

Abstract
Chimeric antigen receptor (CAR)-modified T cells have demonstrated efficacy against B cell leukemias/lymphomas. However, redirecting CAR T cells to malignant T cells is more challenging due to product-specific cis- and trans-activation causing fratricide. Other challenges include the potential for product contamination and T cell aplasia. We expressed non-signaling CARs (NSCARs) in γδ T cells since donor-derived γδ T cells can be used to prevent product contamination, and NSCARs lack signaling/activation domains, but retain antigen-specific tumor cell-targeting capability. As a result, NSCAR targeting requires an alternative cytotoxic mechanism, which can be achieved through utilization of γδ T cells that possess major histocompatibility complex (MHC)-independent cytotoxicity. We designed two distinct NSCARs and demonstrated that they do not enhance tumor-killing by αβ T cells, as predicted. However, both CD5-NSCAR- and CD19-NSCAR-modified γδ T cells enhanced cytotoxicity against T and B cell acute lymphoblastic leukemia (T-ALL and B-ALL) cell lines, respectively. CD5-NSCAR expression in γδ T cells resulted in a 60% increase in cytotoxicity of CD5-expressing T-ALL cell lines. CD19-NSCAR-modified γδ T cells exhibited a 350% increase in cytotoxicity against a CD19-expressing B-ALL cell line compared to the cytotoxicity of naive cells. NSCARs may provide a mechanism to enhance antigen-directed anti-tumor cytotoxicity of γδ T cells through the introduction of a high-affinity interaction while avoiding self-activation.
Keywords: chimeric antigen receptor, CAR, non-stimulating chimeric antigen receptor, NSCAR, anti-CD5CAR, gamma delta T cells, CD19 nonstimulating CAR
Graphical Abstract
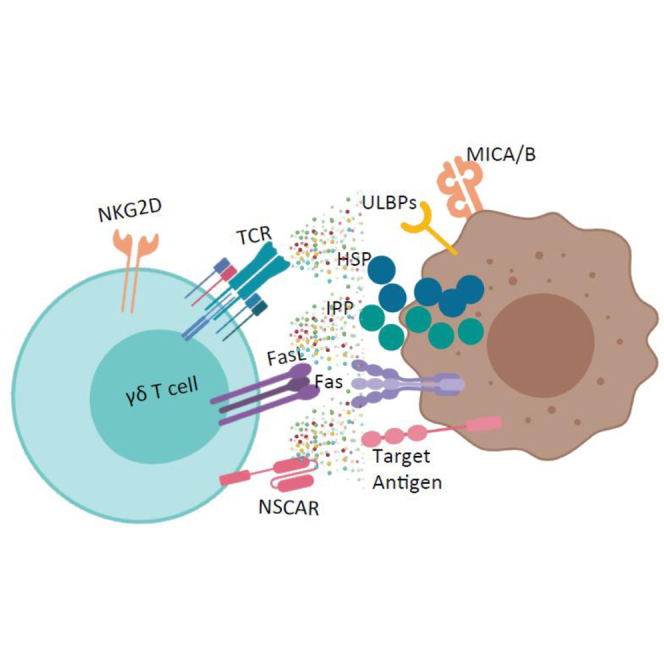
Chimeric antigen receptors (CARs) efficiently direct T cell immunity to a variety of cancers. It is problematic to develop CARs against T cell leukemia because of self-activation of transduced cells. However, unlike αβ T cells, introducing non-stimulating CARs to γδ T cells enhances their killing ability, as these cells recognize and kill through innate mechanisms.
Introduction
Currently, the FDA has approved the use of two chimeric antigen receptor (CAR) T cell therapies, Kymriah1 and Yescarta.2 These therapies are approved to treat adult diffuse large B cell lymphoma3,4 and Kymriah is also approved for pediatric B cell acute lymphoblastic leukemia (B-ALL).5 While these therapies have been successful in treating B cell malignancies, there are additional challenges to translating CAR therapy for the treatment of T cell malignancies. Many pre-clinical studies have developed strategies to treat T cell malignancies, including CARs targeting antigens such as CD5,6, 7, 8, 9, 10 CD7,11, 12, 13 CD4,14,15 and CD3.16,17 However, shared expression of these antigens on the CAR T cells, as well as cancer cells, can result in fratricide, or CAR T cells killing other CAR T cells.6,9,11,17,18 Additionally, a recent report demonstrated evidence of product contamination resulting in clonal expansion of a single leukemic blast that had been modified with the CD19-CAR. The CD19-CAR masked the CD19 antigen from CAR T cells, causing resistance to the therapy.19 Furthermore, a memory response against T cell antigens resulting in T cell aplasia is lethal and is therefore not an option. While therapies targeting B cell malignancies, such as Kymriah and Yescarta, result in potentially lifelong B cell aplasia due to a memory response against the targeted antigen,20,21 these patients can be treated with intravenous immunoglobulin (IVIG) to overcome this condition.22 However, due to increased demand for IVIG over recent years, the United States is currently experiencing a shortage of immunoglobulin.
Many groups have developed solutions to overcome these challenges to treating T cell malignancies using CAR therapy. The simplest option is targeting an antigen that is absent or expressed at low levels on normal T cells such as CD30,23, 24, 25, 26, 27 CD37,28 or TRBC1.29 Unfortunately, the majority of T cell malignancies do not have high expression of these antigens, which limits their usefulness. An alternative strategy is to utilize donor-derived cells, which eliminates the risk of product contamination, as isolating normal T cells from malignant T cells is a significant obstacle. Natural killer (NK) cells and γδ T cells are non-alloreactive and can be used in an allogeneic setting without additional modifications. Additionally, the NK-derived lymphoma cell line, NK-92 cells, can be used as an alternative to T cells for CAR therapy.6, 7, 8,10,15,16 However, the expansion of NK or NK-92 cells is time-consuming, genetic engineering can be challenging, and they are particularly sensitive to cryopreservation.30 Strategies to avoid T cell aplasia have included incorporation of suicide genes and switches into CAR constructs to regulate their expression, provide control over robust responses, and prevent memory cell formation,31, 32, 33, 34, 35, 36, 37 but they are not uniformly effective, and escape of a modified cancer clone could be problematic.
Few strategies that address all three challenges have been evaluated. Therefore, we generated non-signaling CARs (NSCARs) that, when introduced into γδ T cells, enhance target cell killing while sparing the healthy, engineered cells. NSCARs lack the intracellular signaling domains typically present in a CAR (Figure 1A). As a result, NSCARs are non-activating. While expression of a non-signaling CAR is not expected to affect αβ T cell cytotoxicity against tumor cells, we hypothesize NSCARs can enhance γδ T cell cytotoxicity because, in contrast to αβ T cells, γδ T cells possess alternative mechanisms of cytotoxicity and do not require stimulation through CD3ζ in order to initiate target cell killing.38, 39, 40, 41, 42 In addition, ex vivo expanded γδ T cells are relatively short-lived with little expansion in vivo, which can help control cytokine release syndrome (CRS) and other adverse events resulting from CAR T cell therapy. Furthermore, γδ T cells are unlikely to cause GvHD as they interact with antigen independent of major histocompatibility complex (MHC)-recognition, permitting use in an allogeneic setting.43,44 We hypothesize NSCARs can act as anchors to tether the γδ T cells to tumor cells expressing the targeted antigen. While the cells are in close proximity, the cytotoxic mechanisms endogenous to γδ T cells can engage, ultimately resulting in tumor cell death.
Figure 1.

Schematic of CD5-Based and CD19-Based NSCAR Constructs
(A) NSCAR structure with CD28 transmembrane domain, truncated after two amino acids on the intracellular tail. (B and C) Bicistronic NSCAR transgenes in lentiviral vectors expressing enhanced green fluorescent protein (eGFP) and the NSCARs through the inclusion of a p2a sequence. Expression of both sequences are driven by the human ubiquitin C promoter (hUBC) with an interleukin-2 signal peptide (IL-2-SP). The CD5-NSCAR (B) includes a myc epitope tag, whereas the CD19-NSCAR (C) includes the CD8α hinge.
Herein, we design two distinct NSCARs: CD5-NSCAR (Figure 1B) and CD19-NSCAR (Figure 1C). We compare γδ T cell expansion in naive and NSCAR-modified populations and assess the cytotoxicity of NSCAR-modified γδ T cells against T-ALL and B-ALL cell lines. Additionally, we evaluate the effect of CD5-NSCAR expression on the cytotoxicity of αβ T cells. We further compared the CD19-NSCAR to the more traditional CD19-CAR. The results described herein demonstrate proof-of-concept that NSCAR expression in γδ T cells enhances antigen-directed killing, and the mechanisms involved are fundamentally and biologically different in αβ T cells.
Results
CD5 Antigen and CD5-NSCAR Are Downregulated in CD5-NSCAR-Modified Jurkat T Cells without Altering Activation
We and others have previously shown CD5-CAR expression on CD5-positive cells results in the downregulation of the CD5 antigen from the cell surface.6,9 To determine whether CD5 downregulation occurs upon CD5-NSCAR expression, we transduced Jurkat T cells with the CD5-NSCAR at MOIs 0.5 and 1 and CD5 expression was measured by flow cytometry. We detected a significant reduction in the percentage of CD5-positive Jurkat T cells, likely due to interactions with CD5-NSCAR on self and neighboring cells. As NSCARs do not contain a signaling cytoplasmic tail, we determined that these interactions causing CD5 downregulation were not coupled with intracellular signaling. Even at low MOIs, detection of CD5 expression was reduced in transduced cells (MOI 0.5 and MOI 1: p < 0.001). At MOI 1, <5% of the cells remained CD5-positive (Figure 2A).
Figure 2.

CD5 Expression and Activation of NSCAR-Modified Jurkat T Cells
Flow cytometry was performed to measure CD5 and CD69 expression on Jurkat T cells 5 to 6 days post-transduction. In all figures, a representative flow cytometry overlay is illustrated on the right. Black curve, naive; gray curve, MOI 0.5; light gray curve, MOI 1. (A) CD5 expression in naive and CD5-NSCAR-modified Jurkat T cells. Naive and MOI 1, n = 4; MOI 0.5, n = 3. (B) CD69 expression in naive and CD5-NSCAR-modified Jurkat T cells. Naive and MOI 1, n = 5; MOI 0.5, n = 3. (C) CD5 expression in naive, CD19-CAR-, and CD19-NSCAR-modified Jurkat T cells; n = 3. (D) CD69 expression in naive, CD19-CAR-, and CD19-NSCAR-modified Jurkat T cells; n = 3. Statistics were performed using a one-way ANOVA with Dunnett’s method to compare to the naive control group.
We previously demonstrated CD5-CAR expression on CD5-positive Jurkat T cells results in increased activation, as measured by CD69, due to interactions between the CAR and the CD5 antigen.6 However, we hypothesized the CD5-NSCAR would not affect the activation levels of the cells since the NSCAR lacks the intracellular signaling domains typically found in a CAR construct. By flow cytometry, we determined there is no change in CD69 expression in CD5-NSCAR-modified Jurkat T cells compared to the levels of CD69 in naive Jurkat T cells (Figure 2B).
We performed similar experiments with CD19-CAR- and CD19-NSCAR-modified Jurkat T cells. Jurkat T cells modified with the CD19-CAR or CD19-NSCAR did not demonstrate any change in detection of CD5 expression, with 95% of the cells expressing CD5, suggesting the downregulation observed in CD5-NSCAR-modified Jurkat T cells is due to interactions between the NSCAR and cognate antigen (Figure 2C). Jurkat T cells do not express CD19 and, as expected, there is no change in Jurkat T cell activation, as measured by CD69 by flow cytometry, when modified with either a CD19-CAR or CD19-NSCAR (Figure 2D).
The CD5-NSCAR-modified Jurkat T cells and CD5-edited Jurkat T cells were analyzed for CD5-Fc surface expression using flow cytometry. CD5-edited Jurkat T cells were developed in our laboratory using CRISPR-Cas9 genome editing. The CD5-negative fraction of cells were isolated using FACS with >98% purity and expanded under standard Jurkat T cell culture conditions, as described previously.6 Jurkat T cells transduced at an MOI 0.5 were, on average, 25% NSCAR-positive, whereas Jurkat T cells transduced at an MOI 1 were, on average, 70% NSCAR-positive. However, CD5-edited Jurkat T cells have a much higher percentage of NSCAR-expressing cells detected by flow cytometry when transduced with the CD5-NSCAR at the same MOIs. At MOIs 0.5 and 1, ~65% and ~90%, respectively, of CD5-edited Jurkat T cells were NSCAR-positive (Figure 3A). We see the emergence of a population of GFP-positive, CD5-NSCAR-negative Jurkat T cells following transduction of CD5-expressing cells; however, this population is substantially reduced in CD5-NSCAR-modified, CD5-edited Jurkat T cells (Figure S1A). This suggests that CD5 expression on Jurkat T cells blocks or reduces expression of the CD5-NSCAR. These results are consistent with our previous findings using CD5-CAR-modified Jurkat T cells.6
Figure 3.

Effects of CD5-NSCAR Expression in Jurkat T Cells
(A) CD5-NSCAR expression in non-edited (left) or CD5-edited (right) Jurkat T cells 5 days post-transduction. MOI 0.5, n = 3; naive and MOI 1, n = 6 (non-edited Jurkat T cells), n = 5 (CD5-edited Jurkat T cells). (B and C) CD5-NSCAR-modified Jurkat T cells were cultured with naive (black curve) or CD5-edited (gray curve) Jurkat T cells at 1:1 or 1:3 modified to non-modified ratios. (B) CD5-NSCAR expression at each ratio. (C) CD5 expression in non-modified Jurkat T cells following co-culture with CD5-NSCAR-modified Jurkat T cells at each ratio. Flow cytometry was performed to measure CD5-Fc and CD5 antigen expression on Jurkat T cells. Statistics were performed using a two-tailed Student’s t test and one-way ANOVA with Dunnett’s method to compare to the naive group.
To determine whether the expression of the CD5-NSCAR and CD5 antigen in Jurkat T cells vary over time, we measured NSCAR and CD5 expression on non-edited and CD5-edited Jurkat T cells by flow cytometry on days 5 and 15 post-transduction. On day 5, we observed approximately 20% NSCAR-positive cells at MOI 0.5 and approximately 50% NSCAR-positive cells at MOI 1, as previously noted. However, by day 15, the percentage of NSCAR-expressing Jurkat T cells was reduced to ~5% (MOI 0.5) and ~20% (MOI 1) (Figure S1B). Nevertheless, the percentage of GFP-positive cells remained unchanged, suggesting the transduced cells were not dying or diluted in the culture (data not shown). Furthermore, while the CD5 expression levels on Jurkat T cells 5 days post-transduction were very low, such a drastic downregulation was not observed 10 days later, suggesting the balance between CD5 expression and CD5-NSCAR expression shifts over time (Figure S1C). The increase in CD5 antigen expression correlates with a decrease in CD5-NSCAR expression. In contrast, CD5-NSCAR expression on CD5-edited Jurkat T cells was much less variable between days 5 and 15, decreasing from 65% and 80% to 60% and 77%, at MOIs 0.5 and 1, respectively. To confirm the flow cytometry data, we performed western blot analysis using an anti-CD5 antibody with whole cell lysates from Jurkat T cells or CD5-edited Jurkat T cells modified with the CD5-NSCAR. Whole cell lysates were collected on day 15 post-transduction. Western blot and densitometry revealed only slightly lower levels of CD5 protein in whole cell lysates of CD5-NSCAR-modified Jurkat T cells compared to CD5 protein levels in naive Jurkat T cells (Figure S1D). Non-modified and CD5-NSCAR-modified, CD5-edited Jurkat T cells displayed no signs of CD5 protein expression, as expected (data not shown).
Co-culture of CD5-NSCAR-Modified Jurkat T Cells with Non-modified Jurkat T Cells Leads to CD5 Antigen Downregulation in Non-modified Cells and CD5-NSCAR Downregulation in Modified Cells
We hypothesize that the CD5-NSCAR expressed on Jurkat T cells can interact with the CD5 antigen on self and neighboring cells, resulting in downregulation of both proteins. To explore this further, we established a 14-h co-culture to observe changes in CD5-NSCAR expression in Jurkat T cells when cultured with non-modified Jurkat T cells, as well as changes in CD5 antigen expression in the non-modified Jurkat T cells. We cultured CD5-NSCAR-modified and non-modified Jurkat T cells at 1:1 and 1:3 modified to non-modified ratios. After 14 h, we observed a significant downregulation in CD5-NSCAR expression when the cells were cultured at a low ratio of 1:3 with Jurkat T cells (p < 0.001). Despite a lack of statistical significance at the 1:1 ratio, the same trend was observed (p = 0.078). However, when CD5-NSCAR modified cells were cultured with non-modified, CD5-edited Jurkat T cells, there was no change in CD5-NSCAR expression at either ratio (Figure 3B). We conclude the CD5 antigens on non-modified Jurkat T cells can interact with the CD5-NSCAR on the modified Jurkat T cells, resulting in NSCAR-downregulation. Therefore, there is a greater reduction in CD5-NSCAR expression in cultures with a higher percentage of non-modified, CD5-expressing cells. Transduction of CD5-edited Jurkat T cells with the CD5-NSCAR produced similar results to those described above when cultured with non-edited Jurkat T cells or CD5-edited Jurkat T cells (at 1:3, p < 0.001; at 1:1, p = 0.058; Figure S2A).
Additionally, we measured the CD5 expression on the non-modified Jurkat T cells in the co-culture. The data demonstrated a significant decline in CD5 expression as the percentage of CD5-NSCAR-modified Jurkat T cells in the culture increased (at 1:3, p = 0.097; at 1:1, p < 0.001), with fewer than 20% of the cells expressing CD5 on the cell surface at the 1:1 ratio (Figure 3C). This suggests that when there are more CD5-NSCAR-expressing Jurkat T cells in the culture, there is an overall increase in the interactions between the CD5-NSCAR and CD5 antigen, resulting in greater downregulation of the CD5 antigen on non-modified cells. Similar results were obtained when culturing CD5-edited, CD5-NSCAR-modified Jurkat T cells with non-modified Jurkat T cells. However, the CD5 on the non-modified Jurkat T cells downregulated to a greater degree when they were cultured with CD5-edited, CD5-NSCAR-modified Jurkat T cells (95% reduction at the 1:1 ratio) compared to when they were in culture with non-edited, CD5-NSCAR-modified Jurkat T cells (80% reduction at the 1:1 ratio; Figure S2B).
NSCAR Modification Does Not Impede γδ T Cell Expansion and, Contrary to CD19-NSCAR Expression, CD5-NSCAR Expression Downregulates CD5 Antigen Expression
γδ T cells were expanded in serum-free conditions from healthy donor blood using interleukin-2 (IL-2) and zoledronate. On days 7–9 of expansion, flow cytometry was performed to determine the percentage of γδ T cells and CD5 expression within the γδ T cell population. For each expansion, γδ T cells were plated for lentiviral vector transduction and a non-transduced well was plated simultaneously. The expansion of naive and NSCAR-modified γδ T cells was monitored through day 12. The percentage of γδ T cells in the population expanded consistently in both the naive and CD5-NSCAR-modified cultures, with no significant differences in expansion (p = 0.353; Figures 4A and 4B). Both populations of cells expanded ~2.5-fold in the 4–5 days post-transduction suggesting that expression of the CD5-NSCAR does not hinder γδ T cell expansion nor overall proliferation of the culture, despite the presence of CD5 antigen (Figure 4C). Similarly, expansion of γδ T cells modified with the CD19-NSCAR or GFP control lentiviral vectors on days 7–9 was evaluated for 4–5 days post-transduction. The control lentiviral vector encodes eGFP driven by the EF1α promoter, as previously described.45 CD19-NSCAR- and GFP-modified γδ T cells expanded comparable to naive γδ T cells (~2-fold; Figure 4C). While γδ T cells do not express CD19, these data provide evidence for the hypothesis that transduction alone does not affect γδ T cell expansion.
Figure 4.

NSCAR-Modified γδ T Cell Expansion and CD5 Downregulation
(A) Representative flow cytometry plots of γδ T cell expansion. The percentage of γδ T cells on day 7 (left) is compared to the percentage of γδ T cells on day 12 in naive cells (middle) and CD5-NSCAR-modified γδ T cells (right). (B) Percentage of γδ T cells in a population of naive (black curve) and CD5-NSCAR-modified γδ T cells (gray curve) between day of transduction (7–9) and day of cytotoxicity assay (12–13); n = 3. (C) Fold expansion of naive, GFP-modified, CD5-NSCAR-modified and CD19-NSCAR-modified γδ T cells. Naive, n = 5; GFP and CD5-NSCAR, n = 3; CD19-NSCAR, n = 2. (D) Representative flow cytometry plots of CD5 expression in CD5-NSCAR-modified (left) and CD19-NSCAR-modified γδ T cells (right). Black curve, naive; gray curve, NSCAR-modified. (E) Graphical representation of CD5 expression in naive and modified γδ T cells. Naive, n = 8; GFP and CD19-NSCAR, n = 2; CD5-NSCAR, n = 5. Statistics were performed using two-tailed Student’s t test and one-way ANOVA with Dunnett’s method to compare to the naive control group. Each replicate represents an independent donor.
As the studies in Jurkat T cells indicate, interactions between CD5 antigen and CD5-NSCAR result in the apparent downregulation of CD5. To determine whether this occurs in γδ T cells, CD5 expression on the cell surface of naive and CD5-NSCAR-modified γδ T cells was measured by flow cytometry. A significant decrease in the detection of CD5-expressing, CD5-NSCAR-modified γδ T cells was observed compared to the detection of CD5-positive naive γδ T cells, with fewer than 10% of the cells expressing CD5 on the cell surface; p < 0.001. However, there was no significant downregulation of CD5 expression in γδ T cells modified with the CD19-NSCAR or GFP lentiviral vectors (p > 0.05; Figures 4D and 4E).
NSCAR-Modified γδ T Cells Exhibit Enhanced Antigen-Directed Cytotoxicity
To determine whether the CD5-NSCAR enhances the cytotoxicity of γδ T cells, we prepared a cytotoxicity assay with Jurkat T cells and Molt-4 T cells, two CD5-positive/CD19-negative T cell lines. Cytotoxicity assays were also performed using CD19-NSCAR-modified cells and 697 target cells, which is a CD19-positive/CD5-negative B-ALL cell line. Co-cultures were established at 3:1 or 5:1 effector to target (E:T) ratios and incubated for 4 h at 37°C. The percent increase in cytotoxicity compared to non-modified γδ T cells is shown in Figure 5. There was an increase in the cytotoxicity by CD5-NSCAR-modified γδ T cells against both CD5-positive target cell lines compared to non-modified cells (Figures 5A and 5B). Additionally, we measured the cytotoxicity of GFP-modified γδ T cells against Jurkat T cells. The data demonstrated donor variability, resulting in cells from half the donors exhibiting a decrease or no change in cytotoxicity upon GFP-modification, while the other half exhibited enhanced cytotoxicity. The greatest change in cytotoxicity was a 75% increase, however, the percentage of dead Jurkats only increased from 6% to 10.5% (data not shown). On average, at the 5:1 E:T ratio, the CD5-NSCAR-modified γδ T cells cultured with Jurkat T cells or Molt-4 cells resulted in 40% and 35% dead target cells, respectively, both of which correspond to a 50%–60% increase in cytotoxicity compared to that of naive γδ T cells. Furthermore, the CD19-NSCAR enhanced cytotoxicity against 697 cells compared to that of naive γδ T cells, killing on average 32% of the target cells at the 5:1 E:T ratio, which was a 450% increase in killing compared to that of non-modified cells (Figure 5C). These data validate two NSCARs targeting different tumor-cell antigens demonstrating that they can increase γδ T cell anti-tumor cytotoxicity in vitro. Moreover, the CD19-NSCAR expressed on γδ T cells demonstrates similar cytotoxicity against 697 cells as compared to CD19-CAR-modified γδ T cells (p = 0.905 and p = 0.857 at 3:1 and 5:1 E:T ratios, respectively; Figure S3). There was a high degree of donor variability in baseline cytotoxicity, consistent with previous findings;46, 47, 48 however, an increase in cytotoxicity by NSCAR-modified γδ T cells was routinely observed.
Figure 5.

NSCAR-Modified γδ T Cell Cytotoxicity against T-ALL and B-ALL Cell Lines
Effector cells and target cells were cultured at 3:1 (black bars) and 5:1 (white bars) effector to target (E:T) ratios for 4 h. The percent increase in cytotoxicity by modified γδ T cells compared to that of naive γδ T cells is graphed to account for donor variability in baseline cytotoxicity. The baseline is represented as the cytotoxicity of naive γδ T cells. Flow cytometry was used to measure eFluor780, VPD450, and GFP. (A) γδ T cell cytotoxicity against CD5-positive Jurkat cells. Three different donors modified with the CD5-NSCAR are shown separately, including the overall average cytotoxicity. One donor was repeated. (B) γδ T cell cytotoxicity against CD5-positive Molt-4 cells. Cells from two donors were modified with CD5-NSCAR lentiviral vector. (C) CD19-NSCAR-modified γδ T cell cytotoxicity against CD19-positive 697 cells. Cells from two donors were assessed. One donor was repeated. (D) 12-h co-culture of CD19-NSCAR-modified γδ T cells with 697 cells. CD107a expression was measured by flow cytometry 6 days post-transduction (left). ELISA was used to quantify IFN-γ secretion by CD19-CAR- and CD19-NSCAR-modified γδ T cells 6 days post-transduction (right). This experiment was performed in triplicate. Statistics were performed using a two-tailed Student’s t test to compare CD19-NSCAR degranulation or IFN-γ secretion in co-culture with 697 cells compared to that of naive cells cultured with 697 cells.
We hypothesized the NSCAR-modified γδ T cells exhibit their cytotoxic activity through mechanisms endogenous to the γδ T cell, specifically through the release of perforin and granzyme B, as well as interferon-γ (IFN-γ). To evaluate this further, we cultured CD19-NSCAR-modified γδ T cells with 697 target cells at a 5:1 E:T ratio and incubated the cells for 12 h at 37°C. Following the incubation period, cells were evaluated for degranulation and supernatants were collected and analyzed for IFN-γ secretion by ELISA. Upon co-culture with CD19-expressing target cells, there is significantly greater degranulation of CD19-NSCAR-modified γδ T cells compared to degranulation of naive γδ T cells (p = 0.0182). The IFN-γ ELISA demonstrates a trend toward increased IFN-γ secretion by CD19-NSCAR-modified γδ T cells in co-culture with 697 cells compared to secretion by control cells; however, this data was not statistically significant (p = 0.101; Figure 5D).
NSCAR-Modified αβ T Cells Do Not Have Enhanced Anti-Tumor Cytotoxicity
To test our hypothesis that NSCAR expression requires MHC-independent mechanisms of cytotoxicity in order to affect cellular killing in an antigen-specific manner, we performed a cytotoxicity assay culturing CD5-NSCAR-modified αβ T cells with Jurkat target cells at 3:1 and 5:1 E:T ratios. We predicted the CD5-NSCAR would not affect αβ T cell cytotoxicity. Others have previously published studies using constructs similar to the NSCAR and demonstrated that the truncated CAR does not increase T cell activation as measured by CD25,49 nor does it affect cellular proliferation or viability.11 Our data demonstrate that there was no difference in naive αβ T cell cytotoxicity against Jurkat T cells compared to the cytotoxicity of CD5-NSCAR-modified αβ T cells against Jurkat T cells, with both resulting in 40%–45% dead targets at each E:T ratio (3:1 E:T ratio: p = 0.618; 5:1 E:T ratio: p = 0.639; Figure 6). Both donors were transduced equally by the CD5-NSCAR lentiviral vector and one donor was additionally modified with the CD5-CAR (Figure S4A). CD5-CAR-modified αβ T cells killed 80% of the Jurkat target cells (Figure S4B), a 78% increase in cytotoxicity compared to that of naive αβ T cells.
Figure 6.

CD5-NSCAR-Modified αβ T Cell Cytotoxic Activity
Effector cells and target cells were cultured at 3:1 (black bars) and 5:1 (white bars) effector to target (E:T) ratios for 12 h. Cytotoxicity of naive and CD5-NSCAR-modified αβ T cells in culture with Jurkat T cells was determined by flow cytometry measuring eFluor780, VPD450, and GFP. Solid bars represent donor 1 and slashed bars represent donor 2. Statistics were performed using a two-tailed Student’s t test to compare cytotoxicity at each E:T ratio among donors.
NSCAR Shed from the Cell Surface into the Supernatant Can Interact with Target Cells
We hypothesized that the apparent downregulation of the NSCAR may be due, in part, to protein shedding from modified γδ T cells resulting in lower NSCAR on the cell surface. To determine whether shedding was occurring, we cultured γδ T cells in fresh media on day 1 post-transduction. Non-modified cells were cultured under the same conditions and 48 h later, the supernatants were collected and filtered. Jurkat T cells were cultured in the γδ T cell supernatant for 4 h. Flow cytometry was performed to determine the CD5 expression levels on Jurkat T cells following culture in γδ T cell supernatant. Jurkat T cells cultured in their own media, or supernatant from naive γδ T cells, GFP-transduced γδ T cells, or CD19-CAR-transduced γδ T cells all expressed high levels of CD5 as measured by flow cytometry. However, Jurkat T cells cultured in the supernatant of CD5-CAR- or CD5-NSCAR-modified γδ T cells demonstrated a significant reduction in CD5 antigen detection to ~25%. This suggests there was a factor in the supernatant of both CD5-CAR- and CD5-NSCAR-modified γδ T cells that interacted with the Jurkat T cells, resulting in CD5 downregulation or blocking of anti-CD5 antibody from binding CD5 on the T cell surface (CAR and NSCAR: p < 0.001; Figure S5A). We hypothesized that the extracellular portion of the CAR/NSCAR was cleaved from the cell surface and interacting with its cognate antigen. To test this, we pre-incubated the γδ T cell supernatant for 30 min with CD5-Fc, which is a soluble CD5 fused to the Fc portion of an immunoglobulin G (IgG), prior to culturing the Jurkat T cells in the supernatants. Jurkat T cells cultured in the pre-incubated CD5-CAR- or CD5-NSCAR-modified γδ T cell supernatant no longer exhibited decreased detection of CD5 (p = 0.240 and p = 0.402, respectively). CD5 expression was measured at 60% and 70% of the population, respectively. Additionally, the pre-incubation did not affect the percentage of CD5-positive Jurkat T cells cultured in naive γδ T cell supernatant (p = 0.956). Furthermore, upon CD5-Fc pre-incubation, the percentage of CD5-expressing Jurkat T cells cultured in supernatants of CD5-CAR- or CD5-NSCAR-modified γδ T cells did not significantly differ from that of cells cultured in pre-incubated naive γδ T cell supernatants (p = 0.407 and p = 0.584, respectively; Figure S5B).
Similar experiments were performed to determine whether this effect was CD5-NSCAR-specific or whether the CD19-NSCAR behaved similarly. γδ T cells transduced with a CD19-NSCAR were cultured for 24–48 h and the supernatants were then used to culture 697 cells for 4 h as previously described. Following the 4-h incubation, CD19-positive 697 cells were measured by flow cytometry. 697 cells cultured in their own media or supernatant from naive or GFP-modified γδ T cells demonstrated no change in CD19 detection. However, there was a significant decrease in CD19 detection when 697 cells were cultured in supernatant from CD19-NSCAR-modified γδ T cells (p = 0.048), suggesting this effect is not specific to the CD5-NSCAR, nor to T cell antigens (Figure S5C). As described, reduction in CD19 detection could be due to downregulation or blockade of antibody-binding due to CD19-NSCAR interactions with the CD19 antigen. CD19 expression had been reduced to 40% of 697 cells cultured in supernatant from CD19-NSCAR-modified γδ T cells. Furthermore, pre-incubation of γδ T cell supernatant with soluble CD19-Fc under the conditions previously described prevented this reduction in CD19-expressing 697 cells. CD19 was detected in ~80% of the cells cultured in CD19-Fc pre-incubated supernatant from CD19-NSCAR-modified γδ T cells. There is no difference between the percentage of CD19-expressing 697 cells cultured in the pre-incubated naive γδ T cell supernatant compared to that of 697 cells cultured in the pre-incubated CD19-NSCAR-modified γδ T cell supernatant (Figure S5D).
Discussion
γδ T cell therapy provides an alternative cellular vehicle for CAR therapy that may prove advantageous in particular settings, such as for the treatment of T cell malignancies. We have developed a serum-free protocol for ex vivo expansion of Vγ9Vδ2 T cells50,51 and are testing the effectiveness of CAR-modified cells. During these studies, we found that CARs lacking a stimulating domain (i.e., NSCARs) retained their ability to enhance γδ T cell-directed killing and that NSCARs can be valuable in a γδ T cell setting due to their non-stimulating properties. NSCARs prevent strong activation of γδ T cells upon antigen stimulation and act as an anchor to tether the γδ T cells to the tumor cells. We hypothesize that this high-affinity interaction facilitates the engagement of natural, MHC-independent mechanisms of cytotoxicity. We demonstrated that expression of a NSCAR targeting a T cell antigen in γδ T cells does not hinder their expansion, whereas a functional, signaling CAR targeting a T cell antigen results in fratricide and hinders proliferation,11 with the exception being antigens that downregulate rapidly.9 Additionally, we’ve shown CD5 antigen downregulation in γδ T cells modified with the CD5-NSCAR and that CD5 downregulation is specific to expression of the CD5-NSCAR, as it is not observed in CD19-NSCAR-modified γδ T cells. These results are similar to those we and others have shown using anti-CD5 CARs.6,9
We observed donor variability in both naive αβ and γδ T cell cytotoxicity against various cancer cell lines. However, despite the variability, NSCARs consistently enhanced γδ T cell cytotoxicity against cells expressing the targeted antigen. In contrast, NSCAR-modification of αβ T cells did not affect antigen-directed cytotoxicity. We hypothesize that this observed anti-cancer activity is due to the engagement of receptors on the γδ T cells with their ligands on the leukemia cell lines. It was shown that the release of perforin and granzyme may facilitate NSCAR-mediated γδ T cell cytotoxicity and the release of these factors is likely downstream of NKG2D signaling. Additionally, our results indicate the secretion of IFN-γ by CAR T cells does not significantly increase upon co-culture with target cells. Therefore, we predict the predominant mechanisms of action include NKG2D engagement. However, it is possible that additional γδ T cell mechanisms of cytotoxicity, such as Fas-FasL interactions, are involved. Future studies could clarify whether this mechanism is important to NSCAR-mediated γδ T cell cytotoxicity.
A primary advantage to γδ T cell therapy is the inherent anti-tumor cytotoxicity of γδ T cells. We demonstrate NSCAR interactions with the cognate antigen enhance γδ T cell cytotoxicity. However, target antigen downregulation is a known mechanism of tumor-cell escape from CAR-directed killing, and we show that similar resistance mechanisms may occur with NSCARs. Unlike αβ T cells, γδ T cells have endogenous pathways leading to multiple potential mechanisms of cytotoxicity, which are independent of CAR expression. Therefore, in the event of antigen-downregulation in subjects treated with NSCAR-modified γδ T cells, these natural mechanisms of anti-tumor cytotoxicity can prevail, with continued killing of tumor cells. While targeting CD5 with a CAR has been shown to result in transient fratricide, the targeting of other T cell antigens, such as CD7, has demonstrated persistent fratricide and prevention of CAR T cell expansion. However, the degree to which the partial downregulation of CD5 affects manufacturing is currently unknown. Therefore, anti-CD5 strategies may still benefit from NSCAR T cell therapy. We propose that NSCAR-modified γδ T cells can be advantageous particularly in settings of complete antigen downregulation where the use of a CAR results in inhibition of proliferation. Many groups using CAR T cell therapy for the treatment of B cell malignancies have reported numerous cases of antigen-negative relapse.52 The tumor cells downregulate the targeted antigen as a mechanism of escaping CAR T cell killing. Naive γδ T cell infusion into patients has demonstrated some anti-tumor activity53, 54, 55 and therefore we hypothesize in the event of antigen downregulation rendering NSCARs/CARs ineffective, γδ T cells may still demonstrate anti-tumor activity. Therefore, CAR-modified γδ T cells may add a significant benefit for the treatment of B cell malignancies. However, NSCAR γδ T cell therapy may not further increase efficacy in this clinical setting. Furthermore, NSCAR transgenes are substantially shorter than CAR transgenes and multiple NSCARs can be expressed from a single vector, thereby reducing the possibility of antigen escape.
Additionally, we showed that NSCARs were shed from the surface of γδ T cells into the supernatant and that shedding is not unique to NSCARs, as the results are consistent with those using a similar CD5-CAR sequence. Decreased expression of the NSCAR on the cell surface can result in decreased observed cytotoxicity. The mechanism of shedding is not well understood, but it is noteworthy that we engineered the CD5-NSCAR and CD19-NSCAR with different hinge regions. The CD5-NSCAR includes a myc tag while the CD19-NSCAR contains the CD8α hinge, however, NSCAR-shedding is observed with both. Each NSCAR contains a 29 amino acid extracellular CD28 sequence, which may play a role in shedding, as CD28 shedding has been reported.56 There is an observed correlation between the CAR/NSCAR shedding and the transduction efficiency. Donors that yielded a greater percentage of cells expressing the transgene demonstrated a greater reduction in CD5 or CD19 detection on the target cells following culture in the γδ T cell supernatant. Studies to determine the mechanism of shedding and to characterize the protein in the supernatant are required to fully comprehend these observations. Identification of the mechanisms involved can lead to the redesign of CARs and prevention of shedding, which can potentially result in greater antigen-directed cytotoxicity.
NSCARs have the potential to represent an alternative to CAR therapy, particularly in settings of T cell malignancies using donor-derived cells, due to their ability to enhance γδ T cell cytotoxicity in an antigen-directed manner, without self-activating and hindering cellular proliferation. Understanding the mechanism of NSCAR shedding could result in a second generation of NSCARs that are resistant to shedding, a characteristic likely to enhance NSCAR-mediated cytotoxicity. Furthermore, studies assessing the role of NSCARs in additional aspects of γδ T cell tumor killing, such as trafficking to the tumor microenvironment, can influence the generation of another class of NSCARs that are superior to CARs in γδ T cells. In the appropriate clinical setting, NSCARs have the potential to surpass CARs as a viable therapy, increasing anti-tumor efficacy and minimizing off-tumor cytotoxicity.
Materials and Methods
Cell Lines
The Jurkat cell line clone E6-1 was purchased from American Type Culture Collection (ATCC, Manassas, VA). As previously described, the Molt-4 and 697 cell lines were gifted by Dr. Douglas Graham (Emory University).6 CD5-edited Jurkat T cells were generated as previously described.6 All cell lines were cultured in RPMI (Corning, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
Engineering the NSCAR Sequences
The CD5-CAR sequence, as previously described,6 was truncated to remove the CD3ζ signaling domain, as well as the intracellular portion of CD28. The entire transmembrane domain of CD28, as well as two intracellular amino acids, remain. Additionally, we included a unique 21 base-pair sequence on the cytoplasmic end of the truncated CD28 for genetic determination of the proviral sequence. The vector is a bicistronic lentiviral construct, facilitating dual expression of enhanced green fluorescent protein (eGFP) and the NSCAR transgene using a p2a peptide sequence. The CD19-NSCAR was similarly generated by truncation of the CD19-CAR (unpublished data) after the first two intracellular amino acids of CD28. Similar to the CD5-NSCAR, this vector is a bicistronic lentiviral construct, expressing eGFP and the NSCAR transgene using a p2a peptide sequence. However, the CD19-NSCAR has the CD8α hinge where the CD5-NSCAR has the myc tag. The CD19-scFv sequence was generated from codon optimization of a published CD19-scFv sequence produced in a mouse hybridoma cell line.57
Generation of CAR- and NSCAR-Encoding Lentiviral Vectors
HIV-1-based recombinant lentiviral vectors for all CAR and NSCAR constructs were produced and titered, as previously described.6
Lentiviral Vector Transduction of Cell Lines
Lentiviral vector transduction was carried out as previously described using 6 μg/mL polybrene (EMD Millipore, Billerica, MA).6 The transduced cells were cultured for at least 5 days prior to being used for downstream applications. Jurkat T cells were transduced at multiplicity of infection (MOI) of 0.5 or 1.
Expansion of γδ T Cells from Healthy Donor Blood
Blood was obtained from consented, healthy adults with the assistance of the Emory Children’s Clinical and Translational Discovery Core. PBMCs were isolated from 30–50 mL healthy donor blood using Ficoll-Paque density gradient and centrifugation following the manufacturer’s protocol. PBMCs were expanded in serum-free conditions as previously described50 for up to 13 days in vitro. On days 0 and 3, 5 ug/mL zoledronic acid and 500 IU/mL IL-2 were added to the culture. Beginning on day 6, 1,000 IU/mL IL-2 was added to the culture medium. Cells were cultured at 1.5 × 106 cells/mL.
Expansion of αβ T Cells from Healthy Donor Blood
PBMCs were isolated from healthy donor blood as described above. A Pan T cell isolation was performed using Miltenyi’s Pan T cell Isolation kit (Miltenyi Biotech, Germany) and the T cells were expanded in X-VIVO 15 media (Lonza, Switzerland) supplemented with 10% FBS, 1% penicillin/streptomycin, 50 ng/mL IL-2, and 5 ng/mL IL-7. Following T cell isolation, cells were stimulated with CD3/CD28 Dynabeads at a 1:1 ratio for 24 h (Thermo Fisher Scientific, Waltham, MA). Cells were cultured at 1 × 106 cells/mL.
Lentiviral Vector Transduction of γδ T Cells
Lentiviral vector transduction was carried out between days 7 and 9 of expansion. Cells were incubated with 60% vector in culture medium supplemented with 6 μg/mL polybrene for 18–24 h, at which point culture medium was replaced with fresh medium. The transduced cells were cultured for 3–5 days before being used for downstream applications.
Lentiviral Vector Transduction of αβ T Cells
Lentiviral vector transduction was carried out immediately upon removal of the CD3/CD28 Dynabeads. Cells were incubated with 60% vector in culture medium supplemented with 6 μg/mL polybrene for 18–24 h, at which point culture medium was replaced with fresh medium. The transduced cells were cultured for 6 days before being used for downstream applications.
Flow Cytometry Analysis
Analysis was performed using a BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FCS Express 6 software. Antibodies used included anti-CD5 PerCP/Cy5.5, anti-CD3 BV421, anti-γδ T cell receptor (TCR) phycoerythrin (PE) and anti-CD69 APC-Cy7 (BD Biosciences, San Jose, CA). CD5-Fc fusion protein (G&P Biosciences, Santa Clara, CA) and CD19-Fc fusion protein (ACROBiosystems, Newark, DE) were used to detect anti-CD5 constructs and anti-CD19 constructs, respectively, with a secondary anti-IgG Fc antibody (Jackson Immunoresearch Laboratories, West Grove, PA), as previously described.6 Violet Proliferation Dye 450 (VPD450) was used to label the target cells in the cytotoxicity and co-culture studies, and cell death was assessed using eFluor 780 (described below). Degranulation of γδ T cells was detected using anti-CD107a APC (BD Biosciences, San Jose, CA).
Western Blotting
Jurkat T cells were lysed using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO) and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Quantification of protein, separation by SDS-PAGE, and transfer to a nitrocellulose membrane were performed as previously described.6 The blocked membrane was incubated with an anti-CD5 mAb and horseradish peroxidase (HRP)-labeled secondary antibody as previously described.6 Densitometry was performed using ImageJ.
Co-culture Assay Using NSCAR-Modified Jurkat T Cells and CD5-Edited Jurkat T Cells
Naive and CD5-edited Jurkat T cells were transduced with the bicistronic lentiviral vector encoding CD5-NSCAR at MOI 1. After 18–24 h, culture medium was replaced with fresh medium and on day 5, flow cytometry using BD LSRII Flow Cytometer (BD Biosciences, San Jose, CA) confirmed transduction by both eGFP and CD5-Fc binding. Transduced cells were cultured with naive or CD5-edited Jurkat T cells previously labeled with VPD450 at modified to non-modified ratios of 1:1 and 1:3. Non-modified cells were labeled according to the manufacturer’s protocol (BD Biosciences, San Jose, CA). The cells were cultured for 14 h at final concentrations of 5 × 105 cells/mL. Changes in NSCAR expression on modified cells and CD5 expression on non-modified cells were assessed by flow cytometry.
Cytotoxicity Assay
Cytotoxicity assays were performed on days 12 or 13 of γδ T cell expansion, or on day 6 post-αβ T cell transduction. Target cells were labeled with VPD450 using the manufacturer’s protocol (BD Biosciences, San Jose, CA). Effector cells remained unstained. Effector (E) and target (T) cells were mixed in 12 × 75 mm FACS tubes at E:T ratios of 3:1 and 5:1 in a total volume of 250 μL. γδ T cell cytotoxicity assays were incubated for 4 h at 37°C in 5% CO2 and αβ T cell cytotoxicity assays were incubated for 12 h at 37°C in 5% CO2. Following incubation, the cells were washed and stained with eFluor 780 (Thermo Fisher Scientific, Waltham, MA). The double positive eFluor 780 and VPD450 cells were assessed using flow cytometry.
Protein Shedding Assay
On day 1 post-transduction, culture medium was changed on γδ T cells and they were cultured for 48 h under standard conditions as described above. After 48 h, the supernatants were collected and filtered through a 0.22 micron, low-protein binding polyvinylidene fluoride (PVDF) filter (MilliporeSigma, Burlington, MA). Jurkat T cells or 697 cells were then cultured for 4 h in the filtered γδ T cell supernatants. Conditions involving incubation of Jurkat T cells and 697 cells in complete RPMI were included. Additional experiments were performed pre-incubating the γδ T cell supernatant with CD5-Fc or CD19-Fc for 30 min prior to using it to culture the cell lines. Following 4 h, Jurkat T cells and 697 cells were washed to remove free proteins and stained with anti-CD5 or anti-CD19 antibodies, respectively, for flow cytometry.
Degranulation Assay
CD19-CAR- and CD19-NSCAR-modified γδ T cells were cultured with 697 cells in 12 × 75 mm FACS tubes at an E:T ratio of 5:1 in a total volume of 250 μL and incubated for 12 h at 37°C in 5% CO2. 697 cells were labeled with VPD450 using the manufacturer’s protocol prior to co-culture. Following the incubation, cells were stained for flow cytometry to analyze cell surface expression of CD107a using antibodies including anti-CD3 BV421, anti-γδ TCR PE, anti-CD107a APC (BD Biosciences, San Jose, CA), and viability dye eFluor 780 (Thermo Fisher Scientific, Waltham, MA).
IFN-γ ELISA
CD19-NSCAR-modified γδ T cells were cultured with 697 cells as described above for the degranulation assay. Following the 12-h incubation, cell culture supernatants were collected and stored at −80°C for 48 h. IFN-γ secretion was quantified by ELISA (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol.
Statistical Analysis
Statistical significance was determined using unpaired two-tailed Student’s t test and one-way ANOVA. All p values were calculated with SigmaPlot, version 14.0 (Systat Software, Chicago, IL), and p < 0.05 was considered statistically significant.
Author Contributions
L.C.F., S.A.B., R.E.R., A.F., C.B.D., and H.T.S. contributed to the conception, design of experiments and data analysis. L.C.F., S.A.B., R.E.R., A.F. acquired the data. L.C.F., C.B.D., and H.T.S. drafted the manuscript and the revisions. All authors provided their approval of the final version.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by grants from the National Institutes of Health (F31 CA221002-03) and Curing Kids Cancer. MOLT-4 and 697 cells were generously donated by Dr. Douglas Graham’s laboratory at Emory University. All healthy donor blood samples were obtained through the Emory’s Clinical Translational and Discovery Core.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.003.
Supplemental Information
References
- 1.Sheridan C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017;35:691–693. doi: 10.1038/nbt0817-691. [DOI] [PubMed] [Google Scholar]
- 2.Mullard A. Second anticancer CAR T therapy receives FDA approval. Nat. Rev. Drug Discov. 2017;16:818. doi: 10.1038/nrd.2017.249. [DOI] [PubMed] [Google Scholar]
- 3.Bouchkouj N., Kasamon Y.L., de Claro R.A., George B., Lin X., Lee S., Blumenthal G.M., Bryan W., McKee A.E., Pazdur R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019;25:1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 4.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., JULIET Investigators Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 5.Bach P.B., Giralt S.A., Saltz L.B. FDA Approval of Tisagenlecleucel: Promise and Complexities of a $475 000 Cancer Drug. JAMA. 2017;318:1861–1862. doi: 10.1001/jama.2017.15218. [DOI] [PubMed] [Google Scholar]
- 6.Raikar S.S., Fleischer L.C., Moot R., Fedanov A., Paik N.Y., Knight K.A., Doering C.B., Spencer H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. OncoImmunology. 2017;7:e1407898. doi: 10.1080/2162402X.2017.1407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moot R., Raikar S.S., Fleischer L., Querrey M., Tylawsky D.E., Nakahara H., Doering C.B., Spencer H.T. Genetic engineering of chimeric antigen receptors using lamprey derived variable lymphocyte receptors. Mol. Ther. Oncolytics. 2016;3:16026. doi: 10.1038/mto.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gust J., Hay K.A., Hanafi L.A., Li D., Myerson D., Gonzalez-Cuyar L.F., Yeung C., Liles W.C., Wurfel M., Lopez J.A. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mamonkin M., Rouce R.H., Tashiro H., Brenner M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y., Liu Q., Zhong M., Wang Z., Chen Z., Zhang Y., Xing H., Tian Z., Tang K., Liao X. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019;12:49. doi: 10.1186/s13045-019-0732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomes-Silva D., Srinivasan M., Sharma S., Lee C.M., Wagner D.L., Davis T.H., Rouce R.H., Bao G., Brenner M.K., Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Png Y.T., Vinanica N., Kamiya T., Shimasaki N., Coustan-Smith E., Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1:2348–2360. doi: 10.1182/bloodadvances.2017009928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You F., Wang Y., Jiang L., Zhu X., Chen D., Yuan L., An G., Meng H., Yang L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019;9:64–78. [PMC free article] [PubMed] [Google Scholar]
- 14.Ma G., Shen J., Pinz K., Wada M., Park J., Kim S., Togano T., Tse W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. Rep. 2019;15:443–447. doi: 10.1007/s12015-019-09876-5. [DOI] [PubMed] [Google Scholar]
- 15.Pinz K.G., Yakaboski E., Jares A., Liu H., Firor A.E., Chen K.H., Wada M., Salman H., Tse W., Hagag N. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8:112783–112796. doi: 10.18632/oncotarget.22626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen K.H., Wada M., Firor A.E., Pinz K.G., Jares A., Liu H., Salman H., Golightly M., Lan F., Jiang X., Ma Y. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. 2016;7:56219–56232. doi: 10.18632/oncotarget.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasaiyaah J., Georgiadis C., Preece R., Mock U., Qasim W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. 2018;3:99442. doi: 10.1172/jci.insight.99442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamonkin M., Mukherjee M., Srinivasan M., Sharma S., Gomes-Silva D., Mo F., Krenciute G., Orange J.S., Brenner M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2018;6:47–58. doi: 10.1158/2326-6066.CIR-17-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maude S.L., Teachey D.T., Porter D.L., Grupp S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan C., Barry A., Fooks-Parker S., Smith L., Baniewicz D., Hobbie W. Pediatric Survivorship: Considerations Following CAR T-Cell Therapy. Clin. J. Oncol. Nurs. 2019;23:35–41. doi: 10.1188/19.CJON.S1.35-41. [DOI] [PubMed] [Google Scholar]
- 23.Hombach A., Heuser C., Sircar R., Tillmann T., Diehl V., Pohl C., Abken H. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res. 1998;58:1116–1119. [PubMed] [Google Scholar]
- 24.Hombach A., Heuser C., Sircar R., Tillmann T., Diehl V., Pohl C., Abken H. Characterization of a chimeric T-cell receptor with specificity for the Hodgkin’s lymphoma-associated CD30 antigen. J. Immunother. 1999;22:473–480. doi: 10.1097/00002371-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Ramos C.A., Ballard B., Zhang H., Dakhova O., Gee A.P., Mei Z., Bilgi M., Wu M.F., Liu H., Grilley B. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Invest. 2017;127:3462–3471. doi: 10.1172/JCI94306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park S.I., Serody J.S., Shea T.C., Grover N.S., Ivanova A., Morrison K., Eldridge P., Mckay K., Cheng C.J., Covington D. A phase 1b/2 study of CD30-specific chimeric antigen receptor T-cell (CAR-T) therapy in combination with bendamustine in patients with CD30+ Hodgkin and non-Hodgkin lymphoma. J. Clin. Oncol. 2017;35 TPS3095-TPS3095. [Google Scholar]
- 27.Wang C.M., Wu Z.Q., Wang Y., Guo Y.L., Dai H.R., Wang X.H., Li X., Zhang Y.J., Zhang W.Y., Chen M.X. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2017;23:1156–1166. doi: 10.1158/1078-0432.CCR-16-1365. [DOI] [PubMed] [Google Scholar]
- 28.Scarfò I., Ormhøj M., Frigault M.J., Castano A.P., Lorrey S., Bouffard A.A., van Scoyk A., Rodig S.J., Shay A.J., Aster J.C. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood. 2018;132:1495–1506. doi: 10.1182/blood-2018-04-842708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maciocia P.M., Wawrzyniecka P.A., Philip B., Ricciardelli I., Akarca A.U., Onuoha S.C., Legut M., Cole D.K., Sewell A.K., Gritti G. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017;23:1416–1423. doi: 10.1038/nm.4444. [DOI] [PubMed] [Google Scholar]
- 30.Klingemann H. Challenges of cancer therapy with natural killer cells. Cytotherapy. 2015;17:245–249. doi: 10.1016/j.jcyt.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Hoyos V., Savoldo B., Quintarelli C., Mahendravada A., Zhang M., Vera J., Heslop H.E., Rooney C.M., Brenner M.K., Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duong M.T., Collinson-Pautz M.R., Morschl E., Lu A., Szymanski S.P., Zhang M., Brandt M.E., Chang W.C., Sharp K.L., Toler S.M. Two-Dimensional Regulation of CAR-T Cell Therapy with Orthogonal Switches. Mol. Ther. Oncolytics. 2018;12:124–137. doi: 10.1016/j.omto.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Budde L.E., Berger C., Lin Y., Wang J., Lin X., Frayo S.E., Brouns S.A., Spencer D.M., Till B.G., Jensen M.C. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koneru M., O’Cearbhaill R., Pendharkar S., Spriggs D.R., Brentjens R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015;13:102. doi: 10.1186/s12967-015-0460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu C.Y., Roybal K.T., Puchner E.M., Onuffer J., Lim W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Philip B., Kokalaki E., Mekkaoui L., Thomas S., Straathof K., Flutter B., Marin V., Marafioti T., Chakraverty R., Linch D. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 38.Born W.K., Reardon C.L., O’Brien R.L. The function of gammadelta T cells in innate immunity. Curr. Opin. Immunol. 2006;18:31–38. doi: 10.1016/j.coi.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Urban E.M., Chapoval A.I., Pauza C.D. Repertoire development and the control of cytotoxic/effector function in human gammadelta T cells. Clin. Dev. Immunol. 2010;2010:732893. doi: 10.1155/2010/732893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallucci S., Matzinger P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 41.Rincon-Orozco B., Kunzmann V., Wrobel P., Kabelitz D., Steinle A., Herrmann T. Activation of V gamma 9V delta 2 T cells by NKG2D. J. Immunol. 2005;175:2144–2151. doi: 10.4049/jimmunol.175.4.2144. [DOI] [PubMed] [Google Scholar]
- 42.Lança T., Correia D.V., Moita C.F., Raquel H., Neves-Costa A., Ferreira C., Ramalho J.S., Barata J.T., Moita L.F., Gomes A.Q., Silva-Santos B. The MHC class Ib protein ULBP1 is a nonredundant determinant of leukemia/lymphoma susceptibility to gammadelta T-cell cytotoxicity. Blood. 2010;115:2407–2411. doi: 10.1182/blood-2009-08-237123. [DOI] [PubMed] [Google Scholar]
- 43.Deniger D.C., Moyes J.S., Cooper L.J. Clinical applications of gamma delta T cells with multivalent immunity. Front. Immunol. 2014;5:636. doi: 10.3389/fimmu.2014.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morita C.T., Beckman E.M., Bukowski J.F., Tanaka Y., Band H., Bloom B.R., Golan D.E., Brenner M.B. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human γ δ T cells. Immunity. 1995;3:495–507. doi: 10.1016/1074-7613(95)90178-7. [DOI] [PubMed] [Google Scholar]
- 45.Doering C.B., Denning G., Shields J.E., Fine E.J., Parker E.T., Srivastava A., Lollar P., Spencer H.T. Preclinical Development of a Hematopoietic Stem and Progenitor Cell Bioengineered Factor VIII Lentiviral Vector Gene Therapy for Hemophilia A. Hum. Gene Ther. 2018;29:1183–1201. doi: 10.1089/hum.2018.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine B.L., Miskin J., Wonnacott K., Keir C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2016;4:92–101. doi: 10.1016/j.omtm.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salmikangas P., Kinsella N., Chamberlain P. Chimeric Antigen Receptor T-Cells (CAR T-Cells) for Cancer Immunotherapy - Moving Target for Industry? Pharm. Res. 2018;35:152. doi: 10.1007/s11095-018-2436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aleksandrova K., Leise J., Priesner C., Melk A., Kubaink F., Abken H., Hombach A., Aktas M., Essl M., Bürger I. Functionality and Cell Senescence of CD4/ CD8-Selected CD20 CAR T Cells Manufactured Using the Automated CliniMACS Prodigy® Platform. Transfus. Med. Hemother. 2019;46:47–54. doi: 10.1159/000495772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe N., Bajgain P., Sukumaran S., Ansari S., Heslop H.E., Rooney C.M., Brenner M.K., Leen A.M., Vera J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology. 2016;5:e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutton K.S., Dasgupta A., McCarty D., Doering C.B., Spencer H.T. Bioengineering and serum free expansion of blood-derived γδ T cells. Cytotherapy. 2016;18:881–892. doi: 10.1016/j.jcyt.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Zoine J.T., Knight K.A., Fleischer L.C., Sutton K.S., Goldsmith K.C., Doering C.B., Spencer H.T. Ex vivo expanded patient-derived γδ T-cell immunotherapy enhances neuroblastoma tumor regression in a murine model. OncoImmunology. 2019;8:1593804. doi: 10.1080/2162402X.2019.1593804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gardner R., Wu D., Cherian S., Fang M., Hanafi L.A., Finney O., Smithers H., Jensen M.C., Riddell S.R., Maloney D.G., Turtle C.J. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennouna J., Bompas E., Neidhardt E.M., Rolland F., Philip I., Galéa C., Salot S., Saiagh S., Audrain M., Rimbert M. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2008;57:1599–1609. doi: 10.1007/s00262-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakajima J., Murakawa T., Fukami T., Goto S., Kaneko T., Yoshida Y., Takamoto S., Kakimi K. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous gammadelta T cells. Eur. J. Cardiothorac. Surg. 2010;37:1191–1197. doi: 10.1016/j.ejcts.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 55.Meraviglia S., Eberl M., Vermijlen D., Todaro M., Buccheri S., Cicero G., La Mendola C., Guggino G., D’Asaro M., Orlando V. In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 2010;161:290–297. doi: 10.1111/j.1365-2249.2010.04167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hebbar M., Jeannin P., Magistrelli G., Hatron P.Y., Hachulla E., Devulder B., Bonnefoy J.Y., Delneste Y. Detection of circulating soluble CD28 in patients with systemic lupus erythematosus, primary Sjögren’s syndrome and systemic sclerosis. Clin. Exp. Immunol. 2004;136:388–392. doi: 10.1111/j.1365-2249.2004.02427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicholson I.C., Lenton K.A., Little D.J., Decorso T., Lee F.T., Scott A.M., Zola H., Hohmann A.W. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 1997;34:1157–1165. doi: 10.1016/s0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.