

Abstract
Cardiac arrhythmias are common, often the first, and sometimes the life-threatening manifestations of hereditary cardiomyopathies. Pathogenic variants in several genes known to cause hereditary cardiac arrhythmias have also been identified in the sporadic cases and small families with cardiomyopathies. These findings suggest a shared genetic aetiology of a subset of hereditary cardiomyopathies and cardiac arrhythmias. The concept of a shared genetic aetiology is in accord with the complex and exquisite interplays that exist between the ion currents and cardiac mechanical function. However, neither the causal role of cardiac arrhythmias genes in cardiomyopathies is well established nor the causal role of cardiomyopathy genes in arrhythmias. On the contrary, secondary changes in ion currents, such as post-translational modifications, are common and contributors to the pathogenesis of arrhythmias in cardiomyopathies through altering biophysical and functional properties of the ion channels. Moreover, structural changes, such as cardiac hypertrophy, dilatation, and fibrosis provide a pro-arrhythmic substrate in hereditary cardiomyopathies. Genetic basis and molecular biology of cardiac arrhythmias in hereditary cardiomyopathies are discussed.
Keywords: Genetics • Cardiomyopathy • Arrhythmias • Ion channels • Electrophysiology • Sudden death
This article is part of the Spotlight Issue on Inherited Conditions of Arrhythmia.
1. Introduction
Cardiac arrhythmias are common and central features of hereditary cardiomyopathies, regardless of whether the cardiomyopathy is caused by a genetic mutation (or pathogenic variant) affecting myocyte structural proteins or is secondary to a defect in another component of myocytes. Throughout this review, the term mutation is used when evidence for causality is robust. Otherwise, the term pathogenic variant is used to describe genetic variants that are associated with the phenotype. Hereditary cardiomyopathies by definition are considered primary diseases of the myocardium and more specifically, cardiac myocytes. The term cardiomyopathy, however, is loosely applied to various forms of advanced heart failure resulting from secondary causes, such as coronary artery disease (CAD). For clarity, throughout this review, the term cardiomyopathy is used to describe primary diseases of cardiac myocytes and not heart failure secondary to external causes, such as CAD or loading conditions. Likewise, primary ion channel disorders or channelopathies are not considered as cardiomyopathies and are not covered in this review. Primary arrhythmogenic diseases are discussed in other articles in the spotlight issue. To avoid ambiguity in nomenclature, HUGO nomenclature is used to describe gene names (human genes all in capital letters and italics). Proteins are named as their corresponding genes in capital letters but not in italics.
2. An overview of hereditary cardiomyopathies
Common forms of hereditary cardiomyopathies are categorized according to their phenotypic features, as hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (ACM). Other uncommon forms of hereditary cardiomyopathies include restrictive cardiomyopathy (RCM), left ventricular non-compaction cardiomyopathy (LVNC), amyloid cardiomyopathy, among others (Figure 1).1 Only common forms are discussed.
Figure 1.
Classification of hereditary cardiomyopathies. Major forms for hereditary cardiomyopathies, which are classified based on their morphological and physiological features, are depicted. Adult cardiac myocytes are shown in the background to indicate that hereditary cardiomyopathies are primary disorders of cardiac myocytes.
2.1 Hypertrophic cardiomyopathy
HCM is characterized by unexplained cardiac hypertrophy, a small left ventricular (LV) cavity, and a preserved or increased LV ejection fraction (EF) (reviewed in Ref.2). A notable phenotypic feature of HCM is the presence of LV outflow tract obstruction, which is detected at rest in approximately a quarter of patients and could be provoked with exercise or adrenergic stimulation in one-third of the patients. HCM is a major cause of sudden cardiac death (SCD) in the young.3–5 The death is typically due to ventricular fibrillation.
Mutations in genes encoding sarcomere proteins are the main causes, while mutations in another two dozen genes have been associated with HCM. MYH7 and MYBPC3, coding for myosin heavy chain 7 (or β myosin heavy chain) and myosin-binding protein C, respectively, are the two most common causal genes for HC (reviewed in Ref.2). Approximately 40% of HCM is caused by mutations in one of these two genes. TNNT2 gene-encoding cardiac troponin T is the third most common causal gene but is responsible for <5% of the HCM cases. Other uncommon causal genes include TPM1 (a-tropomyosin), TNNI3 (cardiac troponin I), ACTC1 (cardiac α-actin), MYL2 (myosin light chain), MYL3 (myosin light chain 3), and CSRP3 (muscle LIM protein). Collectively, all known causal genes account for ∼50–60% of the HCM cases. The remaining causal genes have not been identified yet. Overall, the prevalence of each specific mutation in HCM is low, as the mutations are rare and often private. There is no clear genotype–phenotype correlation in HCM.
2.2 Dilated cardiomyopathy
DCM is characterized by LV dilatation with an end-diastolic diameter of >2.7 cm/m2 and a reduced LVEF of <45% (reviewed in Ref.6). Patients with DCM typically present with symptoms of heart failure but are often asymptomatic or exhibit reduced exercise tolerance in the early stages of the disease. Cardiac arrhythmias and SCD are typically the late manifestations of the disease and are absent in the early stages of DCM. Clinical manifestations of DCM commonly present in the third and fourth decades of life.
DCM is familial in about half of the patients and exhibits an autosomal-dominant mode of inheritance. DCM occasionally manifests as an X-linked disease, such as in Duchenne and Becker muscular dystrophies and Emery-Dreifuss syndrome. DCM is a genetically heterogeneous disease and mutations in over 50 genes have been associated with DCM.6TTN, encoding the giant sarcomere protein titin, is the most common causal gene being responsible for ∼20% of the DCM cases.7 Mutations in MYH7, TNNT2, ACTC1 are also important, albeit uncommon, causes of DCM, indicating a partially shared genetic basis for the two common forms of hereditary cardiomyopathies, namely HCM and DCM. Overall, the majority of the known DCM genes code for cytoskeletal proteins.
A subset of DCM is caused by mutations in the LMNA gene, which encodes the nuclear inner membrane protein lamin A/C (LMNA). The phenotype, in addition to DCM, is characterized by chronotropic insufficiency, cardiac conduction defects, bradycardia, atrial fibrillation, and ventricular arrhythmias.8 Another subset of DCM is caused by mutations leading to cytoplasmic protein aggregation (proteotoxicity), including mutations in DES, encoding intermediary filament desmin, and CRYAB, encoding chaperon protein α/B-crystallin.9 More recently, RBM20 coding for RNA-binding motif protein 20 has emerged as an important cause of DCM.10 RBM20 protein regulates RNA splicing and targets sarcomere genes among others, leading to their dysregulated expressions.
2.3 Arrhythmogenic cardiomyopathy
ACM is an enigmatic form of hereditary cardiomyopathies, whose cardinal manifestations are ventricular arrhythmias, which often are the first manifestation of the disease preceding pathological and functional abnormalities; SCD, and refractory heart failure (reviewed in Ref.11). ACM is an important cause of SCD in the young.12–14 Both ventricles are typically involved but a subgroup of ACM classically manifests with predominant involvement of the right ventricle, particularly in the early stages, manifesting with cardiac arrhythmias originating from the right ventricle and fibro-fatty infiltration of the right ventricular myocardium. This subgroup is referred to arrhythmogenic right ventricular cardiomyopathy (ARVC).13 An LV-dominant form of ACM also has been described.15 The clinical phenotype of ARVC is also notable for the characteristic electrocardiographic findings including an epsilon wave, depolarization, and repolarization abnormalities in the right precordial leads, which are present in a subset of patients with ACM.16,17
The causal genes for ACM have been partially identified and code for protein constituents of desmosomes, members of the intercalated discs (IDs) (reviewed in Ref.11). IDs are responsible for maintaining mechanical integrity of the myocardium and are signalling hubs for mechano-sensing, such as the Hippo and the canonical WNT signalling pathways, which are dysregulated in ACM.18,19 Mutations in PKP2 gene coding for desmosome protein plakophilin 2 are the most common causes of ACM. Likewise, mutations in DSP-encoding desmoplakin, JUP-encoding junction protein plakoglobin, DSC2-encoding desmocllin 2, and DSG2-encoding desmoglein 2 are known to cause ACM. Mutations in several other genes are also implicated in ACM, including TMEM43 gene coding for transmembrane protein 43, PLN coding for phospholamban, FLNC-encoding filamin C, and TTN coding for titin. Mutations in the known genes are found in ∼40–50% of the ACM cases.
2.4 Others
Genetic basis of other forms of hereditary cardiomyopathies, such as RCM and LVNC is not discussed, suffice it to state that there is a partially shared genetic aetiology among hereditary cardiomyopathies, as mutations in sarcomere genes are also associated with RCM and LVNC.
2.5 Pathogenesis of hereditary cardiomyopathies
The primary defect in hereditary cardiomyopathies, namely the mutation, provides the stimulus for phenotypic expression of the disease. The mutation often affects interactions of the mutant protein with its interactome. The interactome might include proteins that are involved in cardiac conduction and arrhythmias. The early phenotypic responses to altered protein function commonly entails dysregulation of gene expression, altering various biological and functional pathways within the cardiac myocytes. A subset of the dysregulated genes code for proteins that are secreted from cells (secretome) and function as paracrine factors. These paracrine factors, emanating from cardiac myocytes, not only affect cardiac myocytes (through autocrine mechanisms) but also other cell types in the heart, such as fibroblasts and endothelial cells, through paracrine mechanisms. Collectively, the molecular phenotypes lead to functional and structural dysregulation of multiple cell types in cardiomyopathies, particularly in advanced stages of the disease. Therefore, although cardiomyopathies are primary diseases of cardiac myocytes, the phenotype is the consequences of complex interactions among multiple cardiac cell types, through paracrine effects, protein–protein interactions, and other mechanisms. As regards cardiac conduction and arrhythmias, the prevailing data point to cardiac myocytes and cardiac conduction cells as the cell sources of cardiac conduction defects and arrhythmias in cardiomyopathies.
3. Prevalence of cardiac arrhythmias in hereditary cardiomyopathies
3.1 Arrhythmogenic cardiomyopathy
Cardiac arrhythmias are relatively common and often the dangerous manifestations of cardiomyopathies. ACM is the prototypic form of cardiomyopathies, whose cardinal and typically the first manifestation is ventricular arrhythmia.20,21 Ventricular arrhythmias in ACM occur early, prior to, and typically in the absence of discernible cardiac dysfunction. This is in contrast to DCM or ischaemic heart disease, wherein ventricular arrhythmias typically manifest late in the course of the disease and often in the context of heart failure. While early ventricular arrhythmias are the common features of ACM, ventricular arrhythmias also occur late in advanced stages of ACM in conjunction with cardiac dysfunction, as in DCM. Approximately half of the patients with the classic form of ACM, namely ARVC, exhibit non-sustained ventricular tachycardia (NSVT) and approximately one-third show sustained ventricular tachycardia (VT) upon initial evaluation.22 The incidence of sustained ventricular arrhythmias in patients with ACM is ∼5% per year.22 Male patients are at a higher risk of future ventricular arrhythmias, as are those with a history of syncope and prior ventricular arrhythmias as well as cardiac dysfunction.22 Atrial arrhythmias are also common and present in up to half of the patients with ACM, typically in those with atrial and right ventricular enlargement.23,24
3.2 Hypertrophic cardiomyopathy
Cardiac arrhythmias are also common in HCM, typically occur in the context of clinical and pathological phenotypes of HCM and seldom are the sole manifestations. Approximately a quarter of patients with HCM exhibit NSVT in initial evaluation.25,26 Sustained VT is less common and a major cause of syncope and sudden cardiac arrest in patients with HCM. The incidence of atrial fibrillation is ∼2–3% per year and ∼25% of patients with HCM develop atrial fibrillation during the course of the disease.27–30 Atrial fibrillation with rapid ventricular rate is poorly tolerated in patients with HCM due to loss of atrial kick and diastolic dysfunction.27,30,31 Factors associated with increased risk of cardiac arrhythmias in HCM include cardiac hypertrophy, myocardial fibrosis, LVOT obstruction, and left atrial size, the latter for atrial fibrillation.29
3.3 Dilated cardiomyopathy
DCM comprises a heterogeneous group of myocardial diseases characterized by LV dilatation and dysfunction.6 DCM caused by mutations in the LMNA gene is a prototypic form of DCM that exhibits a high incidence of conduction defect and cardiac arrhythmias.32 Conduction defects, such as atrioventricular block, sinus node dysfunction, and atrial fibrillation are common and often the early manifestations of DCM, leading to placement of a pacemaker in approximately one-third of the patients.32 Likewise, ventricular arrhythmias develop in the majority of patients and are responsible for SCD in about half of the DCM patients with mutations in the LMNA gene.32–34 DCM caused by truncating mutations in the TTN gene, encoding titin protein, is also associated with an increased incidence of ventricular arrhythmias, although this has not been consistently observed.35,36
A subset of DCM has been associated with pathogenic variants in the SCN5A gene, which encodes sodium voltage-gated channel alpha subunit 5 (SCN5A), commonly known as Nav1.5, responsible for the fast depolarization phase (Phase 0) of the action potential in cardiac myocytes.37 Mutations in the SCN5A gene are associated with a high burden of atrial and ventricular arrhythmias, cardiac conduction disease, and risk of SCD.37–39 While mechanisms underlying different electrical phenotypes have been extensively studied using in vitro and in vivo approaches, little is known about whether and how sodium channel malfunction leads to ventricular dysfunction and dilation.38
4. Possible shared genetic basis of cardiac arrhythmia and hereditary cardiomyopathies
Shared genetic aetiology has been described for cardiomyopathies, particularly DCM and cardiac arrhythmias. Several genes implicated in cardiomyopathies and arrhythmias are discussed in this section.
4.1 SCN5A
The gene codes for SCN5A protein (also known as Nav1.5), which is predominantly expressed in cardiac myocytes and localizes to plasma membrane at the IDs. It forms a voltage-sensitive channel that undergoes conformational changes in response to a shift in transmembrane voltage. The pore opens in response to rising transmembrane voltage in the early phase of the action potential, enabling influx of sodium per electrochemical gradient. The consequent rise in the transmembrane potential results in closure of the pore, which is then followed by activation of NCX1 to normalize intracellular Na+ concentration. Thus, this channel is responsible for Phase 0 of the action potential.40
Mutations in SCN5A are major causes of inherited arrhythmia syndromes. Gain-of-function (GoF) mutations in SCN5A cause long QT syndrome type 3 whereas loss-of-function (LoF) mutations are major causes of Brugada syndrome.39,41SCN5A mutations are also associated with atrial fibrillation, atrial standstill, cardiac conduction defects, sick sinus syndrome, idiopathic ventricular fibrillation, and multifocal ectopic Purkinje-related premature contractions.41–46
An arrhythmogenic phenotype as a consequence of SCN5A mutations is in accord with the known biological functions of the sodium channel in the generation of the action potential. However, unexpectedly, SCN5A mutations are also associated with DCM.42,47SCN5A mutations have been identified in ∼1.7% of families with DCM.48 Despite the existing data from several independent studies implicating SCN5A variants as causes of DCM, the causality of the SCN5A variants in DCM remains unsettled for various reasons. First, robust genetic evidence, such as co-segregation data in large DCM families, is lacking. Second, studies performed before the era of large-scale DNA sequencing were not designed to exclude the presence of concomitant pathogenic variants in other genes that might cause or contribute to the DCM phenotype. Thirdly, earlier studies included a rather small number of a control population, and therefore, any uncommon or rare variant identified in the cases was considered pathogenic. Fourth, the phenotype of DCM associated with the SCN5A mutations is typically reported in conjunction with conduction defects, supraventricular and ventricular arrhythmias, raising the possibility that the SCN5A variants were the susceptibility pathogenic variants for the arrhythmic phenotype and an undetected variant in another gene was responsible for DCM. Finally, both GoF and LoF SCN5A variants have been associated with DCM, rendering mechanistic explanation challenging.49 Thus, despite the existing data suggesting SCN5A variants as the pathogenic variants in DCM, likely with low penetrance, it is plausible that SCN5A pathogenic variants predispose to cardiac arrhythmias and/or conduction defects in patients with DCM but are not the direct causes of DCM.
4.2 LMNA
A characteristic phenotypic feature of LMNA mutations is cardiac conduction defects, supraventricular and ventricular arrhythmias.8,32 Electric abnormalities typically occur in the background of DCM but could occur in isolation and often as the initial manifestations of laminopathies, which comprise a group of distinct phenotypes associated with the LMNA mutations.32,50,51 In a subset of laminopathies, neuromuscular involvement precedes cardiac manifestations by a decade.52
LMNA is a ubiquitously expressed nuclear envelope protein that interacts with chromatin through lamin-associated domains (LADs) to regulate gene expression.53–55 In human cardiac myocytes, LMNA interacts with ∼20% of the genome and affects expression of several thousand genes.56 LADs have suppressive effects on gene expression, partly through increased CpG methylation and partly through recruitment of suppressive epigenetic markers.56 Consequently, mutations in the LMNA gene, by shifting LADs to different genomic regions, could suppress expression of genes involved in cardiac arrhythmias, such as SCN5A.57 Consequently, phenotypic pleiotropy of LMNA mutations is rather expected, even though specific mechanisms involved have yet to be delineated. Molecular basis of cardiac conduction defects and arrhythmias in DCM associated with the LMNA mutation is not known, suffice it to state that apoptosis is implicated in mice heterozygous for the Lmna gene.58
4.3 TTN
Mutations in the TTN gene are major causes of DCM and responsible for 15–20% of the DCM cases.7,59,60 Because of its enormous size, the TTN gene carries a very large number of variants, including truncating variants (TTNtv), and multiple isoforms in the general population.61 Genetic complexity of the TTN gene poses significant challenges in identification of the pathogenic variants. Despite such complexity, TTNtv have been associated with increased risk of ventricular arrhythmias and atrial fibrillation in patients with DCM.35,62,63 In addition, TTNtv as well as LoF TTN variants have been associated with early-onset familial atrial fibrillation.64,65 Similarly, genome-wide association studies have linked TTN gene variants with atrial fibrillation.66 Along with these findings, rare coding TTN variants are associated with prolongation of the QT interval in the general population.67 Furthermore, in the context of DCM, the association of TTNtv with cardiac arrhythmias seems to be independent of cardiac function.62,63,68 Collectively, the existing data suggest a pro-arrhythmic effect of the pathogenic variants in the TTN gene, particularly in susceptibility to atrial fibrillation. The mechanism(s) responsible for susceptibility to cardiac arrhythmias, independent of structural remodelling, is unknown.
4.4 FLNC
Filamin C (FLNC) protein is a striated muscle-specific member of a family of actin-binding proteins. FLNC along with filamin A and filamin B are involved in multiple cellular processes, including cross-linking of cytoskeletal actin, mechano-transduction, and mechanical stability of sarcomeres, the latter through linking Z disc proteins to the dystrophin-associated-glycoprotein complex and integrins at the cytoplasmic cell membrane.69,70
Pathogenic variants in the FLNC gene were first shown to cause myofibrillar myopathy.71 Although cardiac involvement in myofibrillar myopathy is not uncommon, the first direct evidence linking FLNC mutations to cardiomyopathies emerged through identification of missense mutations in multiple small families with HCM and detection of protein aggregation in the heart.72FLNC mutations also have been linked to RCM, DCM, and ACM.73,74 The existing data suggest a high burden of cardiac arrhythmias in cardiomyopathies associated with the FLNC mutations.75 In addition, a deletion mutation in the FLNC gene has been associated with familial ventricular arrhythmias and SCD in a sibling with normal cardiac function.76 The mechanisms underlying predisposition to cardiac arrhythmias with the FLNC mutations remain unknown and expected to be complex given the diversity of its biological functions. Abnormal trafficking of ion channel proteins is an unexplored proposed mechanism of cardiac arrhythmias observed in cardiomyopathies associated with FLNC mutations.77
4.5 RYR2
The ryanodine receptor 2 (RYR2), encoded by the RYR2 gene, localizes to sarcoplasmic reticulum (SR) and plays an essential role in excitation and contraction coupling in cardiac myocytes.78 During the plateau phase of the action potential, Ca2+ enters the cell via the L-type voltage-dependent calcium channel CACNA1C (Cav1.2), which is located on the transverse tubules. Increased intracellular calcium triggers opening of the RYR2 channels and release of calcium from SR, a process that is referred to as Ca2+-induced-Ca2+ release (CICR). The released calcium binds to troponin C, which induces conformational changes in the troponin complex resulting in shifting the inhibitory arm of troponin I away from the acto-myosin complex. The process leads to flexion of the global head of the myosin over thin actin filament, resulting in displacement of the actin filament by the myosin head and muscle contraction. Excess intracellular calcium is then pumped back from the cytosol into SR by SR Ca2+ ATPase 2 (SERCA2) encoded by ATP2A2 and pumped out of the cell by cell membrane NCX1, re-establishing the homoeostasis.79
Mutations in the RYR2 gene are implicated in cardiac arrhythmias as well as cardiomyopathies, in accord with the prominent role of RYR2 in the intracellular Ca2+ release and the fundamental role of Ca2+ in regulating cardiac contraction and arrhythmogenesis.80–83 Specifically, RYR2 mutations are established causes of familial catecholaminergic polymorphic ventricular tachycardia (CPVT).80,81 Pathogenic variants in the RYR2 gene also have been reported in patients with cardiomyopathies, including ACM and LVNC.82,83 As typical to all RYR2-mediated phenotypes, pathogenic variants in the RYR2 gene confer increased risk of cardiac arrhythmias prior to and in the absence of measurable structural cardiac phenotype. A large genomic rearrangement in the RYR2 gene that leads to loss of exon 3 has been associated with a compound phenotype of sinoatrial node and atrioventricular node dysfunction, atrial fibrillation, atrial standstill, and DCM.84 Deletion and pathogenic variants involving exon 3 have been associated with exercise or stress-induced VT tachycardia as well as LVNC in independent probands and families, supporting the role of this gene not only in arrhythmogenesis but also in LV compaction during embryonic development.85–87 The pathogenic role of RYR2 variants in CPVT is well established, however, their role as causes of cardiomyopathies, perhaps with the exception of LVNC, is less certain. In accord with this notion, knock-in mouse models of RYR2 pathogenic variants identified in CPVT or ACM patients do not exhibit structural remodelling reminiscent of ACM.88,89 In addition to the genetic variants, various functional alterations in RYR2 channels have been identified as contributing to cardiac arrhythmias and dysfunction, as discussed later.
4.6 JPH2
Junctophilin-2 (JPH2) is predominantly expressed in the heart and acts as a structural protein within the junctional membrane complex that physically approximates the plasmalemmal L-type Ca2+ channel and RYR2 on the SR (reviewed in Ref.90). Rare variants in the JPH2 gene have been identified in patients with HCM and have been shown to impart functional effects, such as cardiac hypertrophy and atrial fibrillation in in vitro and in vivo studies.91–94 Likewise, a homozygous deletion variant in the JPH2 gene has been reported in a patient with autosomal-recessive DCM.95 A proposed mechanism pertains to impaired interaction of the mutant JPH2 with RYR2 proteins and consequent destabilization of Ca2+ release from the SR.93
4.7 PLN
PLN codes for a homopentameric protein phospholamban (PLN), which regulates cardiac contractility/relaxation by targeting ATP2A2 (SERCA2) in cardiac SR. Unphosphorylated PLN inhibits SERCA2, where its phosphorylation by protein kinase A (PKA) in response to cAMP of the adrenergic system releases it from SERCA2. The release removes the inhibitory effect of PLN on SERCA2, thereby facilitating removal of Ca2+ from the cytosol to SR and enhanced cardiac relaxation or lusitropy, as observed upon stimulation of the heart with catecholamines.
Mutations in the PLN gene cause DCM and ACM.96–98 The phenotype is characterized by refractory heart failure and a relatively high prevalence of ventricular arrhythmias.96–99 The molecular basis of cardiac arrhythmias in cardiomyopathies associated with the PLN mutations is largely unknown. Given the major role of PLN protein in regulating intracellular calcium concentration through inhibition of ATP2A2, mutations in the PLN gene are expected to predispose to cardiac arrhythmias through affecting calcium cycling in cardiac myocytes as in other forms of heart failure.100
4.8 PKP2
Plakophilin 2 (PKP2) is a desmosome protein located predominantly in the IDs and involved in mechano-sensing and stabilization of other ID proteins, including SCN5A and GJA1 (gap junction protein alpha 1 or connexin 43). Mutations in the PKP2 gene are the most common causes of ACM.101 Among genes coding for the desmosome proteins, only the PKP2 gene has been associated with cardiac arrhythmias, independent of cardiomyopathy, albeit the data are not compelling.102,103 Given co-localization of PKP2, SCN5A, and GJA1 proteins to the IDs, it is plausible that PKP2 mutations de-stabilize localization and function of its binding partners and hence, predispose to cardiac arrhythmias, partially independent of cardiac contractile function.19,104,105 Experimental data in murine models suggest that PKP2 deficiency leads to reduced transcript levels of genes involved in intracellular calcium handling/cycling, thereby disrupting intracellular calcium homoeostasis with a consequent increase in arrhythmogenesis.106
4.9 RBM20
Ribonucleic acid-binding motif protein 20 (RBM20) is a pre mRNA splicing factor that is highly expressed in striated muscle, in particular, cardiac muscle and regulates splicing of cardiac genes.10 Pathogenic variants in RBM20 are associated with DCM and ACM.107–109 RBM20 alters splicing of several genes in cardiac myocytes, in particular, TTN and RYR2, which have been associated with cardiomyopathies as well as cardiac arrhythmias.10,109 Differential splicing of calcium-handling genes CACNA1C and CAMK2D have been observed in iPSC-derived cardiac myocytes from patients with RBM20-associated DCM.110 Data in the Rbm20 knock-out mice indicate aberrant splicing of Ryr2 and Camk2d genes resulting in increased intracellular Ca2+ overload as a mechanism for the pathogenesis of ventricular arrhythmias.109 The findings suggest that treatment with an L-type Ca2+ channel blocker might reduce ventricular arrythmias in this particular form of cardiomyopathy.109
4.10 HCN4
Hyperpolarization and cyclic nucleotide 4 channel (HCN4) encoded by the HCN4 gene, transports positively charged ions to cardiac cells (Figure 2). It is expressed mainly in the sinoatrial node in the adult heart but during murine embryonic development it is also detectable at a lower level in the trabecular (subendocardial) layer of myocardium.111,112 The HCN4 channels play a crucial role in the automaticity of the sinus node through the generation of a slow diastolic depolarization during the Phase 4 of the cardiac action potential in response to increased intracellular cAMP concentration.113 Expression level of Hcn4 gene is downregulated in response to exercise training in mice, which may account for training-induced bradycardia.114 Mutations in the HCN4 gene are associated with sinus node dysfunction, sick sinus syndrome, and inappropriate sinus tachycardia, as well as exercise-induced ventricular ectopic beats.115,116 In addition, HCN4 mutations have been linked to LVNC and susceptibility to ventricular fibrillation occurring in conjunction with sinus bradycardia.117,118 The putative mechanism(s) of LVNC caused by the HCN4 mutations relates to expression of this gene in the early cardiac progenitor cells that give rise to both compact and trabecular layers of the left ventricle as well as its role in normal compaction of the human foetal ventricles.111,112
Figure 2.
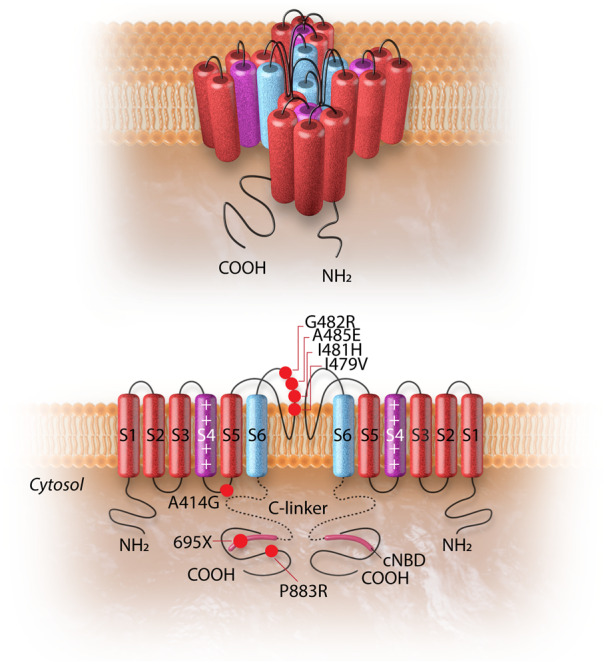
The HCN4 channel and the topology of the HCN4 mutations associated with left ventricular non-compaction cardiomyopathy. The upper panel shows the 3D conformational structure of the HCN4 channel. The lower panel shows the liner structure of the HCN4 channel (two out of four subunits are shown). Each alpha-subunit is composed of six transmembrane helixes (S1–S6), a pore-forming loop, and intracellular N- and C-termini. The positively charged S4 helix (shown in purple) is the voltage sensor of the channel. The four S6 segments (shown in blue) of four-channel monomers together form the ion-conducting passage of the HCN4 channel. The C-terminus comprises of the C-linker (dotted line) and the cyclic nucleotide-binding domain (cNBD), which is connected to the channel core and mediates cyclic AMP-dependent changes in HCN channel gating. cAMP binding causes a conformation change that leads to the assembly of an active tetramer and channel opening. Red dots on the alpha-subunit shown on left indicate the sites of mutation associated with left ventricular non-compaction. The dot with a black dash indicates a truncation resulting from an insertion mutation leading to premature truncation of the protein at amino acid 695.
4.11 PRKAG2
The gene codes for gamma 2 subunit of AMP-activate protein kinase, which sustains metabolism through regulating intracellular ATP concentration.119 Mutations in the PRKAG2 gene lead to glycogen storage in the heart mimicking HCM and Wolff–Parkinson–White syndrome, atrial fibrillation, and progressive infra-His conduction defect.120–122 Pathological examinations of affected human hearts show vacuoles containing amylopectin, a glycogen-related substance.123 Unlike the classic Wolff–Parkinson–White syndrome, the pre-excitation phenotype and cardiac arrhythmias in these patients likely result from structural disruption of annulus fibrosus by excess glycogen-laden cells. Experimental data suggest that PRKAG2 regulates voltage-gated sodium channels in rat ventricular myocytes, leading to prolonging action potential duration and production of arrhythmogenic early after depolarizations (EAD).124 However, data in genetically modified mouse models suggest that pre-excitation and arrhythmias are direct consequences of glycogen storage in cardiac myocytes.125
4.12 ABCC9
The encoded ATP-binding cassette subfamily C member 9 (aka sulfonylurea receptor subunit 2A or SUR2) protein is a regulatory subunit of ATP-sensitive K+ channel. Mutations in the ABCC9 gene are known to cause Cantu syndrome, a disease characterized by a number of developmental abnormalities and congenital heart disease.126ABCC9 mutations also have been identified in patients with DCM and paroxysmal atrial fibrillation.127,128 The causal role of ABCC9 mutations in these phenotypes remain to be established.
4.13 Other genes
Rare variants in CACNA1C and CALM2, encoding the voltage-dependent L-type Ca2+ channel subunit α1C and calmodulin 2, respectively, have been reported in patients with cardiac arrhythmias and cardiomyopathies.129,130 However, the evidence to support the pathogenic role of these variants in cardiomyopathies is not compelling.
5. Risk factors predisposing to cardiac arrhythmias in cardiomyopathies
5.1 Causal genes/mutations
Genotype–phenotype correlation studies in cardiomyopathies have been compounded by the small sample size of the studies as well as genetic and phenotypic heterogeneity of the disease, rendering firm conclusions challenging. Despite these limitations, specific phenotypic characteristics hint to the underlying causal gene(s) in cardiomyopathies. For example, conduction defects, including atrial, atrioventricular, and left bundle branch blocks, supraventricular tachycardia, and ventricular arrhythmias are often observed in patients with DCM caused by the LMNA mutations.131,132 Such phenotypic features, however, are not specific to the LMNA mutations, as they are also observed in Anderson Fabry disease, amyloidosis, and mitochondrial defects, among others. Mutations in TTN and desmosome genes are associated with a high incidence of atrial and ventricular arrhythmias as well as poor prognosis as opposed to those in cytoskeletal protein genes.33,35,62,64,65,68 However, there is considerable phenotypic variability, as truncating TTN mutations also have been associated with a relatively mild form of DCM.36 Several genes are implicated in susceptibility to cardiomyopathies as well as arrhythmogenic syndromes, as discussed earlier under possible shared genetic aetiology. Finally, functional genetic variants in genes encoding protein constituents of ion channels might predispose to cardiac arrhythmias in patients with hereditary cardiomyopathies.
5.2 Biological sex
Patient’s biological sex, partly through the effects of sex hormones on gene expression and partly because of the differences in external factors, such as physical activity, between males and females, is an important determinant of phenotypic expression of various cardiovascular diseases, including cardiomyopathies and arrhythmias. Sex-dependent differences in the phenotypic expression of disease are not because of differences in the genetic aetiology of cardiomyopathies, as the vast majority of cardiomyopathies are autosomal and not sex-linked diseases. In general, phenotypic expression of cardiomyopathies seems to be more pronounced in male than female patients, albeit there is considerable variability among cardiomyopathies as well as inconsistent findings among different studies.7,133–135 The inconsistencies in findings also seem to be relevant to sex-dependent predisposition to cardiac arrhythmias in patients with cardiomyopathies.133,136–138 Overall, ventricular arrhythmias seem to be more common in male patients with cardiomyopathies and Brugada syndrome, whereas female preponderance is well documented in patients with long QT syndromes.136,137,139,140 In the context of specific cardiomyopathy genes, ventricular arrhythmias seem to be more common in male patients who carry mutations in the LMNA or RBM20 gene.34,136,141
5.3 Age
Association of cardiac arrhythmias with age in hereditary cardiomyopathy is influenced by the presence of concomitant conditions as well as age-dependent expression of other cardiac phenotypes, such as myocardial fibrosis, cardiac hypertrophy, and chamber dilatation, among others. In addition, a number of molecular and cellular changes occur with advancing age, such as cell death and inflammation, which affect susceptibility to cardiac arrhythmias in hereditary cardiomyopathies.
Certain cardiac arrhythmias, such as atrial fibrillation exhibit age-dependent penetrance, typically occurring in older age, whether in the context of cardiomyopathies or otherwise.28 In contrast, ventricular arrhythmias in cardiomyopathies and certain channelopathies typically occur at a young age and are major causes of SCD in individuals <40 years of age.142,143 A notable example is HCM, which is considered among the most common causes of SCD in the young.142 In contrast, atrial fibrillation, which occurs in about a quarter of the HCM patients, typically presents late in the course of the disease.28 Ventricular arrhythmias are often the first manifestations of ACM and might indicate disturbed ion channel functions in cardiac myocytes in the presence of desmosome mutations.103,144,145 The early presentation of cardiac arrhythmias in ACM, that is cardiac arrhythmias manifesting in the absence of cardiac dysfunction, suggests a lower threshold for dysregulation of cardiac myocyte electric activity than mechanical function in the presence of mutations in the desmosome proteins. Alternatively, it is also possible that early cardiac arrhythmias originate from the non-myocyte cells in the heart that express the mutant desmosome proteins, such as the conduction system cells, as suggested by the experimental data.146 Cardiac arrhythmias also occur late in the course of ACM and typically in the context of cardiac dysfunction, as in other forms of heart failure.
Cardiac arrhythmias in patients with DCM typically occur late and in the presence of cardiac dysfunction. However, DCM associated with LMNA mutations often exhibits early onset of cardiac conduction defects and supraventricular arrhythmias at a younger age, preceding the onset of cardiac dysfunction.8 Likewise, ventricular arrhythmias occur at a relatively young age in patients with DCM associated with PLN, FLNC, and RBM20 mutations.74,98,108 Because of a relatively high prevalence of ventricular arrhythmias there is considerable phenotypic overlaps between DCM and ACM in patients with mutations in PLN, FLNC, and RBM20 genes.
5.4 Ethnic background
The prevalence of the underlying causal mutations is not known to differ significantly among populations with different ethnic backgrounds, whereas phenotypic expression of cardiomyopathies could vary because of the modifier effects of the genetic backgrounds as well as environmental factors. Phenotypic expression of cardiac hypertrophy is more pronounced and the risk of heart failure is higher in African–American patients with HCM, as compared to Caucasians.147 In contrast, the incidence of atrial fibrillation is lower among the African–American patients with HCM as compared to Caucasians and that of ventricular arrhythmias does not seem to be different.147
BAG3 mutations have been associated with worse clinical outcomes in African–American patients with DCM.148 However, association of cardiac arrhythmias with ethnic background in patients with DCM caused by BAG3 mutations remains unknown. Likewise, there are ethnic differences in the prevalence of ACM and SCD in patients with ACM.11,149 Accordingly, ACM is more common in certain regions of Italy where it accounts for up to 20% of the cases of SCD in athletes.149 It is unclear whether the differences are reflective of differences in the genetic backgrounds of the individuals or are secondary to factors that facilitate diagnosis of the disease.
5.5 Physical exercise
Observational studies suggest an association between sport activities and SCD, particularly in young patients with HCM.4 However, a recent randomized prospective study showed that patients with HCM tolerate moderate-intensity exercise well without occurrence of significant arrhythmias.150 Overall, the incidence of serious cardiac arrhythmias or SCD during physical exercise seems to be relatively low, suggesting that exercise may not be a major risk factor for induction of serious ventricular arrhythmias or SCD in HCM.151
Physical exercise is considered a risk factor for cardiac arrhythmias in ACM, a disease caused primarily by mutations in genes encoding desmosome proteins.152–154 In the context of specific mutations, intense exercise in patients with the TMEM43 p. Ser358Leu mutation is associated with a marked increase in the number of appropriate defibrillator shock, which is a surrogate indicator of malignant ventricular arrhythmias.154 Multiple mechanisms are likely to be involved in the pathogenesis of exercise-induced cardiac arrhythmias in cardiomyopathies. For example, physical exercise by provoking the adrenergic system could activate cAMP-dependent PKA and subsequent phosphorylation of ion channels, affecting their function and leading to arrhythmias during exercise.155–157 Likewise, exercise could induce cardiac arrhythmias by activating the stretch responsive ion channels. Moreover, exercise could alter interaction of the desmosome proteins with a subset of ion channels, such as SCN5A (Nav1.5) that located at the IDs.158 Chronic exercise could affect susceptibility to cardiac arrhythmias by inducing molecular and structural remodelling in the heart. In keeping with the beneficial effects of exercise in other cardiovascular diseases, a recent study in a mouse model suggested beneficial effects of exercise in reversal of dysregulated transcriptional pathways in ACM.159
5.6 Other susceptibility factors
Molecular and cellular inflammatory responses are observed in the myocardium of patients with ACM.160 The inflammatory response is likely due to internal factors but could also be triggered by external factors, such as viruses. Cardiotropic viruses have been detected in the myocardium of patients with ACM.161 It is unclear whether viruses contribute to the pathogenesis of cardiac arrhythmias in ACM, although proteolytic cleavage of dystrophin by coxsackieviruses has been reported to lead to cardiac dilatation and dysfunction.162
Although the presence of concomitant CAD and myocardial ischaemia in patients with HCM is associated with adverse outcomes, its role in predisposition to cardiac arrhythmias is unclear.163 Finally, presence of LV systolic dysfunction, represented as elevated plasma levels of biomarkers of cardiac dysfunction, increases the risk of cardiac arrhythmias in patients with cardiomyopathies, as in other forms of heart failure with reduced EF.164,165
6. Mechanisms of cardiac arrhythmias in hereditary cardiomyopathies
The underpinning mechanisms of cardiac arrhythmias in hereditary cardiomyopathies are not distinct from those involved in primary arrhythmia syndromes, suffice it to state that the arrhythmogenic substrates differ. Mechanisms of cardiac arrhythmias in primary arrhythmia disorders have been covered in other articles in the Spotlight issue. Therefore, the mechanisms are only briefly mentioned.
Re-entrant circuits and abnormal impulse formation can both contribute to arrhythmias.166,167 Re-entrant arrhythmias are more likely to occur in the presence of extensive fibrosis, which contributes to heterogeneous conduction with slow or discontinuous electrical propagation. This may serve as a substrate for re-entrant ventricular arrhythmias that can originate during normal sinus rhythm.168 In addition to fibrosis, downregulation of connexins may contribute to conduction delays, increased heterogeneity in impulse propagation, and an increased dispersion of action potential durations.169
Abnormal impulse formation represents the second major arrhythmia mechanism associated with cardiomyopathies, and can be caused by enhanced automaticity or triggered activity. Enhanced automaticity is the result of diastolic depolarizations due to net inward current during Phase 4 of the action potential. Abnormal automaticity is enhanced by β-adrenergic stimulation, which is consistent with clinical studies in patients with HCM showing that greater than half of all ventricular arrhythmias were associated with moderate to intense physical activity.170 In addition, triggered activity can result from depolarizations that occur during or following cardiac action potentials (APs). EADs occur during Phase 2 or 3 of the AP, whereas delayed afterdepolarizations (DADs) occur after repolarization has been completed. When the amplitude of an EAD or DAD research membrane threshold potential, a spontaneous action potential referred to as triggered activity can occur.167 The development of EADs often depends on the reduced availability of repolarizing K+ current.171 DADs on the other hand are mostly observed under conditions that augment intracellular Ca2+ release and increased sympathetic activity, as seen in hypertrophied and failing hearts.172 Interestingly, frequent ventricular premature depolarizations have been associated with cardiomyopathy adding more complexity to our understanding of arrhythmias associated with cardiomyopathy.173
7. Pathogenesis of cardiac arrhythmias in hereditary cardiomyopathies
The major components involved in the pathogenesis of cardiac arrhythmias in hereditary cardiomyopathy entail two intertwined components of ion channel abnormalities and pro-arrhythmic structural remodelling, which are discussed.
7.1 Pro-arrhythmic structural remodelling in cardiomyopathies
Whereas early cardiac arrhythmias in hereditary cardiomyopathies are typically due to changes in ion channel functions, as discussed earlier, late arrhythmias are generally associated with structural remodelling of the cardiac chambers.
7.1.1 Cardiac hypertrophy
Severe cardiac hypertrophy is considered a risk factor for cardiac arrhythmias and SCD in patients with HCM.26,174 Cardiac hypertrophy affects myocardial electric properties, such as voltage amplitude and conduction, producing an arrhythmogenic substrate.175 At the molecular level, increased cytosolic Ca2+ resulting from increased Ca2+ entry through the L-type calcium channels and reduced exchange through NCX1 could lead to DADs. The latter is implicated as a mechanism of cardiac arrhythmias in cardiac hypertrophy associated with HCM.176
7.1.2 Cardiac dilatation
Progressive cardiac dilatation and remodelling in cardiomyopathies is commonly associated with complex electric remodelling in the heart, involving K+, Na+, and Ca2+ currents, as well as NCX electric properties, which collectively lead to altered spatiotemporal gradient of repolarization, prolongation of the action potential duration (APD), and increased excitability of atrial and ventricular myocytes (reviewed in Ref.177). Molecular changes, which are mainly documented in heart failure with reduced EF and in model organisms but largely are expected to pertain to human DCM as well. Specifically, transcript levels of several genes coding for the components of Na+, K+, and Ca2+ channels are reduced in the endomyocardial biopsy samples from patients with DCM and in model organisms.178–180 In addition, cardiac dilatation and failure is associated with activation of a number of kinases such as PKA and CAMK2, which target ion channel proteins for phosphorylation and affect their functional properties.181,182
Loss of inactivation of SCN5A (Nav1.5) channels leads to increased late inward Na+ current, whereas K+ currents, including Ito, are downregulated, and Ca2+ currents are upregulated in dilated hearts.183–186 The net result of these alterations is the prolongation of the APD. Additionally, Calcium calmodulin-dependent protein kinase II (CAMK2)-mediated phosphorylation of RYR2 in the failing hearts leads to calcium leak from the SR in the setting of downregulated ATP2A2 protein levels and reduced Ca2+-uptake into the SR.187,188 In addition to ion channels and calcium-handling proteins, disorganization of GJA1 (Cx43), a protein critical for the normal impulse conduction in the heart, can contribute to the arrhythmogenicity of failing hearts.189
7.1.3 Myocardial fibrosis
Data on the association of myocardial disarray and fibrosis with cardiac arrhythmias in HCM are suggestive but not conclusive. Myocardial fibrosis, assessed by late gadolinium enhancement (LGE), as a surrogate marker, has been shown to be a modest risk factor for SCD, and hence, by inference cardiac arrhythmias.190–192 While some studies suggest the extent of LGE correlates with the risk of SCD in HCM, others suggest the simple detection of LGE is a risk factor for SCD.190–192
7.1.4 Myocyte disarray
Myocardial disarray, as assessed by diffusion tensor, cardiac magnetic resonance imaging has been associated with an increased risk of ventricular arrhythmias in HCM.193 Experimentally myocardial disarray has been linked to altered transmural distribution of connexin 43, providing a substrate for cardiac arrhythmias in HCM.194
7.2 Functional defects in ion channels in cardiomyopathies
An exquisite ionic homoeostasis is maintained by a large set of molecules in cardiac myocytes to orchestrate orderly cycles of excitation and contraction throughout life (Figure 3). Consequently, a large array of disturbances, whether genetic or otherwise, could disrupt various components of this delicate balance and lead to dysregulation of excitation-contraction cycles, affecting both cardiac rhythm as well as mechanical function. In addition to genetic mutations, which could directly affect structure and function of the ion channels and therefore, contribute to cardiac arrhythmias (as discussed in this spotlight issue), a number of secondary changes, resulting from impaired cardiac function or altered signalling pathways could also affect ion channel function and contribute to arrhythmogenesis in cardiomyopathies. Some of the secondary changes in ion channels, occurring in the context of hereditary cardiomyopathies, are discussed.
Figure 3.

Schematic representation of the structural elements of cardiomyocytes implicated in the pathogenesis of arrhythmias in inherited cardiomyopathies. Protein constituents of cardiac myocyte structural proteins and ion channels are depicted to reflect the complexity of the interactions that mediate maintenance of normal cyclic excitation and contraction cycles throughout life.
7.2.1 Functional defects in cardiac sodium current
The mechanisms underlying SCN5A-mediated cardiac arrhythmias in DCM patients are not known. It is plausible that SCN5A LoF variants promote arrhythmias through slowing the conduction and initiating a re-entry, whereas GoF mutations induce triggered activity during the repolarization or diastolic phase of cardiac cycle.39,41 Whereas the mechanisms responsible for Long QT and Brugada syndromes or conduction defects caused by SCN5A mutations are partially understood, it is unclear whether similar or distinct mechanisms are also involved in the pathogenesis of cardiac arrhythmia occurring in the context of DCM in patients carrying pathogenic variants in the SCN5A gene.
As discussed earlier, it is unclear whether pathogenic variants in SCN5A cause DCM and if so, how they lead to cardiac dilatation and dysfunction. Several hypotheses have been proposed and discussed recently (Figure 4).195 On one end of the spectrum, one may posit that DCM is a consequence of long-standing cardiac conduction abnormalities and arrhythmias resulting from SCN5A protein functional abnormalities, as in ‘tachycardia-induced cardiomyopathy’.195 This hypothesis is supported by the concomitant presence of cardiac conduction abnormalities and arrhythmias in the majority of DCM patients carrying pathogenic variants in the SCN5A gene.47,48 On the opposite end of the spectrum, it is postulated that DCM is a direct consequence of SCN5A channel dysfunction, meaning that the structural phenotype is primarily driven by electrical abnormalities. Accordingly, increased Na+ influx into cardiac myocyte caused by SCN5A GoF mutations could lead to compensatory activation of the NCX1 or the Na+/H+ exchanger, resulting in intracellular Ca2+ overload or acidification, respectively, and consequent impaired excitation–contraction coupling.196–198 A third hypothesis implicates impaired subcellular localization and interaction of SCN5A with other structural proteins, such as PKP2, DSG2, dystrophin, Lim domain binding 3, and caveolin 3, as the putative mechanism for DCM (reviewed in Ref.199). Several other putative mechanisms are also considered, including presence of concomitant pathogenic variants in other genes being responsible for DCM, effects of mutations on dimerization and post-translational modifications of SCN5A, such as phosphorylation, acetylation, ubiquitination, and nitrosylation; as well as disruption of SCN5A trafficking and function (reviewed in Ref.200).201–205 Given the heterogeneity of the SCN5A mutations associated with DCM, one could envisage multiple mechanisms contributing to the pathogenesis of cardiac dilatation and dysfunction.
Figure 4.

Graphic illustration of putative mechanisms underlying dilated cardiomyopathy associated with SCN5A mutations. Gain-of-function (GoF) SCN5A variants are known to cause long QT3 syndrome. GoF variants could also increase INa current and to frequent premature ventricular contractions or ventricular tachycardia, which might contribute to left ventricular dilatation and dysfunction. Likewise, GoF variants could induce compensatory activation of the Na+/Ca2+ exchange protein, resulting in intracellular Ca2+ overload and consequently impaired excitation–contraction coupling and contractile dysfunction. Loss-of-function SCN5A variants are known to cause Brugada syndrome and cardiac conduction defects, the latter could lead to cardiac structural remodelling. SCN5A pathogenic variants could also result in proton leak into the cytosol, acidify the cytosol, and myocardial dysfunction. Experimental data to support these hypotheses are scant. INa, inward depolarizing sodium current; NaV1.5, voltage-gated sodium channel α-subunit; NCX, Na+/Ca2+ exchanger; PVC, premature ventricular contractions.
The interplay between SCN5A and DCM is further compounded by the effects of heart failure on SCN5A functions.206 Reduced peak Na+ current and prolongation of APD in cardiac myocytes are observed in heart failure models.207–210 Likewise, increased late Na+ current (INaL) during the plateau phase of action potential resulting from impaired inactivation of the channel is implicated in the prolongation of action potential and arrhythmogenesis.183 The putative underpinning mechanisms responsible for altered Na+ current are reduced level of SCN5A protein, disrupted subcellular localization, deficient glycosylation of SCN5A, and altered phosphorylation of SCN5A.183,207,209,211,212 Moreover, epigenetic suppression of SCN5A gene expression is implicated in reduced peak INa in an iPSC-cardiomyocyte model of DCM associated with an LMNA mutation.57 Furthermore, reduced levels of full-length SCN5A transcript due to aberrant splicing have been observed in the myocardium of patients with HCM caused by the MYPBC3 mutations.213 Moreover, increased INaL has been implicated in the pathogenesis of HCM.214 However, a clinical trial with ranolazine in humans, an inhibitor of INaL, did not show significant effect on exercise performance, indices of diastolic function, NT-proBNP levels, or quality of life, casting doubt about the pathogenic role of this current in HCM.215 Finally, PKP2 LoF variants are associated with decreased INa density and voltage-dependent inactivation properties of SCN5A, providing a potential mechanism for arrhythmogenesis in ACM.103,216 Mechanistically, reduced peak INa current increases the susceptibility to re-entry by slowing impulse conduction. Increased INaL because of slower rate of inactivation antagonizes repolarization, causes APD prolongation, and increases susceptibility to EADs, mechanisms similar to those observed in long QT syndrome type 3. Thus, in cardiomyopathies, structural and functional modification of SCN5A secondary to cardiac dysfunction could be responsible for cardiac arrhythmias.
7.2.2 Functional defects in cardiac calcium current
Cardiac contraction and relaxation are regulated by a delicate set of channels and pumps that enable balanced influx and efflux of calcium during a cardiac cycle. Voltage-dependent L-type Ca2+ channels are activated during depolarization phase of the action potential to enable influx of Ca2+ into the cytosol. The latter triggers CICR through the RYR2 from the SR, which initiates acto-myosin contraction. This is followed by efflux of Ca2+ from the cytosol, which is carried out by the NCX1 as well as sequestration of Ca2+ back into the SR by the ATP2A2 (SERCA2a) pump, mediating dissociation of actin and myosin and muscle relaxation.79
The role of genetic variants in the components of Ca2+ cycling in cardiac muscle in susceptibility to cardiac arrhythmias in cardiomyopathies was discussed earlier. Secondary changes in the component of Ca2+ handling channels and transporters are also implicated in arrhythmogenesis associated with inherited cardiomyopathies (reviewed in Refs217,218). Mouse models of cardiomyopathies exhibit altered function of the L type Ca2+ channels leading to excess intracellular Ca2+ load.109,219 The components of the channels and pumps involved in maintaining Ca2+ homoeostasis, including the RYR2, are modified upon phosphorylation by various kinases and oxidative stress, which are often altered in cardiomyopathies220,221 (reviewed in Ref.222).
CAMK2, a major calcium-handling protein, is activated and contributes to arrhythmogenesis in cardiomyopathies. Enhanced activation of results in increased phosphorylation of RYR2 and increased diastolic RYR2-mediated Ca2+ leak, which are implicated in arrhythmogenic Ca2+ waves and VT in the mdx mouse model of DCM.223 Likewise, increased CAMK2 activity, hyper-phosphorylation of the RYR2, super inhibition of ATP2A2, and reduced Ca2+ transients are implicated as the mechanisms responsible for cardiac arrhythmias in DCM caused by the PLN mutations.224 Moreover, increased production of reactive oxygen species, commonly observed in cardiomyopathies, promotes both CAMK2 activation and enhanced RYR2-mediated Ca2+ leak, promoting arrhythmogenesis.225 Enhanced CAMK2 activity might induce pro-arrhythmic changes by targeting SCN5A upon phosphorylation, which could result in enhanced late depolarization current and prolongation of action potential duration.226
Cardiac myocytes isolated from human hearts of patients with HCM exhibit increased late Na+ current, increased L-type Ca2+ current, prolonged Ca2+ transients, and higher diastolic Ca2+ concentrations.227 In accord with these observations, CAMK2 activity and enhanced CAMK2-mediated phosphorylation of the RYR2 are implicated in an aberrant intracellular Ca2+ handling in a mouse model of HCM caused by a mutation in the TNNT2 gene.228
PKP2, a major causal gene for ACM in humans, is implicated in regulating expression of several genes involved in Ca2+ homoeostasis, as deletion of Pkp2 gene in mice is associated with downregulation of expression of Ryr2, Ank2 (encoding ankyrin B), Cacna1c, Trdn (triadin), and Casq2 (calsequestrin).106 Downregulation of expression of these genes is associated with a number of changes in Ca2+ homoeostasis, leading to increase Ca2+ transient amplitude and duration, and early as well as delayed after transients.106 The data are insufficient to conclude that the changes are reflective of transcriptional activity of the PKP2 protein.229
Expression level of JPH2, a key junctional membrane protein that is essential for proper intracellular Ca2+ handling in cardiomyocytes, is reduced in human heart biopsies obtained from patients with HCM.230 Functionally, experimental downregulation of JPH2 using an RNA interference is associated with reduced Ca2+ transient, increased Ca2+ store, impaired cardiac contractility, and increased mortality in a cardiac-specific Jph2 knockdown mice.230 Whether these changes contribute to arrhythmogenesis in cardiomyopathies, while expected, remain to be shown.
Finally, auto-antibodies against the L-type Ca2+ channel have been identified in a subset of patients with DCM and their presence has shown to be and independent predictor of VT and SCD.231,232 The findings are in accord with data showing that anti-Ro antibodies, which are linked to complete heart block in auto-immune diseases, inhibit L- and T-type calcium channels (reviewed in Ref.233). Observational data suggest prolongation of the QT interval and increased frequency of ventricular ectopic beats in patients with anti-Ro antibodies.234 Likewise, ex vivo experimental data show that affinity-purified auto-antibodies prolong the APD and promote triggered activity due to early afterdepolarizations leading to VT.231 Whereas the role of auto-antibodies in conduction defects, particularly congenital AV block, is well established, their role and the pertinent mechanisms in the pathogenesis of cardiac arrhythmias in human hereditary cardiomyopathies remain to be established.
7.2.3 Functional defects in cardiac K+ current
Potassium channels are comprised of tetrameric transmembrane proteins that enable specific permeation of the K+ ions, but not other ions, across the cytoplasmic membrane. The K+ channel units are encoded by over two dozen genes in the genome, which are ubiquitously expressed in various cell types, including cardiac myocytes. The primary function of the K+ in cardiac myocytes is to restore the membrane potential to its resting level, that is regulate repolarization (reviewed in Ref.235). The rapid and slow rectifier K+ channels, referred to as IKr and IKs, regulate efflux of K+ during Phase 3 of the action potential in ventricular cardiac myocytes and are the main determinants of the refractory period and therefore, the APD. In addition, the inward rectifier channels (IK1), fast transient outward current (Itof), and slow transient outward current (Itos) contribute to efflux of K+ during various phases of action potential. Genetic variants, transcriptional changes, and post-translational modifications of various proteins involved in the K+ currents are expected to affect susceptibility to cardiac arrhythmias in patients with cardiomyopathies.
There are scant data on K+ channel abnormalities in specific forms of genetic cardiomyopathies. A pathogenic LoF variant in KCNQ1, encoding a subunit of IKs, has been associated with reduced IKs current and VT in a patient with DCM.236 In addition, auto-antibodies against KCNQ1 protein, also known as Kv7.1, have been detected in a subset of patients with DCM and shown to increase IKs current density and shorten the QT interval.237
In the setting of heart failure in patients with cardiomyopathies, the changes are expected to be largely similar to those observed in other forms of heart hypertrophy and failure. Notably, K+ currents are reduced in the failing myocardium, resulting in slow repolarization and prolongation of the APD, setting a pro-arrhythmic substrate.238–241 Likewise, ventricular myocytes isolated from patients with DCM show prolongation of the APD duration and slower repolarization phase.242 At the current level, IKs,IK1, and Ito are generally suppressed whereas small calcium-activated K+ current is increased in heart failure (reviewed in Ref.238). The changes in the expression level of small calcium-activated K+ current proteins have not been consistently observed.243 In conjunction with suppressed K+ currents and prolongation of APD, transcript levels of several genes encoding K+ channel protein subunits are also reduced in cardiac myocytes isolated from experimental models of cardiomyopathies.179,244 Moreover, changes in K+ channels in iPSC-cardiomyocytes generated from patients with cardiomyopathies also have been reported, albeit the findings are preliminary.245,246
7.2.4 Functional defects in cardiac Cl− current
Several chloride channels are expressed in the heart including, CFTR, CIC2, CIC3, CLCA, TMEM16A, and BEST1; and implicated in the regulation of both repolarization and depolarization (reviewed in Ref.247). Alterations in cardiac chloride channels have been implicated in cardiac arrhythmia, myocardial hypertrophy, and heart failure.247 For example, swelling-sensitive Cl− channel (IClswell) is activated in cardiac hypertrophy and failure and contributes to shortening of the APD, depolarization of the resting membrane potential, and induction of arrhythmias.248
7.3 Functional defects in cardiac connexins
Gap junctions are comprised of clusters of connexin proteins, which span the cytoplasmic membrane, form channels, and regulate cell-to-cell electrical and metabolic coupling (reviewed in Ref.249). Six connexin molecules assemble to form a connexon (hemichannel), which connects with the connexon of the neighbouring cardiac myocyte to form a gap junction in the IDs.249 The human genome codes for 21 connexin proteins with similar structural topology.250 Connexins show tissue and cell type-specific expression, enabling unique permeability and gating properties as well as ability to interact with regulatory partners in different tissues.251 GJA1 (gap junction protein alpha 1 or Cx43) is the most abundantly expression connexin in cardiac myocytes, followed by GJA5 (gap junction protein alpha 5 or Cx40), and GJC1 (gap junction protein gamma 1 or Cx45).249 In addition, GJB2 (gap junction protein beta 2 or Cx26) and GJD3 (gap junction protein delta 3 or Cx31.9) are also expressed in cardiac myocytes, albeit at low levels. GJA1 and GJA5 are predominantly expressed in ventricular (>90%) and atrial cardiac myocytes, respectively, and less in the cardiac conduction system, whereas GJC1 expression is found mainly in the cardiac conduction system.249,252,253 Diversity in the expression levels and composition of cardiac connexins leads to homomeric and heteromeric constitution of connexons with variable biological properties. In addition, connexins undergo considerable remodelling in the heart during cardiac development and under pathological conditions, both in terms of expression levels as well as post-translational modifications, including phosphorylation, which regulate its gating, trafficking, assembly/disassembly, distribution, and degradation.254,255
There is no compelling human molecular genetic data to link pathogenic variants in genes encoding cardiac connexin with cardiomyopathies and arrhythmias (reviewed in Ref.256). However, molecular remodelling of the IDs in cardiomyopathies is associated with reduced levels, post-translational modifications, and localization of connexins in the heart.19,257–259 The changes are particularly notable for GJA1 (Cx43) in the advanced stages of ACM, wherein the total level and localization of GJA1 to IDs are markedly reduced.19,257–259 However, altered expression and localization of GJA1 are not specific to ACM but are also observed in other forms of heart failure and in cardiomyopathies, including DCM, HCM, and model organisms of cardiomyopathies.194,243,260–262
The precise functional impacts of altered GJA1 expression and localization on susceptibility to cardiac arrhythmias are not well established. Conventionally, alterations in are thought to enhance susceptibility to cardiac arrhythmias by altering action potential conduction in the heart. However, part if not the whole effects of connexins might be mediated through their interactions with SCN5A and electrotonic/ephaptic conduction (reviewed in Ref.263).
Atrial fibrillation is associated with changes in the expression level and location of GJA5 (Cx40) in the atrial tissue and pulmonic veins (reviewed in Ref.264). However, in the context of cardiomyopathies, the role of GJA5 in susceptibility to atrial arrhythmias remains unclear.
8. Treatment
Conventional approaches, comprised of pharmacological therapy, radiofrequency ablation, and implantable cardioverter/defibrillator (ICD) implantation, are applied for the treatment of cardiac arrhythmias in patients with hereditary cardiomyopathies, and therefore, not discussed. Nevertheless, it merits noting that because of the presence of an arrhythmogenic substrate the threshold for implantation of an ICD in the management of cardiac arrhythmias is relatively low. The notion is partly reinforced by the success of radiofrequency ablation in reducing ventricular arrhythmias in patients with ACM and the effectiveness of ICDs in prevention of SCD in patients with HCM.265–267
9. Concluding remarks
Cardiac arrhythmias are common and dangerous manifestations of hereditary cardiomyopathies. ACM is the prototypic form of hereditary cardiomyopathies wherein ventricular arrhythmias are the cardinal and early manifestations of the disease, typically occurring prior to the onset of cardiac dysfunction. Numerous mechanisms are implicated in predisposition to cardiac arrhythmias in hereditary cardiomyopathies, including a shared genetic aetiology, pro-arrhythmic structural and molecular myocardial remodelling, altered mRNA splicing of genes regulating ion currents, post-translational modifications of ion channels, and trafficking of proteins involved in electric activity of the heart. Elucidation of the genetic and non-genetic basis of cardiac arrhythmias in hereditary cardiomyopathies might pave the way for identification of new drug targets and development of new therapies.
Conflict of interest: none declared.
Funding
This work was supported in part by grants from National Institutes of Health (NIH), National Heart, Lung and Blood Institute (NHLBI, R01 HL151737, 1R01HL132401, S10 OD018135), Leducq Foundation (14 CVD 03), The Ewing Halsell Foundation, George and Mary Josephine Hamman Foundation, and TexGen Fund from Greater Houston Community Foundation.
This article was guest edited by Carol Ann Remme, The Netherlands and Silvia Priori, Italy.
References
- 1. McKenna W, Maron J, Barry J, Thiene G.. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res 2017;121:722–730. [DOI] [PubMed] [Google Scholar]
- 2. Marian AJ, Braunwald E.. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, Davis AM, Thompson T, Connell V, Wallace J, Naylor C, Crawford J, Love DR, Hallam L, White J, Lawrence C, Lynch M, Morgan N, James P, Du Sart D, Puranik R, Langlois N, Vohra J, Winship I, Atherton J, McGaughran J, Skinner JR, Semsarian C.. A prospective study of sudden cardiac death among children and young adults. N Engl J Med 2016;374:2441–2452. [DOI] [PubMed] [Google Scholar]
- 4. Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO.. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980. Circulation 2009;119:1085–1092. [DOI] [PubMed] [Google Scholar]
- 5. O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ, Elliott PM, Hypertrophic Cardiomyopathy Outcomes Investigators. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014;35:2010–2020. [DOI] [PubMed] [Google Scholar]
- 6. McNally EM, Mestroni L.. Dilated cardiomyopathy. Circ Res 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE.. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B.. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715–1724. [DOI] [PubMed] [Google Scholar]
- 9. Arbustini E, Morbini P, Grasso M, Fasani R, Verga L, Bellini O, Dal Bello B, Campana C, Piccolo G, Febo O, Opasich C, Gavazzi A, Ferrans VJ.. Restrictive cardiomyopathy, atrioventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J Am Coll Cardiol 1998;31:645–653. [DOI] [PubMed] [Google Scholar]
- 10. Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M.. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corrado D, Basso C, Judge DP.. Arrhythmogenic cardiomyopathy. Circ Res 2017;121:784–802. [DOI] [PubMed] [Google Scholar]
- 12. Groeneweg JA, Bhonsale A, James CA, Te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AC, Kassamali B, Atsma DE, Volders PG, de Groot NM, de Boer K, Zimmerman SL, Kamel IR, van der Heijden JF, Russell SD, Jan Cramer M, Tedford RJ, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Hauer RN, Calkins H.. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet 2015;8:437–446. [DOI] [PubMed] [Google Scholar]
- 13. Thiene G. The research venture in arrhythmogenic right ventricular cardiomyopathy: a paradigm of translational medicine. Eur Heart J 2015;36:837–846. [DOI] [PubMed] [Google Scholar]
- 14. Corrado D, Basso C, Schiavon M, Thiene G.. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364–369. [DOI] [PubMed] [Google Scholar]
- 15. Mast TP, Teske AJ, vd Heijden JF, Groeneweg JA, Te Riele AS, Velthuis BK, Hauer RN, Doevendans PA, Cramer MJ.. Left ventricular involvement in arrhythmogenic right ventricular dysplasia/cardiomyopathy assessed by echocardiography predicts adverse clinical outcome. J Am Soc Echocardiogr 2015;28:1103–1113.e1109. [DOI] [PubMed] [Google Scholar]
- 16. Marcus FI. Epsilon waves aid in the prognosis and risk stratification of patients with ARVC/D. J Cardiovasc Electrophysiol 2015;26:1211–1212. [DOI] [PubMed] [Google Scholar]
- 17. Platonov PG, Calkins H, Hauer RN, Corrado D, Svendsen JH, Wichter T, Biernacka EK, Saguner AM, Te Riele AS, Zareba W.. High interobserver variability in the assessment of epsilon waves: implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2016;13:208–216. [DOI] [PubMed] [Google Scholar]
- 18. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ.. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006;116:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ.. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res 2014;114:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corrado D, Link MS, Calkins H.. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2017;376:61–72. [DOI] [PubMed] [Google Scholar]
- 21. Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R.. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol 2018;72:784–804. [DOI] [PubMed] [Google Scholar]
- 22. Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, Bourfiss M, Fortier A, Lie OH, Saguner AM, Svensson A, Andorin A, Tichnell C, Murray B, Zeppenfeld K, van den Berg MP, Asselbergs FW, Wilde AAM, Krahn AD, Talajic M, Rivard L, Chelko S, Zimmerman SL, Kamel IR, Crosson JE, Judge DP, Yap SC, van der Heijden JF, Tandri H, Jongbloed JDH, Guertin MC, van Tintelen JP, Platonov PG, Duru F, Haugaa KH, Khairy P, Hauer RNW, Calkins H, Te Riele A, James CA.. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2019;40:1850–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu AF, Zado E, Marchlinski FE.. Atrial arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and ventricular tachycardia. Am J Cardiol 2010;106:720–722. [DOI] [PubMed] [Google Scholar]
- 24. Wu L, Guo J, Zheng L, Chen G, Ding L, Qiao Y, Sun W, Yao Y, Zhang S.. Atrial remodeling and atrial tachyarrhythmias in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 2016;118:750–753. [DOI] [PubMed] [Google Scholar]
- 25. Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ.. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol 2003;42:873–879. [DOI] [PubMed] [Google Scholar]
- 26. Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ.. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol 2005;45:697–704. [DOI] [PubMed] [Google Scholar]
- 27. Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ.. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001;104:2517–2524. [DOI] [PubMed] [Google Scholar]
- 28. Siontis KC, Geske JB, Ong K, Nishimura RA, Ommen SR, Gersh BJ.. Atrial fibrillation in hypertrophic cardiomyopathy: prevalence, clinical correlations, and mortality in a large high-risk population. J Am Heart Assoc 2014;3:e001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Debonnaire P, Joyce E, Hiemstra Y, Mertens BJ, Atsma DE, Schalij MJ, Bax JJ, Delgado V, Marsan NA.. Left atrial size and function in hypertrophic cardiomyopathy patients and risk of new-onset atrial fibrillation. Circ Arrhythm Electrophysiol 2017;10. pii: e004052. [DOI] [PubMed] [Google Scholar]
- 30. Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM.. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014;100:465–472. [DOI] [PubMed] [Google Scholar]
- 31. Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, Rakowski H.. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;39:638–645. [DOI] [PubMed] [Google Scholar]
- 32. van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, Crijns HJGM, Pinto YM.. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med 2005;83:79–83. [DOI] [PubMed] [Google Scholar]
- 33. Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, Ramani F, Brun F, Cocciolo A, Puggia I, Morea G, McKenna WJ, La Rosa FG, Taylor MRG, Sinagra G, Mestroni L.. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol 2019;74:1480–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AA, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM.. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol 2012;59:493–500. [DOI] [PubMed] [Google Scholar]
- 35. Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiaran Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner-La Rocca HP, Brunner HG, Krapels IPC, Heymans S.. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J 2018;39:864–873. [DOI] [PubMed] [Google Scholar]
- 36. Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, Biagini E, van Haelst PL, van Wijngaarden J, van den Berg MP, Wilde AA, Mannens MM, de Boer RA, van Spaendonck-Zwarts KY, van Tintelen JP, Pinto YM.. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail 2017;19:512–521. [DOI] [PubMed] [Google Scholar]
- 37. Asatryan B. Cardiac sodium channel dysfunction and dilated cardiomyopathy: a contemporary reappraisal of pathophysiological concepts. J Clin Med 2019;8. pii: E1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilde AAM, Amin AS.. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin Electrophysiol 2018;4:569–579. [DOI] [PubMed] [Google Scholar]
- 39. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT.. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–811. [DOI] [PubMed] [Google Scholar]
- 40. Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 2010;67:915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q.. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293–296. [DOI] [PubMed] [Google Scholar]
- 42. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL.. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005;293:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H.. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001;104:3081–3086. [DOI] [PubMed] [Google Scholar]
- 44. Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr,. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 2003;112:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AAA.. cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res 2003;92:14–22. [DOI] [PubMed] [Google Scholar]
- 46. Laurent G, Saal S, Amarouch MY, Beziau DM, Marsman RF, Faivre L, Barc J, Dina C, Bertaux G, Barthez O, Thauvin-Robinet C, Charron P, Fressart V, Maltret A, Villain E, Baron E, Merot J, Turpault R, Coudiere Y, Charpentier F, Schott JJ, Loussouarn G, Wilde AA, Wolf JE, Baro I, Kyndt F, Probst V.. Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol 2012;60:144–156. [DOI] [PubMed] [Google Scholar]
- 47. McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L, Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004;110:2163–2167. [DOI] [PubMed] [Google Scholar]
- 48. McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L, Familial Cardiomyopathy Registry Research Group. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol 2011;57:2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nguyen TP, Wang DW, Rhodes TH, George AL Jr.. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res 2008;102:364–371. [DOI] [PubMed] [Google Scholar]
- 50. Marsman RF, Bardai A, Postma AV, Res JC, Koopmann TT, Beekman L, van der Wal AC, Pinto YM, Lekanne Deprez RH, Wilde AA, Jordaens LJ, Bezzina CR.. A complex double deletion in LMNA underlies progressive cardiac conduction disease, atrial arrhythmias, and sudden death. Circ Cardiovasc Genet 2011;4:280–287. [DOI] [PubMed] [Google Scholar]
- 51. Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, Garg V.. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm 2009;6:707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peretto G, Di Resta C, Perversi J, Forleo C, Maggi L, Politano L, Barison A, Previtali SC, Carboni N, Brun F, Pegoraro E, D’Amico A, Rodolico C, Magri F, Manzi RC, Palladino A, Isola F, Gigli L, Mongini TE, Semplicini C, Calore C, Ricci G, Comi GP, Ruggiero L, Bertini E, Bonomo P, Nigro G, Resta N, Emdin M, Favale S, Siciliano G, Santoro L, Sinagra G, Limongelli G, Ambrosi A, Ferrari M, Golzio PG, Bella PD, Benedetti S, Sala S, Italian Network for Laminopathies (NIL). Cardiac and neuromuscular features of patients with LMNA-related cardiomyopathy. Ann Intern Med 2019;171:458. [DOI] [PubMed] [Google Scholar]
- 53. Rober RA, Weber K, Osborn M.. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 1989;105:365–378. [DOI] [PubMed] [Google Scholar]
- 54. Schreiber KH, Kennedy BK.. When lamins go bad: nuclear structure and disease. Cell 2013;152:1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Steensel B, Belmont AS.. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 2017;169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, Gurha P, Marian AJ.. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res 2019;124:1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Salvarani N, Crasto S, Miragoli M, Bertero A, Paulis M, Kunderfranco P, Serio S, Forni A, Lucarelli C, Dal Ferro M, Larcher V, Sinagra G, Vezzoni P, Murry CE, Faggian G, Condorelli G, Di Pasquale E.. The K219T-lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat Commun 2019;10:2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner DA, Berul CI, Seidman CE, Seidman JG.. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 2008;44:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O’Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA.. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 2015;7:270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fatkin D, Huttner IG.. Titin truncating mutations in dilated cardiomypathy: the long and short of it. Curr Opin Cardiol 2017;32:232–238. [DOI] [PubMed] [Google Scholar]
- 61. Ware JS, Cook SA.. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol 2018;15:241–252. [DOI] [PubMed] [Google Scholar]
- 62. Corden B, Jarman J, Whiffin N, Tayal U, Buchan R, Sehmi J, Harper A, Midwinter W, Lascelles K, Markides V, Mason M, Baksi J, Pantazis A, Pennell DJ, Barton PJ, Prasad SK, Wong T, Cook SA, Ware JS.. Association of titin-truncating genetic variants with life-threatening cardiac arrhythmias in patients with dilated cardiomyopathy and implanted defibrillators. JAMA Netw Open 2019;2:e196520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, Kember RL, Lester Kirchner H, Leader JB, Liang L, McDermott-Roe C, Babu A, Morley M, Nealy Z, Person TN, Pulenthiran A, Small A, Smelser DT, Stahl RC, Sturm AC, Williams H, Baras A, Margulies KB, Cappola TP, Dewey FE, Verma A, Zhang X, Correa A, Hall ME, Wilson JG, Ritchie MD, Rader DJ, Murray MF, Fornwalt BK, Arany Z; DiscovEHR and Penn Medicine Biobank Studies. Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation 2019;140:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ahlberg G, Refsgaard L, Lundegaard PR, Andreasen L, Ranthe MF, Linscheid N, Nielsen JB, Melbye M, Haunso S, Sajadieh A, Camp L, Olesen SP, Rasmussen S, Lundby A, Ellinor PT, Holst AG, Svendsen JH, Olesen MS.. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun 2018;9:4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choi SH, Weng LC, Roselli C, Lin H, Haggerty CM, Shoemaker MB, Barnard J, Arking DE, Chasman DI, Albert CM, Chaffin M, Tucker NR, Smith JD, Gupta N, Gabriel S, Margolin L, Shea MA, Shaffer CM, Yoneda ZT, Boerwinkle E, Smith NL, Silverman EK, Redline S, Vasan RS, Burchard EG, Gogarten SM, Laurie C, Blackwell TW, Abecasis G, Carey DJ, Fornwalt BK, Smelser DT, Baras A, Dewey FE, Jaquish CE, Papanicolaou GJ, Sotoodehnia N, Van Wagoner DR, Psaty BM, Kathiresan S, Darbar D, Alonso A, Heckbert SR, Chung MK, Roden DM, Benjamin EJ, Murray MF, Lunetta KL, Lubitz SA, Ellinor PT; DiscovEHR study and the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA 2018;320:2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nielsen JB, Fritsche LG, Zhou W, Teslovich TM, Holmen OL, Gustafsson S, Gabrielsen ME, Schmidt EM, Beaumont R, Wolford BN, Lin M, Brummett CM, Preuss MH, Refsgaard L, Bottinger EP, Graham SE, Surakka I, Chu Y, Skogholt AH, Dalen H, Boyle AP, Oral H, Herron TJ, Kitzman J, Jalife J, Svendsen JH, Olesen MS, Njølstad I, Løchen M-L, Baras A, Gottesman O, Marcketta A, O’Dushlaine C, Ritchie MD, Wilsgaard T, Loos RJF, Frayling TM, Boehnke M, Ingelsson E, Carey DJ, Dewey FE, Kang HM, Abecasis GR, Hveem K, Willer CJ.. Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet 2018;102:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kapoor A, Bakshy K, Xu L, Nandakumar P, Lee D, Boerwinkle E, Grove ML, Arking DE, Chakravarti A.. Rare coding TTN variants are associated with electrocardiographic QT interval in the general population. Sci Rep 2016;6:28356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tayal U, Newsome S, Buchan R, Whiffin N, Walsh R, Barton PJ, Ware JS, Cook SA, Prasad SK.. Truncating variants in titin independently predict early arrhythmias in patients with dilated cardiomyopathy. J Am Coll Cardiol 2017;69:2466–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fujita M, Mitsuhashi H, Isogai S, Nakata T, Kawakami A, Nonaka I, Noguchi S, Hayashi YK, Nishino I, Kudo A.. Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Dev Biol 2012;361:79–89. [DOI] [PubMed] [Google Scholar]
- 70. Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S, Kunkel LM.. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol 2000;148:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vorgerd M, van der Ven PFM, Bruchertseifer V, Löwe T, Kley RA, Schröder R, Lochmüller H, Himmel M, Koehler K, Fürst DO, Huebner A.. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet 2005;77:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C, León D, Martín M, Puente XS, López-Otín C.. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun 2014;5:5326. [DOI] [PubMed] [Google Scholar]
- 73. Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, Gibson WT, McKinnon ML, McGillivray B, Alvarez N, Giuffre M, Schwartzentruber J, Consortium FC, Gerull B, FORGE Canada Consortium. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat 2016;37:269–279. [DOI] [PubMed] [Google Scholar]
- 74. Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padrón-Barthe L, Duro-Aguado I, Jiménez-Jáimez J, Hidalgo-Olivares VM, García-Campo E, Lanzillo C, Suárez-Mier MP, Yonath H, Marcos-Alonso S, Ochoa JP, Santomé JL, García-Giustiniani D, Rodríguez-Garrido JL, Domínguez F, Merlo M, Palomino J, Peña ML, Trujillo JP, Martín-Vila A, Stolfo D, Molina P, Lara-Pezzi E, Calvo-Iglesias FE, Nof E, Calò L, Barriales-Villa R, Gimeno-Blanes JR, Arad M, García-Pavía P, Monserrat L.. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 2016;68:2440–2451. [DOI] [PubMed] [Google Scholar]
- 75. Hall CL, Akhtar MM, Sabater-Molina M, Futema M, Asimaki A, Protonotarios A, Dalageorgou C, Pittman AM, Suarez MP, Aguilera B, Molina P, Zorio E, Hernandez JP, Pastor F, Gimeno JR, Syrris P, McKenna WJ.. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol 2019. [DOI] [PubMed] [Google Scholar]
- 76. Mangum KD, Ferns SJ.. A novel familial truncating mutation in the filamin C gene associated with cardiac arrhythmias. Eur J Med Genet 2019;62:282–285. [DOI] [PubMed] [Google Scholar]
- 77. Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, Gowan K, Jones KL, Brun F, Merlo M, Miani D, Sweet M, Devaraj K, Wartchow EP, Gigli M, Puggia I, Salcedo EE, Garrity DM, Ambardekar AV, Buttrick P, Reece TB, Bristow MR, Saffitz JE, Mestroni L, Taylor M.. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures. JACC Clin Electrophysiol 2018;4:504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kushnir A, Wajsberg B, Marks AR.. Ryanodine receptor dysfunction in human disorders. Biochim Biophys Acta Mol Cell Res 2018;1865:1687–1697. [DOI] [PubMed] [Google Scholar]
- 79. Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR, Xie ZJ. Na+/Ca2+exchange and Na+/K+-ATPase in the heart. J Physiol 2015;593:1361–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA.. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 81. Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K.. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001;103:485–490. [DOI] [PubMed] [Google Scholar]
- 82. Roux-Buisson N, Gandjbakhch E, Donal E, Probst V, Deharo J-C, Chevalier P, Klug D, Mansencal N, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Trapani J, Fouret P, Frank R, Fressart V, Fauré J, Lunardi J, Charron P.. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: results of a systematic screening. Heart Rhythm 2014;11:1999–2009. [DOI] [PubMed] [Google Scholar]
- 83. Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A.. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189–194. [DOI] [PubMed] [Google Scholar]
- 84. Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, Postma AV, van Langen I, Mannens MM, Wilde AA.. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 2007;116:1569–1576. [DOI] [PubMed] [Google Scholar]
- 85. Ohno S, Omura M, Kawamura M, Kimura H, Itoh H, Makiyama T, Ushinohama H, Makita N, Horie M.. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. Europace 2014;16:1646–1654. [DOI] [PubMed] [Google Scholar]
- 86. Campbell MJ, Czosek RJ, Hinton RB, Miller EM.. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am J Med Genet A 2015;167:2197–2200. [DOI] [PubMed] [Google Scholar]
- 87. Roston TM, Guo W, Krahn AD, Wang R, Van Petegem F, Sanatani S, Chen SR, Lehman A.. A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular non-compaction and atypical catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol 2017;50:227–233. [DOI] [PubMed] [Google Scholar]
- 88. Dobrev D, Wehrens X.. Mouse models of cardiac arrhythmias. Circ Res 2018;123:332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pan X, Philippen L, Lahiri SK, Lee C, Park SH, Word TA, Li N, Jarrett KE, Gupta R, Reynolds JO, Lin J, Bao G, Lagor WR, Wehrens X.. In vivo Ryr2 editing corrects catecholaminergic polymorphic ventricular tachycardia. Circ Res 2018;123:953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Beavers DL, Landstrom AP, Chiang DY, Wehrens XH.. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res 2014;103:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XH, Claycomb WC, Ko JK, Hwang M, Pan Z, Ma J, Ackerman MJ.. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol 2007;42:1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, Ikeda A, Kamatani N, Takeshima H, Matsuoka R.. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet 2007;52:543–548. [DOI] [PubMed] [Google Scholar]
- 93. Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ, Wehrens X.. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol 2013;62:2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Quick AP, Landstrom AP, Wang Q, Beavers DL, Reynolds JO, Barreto-Torres G, Tran V, Showell J, Philippen LE, Morris SA, Skapura D, Bos JM, Pedersen SE, Pautler RG, Ackerman MJ, Wehrens XH.. Novel junctophilin-2 mutation A405S is associated with basal septal hypertrophy and diastolic dysfunction. JACC Basic Transl Sci 2017;2:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jones EG, Mazaheri N, Maroofian R, Zamani M, Seifi T, Sedaghat A, Shariati G, Jamshidi Y, Allen HD, Wehrens XHT, Galehdari H, Landstrom AP.. Analysis of enriched rare variants in JPH2-encoded junctophilin-2 among Greater Middle Eastern individuals reveals a novel homozygous variant associated with neonatal dilated cardiomyopathy. Sci Rep 2019;9:9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE.. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003;299:1410–1413. [DOI] [PubMed] [Google Scholar]
- 97. DeWitt MM, MacLeod HM, Soliven B, McNally EM.. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol 2006;48:1396–1398. [DOI] [PubMed] [Google Scholar]
- 98. van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck-Zwarts KY, Lekanne dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP.. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail 2012;14:1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW II, MacLennan DH, Kremastinos DT, Kranias EG.. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 2003;111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kranias EG, Hajjar RJ.. The phospholamban journey 4 decades after setting out for Ithaka. Circ Res 2017;120:781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L.. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004;36:1162–1164. [DOI] [PubMed] [Google Scholar]
- 102. Tester DJ, Ackerman JP, Giudicessi JR, Ackerman NC, Cerrone M, Delmar M, Ackerman MJ.. Plakophilin-2 truncation variants in patients clinically diagnosed with catecholaminergic polymorphic ventricular tachycardia and decedents with exercise-associated autopsy negative sudden unexplained death in the young. JACC Clin Electrophysiol 2019;5:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M.. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, Isom LL, Delmar M.. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res 2009;105:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA.. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2013;10:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cerrone M, Montnach J, Lin X, Zhao YT, Zhang M, Agullo-Pascual E, Leo-Macias A, Alvarado FJ, Dolgalev I, Karathanos TV, Malkani K, Van Opbergen CJM, van Bavel JJA, Yang HQ, Vasquez C, Tester D, Fowler S, Liang F, Rothenberg E, Heguy A, Morley GE, Coetzee WA, Trayanova NA, Ackerman MJ, van Veen TAB, Valdivia HH, Delmar M.. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun 2017;8:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM.. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009;54:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Parikh VN, Caleshu C, Reuter C, Lazzeroni LC, Ingles J, Garcia J, McCaleb K, Adesiyun T, Sedaghat-Hamedani F, Kumar S, Graw S, Gigli M, Stolfo D, Dal Ferro M, Ing AY, Nussbaum R, Funke B, Wheeler MT, Hershberger RE, Cook S, Steinmetz LM, Lakdawala NK, Taylor MRG, Mestroni L, Merlo M, Sinagra G, Semsarian C, Meder B, Judge DP, Ashley E.. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail 2019;12:e005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. van den Hoogenhof MMG, Beqqali A, Amin AS, van der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck-Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE.. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018;138:1330–1342. [DOI] [PubMed] [Google Scholar]
- 110. Wyles SP, Li X, Hrstka SC, Reyes S, Oommen S, Beraldi R, Edwards J, Terzic A, Olson TM, Nelson TJ.. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum Mol Genet 2016;25:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Spater D, Abramczuk MK, Buac K, Zangi L, Stachel MW, Clarke J, Sahara M, Ludwig A, Chien KR.. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat Cell Biol 2013;15:1098–1106. [DOI] [PubMed] [Google Scholar]
- 112. van Eif VWW, Stefanovic S, Mohan RA, Christoffels VM.. Gradual differentiation and confinement of the cardiac conduction system as indicated by marker gene expression. Biochim Biophys Acta Mol Cell Res 2020;1867:118509. [DOI] [PubMed] [Google Scholar]
- 113. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res 2010;106:434–446. [DOI] [PubMed] [Google Scholar]
- 114. D’Souza A, Bucchi A, Johnsen AB, Logantha SJRJ, Monfredi O, Yanni J, Prehar S, Hart G, Cartwright E, Wisloff U, Dobryznski H, DiFrancesco D, Morris GM, Boyett MR.. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat Commun 2014;5:3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D.. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med 2006;354:151–157. [DOI] [PubMed] [Google Scholar]
- 116. Schweizer PA, Schroter J, Greiner S, Haas J, Yampolsky P, Mereles D, Buss SJ, Seyler C, Bruehl C, Draguhn A, Koenen M, Meder B, Katus HA, Thomas D.. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol 2014;64:757–767. [DOI] [PubMed] [Google Scholar]
- 117. Milano A, Vermeer AM, Lodder EM, Barc J, Verkerk AO, Postma AV, van der Bilt IA, Baars MJ, van Haelst PL, Caliskan K, Hoedemaekers YM, Le Scouarnec S, Redon R, Pinto YM, Christiaans I, Wilde AA, Bezzina CR.. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol 2014;64:745–756. [DOI] [PubMed] [Google Scholar]
- 118. Servatius H, Porro A, Pless SA, Schaller A, Asatryan B, Tanner H, de Marchi SF, Roten L, Seiler J, Haeberlin A, Baldinger SH, Noti F, Lam A, Fuhrer J, Moroni A, Medeiros-Domingo A.. Phenotypic spectrum of HCN4 mutations: a clinical case. Circ Genom Precis Med 2018;11:e002033. [DOI] [PubMed] [Google Scholar]
- 119. Steinberg GR, Carling D.. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 2019;18:527–551. [DOI] [PubMed] [Google Scholar]
- 120. Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H.. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001;10:1215–1220. [DOI] [PubMed] [Google Scholar]
- 121. Gollob MH, Green MS, Tang AS-L, Gollob T, Karibe A, Hassan A-S, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Tapscott T, Gonzales O, Begley D, Mohiddin S, Roberts R.. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 2001;344:1823–1831. [DOI] [PubMed] [Google Scholar]
- 122. Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R.. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 2001;104:3030–3033. [DOI] [PubMed] [Google Scholar]
- 123. Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG.. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003;107:2850–2856. [DOI] [PubMed] [Google Scholar]
- 124. Light PE, Wallace CH, Dyck JR.. Constitutively active adenosine monophosphate-activated protein kinase regulates voltage-gated sodium channels in ventricular myocytes. Circulation 2003;107:1962–1965. [DOI] [PubMed] [Google Scholar]
- 125. Kim M, Hunter RW, Garcia-Menendez L, Gong G, Yang YY, Kolwicz SC Jr, Xu J, Sakamoto K, Wang W, Tian R.. Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res 2014;114:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. van Bon BW, Gilissen C, Grange DK, Hennekam RC, Kayserili H, Engels H, Reutter H, Ostergaard JR, Morava E, Tsiakas K, Isidor B, Le Merrer M, Eser M, Wieskamp N, de Vries P, Steehouwer M, Veltman JA, Robertson SP, Brunner HG, de Vries BB, Hoischen A.. Cantu syndrome is caused by mutations in ABCC9. Am J Hum Genet 2012;90:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A.. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet 2004;36:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, Seino S, Asirvatham SJ, Jahangir A, Terzic A.. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Rev Cardiol 2007;4:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Boczek NJ, Ye D, Jin F, Tester DJ, Huseby A, Bos JM, Johnson AJ, Kanter R, Ackerman MJ.. Identification and functional characterization of a novel CACNA1C-mediated cardiac disorder characterized by prolonged QT intervals with hypertrophic cardiomyopathy, congenital heart defects, and sudden cardiac death. Circ Arrhythm Electrophysiol 2015;8:1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zahavich L, Tarnopolsky M, Yao R, Mital S.. Novel association of a de novo CALM2 mutation with long QT syndrome and hypertrophic cardiomyopathy. Circ Genom Precis Med 2018;11:e002255. [DOI] [PubMed] [Google Scholar]
- 131. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L, Familial Dilated Cardiomyopathy Registry Research Group. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771–780. [DOI] [PubMed] [Google Scholar]
- 132. Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L.. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol 2002;39:981–990. [DOI] [PubMed] [Google Scholar]
- 133. Meyer S, van der Meer P, van Tintelen JP, van den Berg MP.. Sex differences in cardiomyopathies. Eur J Heart Fail 2014;16:238–247. [DOI] [PubMed] [Google Scholar]
- 134. Olivotto I, Maron MS, Adabag AS, Casey SA, Vargiu D, Link MS, Udelson JE, Cecchi F, Maron BJ.. Gender-related differences in the clinical presentation and outcome of hypertrophic cardiomyopathy. J Am Coll Cardiol 2005;46:480–487. [DOI] [PubMed] [Google Scholar]
- 135. Bauce B, Frigo G, Marcus FI, Basso C, Rampazzo A, Maddalena F, Corrado D, Winnicki M, Daliento L, Rigato I, Steriotis A, Mazzotti E, Thiene G, Nava A.. Comparison of clinical features of arrhythmogenic right ventricular cardiomyopathy in men versus women. Am J Cardiol 2008;102:1252–1257. [DOI] [PubMed] [Google Scholar]
- 136. van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Grasso M, Serio A, Jenkins S, Rowland C, Richard P, Wilde AA, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Christiaans I, Pinto YM.. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail 2013;15:376–384. [DOI] [PubMed] [Google Scholar]
- 137. Bhonsale A, James CA, Tichnell C, Murray B, Madhavan S, Philips B, Russell SD, Abraham T, Tandri H, Judge DP, Calkins H.. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol 2013;6:569–578. [DOI] [PubMed] [Google Scholar]
- 138. Takahashi A, Shiga T, Shoda M, Tanizaki K, Manaka T, Ejima K, Kasanuki H, Hagiwara N.. Gender difference in arrhythmic occurrences in patients with nonischemic dilated cardiomyopathy and implantable cardioverter-defibrillator. Heart Vessels 2010;25:150–154. [DOI] [PubMed] [Google Scholar]
- 139. Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, Monteforte N, Memmi M, Gambelli P, Novelli V, Bloise R, Catalano O, Moro G, Tibollo V, Morini M, Bellazzi R, Napolitano C, Bagnardi V, Priori SG.. Arrhythmogenic right ventricular cardiomyopathy: clinical course and predictors of arrhythmic risk. J Am Coll Cardiol 2016;68:2540–2550. [DOI] [PubMed] [Google Scholar]
- 140. Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, Towbin JA, Priori SG, Napolitano C, Robinson JL, Andrews M, Timothy K, Hall WJ.. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation 1998;97:2237–2244. [DOI] [PubMed] [Google Scholar]
- 141. Hey TM, Rasmussen TB, Madsen T, Aagaard MM, Harbo M, Molgaard H, Moller JE, Eiskjaer H, Mogensen J.. Pathogenic RBM20-variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ Heart Fail 2019;12:e005700. [DOI] [PubMed] [Google Scholar]
- 142. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F.. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000;102:858–864. [DOI] [PubMed] [Google Scholar]
- 143. Skinner JR, Winbo A, Abrams D, Vohra J, Wilde AA.. Channelopathies that lead to sudden cardiac death: clinical and genetic aspects. Heart Lung Circ 2019;28:22–30. [DOI] [PubMed] [Google Scholar]
- 144. Te Riele ASJM, Agullo-Pascual E, James CA, Leo-Macias A, Cerrone M, Zhang M, Lin X, Lin B, Rothenberg E, Sobreira NL, Amat-Alarcon N, Marsman RF, Murray B, Tichnell C, van der Heijden JF, Dooijes D, van Veen TAB, Tandri H, Fowler SJ, Hauer RNW, Tomaselli G, van den Berg MP, Taylor MRG, Brun F, Sinagra G, Wilde AAM, Mestroni L, Bezzina CR, Calkins H, Peter van Tintelen J, Bu L, Delmar M, Judge DP.. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kim JC, Perez-Hernandez M, Alvarado FJ, Maurya SR, Montnach J, Yin Y, Zhang M, Lin X, Vasquez C, Heguy A, Liang FX, Woo SH, Morley GE, Rothenberg E, Lundby A, Valdivia HH, Cerrone M, Delmar M.. Disruption of Ca(2+)i homeostasis and connexin 43 hemichannel function in the right ventricle precedes overt arrhythmogenic cardiomyopathy in plakophilin-2-deficient mice. Circulation 2019;140:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Karmouch J, Zhou QQ, Miyake CY, Lombardi R, Kretzschmar K, Bannier-Helaouet M, Clevers H, Wehrens XHT, Willerson JT, Marian AJ.. Distinct cellular basis for early cardiac arrhythmias, the cardinal manifestation of arrhythmogenic cardiomyopathy, and the skin phenotype of cardiocutaneous syndromes. Circ Res 2017;121:1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Eberly LA, Day SM, Ashley EA, Jacoby DL, Jefferies JL, Colan SD, Rossano JW, Semsarian C, Pereira AC, Olivotto I, Ingles J, Seidman CE, Channaoui N, Cirino AL, Han L, Ho CY, Lakdawala NK.. Association of race with disease expression and clinical outcomes among patients with hypertrophic cardiomyopathy. JAMA Cardiol 2020;5:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Myers VD, Gerhard GS, McNamara DM, Tomar D, Madesh M, Kaniper S, Ramsey FV, Fisher SG, Ingersoll RG, Kasch-Semenza L, Wang J, Hanley-Yanez K, Lemster B, Schwisow JA, Ambardekar AV, Degann SH, Bristow MR, Sheppard R, Alexis JD, Tilley DG, Kontos CD, McClung JM, Taylor AL, Yancy CW, Khalili K, Seidman JG, Seidman CE, McTiernan CF, Cheung JY, Feldman AM.. Association of variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol 2018;3:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N.. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129–133. [DOI] [PubMed] [Google Scholar]
- 150. Saberi S, Wheeler M, Bragg-Gresham J, Hornsby W, Agarwal PP, Attili A, Concannon M, Dries AM, Shmargad Y, Salisbury H, Kumar S, Herrera JJ, Myers J, Helms AS, Ashley EA, Day SM.. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. Jama 2017;317:1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pelliccia A, Lemme E, Maestrini V, Di Paolo FM, Pisicchio C, Di Gioia G, Caselli S.. Does sport participation worsen the clinical course of hypertrophic cardiomyopathy? Clinical outcome of hypertrophic cardiomyopathy in athletes. Circulation 2018;137:531–533. [DOI] [PubMed] [Google Scholar]
- 152. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H.. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G.. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol 2003;42:1959–1963. [DOI] [PubMed] [Google Scholar]
- 154. Paulin FL, Hodgkinson KA, MacLaughlan S, Stuckless SN, Templeton C, Shah S, Bremner H, Roberts JD, Young TL, Parfrey PS, Connors SP.. Exercise and arrhythmic risk in TMEM43 p. S358L arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2020. pii: S1547-5271(20)30181-8. [DOI] [PubMed] [Google Scholar]
- 155. Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR.. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 2004;109:3208–3214. [DOI] [PubMed] [Google Scholar]
- 156. Chen L, Kurokawa J, Kass RS.. Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J Biol Chem 2005;280:31347–31352. [DOI] [PubMed] [Google Scholar]
- 157. Wu J, Naiki N, Ding WG, Ohno S, Kato K, Zang WJ, Delisle BP, Matsuura H, Horie M.. A molecular mechanism for adrenergic-induced long QT syndrome. J Am Coll Cardiol 2014;63:819–827. [DOI] [PubMed] [Google Scholar]
- 158. Cerrone M, Delmar M.. Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc Med 2014;24:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Cheedipudi SM, Hu J, Fan S, Yuan P, Karmouch J, Czernuszewicz G, Robertson MJ, Coarfa C, Hong K, Yao Y, Moore HC, Wehrens X, Gurha P, Marian AJ.. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc Res 2020;116;1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M.. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996;94:983–991. [DOI] [PubMed] [Google Scholar]
- 161. Bowles NE, Ni J, Marcus F, Towbin JA.. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2002;39:892–895. [DOI] [PubMed] [Google Scholar]
- 162. Lim BK, Peter AK, Xiong D, Narezkina A, Yung A, Dalton ND, Hwang KK, Yajima T, Chen J, Knowlton KU.. Inhibition of Coxsackievirus-associated dystrophin cleavage prevents cardiomyopathy. J Clin Invest 2013;123:5146–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Berger PB, Tajik AJ.. Adverse prognosis of patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation 2003;108:2342–2348. [DOI] [PubMed] [Google Scholar]
- 164. Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, Daubert JP, Higgins SL, Brown MW, Andrews ML, Multicenter Automatic Defibrillator Implantation Trial II Investigators. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002;346:877–883. [DOI] [PubMed] [Google Scholar]
- 165. Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp-Channing N, Davidson-Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH.. Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med 2005;352:225–237. [DOI] [PubMed] [Google Scholar]
- 166. McCauley MD, Wehrens XH.. Animal models of arrhythmogenic cardiomyopathy. Dis Model Mech 2009;2:563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Antzelevitch C, Burashnikov A.. Overview of basic mechanisms of cardiac arrhythmia. Card Electrophysiol Clin 2011;3:23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Tschabrunn CM, Marchlinski FE.. Ventricular tachycardia mapping and ablation in arrhythmogenic right ventricular cardiomyopathy/dysplasia: lessons learned. World J Cardiol 2014;6:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Sato T, Ohkusa T, Honjo H, Suzuki S, Yoshida MA, Ishiguro YS, Nakagawa H, Yamazaki M, Yano M, Kodama I, Matsuzaki M.. Altered expression of connexin43 contributes to the arrhythmogenic substrate during the development of heart failure in cardiomyopathic hamster. Am J Physiol Heart Circ Physiol 2008;294:H1164–H1173. [DOI] [PubMed] [Google Scholar]
- 170. Link MS, Bockstall K, Weinstock J, Alsheikh-Ali AA, Semsarian C, Estes NAM III, Spirito P, Haas TS, Rowin EJ, Maron MS, Maron BJ.. Ventricular tachyarrhythmias in patients with hypertrophic cardiomyopathy and defibrillators: triggers, treatment, and implications. J Cardiovasc Electrophysiol 2017;28:531–537. [DOI] [PubMed] [Google Scholar]
- 171. Aiba T, Tomaselli GF.. Electrical remodeling in the failing heart. Curr Opin Cardiol 2010;25:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Aronson RS. Afterpotentials and triggered activity in hypertrophied myocardium from rats with renal hypertension. Circ Res 1981;48:720–727. [DOI] [PubMed] [Google Scholar]
- 173. Carballeira Pol L, Deyell MW, Frankel DS, Benhayon D, Squara F, Chik W, Kohari M, Deo R, Marchlinski FE.. Ventricular premature depolarization QRS duration as a new marker of risk for the development of ventricular premature depolarization-induced cardiomyopathy. Heart Rhythm 2014;11:299–306. [DOI] [PubMed] [Google Scholar]
- 174. Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, McKenna WJ.. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 2001;357:420–424. [DOI] [PubMed] [Google Scholar]
- 175. Schumacher B, Gietzen FH, Neuser H, SchüMmelfeder J, Schneider M, Kerber S, Schimpf R, Wolpert C, Borggrefe M.. Electrophysiological characteristics of septal hypertrophy in patients with hypertrophic obstructive cardiomyopathy and moderate to severe symptoms. Circulation 2005;112:2096–2101. [DOI] [PubMed] [Google Scholar]
- 176. Coppini R, Ferrantini C, Mugelli A, Poggesi C, Cerbai E.. Altered Ca(2+) and Na(+) homeostasis in human hypertrophic cardiomyopathy: implications for arrhythmogenesis. Front Physiol 2018;9:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Nass RD, Aiba T, Tomaselli GF, Akar FG.. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Rev Cardiol 2008;5:196–207. [DOI] [PubMed] [Google Scholar]
- 178. Kepenek ES, Ozcinar E, Tuncay E, Akcali KC, Akar AR, Turan B.. Differential expression of genes participating in cardiomyocyte electrophysiological remodeling via membrane ionic mechanisms and Ca(2+)-handling in human heart failure. Mol Cell Biochem 2020;463:33–44. [DOI] [PubMed] [Google Scholar]
- 179. Suzuki T, Shioya T, Murayama T, Sugihara M, Odagiri F, Nakazato Y, Nishizawa H, Chugun A, Sakurai T, Daida H, Morimoto S, Kurebayashi N.. Multistep ion channel remodeling and lethal arrhythmia precede heart failure in a mouse model of inherited dilated cardiomyopathy. PLoS One 2012;7:e35353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Bosch RF, Scherer CR, Rub N, Wohrl S, Steinmeyer K, Haase H, Busch AE, Seipel L, Kuhlkamp V.. Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces I(Ca, L) and I(to) in rapid atrial pacing in rabbits. J Am Coll Cardiol 2003;41:858–869. [DOI] [PubMed] [Google Scholar]
- 181. Swaminathan PD, Purohit A, Hund TJ, Anderson ME.. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res 2012;110:1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ednie AR, Parrish AR, Sonner MJ, Bennett ES.. Reduced hybrid/complex N-glycosylation disrupts cardiac electrical signaling and calcium handling in a model of dilated cardiomyopathy. J Mol Cell Cardiol 2019;132:13–23. [DOI] [PubMed] [Google Scholar]
- 183. Undrovinas AI, Maltsev VA, Sabbah HN.. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci 1999;55:494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. KäÄB S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF, Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res 1996;78:262–273. [DOI] [PubMed] [Google Scholar]
- 185. Houser SR, Piacentino V III, Weisser J.. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 2000;32:1595–1607. [DOI] [PubMed] [Google Scholar]
- 186. Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA.. Ca2+/calmodulin-dependent protein kinase II-dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem 2008;283:25524–25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM.. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 188. Movsesian MA, Karimi M, Green K, Jones LR.. Ca(2+)-transporting ATPase, phospholamban, and calsequestrin levels in nonfailing and failing human myocardium. Circulation 1994;90:653–657. [DOI] [PubMed] [Google Scholar]
- 189. Mueller EE, Momen A, Masse S, Zhou YQ, Liu J, Backx PH, Henkelman RM, Nanthakumar K, Stewart DJ, Husain M.. Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc Res 2011;89:623–633. [DOI] [PubMed] [Google Scholar]
- 190. Ismail TF, Jabbour A, Gulati A, Mallorie A, Raza S, Cowling TE, Das B, Khwaja J, Alpendurada FD, Wage R, Roughton M, McKenna WJ, Moon JC, Varnava A, Shakespeare C, Cowie MR, Cook SA, Elliott P, O’Hanlon R, Pennell DJ, Prasad SK.. Role of late gadolinium enhancement cardiovascular magnetic resonance in the risk stratification of hypertrophic cardiomyopathy. Heart 2014;100:1851–1858. [DOI] [PubMed] [Google Scholar]
- 191. Briasoulis A, Mallikethi-Reddy S, Palla M, Alesh I, Afonso L.. Myocardial fibrosis on cardiac magnetic resonance and cardiac outcomes in hypertrophic cardiomyopathy: a meta-analysis. Heart 2015;101:1406–1411. [DOI] [PubMed] [Google Scholar]
- 192. Chan RH, Maron BJ, Olivotto I, Pencina MJ, Assenza GE, Haas T, Lesser JR, Gruner C, Crean AM, Rakowski H, Udelson JE, Rowin E, Lombardi M, Cecchi F, Tomberli B, Spirito P, Formisano F, Biagini E, Rapezzi C, De Cecco CN, Autore C, Cook EF, Hong SN, Gibson CM, Manning WJ, Appelbaum E, Maron MS.. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation 2014;130:484–495. [DOI] [PubMed] [Google Scholar]
- 193. Ariga R, Tunnicliffe EM, Manohar SG, Mahmod M, Raman B, Piechnik SK, Francis JM, Robson MD, Neubauer S, Watkins H.. Identification of myocardial disarray in patients with hypertrophic cardiomyopathy and ventricular arrhythmias. J Am Coll Cardiol 2019;73:2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Ripplinger CM, Li W, Hadley J, Chen J, Rothenberg F, Lombardi R, Wickline SA, Marian AJ, Efimov IR.. Enhanced transmural fiber rotation and connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res 2007;101:1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Bezzina CR, Remme CA.. Dilated cardiomyopathy due to sodium channel dysfunction: what is the connection? Circ Arrhythm Electrophysiol 2008;1:80–82. [DOI] [PubMed] [Google Scholar]
- 196. Moreau A, Gosselin-Badaroudine P, Boutjdir M, Chahine M.. Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front Pharmacol 2015;6:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, Brink M, Chahine M.. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One 2012;7:e38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ge J, Sun A, Paajanen V, Wang S, Su C, Yang Z, Li Y, Wang S, Jia J, Wang K, Zou Y, Gao L, Wang K, Fan Z.. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol 2008;1:83–92. [DOI] [PubMed] [Google Scholar]
- 199. Kyle JW, Makielski JC.. Diseases caused by mutations in Nav1.5 interacting proteins. Card Electrophysiol Clin 2014;6:797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Marionneau C, Abriel H.. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J Mol Cell Cardiol 2015;82:36–47. [DOI] [PubMed] [Google Scholar]
- 201. El Refaey M, Musa H, Murphy NP, Lubbers ER, Skaf M, Han M, Cavus O, Koenig SN, Wallace MJ, Gratz D, Bradley E, Alsina KM, Wehrens XHT, Hund TJ, Mohler PJ.. Protein phosphatase 2A regulates cardiac Na(+) channels. Circ Res 2019;124:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschenes I.. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 2017;8:2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. van Bemmelen MX, Rougier JS, Gavillet B, Apotheloz F, Daidie D, Tateyama M, Rivolta I, Thomas MA, Kass RS, Staub O, Abriel H.. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res 2004;95:284–291. [DOI] [PubMed] [Google Scholar]
- 204. Valdivia CR, Ueda K, Ackerman MJ, Makielski JC.. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 2009;297:H1446–H1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC.. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J Mol Cell Cardiol 2013;61:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Nattel S, Maguy A, Le Bouter S, Yeh YH.. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 2007;87:425–456. [DOI] [PubMed] [Google Scholar]
- 207. Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF.. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem 2001;276:28197–28203. [DOI] [PubMed] [Google Scholar]
- 208. Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC.. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 2005;38:475–483. [DOI] [PubMed] [Google Scholar]
- 209. Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI.. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol 2004;37:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Ke HY, Yang HY, Francis AJ, Collins TP, Surendran H, Alvarez-Laviada A, Firth JM, MacLeod KT.. Changes in cellular Ca(2+) and Na(+) regulation during the progression towards heart failure in the guinea pig. J Physiol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Greer-Short A, Musa H, Alsina KM, Ni L, Word TA, Reynolds JO, Gratz D, Lane C, El-Refaey M, Unudurthi S, Skaf M, Li N, Fedorov VV, Wehrens XHT, Mohler PJ, Hund TJ.. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Rivaud MR, Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Rothenberg E, Bezzina CR, Delmar M, Remme CA.. Sodium channel remodeling in subcellular microdomains of murine failing cardiomyocytes. J Am Heart Assoc 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Noyes AM, Zhou A, Gao G, Gu L, Day S, Andrew Wasserstrom J, Dudley SC.. Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int J Cardiol 2017;249:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Coppini R, Mazzoni L, Ferrantini C, Gentile F, Pioner JM, Laurino A, Santini L, Bargelli V, Rotellini M, Bartolucci G, Crocini C, Sacconi L, Tesi C, Belardinelli L, Tardiff J,, Mugelli A, Olivotto I, Cerbai E, Poggesi C.. Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ Heart Fail 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Olivotto I, Camici PG, Merlini PA, Rapezzi C, Patten M, Climent V, Sinagra G, Tomberli B, Marin F, Ehlermann P, Maier LS, Fornaro A, Jacobshagen C, Ganau A, Moretti L, Hernandez Madrid A, Coppini R, Reggiardo G, Poggesi C, Fattirolli F, Belardinelli L, Gensini G, Mugelli A.. Efficacy of ranolazine in patients with symptomatic hypertrophic cardiomyopathy: the RESTYLE-HCM randomized, double-blind, placebo-controlled study. Circ Heart Fail 2018;11:e004124. [DOI] [PubMed] [Google Scholar]
- 216. Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M.. Interactions between ankyrin-G, plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res 2011;109:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Kranias EG, Bers DM.. Calcium and cardiomyopathies. Subcell Biochem 2007;45:523–537. [DOI] [PubMed] [Google Scholar]
- 218. Hamilton S, Terentyev D.. Proarrhythmic remodeling of calcium homeostasis in cardiac disease; implications for diabetes and obesity. Front Physiol 2018;9:1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Viola H, Johnstone V, Cserne Szappanos H, Richman T, Tsoutsman T, Filipovska A, Semsarian C, Hool L.. The L-type Ca(2+) channel facilitates abnormal metabolic activity in the cTnI-G203S mouse model of hypertrophic cardiomyopathy. J Physiol 2016;594:4051–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, Valdivia HH, Day SM.. Genotype-dependent and -independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation 2016;134:1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Fu Y, Westenbroek RE, Scheuer T, Catterall WA.. Basal and beta-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc Natl Acad Sci U S A 2014;111:16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Goldhaber JI, Qayyum MS.. Oxygen free radicals and excitation-contraction coupling. Antioxid Redox Signal 2000;2:55–64. [DOI] [PubMed] [Google Scholar]
- 223. Ather S, Wang W, Wang Q, Li N, Anderson ME, Wehrens XH.. Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm 2013;10:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Liu GS, Morales A, Vafiadaki E, Lam CK, Cai WF, Haghighi K, Adly G, Hershberger RE, Kranias EG.. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res 2015;107:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wang Q, Quick AP, Cao S, Reynolds J, Chiang DY, Beavers D, Li N, Wang G, Rodney GG, Anderson ME, Wehrens XHT.. Oxidized CaMKII (Ca(2+)/calmodulin-dependent protein kinase II) is essential for ventricular arrhythmia in a mouse model of Duchenne muscular dystrophy. Circ Arrhythm Electrophysiol 2018;11:e005682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ.. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 2012;126:2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A.. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013;127:575–584. [DOI] [PubMed] [Google Scholar]
- 228. Okuda S, Sufu-Shimizu Y, Kato T, Fukuda M, Nishimura S, Oda T, Kobayashi S, Yamamoto T, Morimoto S, Yano M.. CaMKII-mediated phosphorylation of RyR2 plays a crucial role in aberrant Ca(2+) release as an arrhythmogenic substrate in cardiac troponin T-related familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun 2018;496:1250–1256. [DOI] [PubMed] [Google Scholar]
- 229. Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kuhn B.. Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech 2013;6:1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ.. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail 2011;4:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Xiao H, Wang M, Du Y, Yuan J, Cheng X, Chen Z, Zou A, Wei F, Zhao G, Liao YH.. Arrhythmogenic autoantibodies against calcium channel lead to sudden death in idiopathic dilated cardiomyopathy. Eur J Heart Fail 2011;13:264–270. [DOI] [PubMed] [Google Scholar]
- 232. Yu H, Pei J, Liu X, Chen J, Li X, Zhang Y, Li N, Wang Z, Zhang P, Cao K, Pu J.. Calcium channel autoantibodies predicted sudden cardiac death and all-cause mortality in patients with ischemic and nonischemic chronic heart failure. Dis Markers 2014;2014:796075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Qu YS, Lazzerini PE, Capecchi PL, Laghi-Pasini F, El Sherif N, Boutjdir M.. Autoimmune calcium channelopathies and cardiac electrical abnormalities. Front Cardiovasc Med 2019;6:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Lazzerini PE, Capecchi PL, Guideri F, Bellisai F, Selvi E, Acampa M, Costa A, Maggio R, Garcia-Gonzalez E, Bisogno S, Morozzi G, Galeazzi M, Laghi-Pasini F.. Comparison of frequency of complex ventricular arrhythmias in patients with positive versus negative anti-Ro/SSA and connective tissue disease. Am J Cardiol 2007;100:1029–1034. [DOI] [PubMed] [Google Scholar]
- 235. Huang CL. Murine electrophysiological models of cardiac arrhythmogenesis. Physiol Rev 2017;97:283–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Xiong Q, Cao L, Hu J, Marian AJ, Hong K.. A rare loss-of-function SCN5A variant is associated with lidocaine-induced ventricular fibrillation. Pharmacogenomics J 2014;14:372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Li J, Seyler C, Wiedmann F, Schmidt C, Schweizer PA, Becker R, Katus HA, Thomas D.. Anti-KCNQ1 K(+) channel autoantibodies increase IKs current and are associated with QT interval shortening in dilated cardiomyopathy. Cardiovasc Res 2013;98:496–503. [DOI] [PubMed] [Google Scholar]
- 238. Algalarrondo V, Nattel S.. Potassium channel remodeling in heart disease. Card Electrophysiol Clin 2016;8:337–347. [DOI] [PubMed] [Google Scholar]
- 239. Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res 2004;61:208–217. [DOI] [PubMed] [Google Scholar]
- 240. Beuckelmann DJ, Nabauer M, Erdmann E.. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 241. Li GR, Lau CP, Leung TK, Nattel S.. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 2004;1:460–468. [DOI] [PubMed] [Google Scholar]
- 242. Koumi S, Backer CL, Arentzen CE.. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation 1995;92:164–174. [DOI] [PubMed] [Google Scholar]
- 243. Parajuli N, Valtuille L, Basu R, Famulski KS, Halloran PF, Sergi C, Oudit GY.. Determinants of ventricular arrhythmias in human explanted hearts with dilated cardiomyopathy. Eur J Clin Invest 2015;45:1286–1296. [DOI] [PubMed] [Google Scholar]
- 244. Hueneke R, Adenwala A, Mellor RL, Seidman JG, Seidman CE, Nerbonne JM.. Early remodeling of repolarizing K(+) currents in the alphaMHC(403/+) mouse model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 2017;103:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. El-Battrawy I, Zhao Z, Lan H, Li X, Yucel G, Lang S, Sattler K, Schunemann JD, Zimmermann WH, Cyganek L, Utikal J, Wieland T, Bieback K, Bauer R, Ratte A, Pribe-Wolferts R, Rapti K, Nowak D, Wittig J, Thomas D, Most P, Katus HA, Ravens U, Schmidt C, Borggrefe M, Zhou XB, Muller OJ, Akin I.. Ion channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circ Genom Precis Med 2018;11:e001893. [DOI] [PubMed] [Google Scholar]
- 246. Ma D, Liu Z, Loh LJ, Zhao Y, Li G, Liew R, Islam O, Wu J, Chung YY, Teo WS, Ching CK, Tan BY, Chong D, Ho KL, Lim P, Yong RYY, Panama BK, Kaplan AD, Bett GCL, Ware J, Bezzina CR, Verkerk AO, Cook SA, Rasmusson RL, Wei H.. Identification of an INa-dependent and Ito-mediated proarrhythmic mechanism in cardiomyocytes derived from pluripotent stem cells of a Brugada syndrome patient. Sci Rep 2018;8:11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J Physiol 2009;587:2163–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Clemo HF, Stambler BS, Baumgarten CM.. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res 1999;84:157–165. [DOI] [PubMed] [Google Scholar]
- 249. Desplantez T. Cardiac Cx43, Cx40 and Cx45 co-assembling: involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell Biol 2017;18:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Sohl G, Willecke K.. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes 2003;10:173–180. [DOI] [PubMed] [Google Scholar]
- 251. Ribeiro-Rodrigues TM, Martins-Marques T,, Morel S, Kwak BR, Girao H.. Role of connexin 43 in different forms of intercellular communication – gap junctions, extracellular vesicles and tunnelling nanotubes. J Cell Sci 2017;130:3619–3630. [DOI] [PubMed] [Google Scholar]
- 252. Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV.. Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol 2009;47:23–31. [DOI] [PubMed] [Google Scholar]
- 253. Vozzi C, Dupont E, Coppen SR, Yeh HI, Severs NJ.. Chamber-related differences in connexin expression in the human heart. J Mol Cell Cardiol 1999;31:991–1003. [DOI] [PubMed] [Google Scholar]
- 254. Aasen T, Johnstone S, Vidal-Brime L, Lynn KS, Koval M.. Connexins: synthesis, post-translational modifications, and trafficking in health and disease. Int J Mol Sci 2018;19. pii: E1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Alcolea S, Theveniau-Ruissy M, Jarry-Guichard T, Marics I, Tzouanacou E, Chauvin JP, Briand JP, Moorman AF, Lamers WH, Gros DB.. Downregulation of connexin 45 gene products during mouse heart development. Circ Res 1999;84:1365–1379. [DOI] [PubMed] [Google Scholar]
- 256. Delmar M, Makita N.. Cardiac connexins, mutations and arrhythmias. Curr Opin Cardiol 2012;27:236–241. [DOI] [PubMed] [Google Scholar]
- 257. Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE.. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med 2009;360:1075–1084. [DOI] [PubMed] [Google Scholar]
- 258. Tandri H, Asimaki A, Dalal D, Saffitz JE, Halushka MK, Calkins H.. Gap junction remodeling in a case of arrhythmogenic right ventricular dysplasia due to plakophilin-2 mutation. J Cardiovasc Electrophysiol 2008;19:1212–1214. [DOI] [PubMed] [Google Scholar]
- 259. Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE.. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004;1:3–11. [DOI] [PubMed] [Google Scholar]
- 260. Salameh A, Krautblatter S, Karl S, Blanke K, Gomez DR, Dhein S, Pfeiffer D, Janousek J.. The signal transduction cascade regulating the expression of the gap junction protein connexin43 by beta-adrenoceptors. Br J Pharmacol 2009;158:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Sanbe A, James J, Tuzcu V, Nas S, Martin L, Gulick J, Osinska H, Sakthivel S, Klevitsky R, Ginsburg KS, Bers DM, Zinman B, Lakatta EG, Robbins J.. Transgenic rabbit model for human troponin I-based hypertrophic cardiomyopathy. Circulation 2005;111:2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Le Dour C, Macquart C, Sera F, Homma S, Bonne G, Morrow JP, Worman HJ, Muchir A. Decreased WNT/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum Mol Genet 2017;26:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Gourdie RG. The cardiac gap junction has discrete functions in electrotonic and ephaptic coupling. Anat Rec (Hoboken )2019;302:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Chaldoupi SM, Loh P, Hauer RN, de Bakker JM, van Rijen HV.. The role of connexin40 in atrial fibrillation. Cardiovasc Res 2009;84:15–23. [DOI] [PubMed] [Google Scholar]
- 265. Philips B, te Riele AS, Sawant A, Kareddy V, James CA, Murray B, Tichnell C, Kassamali B, Nazarian S, Judge DP, Calkins H, Tandri H.. Outcomes and ventricular tachycardia recurrence characteristics after epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm 2015;12:716–725. [DOI] [PubMed] [Google Scholar]
- 266. Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NA III, Shannon KM, Ashley EA, Day SM, Pacileo G, Formisano F, Devoto E,, Anastasakis A, Bos JM,, Woo A, Autore C, Pass RH, Boriani G, Garberich RF, Almquist AK, Russell MW, Boni L, Berger S,, Maron MS, Link MS.. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:1527–1535. [DOI] [PubMed] [Google Scholar]
- 267. Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA III, Spirito P.. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 2000;342:365–373. [DOI] [PubMed] [Google Scholar]