPS28) or tetrameric (TSG101–VPS28 –VPS37B –MVB12A) ESCRT-I head. Beads were washed and complex formation analyzed by SDS-PAGE. The SDS-PAGE gels shown for the pull-down experiments in (d) and (f) represent two independent biological replicates. Uncropped images for panels (d) and (f) are in Supplementary Figure 1.
Vertebrate TSG101 orthologs contain an internal PTAP motif that resides in the ESCRT-I headpiece. The UEV domain is capable of binding to peptides corresponding to the isolated TSG101 PTAP motif48. The human ESCRT-I headpiece structure revealed that access to the PTAP sequence of TSG101 is occluded by the VPF motif of MVB12A (Fig. 2e). The first Pro and the Thr of the PTAP motif form β-sheet hydrogen bonds with the Pro and Phe of the MVB12A VPF motif. These internal interactions made it seem unlikely that the UEV domain could bind to the PTAP motif in the context of this structure. In order to test this possibility, in vitro pull-down experiments were carried out using GST-tagged UEV as bait and various ESCRT-I head assemblies as prey (Fig. 2f). The UEV domain was capable of pulling down a dimeric version of the ESCRT-I head containing TSG101 and VPS28. This suggests that the PTAP motif of TSG101 is accessible to the UEV domain in the absence of MVB12A and VPS37B. Consistent with the observed sequestration of the PTAP motif in the full headpiece structure, the tetrameric version of the ESCRT-I head cannot bind to the UEV (Fig. 2e, 2f).
Helical assemblies of ESCRT-I substructures in crystals and in vitro
Human ESCRT-I headpiece crystals grow in space group P61 and contain six copies of the complex per asymmetric unit (Fig. 3a). This unusual combination of crystallographic and non-crystallographic symmetry elements corresponds to a crystal consisting of parallel helical tubes (Fig. 3b). Each tube is constructed of a series of three interwoven right-handed helices (Fig. 3b). The helices are built from individual ESCRT-I headpiece protomers that encircle a central lumen devoid of electron density. Each continuous helix consists of 12 ESCRT-I headpiece units per turn. The outer and lumen diameters of the assembly are 17 nm and 6.5 nm, respectively. Analysis of the intra-helical interface shows that it is mainly electrostatic in nature and is composed almost exclusively of residues from VPS28 (Fig. 3c and d). Most of these residues are well conserved from yeast to humans (Extended Data Fig. 2). VPS28 Lys54 and Lys58, located approximately midway along helix α1 of VPS28, form multiple salt bridges with acidic residues from the equivalent helix of a distinct copy of VPS28 (VPS28’) (Fig. 3d). Asp59, situated within a flexible loop linking α1 and α2 of VPS28, also forms multiple electrostatic interactions with Arg72 and Gln76 of VPS28’ α2’.
Fig. 3: Helical ESCRT-I head assemblies.
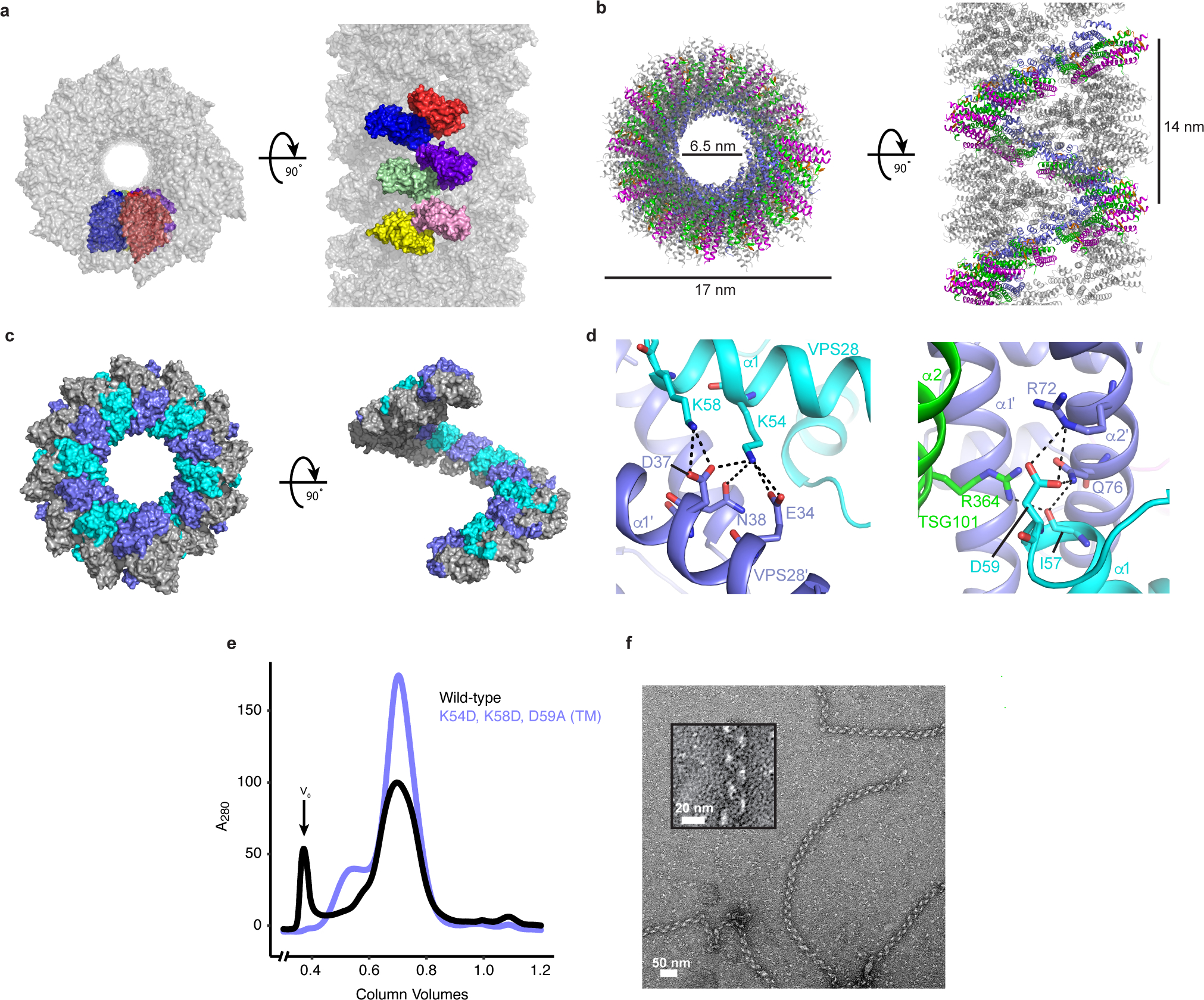
a, The crystal from which the ESCRT-I head structure was determined consists of a series of helical tubes, one of which has been isolated and shown as a surface representation. The crystal exhibits P61 symmetry with 6 copies of the tetrameric ESCRT-I head in the asymmetric unit. An individual asymmetric unit is highlighted and each ESCRT-I head protomer assigned a distinct color for clarity. b, Tube composed of three interleaved ESCRT-I head helices. One continuous helix is colored for clarity. TSG101, VPS28, VPS37B, MVB12A are colored green, purple, magenta and orange respectively. Tube dimensions are labelled. c, Surface representation of a single, continuous ESCRT-I helix. The intra-helical interface is primarily composed of residues from VPS28. Individual VPS28 subunits are colored either purple or cyan in order to clearly distinguish surface boundaries. d, Detailed views of the helical interface highlighting key residues. Inter-protomer bonds depicted with dashed lines. Subunits are colored as in (c). e, Size-exclusion chromatograms of WT and mutant (VPS28 K54D, K58D, D59A) binary (TSG101–VPS28) ESCRT-I head subcomplexes. Proteins were expressed in E.coli and subjected to initial IMAC purification prior to being applied on a size-exclusion column. Void volume (V0) labelled accordingly. Chromatograms are representative of 3 independent biological replicates. f, Electron micrograph of WT ESCRT-I filaments obtained from the pooled V0 fraction shown in (e). Filaments were observed for wild-type but were not observed for the mutant construct in three biological replicates. Zoomed view of a typical WT ESCRT-I filament shown inset.
We noticed that similar interactions occur upon re-examining one of the yeast headpiece structures (PDB 2CAZ) (Extended Data Fig. 3)43. The yeast ESCRT-I crystal is constructed from a series of laterally stacked tubes, each of which is composed of ESCRT-I head protomers arranged in a continuous helix. These tubes exhibit similar dimensions to their human counterparts with an outer diameter of 16 nm, inner diameter of 7.5 nm and an identical number of subunits per turn (12). The inter-protomer interfaces are also remarkably similar, although it was not possible to compare specific side chain interactions at the lower resolution of this yeast headpiece structure. The yeast tubes seen in the crystal contain a single, one-start, helix of ESCRT-I protomers as opposed to the three-start assembly seen in the human ESCRT-I crystal. As a result, the yeast assembly has a reduced helical pitch of 5 nm as compared with 14 nm for the human.
Attempts to purify an ESCRT-I headpiece subcomplex containing TSG101 and VPS28 subunits yielded a high molecular weight species as determined by size-exclusion chromatography (Fig. 3e). Isolation of this fraction and examination by negative stain electron microscopy identifies this higher molecular weight species as long, helical and filamentous in structure (Fig. 3f). These filaments are flexible, which precluded precise calculation of helical parameters and high-resolution cryo-EM structure determination. Nevertheless, the ~20 nm outer diameter of these filaments is consistent, within the uncertainty of the measurement, with that of the human ESCRT-I headpiece filament in the crystal (Fig. 3a–c). In order to confirm that these filaments are analogous to the assemblies observed in the crystal structure, we produced a triple mutant VPS28 construct (K54D, K58D, D59A) designed to disrupt the helical interface, subsequently referred to as ‘VPS28 TM’. These mutants do not affect the ability of VPS28 and TSG101 to heterodimerize as judged by SDS-PAGE. However, the mutant complex failed to produce any higher molecular weight species, suggesting that the helical interface had indeed been disrupted and was the same as in the crystal structure. These results confirm that, like the ESCRT-IIIs, ESCRT-I subunits are capable of forming helical filaments in solution, and that these filaments are formed by electrostatic interactions between VPS28 protomers.
The helical interface of VPS28 stabilizes ESCRT-I accumulation in vivo
To examine the importance of the helical interface in ESCRT-I assembly in vivo, we examined the role of VPS28-mediated ring assembly in ESCRT recruitment to autophagosomes. HA-tagged VPS28 TM and control wild-type VPS28 (VPS28 WT) were lentivirally transduced into U-2 OS human osteosarcoma cells in which endogenous VPS37A is replaced by GFP-VPS37A49. Consistent with in vitro data, exogenously-introduced VPS28 displaced nearly all endogenous VPS28 and successfully incorporated into the ESCRT-I complex regardless of the mutations (Fig. 4a). The mutations also did not interfere with the membrane targeting of the complex as judged by the colocalization with HA-VPS28 MT and GFP-VPS37A on LC3-labelled autophagic structures (Fig. 4b). However, in the mutant-expressing cells, stabilization of ESCRT assembly by depleting the ESCRT-III component CHMP2A failed to induce further accumulation of VPS37A (Fig. 4c) despite an increase of autophagic structures (Fig. 4b, c). These results indicate that the mutations within the helical interface of VPS28 inhibit the accumulation of ESCRT-I without impairing the formation and membrane targeting of individual ESCRT-I tetramers.
Fig. 4: Helical contacts are required for phagophore closure in autophagy.
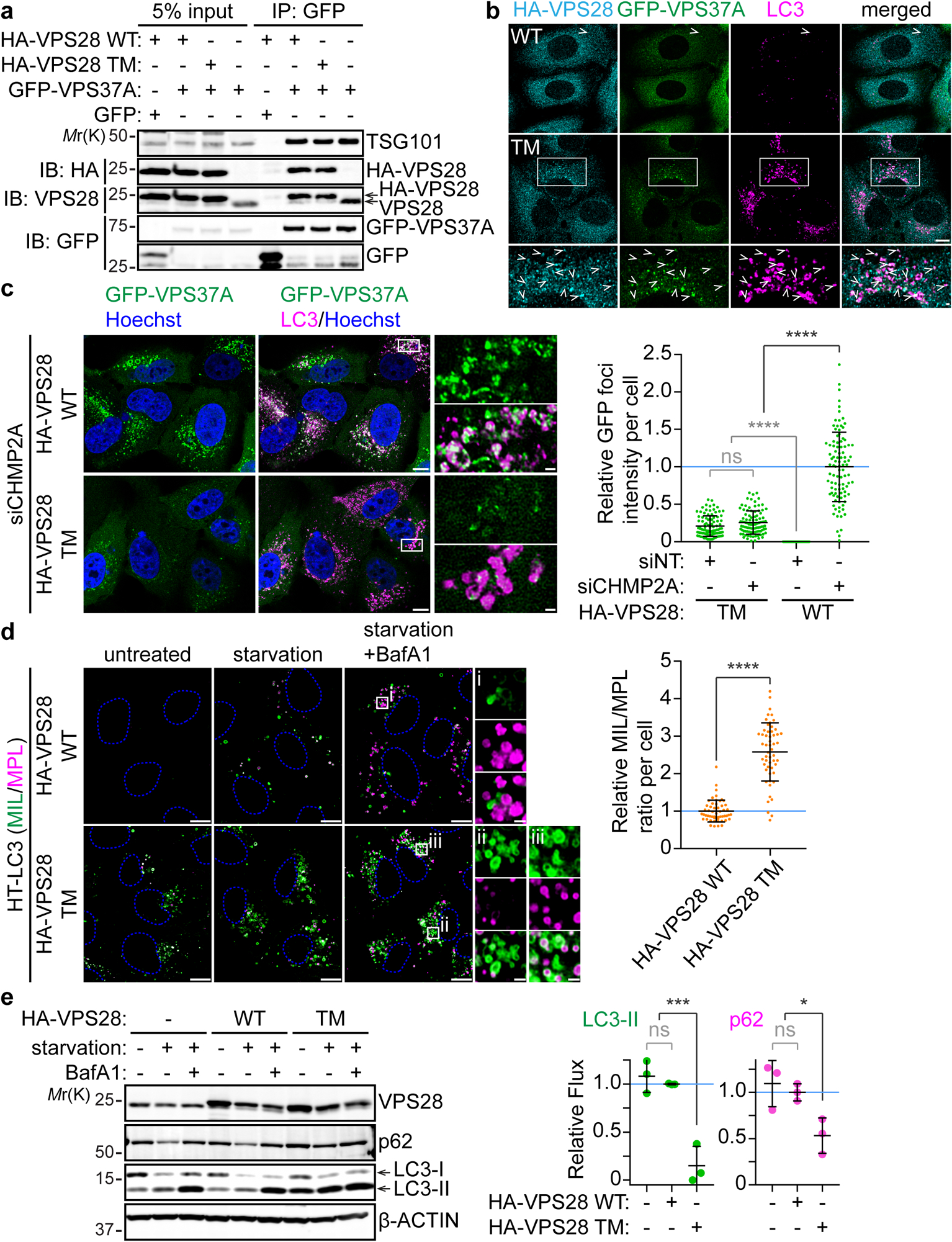
VPS37A-knockout (a-c) and wild-type (d,e) U-2 OS cells stably expressing GFP-VPS37A and HaloTag(HT)-LC3, respectively, were transduced with lentiviruses encoding HA-VPS28 WT or HA-VPS28 TM for 4 days. a, Western blot analysis of whole-cell lysates (input) and immunoprecipitates (IP) from the indicated cells. b,c, Confocal images of cells that were starved for 3h and stained for HA and LC3 (b) or nucleofected with non-targeting siRNA (siNT) or CHMP2A siRNA (siCHMP2A) for 48 h and stained for LC3 and Hoechst 33342 (c). Arrowheads in b indicate colocalization of HA-tagged VPS28 (WT or T-2) with GFP-VPS37A on LC3-positive structures. In c, the fluorescence intensity of GFP-VPS37A foci normalized to total GFP intensity per cell is shown relative to the mean of the control HA-VPS28 WT-expressing cells that were transfected with siCHMP2A (n = 98 cells). d, Confocal images of cells that were starved in the presence of 100 nM Bafilomycin A1 (BafA1) for 3 h and subjected to the HT-LC3 autophagosome completion assay using AlexaFluor 488 (AF488)-conjugated membrane-impermeable ligand (MIL) and tetramethylrhodamine (TMR)-conjugated membrane-permeable ligand (MPL). The MIL/MPL fluorescence intensity ratio for each cell in the starvation plus BafA1 treatment group was calculated and shown relative to the mean of the control HA-VPS28 WT-expressing cells (n = 54 cells). e, Western blot analysis of cells starved in the presence or absence of BafA1 (100 nM) for 3 h. Autophagic flux under starvation conditions was calculated as described in the Methods section and normalized to the mean of the control HA-VPS28 WT-expressing cells (n = 3 independent experiments). Representative blots or images from 2 (a, b) or 3 (c-e) independent experiments are shown. In b, and c and d, magnified images in the boxed areas are shown in the bottom panels and the right panels, respectively. The scale bars represent 10 μm, and 1 μm in the magnified images. Statistical significance was determined by one-way ANOVA followed by Holm-Sidak’s multiple comparison test (c, e) and Mann-Whitney nonparametric t-test (d). All values in the graphs are mean ± SD. ns, not significant; *, p≤0.05; ****, p≤0.0001. Uncropped images for panels (a) and (e) are in Supplementary Figure 1.
The ESCRT-I helical interface is required for autophagosome closure
The effect of ESCRT-I helical assembly disruption on phagophore closure was evaluated using the HaloTag-LC3 (HT-LC3) assay which employs the HaloTag receptor-conjugated LC3 reporter (HT-LC3) in combination with membrane-impermeable ligands (MIL) and -permeable ligands (MPL) labelled with two different fluorescent probes to distinguish membrane-unenclosed and -enclosed HT-LC3-II6. To prevent the degradation of autophagosome-sequestered LC3-II, the experiment was performed in the presence of the lysosomal inhibitor Bafilomycin A1 (BafA1). As expected, the mutant-transduced cells displayed an accumulation of MIL+MPL− unclosed autophagosomes and consequently increased the MIL to MPL ratio (Fig. 4d) indicating a defect in the membrane sealing. The importance of ESCRT-I helical assembly formation in autophagy was further determined by measuring autophagic flux. In the HA-VPS28 WT-transduced cells, starvation increased the amount of LC3-II which was further elevated in the presence of BafA1 similar to parental non-transduced control cells indicating normal autophagy induction and autophagic degradation (Fig. 4e). Consistently, the level of starvation-induced degradation of the autophagic substrate p62 in the WT-expressing cells was comparable to that in the control cells. In contrast, the levels of LC3-II and p62 were increased even under normal growth conditions and starvation-induced turnover of these proteins was significantly suppressed by the mutant expression. Collectively, these results indicate that ESCRT-I filament assembly is required for autophagosome closure and subsequent substrate degradation.
The ESCRT-I helical interface is required for HIV-1 release
The role of the ESCRT-I helical assembly contacts in HIV-1 release was probed using the same triple mutant VPS28-TM described above. We used the previously described LYPxNL mutant (YP-) of the infectious pNL4.3 molecular clone, which lacks the ALIX binding site and is therefore reliant only on the ESCRT-I, to promote virus release50 to directly address the role of ESCRT-I in this process (Fig. 5). Endogenous VPS28 in 293T cells was first depleted by RNAi (Invitrogen) and after 48 h was replaced with either RNAi resistant WT VPS28 (lanes 3–5), an empty plasmid vector control (lane 2), or the TM VPS28 mutant (lanes 6–8) predicted to fail in forming higher order assemblies. As expected, reintroduction of WT but not mutant VPS28 led to a rescue in viral particle production by the YP- HIV-1 virus (lanes 3–5) similar to the non-depleted control (lane 1). Conversely, the VPS28 mutant supported very little to undetectable release of mature virus, indicating that the VPS28-mediated higher order assembly of ESCRT-I is essential for HIV-1 release.
Fig. 5: Helical contacts are required for efficient HIV-1 release.
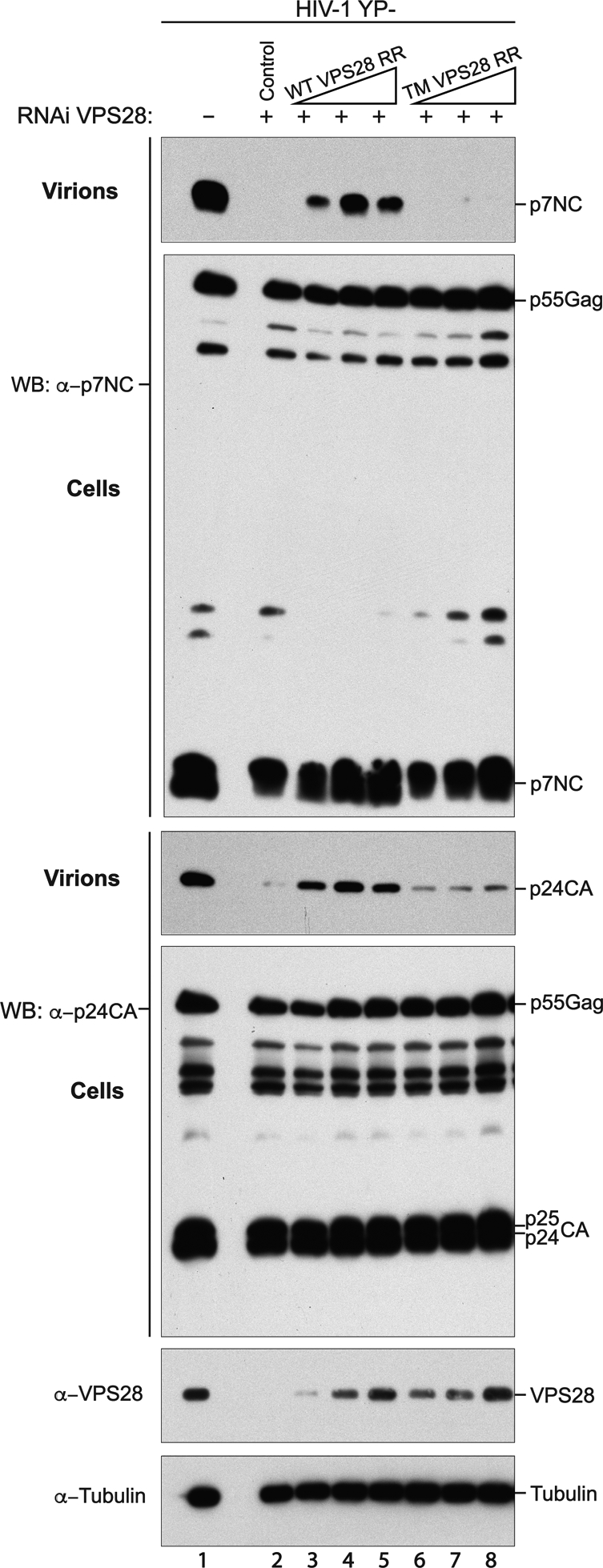
Depletion of endogenous VPS28 with RNAi (lanes 2–8) and reconstitution with empty vector (lane 2), WT VPS28 (lanes 3–5), or VPS28 TM mutant (lanes 6–8) in 293T cells. Cells were co-transfected with an HIV-1 mutant with the LYPnXL L domain abrogated and virus release was compared with a non-depleted control (lane 1). Virion production and Gag expression levels were compared by Western blotting with anti-NC and anti-CA antibodies and cellular protein levels were probed with anti-VPS28 and anti-tubulin antibodies. The images shown are representative of three biological replicates of the experiment. Uncropped images are in Supplementary Figure 1.
Coarse-grained simulation reveals optimal Gag lattice geometry is essential for ESCRT-I oligomerization.
To gain insight into the molecular structures of ESCRT-I assemblies at HIV-1 bud necks, we used coarse-grained (CG) molecular dynamics (MD) study to examine the formation of ESCRT-I rings at the necks of immature Gag lattice at various stages of initial budding. Our objective was to elucidate ESCRT-I assembly dynamics and determine the optimal Gag shell cross-section required for self-assembly of a 12-membered ESCRT-I ring oligomer. We first developed a CG model of ESCRT-I that is derived from the experimental structural data of the human ESCRT-I headpiece, yeast ESCRT-I stalk [PDB 2P22]35 and the ubiquitin E2 variant (UEV) domain of TSG101 [PDB 3OBU]32. The linker connecting the UEV domain of TSG101 and the stalk of ESCRT-I is modeled as a flexible linear polymer. The full-length ESCRT-I CG model used in this study is depicted in Fig. 6a. Our ESCRT-I CG model successfully captures the characteristic rod-like shape of the ESCRT-I core and the rotational flexibility of the ESCRT-I core imparted by the linker. We introduced attractive interactions between specific CG sites at the ESCRT-I head domain required for self-assembly. These CG sites correspond to amino acid residues of human ESCRT-I headpiece involved in direct electrostatic contacts in the helical assembly and predominantly belongs to VPS28. The location of energy minima corresponding to the attractive interactions between specific CG sites was set at the average distance between the same sites measured for the CG-representation of the 12 ESCRT-I headpiece units in the helical assembly. We chose the strength of these pairwise attractive interaction as EE1-E1 = −4 kcal mol−1. This particular value corresponds to the strongest interaction strength that does not lead to significant oligomerization in solution in our simulations (at least 90% of ESCRT-I hetero-tetramers do not oligomerize in solution, Fig. 6b). Finally, to emulate the recognition of the UEV domain of TSG101 by the PTAP motif of HIV-1 Gag, we included attractive interaction between the last bead of Gag and two CG sites at the C-terminal end of UEV domain. We describe the details of the ESCRT-I and Gag CG model in Methods.
Fig. 6: ESCRT-I assembly from CGMD simulations.
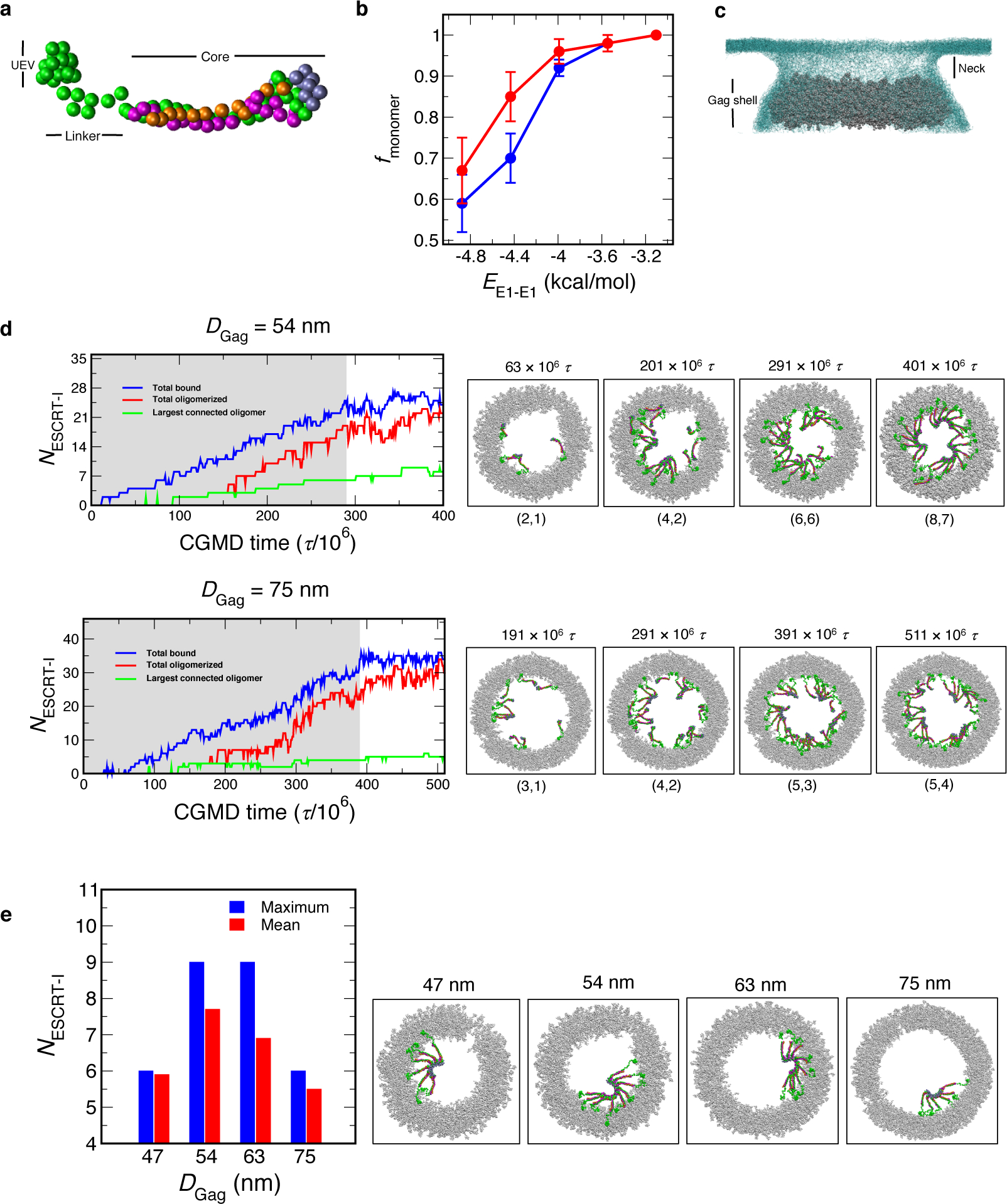
a, CG representation of ESCRT-I. The CG sites representing TSG101, VPS28, VPS37B, MVB12A are colored in green, purple, magenta and orange respectively. b, Weak interactions between ESCRT-I headpiece prevent oligomerization in solution. In a simulation cell containing 32 (red) or 64 (blue) randomly placed ESCRT-I hetero-tetramers, we varied the strength of the ESCRT-I attractive interaction (EE1-E1) from −3.1 to −4.9 kcal/mol. The fraction of ESCRT-I hetero-tetramers remaining as monomer (fmonomer) after 200 × 106 τ monotonically decreases with increasing attractive interaction (EE1-E1). We note that fmonomer sharply decreases for EE1-E1 < −4 kcal/mol. The error bars represent standard deviation computed from the final outcome of 3 independent simulations. c, Bud-like geometry used in the CGMD simulations. The Gag shell is represented in silver balls and the membrane is represented in light blue mesh. In our simulations ESCRT-I is introduced in the neck region. d, ESCRT-I assembly time series profiles as a function of CGMD time step (τ) for Gag shells, DGag = 54 and 75 nm. The timeseries profile depicts (i) total ESCRT-I bound to Gag (blue), (ii) total oligomerized (red), and (iii) largest connected oligomer (green). The shaded region in the time series indicate insertion of ESCRT-I at the membrane neck every 10 × 106 τ. The snapshots show a top view of ESCRT-I assembly at different stages of the time series. The size of the two largest oligomers are mentioned in parentheses for each snapshot. e, Mean largest oligomer and observed maximum oligomer size for each Gag shell calculated from last 50 × 106 τ of annealing CGMD trajectories. The snapshots depict the maximum largest oligomer observed for each Gag shell in the above-mentioned time frame.
To emulate the budding configuration of immature Gag lattice, we created model Gag shells of varying cross-sections representing immature Gag lattice at different stages of budding. The internal diameter (DGag) of these Gag shells range from 47 nm to 75 nm. These Gag shells are wrapped with membrane resembling the budding neck geometry (Fig. 6c). The details of system preparation and equilibration is provided in Methods. To simulate the gradual accumulation of ESCRT-I to the site of Gag assembly we inserted a single ESCRT-I hetero-tetramer every 10 × 106 CGMD time steps (τ) at the membrane neck region (Fig. 6c, Supplementary Video 1). This particular frequency of ESCRT-I insertion was chosen to allow sufficient time for the inserted ESCRT-I to diffuse and reach the Gag shell. We inserted ESCRT-I hetero-tetramer until the lumen of the Gag shell is saturated and ESCRT-I is recruited by the Gag shell. These simulations ranged from 270 × 106 to 420 × 106 τ depending on the cross-section of the shell. The systems were further evolved for 120 × 106 τ to allow for annealing of initial clusters formed during ESCRT-I recruitment and growth of the largest cluster. In what follows we summarize the assembly dynamics of ESCRT-I oligomerization.
To examine the assembly dynamics of ESCRT-I oligomerization we present NESCRT-I time evolution of the simulation trajectory of three parameters: (i) total ESCRT-I bound to Gag, (ii) total oligomerized, and (iii) largest connected oligomer. Fig. 6d shows the NESCRT-I time profile for a Gag shell of diameter (DGag) 54 and 75 nm. In the initial phase of the simulation as ESCRT-I is introduced in the membrane neck we observe continuous recruitment of ESCRT-I by the Gag shell. In the course of the simulation and concurrent to ESCRT-I binding to Gag, ESCRT-I oligomerization progresses via attachment of bound monomers, as illustrated in Fig. 6e. We note that in the simulations, multiple clusters independently grew and persisted over the course of the simulations. These intermediate clusters are typically smaller than the 12-membered ring of ESCRT-I hetero-tetramers observed in experiments. Annealing to a complete 12-membered oligomer requires coalescence or partial disassembly of these intermediate clusters. In the second phase of the simulations and once lumen saturation was achieved, the systems were evolved without further insertion of any ESCRT-I hetero-tetramers. On the time scale of our CGMD simulations and for each Gag shell, we observed growth of the largest oligomer by rearrangement of the intermediate clusters. Overall, our simulations suggest a slow and stepwise pathway of ESCRT-I oligomerization. In what follows, we summarize the outcome of our assembly simulations for Gag shells of different cross-sections and report the average largest oligomer to illustrate the efficacy of ESCRT-I self-assembly for Gag shells of different diameters.
Fig. 6e shows the mean largest ESCRT-I oligomer size and the maximum oligomer size for each Gag shell of diameter DGag = 47, 54, 63 and 75 nm. For each Gag shell, the largest oligomer is calculated over the last 50 × 106 τ of the annealing simulations and averaged over 3 independent simulations. On the time scale accessible to our simulations, we can observe the growth of ESCRT-I oligomer up to a maximum 9-mer for DGag = 54 nm. which is close to the experimental 12-mer ESCRT-I helical assembly. In contrast, Gag shells DGag = 63 and 75 nm exhibit diminished assembly competence – the wider the cross-section of the shell, the less the geometric compatibility with higher-order ESCRT-I oligomers. Similarly, ESCRT-I oligomerization is also hindered by a narrower Gag shell (DGag = 47 nm). Our simulations illustrate that an optimal Gag shell is required to stabilize higher-order ESCRT-I assembly. Based on our simulations, we expect that assembly of the full 12-mer ESCRT-I complex is templated when the opening of the immature Gag virion approaches 50 nm during budding. We note that our CGMD simulations likely cannot access the timescales required for extensive partial disassembly and assembly of intermediate clusters typical for annealing, thereby leading to the perfect 12-ESCRT-I oligomer.
Discussion
The structure of the heterotetrameric human ESCRT-I headpiece sheds light on the organization of ESCRT-I and on how the ESCRT system orchestrates membrane scission. The structure shows how UMA domain proteins are incorporated into the mammalian complex by the VPF motif. The VPF motif is conserved throughout the UMA domain proteins, thus this structure should serve as a prototype for the five different human UMA proteins and the twenty theoretically possible human ESCRT-I complex variants. While it has been reported that some of these theoretical complexes do not actually form39, the VPF motif and the sites that it contacts on TSG101 on VPS37B are well conserved. Subcomplex specificity must therefore be determined by residues in the stalk or elsewhere.
The UEV domain of TSG101 binds to the TSG101 PTAP motif, which could in principle autoinhibit ESCRT-I with respect to cargo recognition48. We now know that while the UEV domain of TSG101 can bind to a dimeric version of the ESCRT-I headpiece consisting of just TSG101 and VPS28, it cannot bind to the tetrameric complex. The subunits of most protein complexes are not synthesized with precisely matching stoichiometry, and partially assembled complexes can act as dominant negatives. Exposure of the PTAP motif in the partially assembled TSG101–VPS28 complex could potentially serve to prevent premature cargo recognition by an immature precursor complex. Upon formation of the active tetrameric complex MVB12A would sequester the internal PTAP motif, dislodge the UEV domain, and so free it up for cargo recognition.
The VPF motif interacts with Lys158, which is highly conserved amongst metazoan VPS37B homologs. Homozygous mutation of the equivalent residue within VPS37A to asparagine has been linked with complex hereditary spastic paraparesis (CHSP) in multiple individuals51. It is interesting that VPS37A is the ortholog uniquely involved in autophagosome closure49. In our pull-down experiments the K158N mutation had little effect on the interaction between MVB12A UMA-N and the trimeric head while K158D abolished the interaction. Complete abolition of the UMA protein incorporation into VPS37A complexes by K158D would have a stronger defect, perhaps resulting in embryonic lethality as seen with TSG10152, as opposed to the CHSP resulting from K158N.
The ESCRTs are an integrated pathway consisting of the upstream complexes ESCRT-I and -II and ALIX, and the downstream membrane-severing ESCRT-III complex and the VPS4 ATPase. Despite that both upstream and downstream components are required for biological function, nearly all structural12 and biophysical11,20 analysis of higher order assemblies of ESCRTs has focused on the downstream components. Structures have been observed in cellular cryo-ET53 and deep etch EM (DEEM)17 that resemble the dimension of a postulated bud neck assembly of ESCRT-I54. However, the resolution of these studies has not been sufficient to positively identify ESCRT-I, nor to design mutational probes.
We have now identified a higher-order, helical ESCRT-I assembly. The pitch of the helix is variable when comparing yeast and human structures. There are no direct contacts between successive turns of the human ESCRT-I helix, there it seems possible, or even likely that in cells, a single ring rather than a helix, is formed. Quantitative imaging of genomically integrated GFP-tagged TSG101 led to an estimate that 3–11 copies of TSG101 were involved in HIV-1 release from HeLa cells55. The upper bound of this estimate essentially coincides with our finding that 12 copies make up a single helical repeat.
Using this assembly as a template upon which to model the full complex, the UEV domains project outward to form a 12-membered ring. This dimension is plausible for many of the cell processes involving ESCRT-I, including MVB budding, autophagosome closure, and HIV-1 release. In HIV-1 release, this dimension is about half of the maximum inner diameter of the immature Gag lattice. Thus it corresponds to an immature lattice has grown past a hemisphere and from there, half-way to complete closure. Electron-microscopy-based imaging studies have identified spoke-like projections of approximately 20 nm in length that appear to localize at the edge of the immature Gag lattice17,53. These assemblies are only observed in cells when the bud neck is more than half the diameter of the bud, suggesting that they assemble during the early stages of bud formation53. In our coarse grained simulations, larger assemblies of ESCRTs are only stable when the diameter of the bud neck is close to the most preferred value. If the ESCRTs were to act at this stage, this would leave a corresponding gap in the immature lattice, consistent with observation56. This would also result in incorporation of a small number of ESCRT-I complexes into the released virion, consistent with reports55,57.
The VPS28 C-terminal domain (CTD) responsible for recruiting downstream factors is connected to the headpiece portion of VPS28 by a short linker such that the CTD should be very close to what is seen here with the headpiece alone. With inner and outer diameters of 6.5 and 17 nm, respectively, the headpiece ring dimensions correspond to the narrow ends of ESCRT-III cone structures12,58. Most tubular ESCRT-III structures are larger, with diameters of 24 nm to 400 nm12,59. The narrowness of the ESCRT-I ring suggests that the ESCRT-III cone grows outward from the ESCRT-I ring, rather than inward1 ,2,60. This might be consistent with the return of most of the ESCRT-III to the cytosol, but with the ESCRT-I retained in the virion2–4.
This leads us to propose the following model for the role of ESCRT-I in HIV-1 release. Prior to HIV scission, single ESCRT-I tetrameric complexes are gradually recruited to Gag61. In analogy to this in autophagy, we observed that VPS28 mutations do not affect recruitment of ESCRTs to LC3 membranes. Without a geometry compatible with 12-membered ring formation, they do not template ESCRT-III spirals, consistent with the observation that ESCRT-III is not stably recruited at this stage62. As the cross-section of the membrane neck narrows to around 55 nm, ESCRT-I at the Gag boundary reaches high local concentration and the geometry is compatible with ring formation, stabilizing the assembly. HIV budding profiles with openings of about these dimensions have been reported to manifest spoke-like assemblies53. This generates a helical or ring-shaped platform, upon which the downstream ESCRTs assemble to drive membrane scission. This model could explain why live cell imaging studies have reported a gradual accumulation of ESCRT-I at budding sites61 followed by a sharp late burst in ESCRT-IIIs immediately prior to scission62. Our finding that the filament contacts are conserved from yeast to humans, are functionally required for autophagosome closure, and explain ESCRT recruitment patterns in both HIV release and autophagosome biogenesis, suggests this mechanism applies widely.
Online methods
Protein expression and purification
The codon-optimized synthetic genes for human TSG101 308–388, VPS37B 97–167, VPS28 1–122 and MVB12A 206–268 were subcloned into cassettes 1–4 of the polycistronic pST39 vector. MVB12A was tagged with an N-terminal GST tag followed by a TEV cleavage site. The plasmid was transformed into E. coli strain BL21(DE3) Star followed by overnight expression at 20 °C. Cells were pelleted and resuspended in buffer (50 mM Tris pH 8.0, 300 mM NaCl, 3 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride) and lysed by sonication. The tetrameric complex was isolated by glutathione affinity chromatography followed by overnight on-resin TEV cleavage. Cleaved product was dialyzed into low salt buffer (50 mM Tris pH 8.0, 50 mM NaCl, 3 mM β-mercaptoethanol), applied to a HiTrap Q-sepharose FF column (GE Healthcare) and eluted with a linear 50 mM to 1 M NaCl gradient. Pooled fractions were passed through nickel-charged agarose to remove residual hexahistidine-tagged TEV protease. Flow-through was concentrated and applied to a Superdex 75 size exclusion column. The fractions containing the complex were concentrated in buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP).
The binary TSG101–VPS28 subcomplex was cloned in a similar manner to the tetrameric complex but VPS37B and MVB12A were omitted and a TEV-cleavable hexahistidine tag was fused to the C-terminus of VPS28. The subcomplex was expressed in BL21(DE3) E. coli cells Star at 20 °C overnight. Cells were resuspended in buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 3 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, 10 mM imidazole) and lysed by sonication. The ESCRT-I head complex was isolated by Ni2+ affinity chromatography, washed with 30 mM imidazole buffer and eluted with 300 mM imidazole. Eluant was concentrated and applied to a Superdex S200 size exclusion column equilibrated in buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP). The subcomplex eluted as a two peaks, each of which was individually pooled.
DNA coding for the human TSG101 UEV domain (residues 2–145) was subcloned into the pGST2 vector. The plasmid was transformed into E. coli strain BL21(DE3) Star and expressed overnight at 30 °C. Cells were pelleted and resuspended in buffer (50 mM Tris pH 8.0, 300 mM NaCl, 3 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride) and lysed by sonication. GST-UEV was isolated using glutathione affinity chromatography, washed, and eluted with buffer containing 30 mM reduced glutathione. Eluate was concentrated and further purified using a Superdex 200 size exclusion column equilibrated in buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.5 mM TCEP).
All mutant constructs used in this study were generated by QuikChange site-directed mutagenesis according to the manufacturers instructions (Agilent).
X-ray crystallography
Crystals of human ESCRT-I head were grown using the hanging drop vapor diffusion method at 18 °C. 2 μL of the protein sample (8.2 mg ml−1) was mixed with 2 μL reservoir solution and suspended over a 500 μL reservoir of 2 % w/v γ-Polyglutamic acid (γ-PGA) (Molecular Dimensions), 100 mM sodium formate, 100 mM sodium acetate pH 5.0. Crystals appeared within 3 days and continued to grow for approximately one week. Crystals were cryoprotected in reservoir solution supplemented with 25 % (v/v) Glycerol. A Native dataset was collected from a single crystal under cryogenic conditions (100 K) at a wavelength of 1.11583 Å using a Dectris Pilatus3 S 6M detector (beamline 8.3.1, ALS). The data was indexed and integrated using XDS63. Integrated reflections were scaled, merged and truncated using the CCP4 software suite64. Initial phases were determined by molecular replacement with the program PHASER65 using the trimeric yeast ESCRT-I head structure (PDB 2F66) as a search model. Iterative rounds of manual model building and refinement were performed using Coot66 and Phenix Refine67 respectively. 99.87% of the residues are in the most favored and additionally allowed regions of the Ramachandran plot. The space group is P61 with 6 molecules per asymmetric unit. Statistics of the final structure are shown in (Table 1). Structural figures were produced using the program PyMOL (W. Delano, https://pymol.org).
Table 1.
Data collection and refinement statistics
| Human ESCRT-I Head (PDB code: 6VME) |
||
|---|---|---|
| Data collection | ||
| Space group | P61 | |
| Cell dimensions | ||
| a, b, c (Å) | 162.58 162.58 139.72 | |
| α, β, γ (°) | 90, 90, 120 | |
| Resolution (Å) | 99.18 – 2.19 (2.269 – 2.19) | |
| Rpim | 0.042 (0.97) | |
| I / σI | 17.22 (0.73) | |
| Completeness (%) | 99.46 (95.09) | |
| Redundancy | 20.5 (20.2) | |
| Refinement | ||
| Resolution (Å) | 99.18 – 2.19 | |
| No. reflections | 106903 | |
| Rwork / Rfree | 0.2071/0.2439 | |
| No. atoms | ||
| Protein | 12478 | |
| Ligand/ion | 0 | |
| Water | 308 | |
| B-factors | ||
| Protein | 57.33 | |
| Ligand/ion | ||
| Water | 52.96 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 0.90 |
Values in parentheses are for highest-resolution shell.
Electron microscopy
The ESCRT-I TSG101–VPS28 head subcomplex was purified as described above. Void peak fractions from the final size-exclusion step were pooled and diluted to 3.5 μM for negative stain electron microscopy. 5 μl of sample was applied to glow-discharged continuous carbon-coated copper grids (CF400-CU, Electron Microscopy Sciences) and negatively stained with 2 % (w/v) uranyl acetate. Samples were inspected using transmission electron microscopy (Tecnai-12, FEI) operated at 120 keV with a magnification of 49,000×. Images were collected with a charge-coupled device (CCD) detector (4k TemCam-F416, TVIPS).
Pull-down assays
Pull-downs were performed to test the effect of specific mutations on the ability of MVB12A to interact with the ESCRT-I head. In the case of both the WT and mutant constructs, GST-MVB12A (206–268) was co-expressed with TSG101 (308–388), VPS37B (97–167) and a C-terminal hexahistidine tagged version of VPS28 (1–122). Cell lysates were applied to either 30 μL of GSH or Ni-NTA resin prewashed with binding buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP, 20 mM imidazole) and incubated at 4 °C for 30 minutes. The beads were washed three times and complex formation analyzed by SDS-PAGE.
For the TSG101 UEV pull-down assays, pre-purified bait (GST-UEV or free GST) was incubated with 30 μL of GSH resin prewashed in binding buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP) for 30 minutes at 4 °C. Beads were washed three times to remove excess bait. ESCRT-I head prey (tetramer or dimer) was added to the beads in 500 μL binding buffer and incubated at 4 °C overnight. Beads were again washed three times to remove unbound prey and bound proteins analyzed by SDS-PAGE.
Antibodies and siRNAs
The following antibodies were used for immunoblotting (IB) and immunofluorescence (IF): β-ACTIN (IB, Sigma-Aldrich, A5441, 1:10,000); GFP (IB, Cell Signaling, 2956, 1:1,000); HA (IB, IF, BioLegend, 901513, 1:2,000 (IB), 1:1,000 (IF)); MAP1LC3B (IB, Novus, NB100–2220, 1:3,000; IF, Cell Signaling, 3868, 1:200); p62 (IB; American Research Products, 03-GP62-C, 1:4,000); TSG101 (IB, Abcam, ab83, 1:1,000); VPS28 (IB, Santa Cruz Biotechnology, sc-166537, 1:100). ON-TARGETplus SMART Pool Non-targeting (D-001810–10) and CHMP2A (L-020247–01) siRNAs were obatined from GE Healthcare Dharmacon. pCDH1-CMV-HA-VPS28(WT)-SV40-hygro and pCDH1-CMV-HA-VPS28(TM[K54D, K58D, D59A])-SV40-hygro were generated using Gibson Assembly. All other reagents were obtained from the following sources: Bafilomycin A1 (LC Laboratories, B-1080); Hoechst 33342 (Invitrogen, NucBlue, R37605); Membrane-impermeable HaloTag Ligand (MIL) (Promega, Alexa Fluor 488-conjugated, G1001); Membrane-permeable HaloTag Ligand (MPL) (Promega, tetramethylrhodamine-conjugated, G8251); normal goat serum (Sigma-Aldrich, G9023); Nucleofector Kit V (Lonza, VCA-1003); paraformaldehyde (Electron Microscopy Sciences, 15710); XF Plasma Membrane Permeabilizer (XF-PMP) (Seahorse Bioscience, 102504–100).
Cell culture
U-2 OS cells were obtained from American Type Culture Collection and maintained in McCoy’s 5A Medium supplemented with 10% FBS. To induce autophagy, cells were rinsed three times with Dulbecco’s Phosphate Buffered Saline (DPBS) and incubated with amino acid-free DMEM (Wako, 048–33575). GFP-VPS37A-expressing VPS37A knockout U-2 OS cells6 and HT-LC3-expressing U-2 OS cells49 were generated as described previously.
Coimmunoprecipitation
Cell lysates were prepared in 0.5% NP-40 lysis buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) containing protease inhibitors and subjected to immunoprecipitation using with anti-GFP-conjugated agarose beads (GFP-Trap beads, Chromotek, gtma). The immunoprecipitates were washed three times with lysis buffer and subjected to immunoblotting.
Immunofluorescence and confocal microscopy
Cells grown on Lab-TekII Chambered Coverglass, Chamber Slide (Nunc, 154941) were fixed and permeabilized in 4% paraformaldehyde-PBS at room temperature (RT) for 3 min followed by methanol at −20°C for 7 min, blocked in 10% normal goat serum for 1 h, and incubated with the primary antibodies overnight at 4°C followed by the secondary antibodies for 1h at RT. All fluorescence images were obtained using a Leica AOBS SP8 laser-scanning confocal microscope (63x oil-immersion [1.2 numerical aperture] lens) with the highly sensitive HyD detectors and the Leica image acquisition software LAX, deconvolved using Huygens deconvolution software (Scientific Volume Imaging), and analyzed using Imaris software (Bitplane) and Volocity software (PerkinElmer) without gamma adjustment.
Autophagy assays
To induce autophagy, cells were rinsed three times with Dulbecco’s Phosphate Buffered Saline (DPBS) and incubated with amino acid-free DMEM (Wako, 048–33575) for 3 h. HT-LC3 autophagosome completion assay was performed as described previously49. For immunoblotting-based autophagic flux assay, cell lysates were prepared in radio-immunoprecipitation assay buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% SDS, 1% Triton X-100, 1% Deoxycholate, 5 mM EDTA, pH 8.0) containing protease inhibitors and subjected to SDS-PAGE followed by immunoblotting with antibodies against LC3, p62 and β-ACTIN. The signal intensities were quantified using the Image Studio version 5 software (LI-COR Biotechnology) and the levels of LC3-II and p62 were normalized to the respective value of β-actin. Autophagic flux was calculated by subtracting normalized LC3-II and p62 values in starvation group from those in starvation plus BafA1 group.
Statistical analyses
Statistical significance was determined using Graph Pad Prism 7.0. Threshold for statistical significance for each test was set at 95% confidence (p<0.05).
HIV-1 release
293T cells (2.2 × 106) were seeded into T25 flasks and transfected with VPS28-specific RNAi or control RNAi (Invitrogen), 1–2 μg of LYPxNL-abrogated (YP-) mutant HIV-1 proviral DNA together with expression vectors for RNAi Resistant (RR) wild-type (WT) or mutant VPS28, (TM) or an empty vector control. Twenty-four hours after transfection, culture supernatants were harvested as previously described in68 and virions were then pelleted through 20% sucrose cushions. Cells were collected and lysed in 0.5% lysis buffer (RIPA Buffer) (140 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% Nonidet P-40 [NP-40], 0.5% sodium deoxycholate, 0.05% sodium dodecyl sulfate [SDS]) and Complete Protease Inhibitor Cocktail (Roche). Isolated virions and viral proteins in cell lysates were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting for viral proteins with a goat anti-HIV NC (a generous gift from Robert Gorelick) and a mouse anti-CA antibody (AIDS Repository cat# clone 183-H12–5C). Host proteins were detected by Western blotting with mouse anti-tubulin and VPS28 expression was verified with a rabbit anti-VPS28 antibody (Sigma).
Eukaryotic Cell Lines
HEK293T and U-2 OS cells were obtained from American Type Culture Collection. All cell lines were passaged for fewer than 2 months before use and periodically authenticated by morphologic inspection and mycoplasma testing.
CG Model Generation of ESCRT-I and Gag.
The CG model of ESCRT-I headpiece and UEV domain of TSG101 was generated from the all-atom trajectories using the Essential Dynamics Coarse-graining (ED-CG) method.69 ED-CG is a bottom-up method that variationally determines CG sites to capture the fluctuations in the essential subspace determined from principal component analysis of the all-atom trajectories. The details of the simulations to generate all-atom trajectories are provided in Supplementary Note 1. The resulting CG model of ESCRT-I headpiece and UEV domain of TSG101 have 33 and 19 CG sites respectively, with an average resolution of ~8 amino acids per CG site. Next, effective interactions between CG sites were represented as a network of effective harmonic spring for all neighboring CG sites within 3 nm of the reference CG site. The spring constants for CG site pairs were derived using the hetero-elastic network (hENM) method.70 The CG model for the ESCRT-I stalk is directly derived from the experimental crystal structure for yeast ESCRT-I available in Protein Data Bank [PDB 2P22]35 maintaining the same average resolution of ~8 amino acids per CG site. Yeast ESCRT-I stalk contains 3 subunits - Vps23, Vps37 and Mvb12. In the case of 4 long α helices constituting the stalk [Vps23 (α1, α2), Mvb12 (α2) and Vps37 (α4)], a single CG site is created every 8 consecutive amino acids. Two additional CG sites were placed to represent Vps37 (α1, α2) at the base of the stalk. The initial position of CG sites was calculated from the center of mass of the Cα atom of the corresponding amino acid residues that the CG site represent. Overall, the CG model of the stalk contains 31 CG sites. Next, all CG sites within 3 nm of a central CG site were connected with effective harmonic springs. In the case of CG sites corresponding to the same subunit (e.g., Vps23 and Vps23) we used a spring constant of 50 kcal mol−1 nm−2, while for CG sites corresponding to different subunits a weaker spring constant of 5 kcal mol−1 nm−2 was used. The stalk and the headpiece were connected by harmonic bonds using the same protocol as above. Finally, inter-domain interactions were represented using a combination of a soft excluded volume (Eexcl) and Gaussian attractive (Egauss) potentials
where rij is the pairwise distance between CG site types i and j. For all CG sites types at headpiece involved in self-assembly Gaussian attractive (Egauss) potentials were used. Using the CG-mapped structure of the 12-mer ESCRT-I headpiece, the mean distance between a pair ij is assigned as rij,h. The rij,h values ranged from 1.28 to 1.8 nm. For these attractive ij pairs σij = 0.09 nm, and Hij = −0.9 nm kcal/mol are used. We note that strength of ESCRT-I attractive interaction (EE1-E1) described in the main text is same as . The particular chosen value of Hij is the maximum value that does not lead to significant oligomerization in ESCRT-I solution in the CGMD simulations (Fig. 6b). This leads to the EE1-E1 value of −4 kJ/mol. Details of the simulations of ESCRT-I solutions are described in the section having the CG simulation details. All pairwise interaction modeled with Egauss used a 30 Å radial cutoff. For the rest of the pairwise CG interactions a soft excluded volume (Eexcl) interaction is used with Aij = 25 kcal/mol, rij,c = 1.5 nm. The linker domain is represented as a linear chain of 9 CG particles that are linked with harmonic bonds of spring constant 50 kcal mol−1 nm−2 and equilibrium distance is maintained at 1.5 nm. The intra- or inter-domain interaction of the linker CG sites is modeled with excluded volume (Eexcl) potential with the same parameters as above. To model the binding of the p6 domain of Gag and UEV domain of TSG101, we first added an extra CG binding site at the end of the NC domain of the CG Gag (the details of the CG Gag model is described in the next section). This CG binding site is linked to three nearest CG beads of NC domain by harmonic bonds of spring constant 50 kcal mol−1 nm−2 and equilibrium distance 1.5 nm. To implicitly allow binding of ESCRT-I and p6 domain, 2 C-terminal CG sites of UEV domain of TSG101 with the binding site. The attractive interaction is modeled with Gaussian attractive (Egauss) potentials, with values rij,h = 1.2nm, σij = 0.09 nm, and Hij = −4.2 nm kcal/mol are used. In a separate publication details of the characterization of the ESCRT-I CG model will be presented.
The CG model of Gag of consists of matrix (MA), capsid/spacer peptide 1 (CA/SP1), and nucleocapsid (NC) domains. The CG model of each of the domains are derived from the all-atom trajectories using ED-CG method, and the intra-domain interactions are represented using the heterogeneous network of springs. Inter-domain interactions are represented using a combination of a soft excluded volume (Eexcl) and Gaussian attractive (Egauss) potentials functions described above. These interaction parameters are found to be sufficient to stabilize the formation of the immature lattice, which we tested by aligning hexamers of our CG model to cryo-electron microscopy maps of immature HIV-1 lattices provided by John Briggs (MRC LMB). Details of the all-atom simulations used for coarse-graining and CG model details are provided in the Supplementary notes.
CG Simulation details and analysis
All CGMD simulations were performed using the LAMMPS MD software71. The equations of motions were integrated with the velocity Verlet algorithm using a time step (τ) of 50 fs. The simulations of ESCRT-I assembly templated by Gag shell were performed under constant NpxyT ensemble. Periodic boundary conditions were implemented in the direction parallel to the membrane plane (xy plane). The simulation cell was nonperiodic in the z direction and terminated by a reflective wall. In the simulation setup with the “budding neck” geometry, the membrane region in contact the Gag shell were kept frozen, while the equations of motion for the rest of the membrane patch was integrated. This allows to maintain the “bud-like” shape of the membrane. Similarly, for the CGMD simulations for ESCRT-I self-assembly, the MA-CA/SP1 domain of each Gag polyprotein is kept frozen to maintain the crystallinity of the relaxed Gag shells. The temperature in the simulations was maintained at 310 K using Langevin dynamics with a damping constant of 200τ72. The pressure in the xy direction was controlled with the Nose-Hoover barostat with a coupling constant of 500τ73. The pressure tensors in the x and y directions were coupled and maintained at 0 bar. The simulations of ESCRT-I in solution were performed in the constant NVT ensemble. The system temperature in these simulations was maintained in the same way as described above, while the Gag and ESCRT-I were modelled with the CG models described earlier. The lipid bilayer was modeled with a 3-site CG model74. Visualization of the simulation trajectory was created using Visual Molecular Dynamics (VMD) software75. We characterized ESCRT-I oligomerization by applying a neighboring criterion: an ESCRT-I hetero-tetramer (i) was considered to be bound to another hetero-tetramer (j) if all the binding CG pairs ij at the headpiece were within a 0.3 nm cutoff.
We prepared model Gag shells used in the simulation of ESCRT-I assembly from cryo-electron microscopy maps of immature HIV-1 lattices, with an approximately hemispherical geometry. These immature Gag lattices were mapped to CG representation with our CG Gag model and relaxed. Using the relaxed structure as our parent lattice, we selected a segment of the hemisphere, typically of height 20 nm. The upper and lower limit of the height used to select a segment was progressively varied to create Gag shells of different diameters (DGag). We note that DGag in this study was defined as the lowest cross-section of the segment. The Gag shells were then placed in a simulation cell of dimensions 152 nm × 152 nm × 56 nm in the x, y, z directions respectively, as depicted in Fig. 6c. Then, the membrane layer resembling a “bud-like” shape was wrapped around the Gag shells. The composite systems were then relaxed for 5 × 106 CGMD timesteps. We performed three independent simulations of ESCRT-I self-assembly for each Gag shell (4 different cross-sections) resulting in 12 total simulations. In each simulation the initial velocity distribution of inserted ESCRT-I was assigned via a random number generator seed from Boltzmann distribution. Simulation trajectories were saved every 1 × 106 CGMD timesteps.
To prepare systems of ESCRT-I solution we first created a simulation cell of dimension 200 nm × 200 nm × 200 nm. Next in the simulation cell ESCRT-I units are evenly spaced in a 3-dimensional grid of (4 × 4 × 4) or (4 × 4 × 8) with a 10 nm separation between every ESCRT-I unit, resulting in two systems containing 32 or 64 ESCRT-I heterotetramers. Then 3 independent simulations were evolved at 310 K for each system (32 or 64 ESCRT-I) for 10 × 106 CGMD timesteps. In these simulations all attractive interactions between ESCRT-I headpiece is turned off to prevent oligomerization. The final state of each independent system is used as initial configuration for simulations of ESCRT-I assembly in solution. For the assembly simulations, we turn on the attractive interactions between ESCRT-I headpiece and vary the value of EE1-E1 from −3.1 to −4.9 kcal/mol (Hij from −0.7 to −1.1 nm kcal/mol). For each EE1-E1, three independent simulations are evolved for 200 × 106 τ resulting in a total of 15 simulations.
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code PDB 6VME. Uncropped images for Figure 1c, 2d, 2f, 4a, 4e, 5 and Extended Data Fig. 1 are available in Supplementary Figure 1; source data for graphs in figure 4 are available online.
Extended Data
Extended Data Fig. 1. Effect of specific mutations on ESCRT-I head complex integrity.
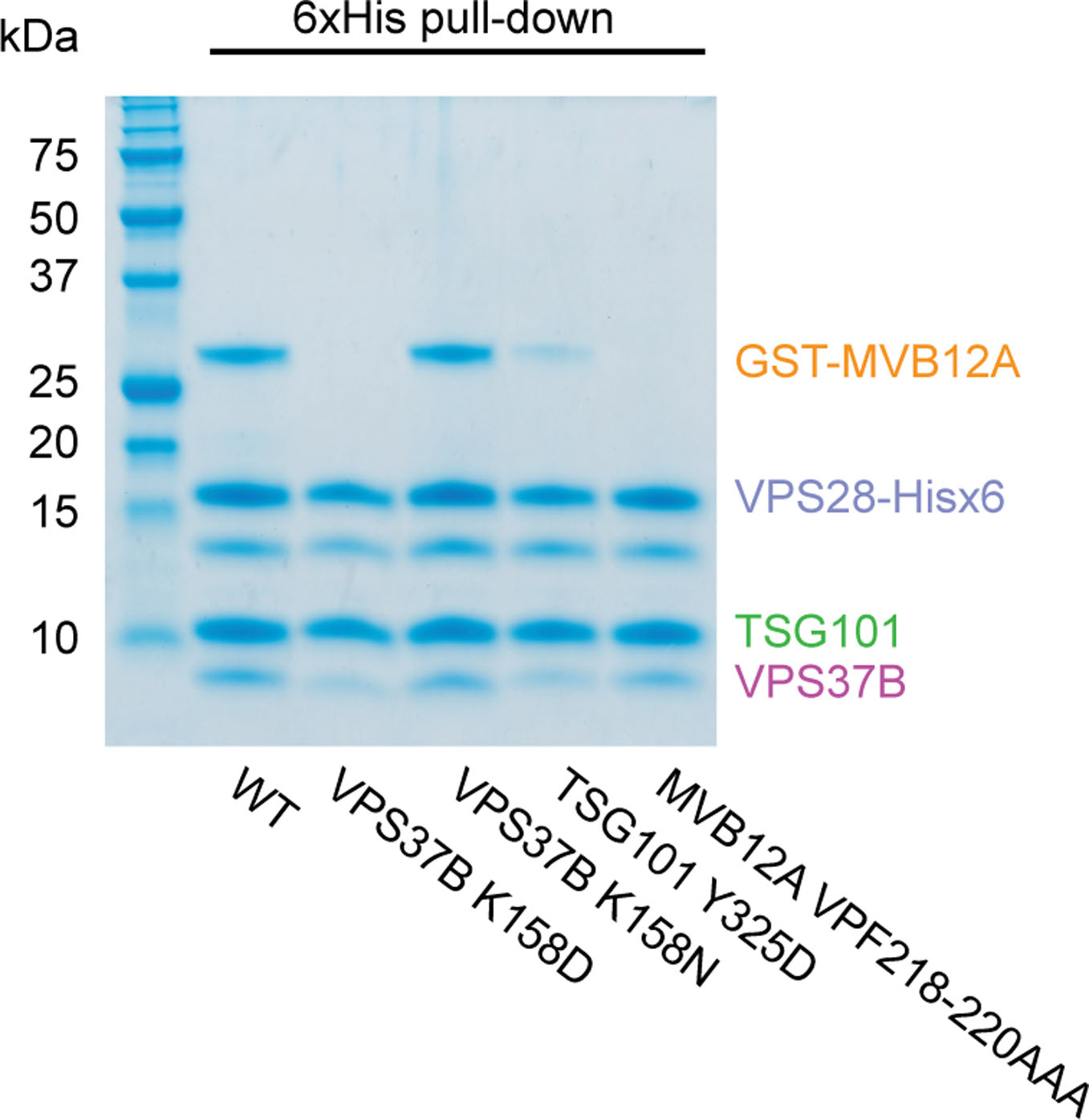
Mutant versions of the ESCRT-I head complex were expressed in E. coli. VPS28 and MVB12A subunits were expressed as C-terminal hexahistidine and N-terminal GST fusions respectively. Lysate was incubated with Ni-NTA agarose beads and complex integrity analyzed by SDS-PAGE following multiple washes. The SDS-PAGE image shown is representative of two independent biological repeats. Uncropped image is in Supplementary Figure 1.
Extended Data Fig. 2. Sequence alignment of VPS28 orthologs.

Secondary structure displayed above the alignment is derived from the human ESCRT-I head structure. Alignment was generated using ClustalW and ESPript.
Extended Data Fig. 3. Helical yeast ESCRT-I head tubes.

The trimeric yeast ESCRT-I head forms helical tubes within a crystal (PDB 2CAZ). The crystal is constructed from a series of laterally stacked tubes where each tube is composed of a single, continuous helix of yeast ESCRT-I head protomers. Vps23, Vps28, Vps37 are colored green, purple and magenta respectively. Tube dimensions are labeled.
Supplementary Material
Acknowledgments
We thank B. Yang for contributing to early stages of this project, S. Fromm, C. Buffalo, and P. Grob for electron microscopy advice and support, K. Larsen for comments on the manuscript, and J. Briggs for providing immature Gag lattice maps. This work was supported by NIH grants R37 AI112442 (J.H.H.), R01 GM127954 (H.G.W), R01 GM128507 (A.H. and G.A.V.) and F32 AI150477 (A.J.P.). Beamline 8.3.1 at the Advanced Light Source is supported by the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc., and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences.
Footnotes
Competing interests
J.H.H. is a founder of Casma Therapeutics.
References
- 1.Schöneberg J, Lee I-H, Iwasa JH & Hurley JH Reverse-topology membrane scission by the ESCRT proteins. Nature Reviews Molecular Cell Biology 18, 5–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCullough J, Frost A & Sundquist WI Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu Rev Cell Dev Biol 34, 85–109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippincott-Schwartz J, Freed EO & van Engelenburg SB A Consensus View of ESCRT-Mediated Human Immunodeficiency Virus Type 1 Abscission. Annu Rev Virol 4, 309–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurley JH & Cada AK Inside job: how the ESCRTs release HIV-1 from infected cells. Biochem Soc Trans 46, 1029–1036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoten CL & Carlton JG ESCRT-dependent control of membrane remodelling during cell division. Semin Cell Dev Biol 74, 50–65 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi Y et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun 9, 2855 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhen Y et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy, 1–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou F et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Biol 218, 1908–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olmos Y & Carlton JG The ESCRT machinery: new roles at new holes. Curr. Opin. Cell Biol 38, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campsteijn C, Vietri M & Stenmark H Novel ESCRT functions in cell biology: spiraling out of control? Curr. Opin. Cell Biol 41, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Schöneberg J et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science 362, 1423–1428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCullough J et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 350, 1548–1551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henne WM, Buchkovich NJ, Zhao Y & Emr SD The Endosomal Sorting Complex ESCRT-II Mediates the Assembly and Architecture of ESCRT-III Helices. Cell 151, 356–371 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Hanson PI, Roth R, Lin Y & Heuser JE Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. The Journal of Cell Biology 180, 389–402 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Q-T et al. Structural analysis and modeling reveals new mechanisms governing ESCRT-III spiral filament assembly. The Journal of Cell Biology 206, 763–777 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiaruttini N et al. Relaxation of Loaded ESCRT-III Spiral Springs Drives Membrane Deformation. Cell 163, 866–879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cashikar AG et al. Structure of cellular ESCRT-III spirals and their relationship to HIV budding. Elife e02184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Effantin G et al. ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cell. Microbiol 15, 213–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lata S et al. Structural Basis for Autoinhibition of ESCRT-III CHMP3. J. Mol. Biol 378, 818–827 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mierzwa BE et al. Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat. Cell Biol 19, 787–798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.VerPlank L et al. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. U. S. A 98, 7724–7729 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrus JE et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107, 55–65 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Martin-Serrano J, Zang T & Bieniasz PD HIV-I and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med 7, 1313–1319 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Demirov DG, Ono A, Orenstein JM & Freed EO Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. U. S. A 99, 955–960 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Schwedler UK et al. The protein network of HIV budding. Cell 114, 701–713 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Strack B, Calistri A, Craig S, Popova E & Gottlinger HG AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114, 689–699 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Ali N et al. Recruitment of UBPY and ESCRT Exchange Drive HD-PTP-Dependent Sorting of EGFR to the MVB. Curr. Biol 23, 453–461 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Loncle N, Agromayor M, Martin-Serrano J & Williams DW An ESCRT module is required for neuron pruning. Scientific Reports 5, doi: 10.1038/srep08461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkinson MDJ et al. A non-canonical ESCRT pathway, including histidine domain phosphotyrosine phosphatase (HD-PTP), is used for down-regulation of virally ubiquitinated MHC class I. Biochem. J 471, 79–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doyotte A, Mironov A, McKenzie E & Woodman P The Bro1-related protein HD-PTP/PTPN23 is required for endosomal cargo sorting and multivesicular body morphogenesis. Proc. Natl. Acad. Sci. U. S. A 105, 6308–6313 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pornillos O, Alam SL, Davis DR & Sundquist WI Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol 9, 812–817 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Im YJ et al. Crystallographic and functional analysis of the ESCRT-1/HIV-1 Gag PTAP interaction. Structure 18, 1536–1547 (2010. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundquist WI et al. Ubiquitin recognition by the human TSG101 protein. Mol. Cell 13, 783–789 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Teo H, Veprintsev DB & Williams RL Structural insights into endosomal sorting complex required for transport (ESCRT-I) recognition of ubiquitinated proteins. J. Biol. Chem 279, 28689–28696 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Kostelansky MS et al. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell 129, 485–498 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katzmann DJ, Babst M & Emr SD Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 106, 145–155 (2001). [DOI] [PubMed] [Google Scholar]
- 37.de Souza RF & Aravind L UMA and MABP domains throw light on receptor endocytosis and selection of endosomal cargoes. Bioinformatics 26, 1477–1480 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morita E et al. Identification of human MVB12 proteins as ESCRT-I subunits that function in HIV budding. Cell Host and Microbe 2, 41–53 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stefani F et al. UBAP1 is a component of an endosome-specific ESCRT-I complex that is essential for MVB sorting. Curr. Biol 21, 1245–1250 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Audhya A, McLeod IX, Yates JR & Oegama K MVB-12, a fourth subunit of metazoan ESCRT-I, functions in receptor downregulation. PLoS One 2, e956 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boura E et al. Solution structure of the ESCRT-I complex by small angle x-ray scattering, EPR, and FRET spectroscopy. Proc. Natl. Acad. Sci. U. S. A 108, 9437–9442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kostelansky MS et al. Structural and functional organization of the ESCRT-I trafficking complex. Cell 125, 113–126 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teo HL et al. ESCRT-I core and ESCRT-II GLUE domain structures reveal role for GLUE in linking to ESCRT-I and membranes. Cell 125, 99–111 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Im YJ & Hurley JH Integrated structural model and membrane targeting mechanism of the human ESCRT-II complex. Dev Cell 14, 902–913 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill DJ et al. Structural insight into the ESCRT-I/-II link and its role in MVB trafficking. Embo J 26, 600–612 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pineda-Molina E et al. The crystal structure of the C-terminal domain of Vps28 reveals a conserved surface required for Vps20 recruitment. Traffic 7, 1007–1016 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Agromayor M et al. The UBAP1 Subunit of ESCRT-I Interacts with Ubiquitin via a SOUBA Domain. Structure 20, 414–428 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pornillos O et al. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J Cell Biol 162, 425–434 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takahashi Y et al. VPS37A directs ESCRT recruitment for phagophore closure. J Cell Biol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dussupt V et al. The Nucleocapsid Region of HIV-1 Gag Cooperates with the PTAP and LYPXnL Late Domains to Recruit the Cellular Machinery Necessary for Viral Budding. PLoS Path. 5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zivony-Elboum Y et al. A founder mutation in Vps37A causes autosomal recessive complex hereditary spastic paraparesis. J Med Genet 49, 462–472 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Ruland J et al. p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc.Natl.Acad.Sci.U.S.A 98, 1859–1864 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ladinsky MS et al. Electron Tomography of HIV-1 Infection in GutAssociated Lymphoid Tissue. PLoS Path. 10, doi: 10.1371/journal.ppat.1003899 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boura E & Hurley JH Structural basis for membrane targeting by the MVB12-associated β-prism domain of the human ESCRT-I MVB12 subunit. Proc. Natl. Acad. Sci. U. S. A 109, 1901–1906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffman HK, Fernandez MV, Groves NS, Freed EO & van Engelenburg SB Genomic tagging of endogenous human ESCRT-I complex preserves ESCRT-mediated membrane-remodeling functions. J Biol Chem (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carlson LA et al. Cryo Electron Tomography of Native HIV-1 Buding Sites. PLoS Path. 6, e1001173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Engelenburg SB et al. Distribution of ESCRT Machinery at HIV Assembly Sites Reveals Virus Scaffolding of ESCRT Subunits. Science 343, 653–656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobro MJ et al. Electron cryotomography of ESCRT assemblies and dividing Sulfolobus cells suggests that spiraling filaments are involved in membrane scission. Molecular Biology of the Cell 24, 2319–2327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bodon G et al. Charged Multivesicular Body Protein 2B (CHMP2B) of the Endosomal Sorting Complex Required for Transport-III (ESCRT-III) Polymerizes into Helical Structures Deforming the Plasma Membrane. J. Biol. Chem 286, 40276–40286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carlson L-A, Shen Q-T, Pavlin MR & Hurley JH ESCRT Filaments as Spiral Springs. Dev. Cell 35, 397–398 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Bleck M et al. Temporal and spatial organization of ESCRT protein recruitment during HIV-1 budding. Proc. Natl. Acad. Sci. U. S. A 111, 12211–12216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson DS, Bleck M & Simon SM Timing of ESCRT-III protein recruitment and membrane scission during HIV-1 assembly. Elife 10.7554/eLife.36221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kabsch WXDS Acta crystallographica. Section D, Biological crystallography 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winn MD et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sette P et al. The Phe105 loop of Alix Bro1 domain plays a key role in HIV-1 release. Structure 19, 1485–1495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z et al. A Systematic Methodology for Defining Coarse-Grained Sites in Large Biomolecules. Biophys. J 95, 5073–5083 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lyman E, Pfaendtner J & Voth GA Systematic Multiscale Parameterization of Heterogeneous Elastic Network Models of Proteins. Biophys. J 95, 4183–4192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Plimpton S Fast Parallel Algorithms for Short-Range Molecular Dynamics. Journal of Computational Physics 117, 1–19 (1995). [Google Scholar]
- 72.Schneider T & Stoll E Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Physical Review B 17, 1302–1322 (1978). [Google Scholar]
- 73.Martyna GJ, Tobias DJ & Klein ML Constant pressure molecular dynamics algorithms. The Journal of chemical physics 101, 4177–4189 (1994). [Google Scholar]
- 74.Grime JMA & Madsen JJ Efficient Simulation of Tunable Lipid Assemblies Across Scales and Resolutions. arXiv:1910.05362 (2019) [Google Scholar]
- 75.Humphrey W, Dalke A & Schulten K VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33-& (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code PDB 6VME. Uncropped images for Figure 1c, 2d, 2f, 4a, 4e, 5 and Extended Data Fig. 1 are available in Supplementary Figure 1; source data for graphs in figure 4 are available online.