

Abstract
The inner-centromere is a region on every mitotic chromosome that enables specific biochemical reactions, which underlie properties such as the maintenance of cohesion, the regulation kinetochores and the assembly of specialized chromatin that can resist microtubule pulling forces. The Chromosomal Passenger Complex (CPC) is abundantly localized to inner centromeres and it is unclear if it contains non-kinase activities that can contributes to generation of these unique chromatin properties. We find that the Borealin subunit of the CPC drives its phase-separation in vitro at concentrations that are below those found on the inner-centromere. We also provide strong evidence that the CPC exists in a phase-separated state at the inner-centromere. CPC phase-separation is required for its inner-centromere localization and function during mitosis. We suggest the CPC combines phase-separation, kinase, and histone code reading activities to enable the formation of a chromatin body with unique biochemical activities at the inner-centromere.
Introduction:
The chromatin region between sister centromeres, the inner-centromere, is the region were cohesion is retained until anaphase and it serves as the platform for mitotic signaling1. The inner-centromere also has to be uniquely organized in order to withstand the pulling forces generated at kinetochores during mitosis2,3. The emergence of such properties requires compartmentalization of multiple biochemical reactions to this chromatin region. How such compartmentalization is achieved is an important unanswered question.
Phase-separation, is an emergent property of multivalent scaffold proteins that are present at high local concentration and display liquid-like behavior. Phase-separation underlies formation of membrane-less organelles capable of compartmentalizing various biochemical processes in both cytoplasm and interphase nuclei4,5–7. We hypothesized that phase-separation underlies compartmentalization at the inner-centromeres. The chromosomal passenger complex (CPC), which contains kinase Aurora-B, and at least three other subunits-INCENP, Survivin, and Borealin, is an abundant component of the inner-centromere (~10 μM)8 and regulates most of its known functions1.
Mitotic signaling is initiated by the concentration of the CPC on inner-centromeric chromatin or at midzone, which enables autoactivation of Aurora-B kinase9–11. During mitosis the CPC is recruited to the inner-centromere by two orthogonal histone marks H3T3ph and H2AT120ph12. Survivin directly binds H3T3ph and Borealin indirectly binds H2AT120ph through its interaction with Sgo113–16,17,18. However, it is unclear how the histone reading subunit Survivin could be an anchor since it turns over at inner-centromeres ~5 times faster than the Aurora-B, INCENP, and Borealin subunits19,20. Furthermore, it is unclear how the weak (~2-4 μM) affinity between Survivin and H3T3ph can stably anchor CPC17,18. While this concern is somewhat alleviated by the involvement of a second histone mark, it is unclear how Sgo1 could anchor most of the CPC since its concentration is ~5 times lower than CPC subunits21.
The kinase activity of Aurora-B has a well-appreciated role in regulating inner-centromere1,22,23. Recent evidence suggests that non-kinase subunits of the CPC protect cohesion at inner-centromeres independent of their role in localizing the Aurora-B24. How the non-kinase subunits of the CPC exert these effects on biophysical organization of the inner-centromere is unclear. Here, we demonstrate that the inner-centromere targeting region of the CPC can phase-separate in vitro and provide strong evidence that the CPC at the inner-centromere exist in a phase-separated state. We also demonstrate that the CPC’s ability to undergo phase-separation is important for its inner-centromere localization and function. We provide a plausible model that explains how combining kinase, histone code reading and phase-separation activities in a single complex enables the formation of an inner-centromere body at a single location of every chromosome at the beginning of mitosis.
Results:
The centromere targeting subunits of the CPC phase-separate in vitro
We tested whether subunits of the CPC could phase-separate in vitro. We expressed Survivin, Borealin, and the N-terminal 58 amino acids of INCENP (ISB) in E. coli (Fig.1a, Supplementary Fig.1a, b). This set of proteins has all the activities required to localize the CPC to the inner-centromere25. We observed spontaneous ISB phase-separation under conditions of high protein concentration, low salt concentration or the presence of the molecular crowding agent PEG-3350 (Fig.1b, c, d). Coacervates formed with GFP-ISB were fluorescent, demonstrating they are composed of CPC proteins (Fig.1d). The homologous complex from Xenopus laevis also phase-separated (Supplementary Fig.2a) demonstrating evolutionary conservation of this property.
Figure 1: The centromere-targeting region of the CPC phase-separates in vitro under physiological conditions.
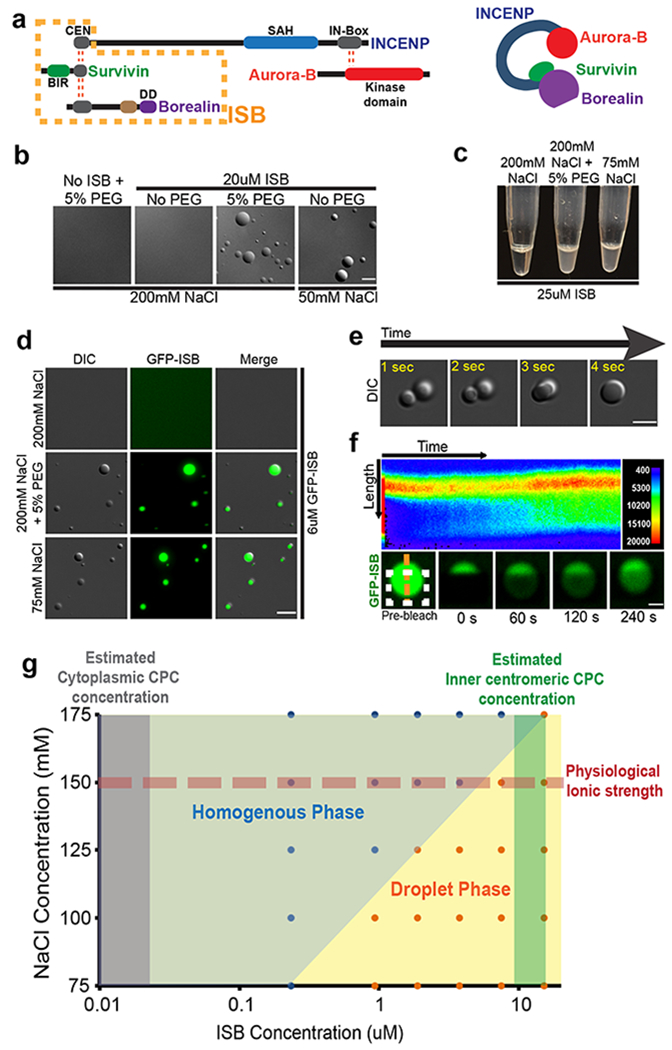
(a) Schematic of the CPC showing various domains in the CPC subunits, where DD is dimerization domain. Red dotted lines indicate interactions between subunits. Orange dotted box indicates the INCENP1-58-Survivin-Borealin (ISB) region used for biochemical analysis in this study. (b) DIC micrographs of the ISB coacervates under indicated conditions. (c) Turbidity generated by the phase-separation of the ISB complex under indicated conditions. (d) The phase-separated droplets of the GFP-ISB complex contain fluorescence. (e) Fusion of ISB coacervates as visualized by time-lapse imaging. (f) FRAP analysis of GFP-ISB coacervates. GFP- INCENP1-58 was photo bleached in the ISB coacervates (white dotted box indicates the bleached area) and recovery of fluorescence was monitored. (Top) Pseudo-colored kymograph of the fluorescent intensity at the orange dotted line (shown in the bottom) of the FRAP experiment. Color corresponds to the fluorescent intensity as shown on the top right. (Bottom) Time-lapse images from the FRAP experiment. Experiment repeated independently 3 times for b-f. (g) Phase diagram of ISB phase-separation as a function of NaCl and ISB concentration. Red dotted line indicates conditions with physiological ionic strength. Blue (homogenous phase) and orange (droplet phase) filled circles show the actual conditions sampled in the experiment. Grey shaded region indicates the estimated cytoplasmic CPC concentration and dark green shaded region indicates estimated centromeric CPC concentration. Scale bar in b and d-f is 5 μm. Source data for g are shown in Supplementary Table 2.
The ISB coacervates display liquid-like properties that are similar to those observed in phase-separating proteins4–6,26. They are highly circular in shape, the larger coacervates undergo shear upon placement of a coverslip, and the coacervates undergo fusion at early time points after induction of phase-separation (Fig.1e, Supplementary Fig.2b, c). Some proteins that phase-separate maintain their liquid-like state whereas others gelate over time4. We hypothesized that ISB formed a gel since no fusion events were seen (as in Fig.1e) after ~1 minute of inducing phase-separation and we observed coacervates that appeared to be arrested mid-fusion (Supplementary Fig.2j, k, l). We tested the material property of the ISB droplet by photo-bleaching a sub-region of GFP-ISB coacervate and saw little internal rearrangement over minute’s time scale. We conclude that ISB coacervates quickly (~1 min) mature to have gel-like properties (Fig.1f).
The size of ISB coacervates were dependent upon concentrations of salt, ISB or molecular crowding agent (Supplementary Fig.2 d–i). These data suggest that charge-based intermolecular interactions between ISB molecules underlie coacervate formation. Interestingly, the ISB phase diagram predicts that the CPC would exist in a phase-separated state at concentrations that are estimated at the inner-centromeres (10 μM), whereas it would exist in a homogeneous state at cytoplasmic CPC concentrations (0.01 μM)8 under physiological conditions (Fig.1g).
The dynamics of CPC subunits are similar in coacervates and at the inner-centromere
To determine whether the ISB in the droplets exchanged with ISB in the surrounding buffer we performed Fluorescence Recovery After Photobleaching (FRAP) of GFP-ISB droplets in vitro. We observed a slow exchange of ISB protein (t1/2-99.71 s, 50.86% mobile fraction) (Fig.2a, b; Supplementary Fig.2m). Interestingly, these exchange dynamics are similar to what has been measured for INCENP at the inner-centromeres in vivo (t1/2 83.2±33.5 s, ~55-60% mobile fraction)27. We also compared the diffusion rates of CPC components inside coacervates in vitro and at inner-centromeres using Fluorescence Correlation Spectroscopy (FCS). GFP-Borealin dynamics in both the cytoplasm and the inner-centromere is best described by two populations fitting model (Dslow=0.1-1 μm2/s, Dfast =10-15 μm2/s) as previously reported (Supplemental Fig.3a–e) 28,29. The slow diffusing population (Dslow) was previously interpreted as the diffusion of Borealin as part of the CPC complex. The CPC in the inner-centromere diffused slower that the population in the cytoplasm of mitotic cells (Fig.2b, c). Interestingly, CPC in the inner-centromere (Dslow=0.1-0.5 μm2/s) had a similar diffusion rate to that measured for GFP-INCENP inside the ISB droplet in vitro (D=0.05-0.3 μm2/s, Fig.2d,e Supplementary Fig.3f). ISB outside the droplet diffused much faster than inside the droplet (Fig.2e), demonstrating that phase-separation changes the hydrodynamic properties of the protein. ISB outside the coacervates diffused faster than the CPC in the cytoplasm (Fig.2c, e), presumably because of the more crowded environment of the cytoplasm. Strong correlation between both FRAP and FCS data, in vitro and in vivo, demonstrates that coacervate formation may explain the reduced dynamics of the CPC components measured at inner-centromeres, and suggests the CPC exists as a coacervate inner-centromeres.
Figure 2. The dynamics of the CPC subunits in coacervates are similar to those measured in the inner-centromere.
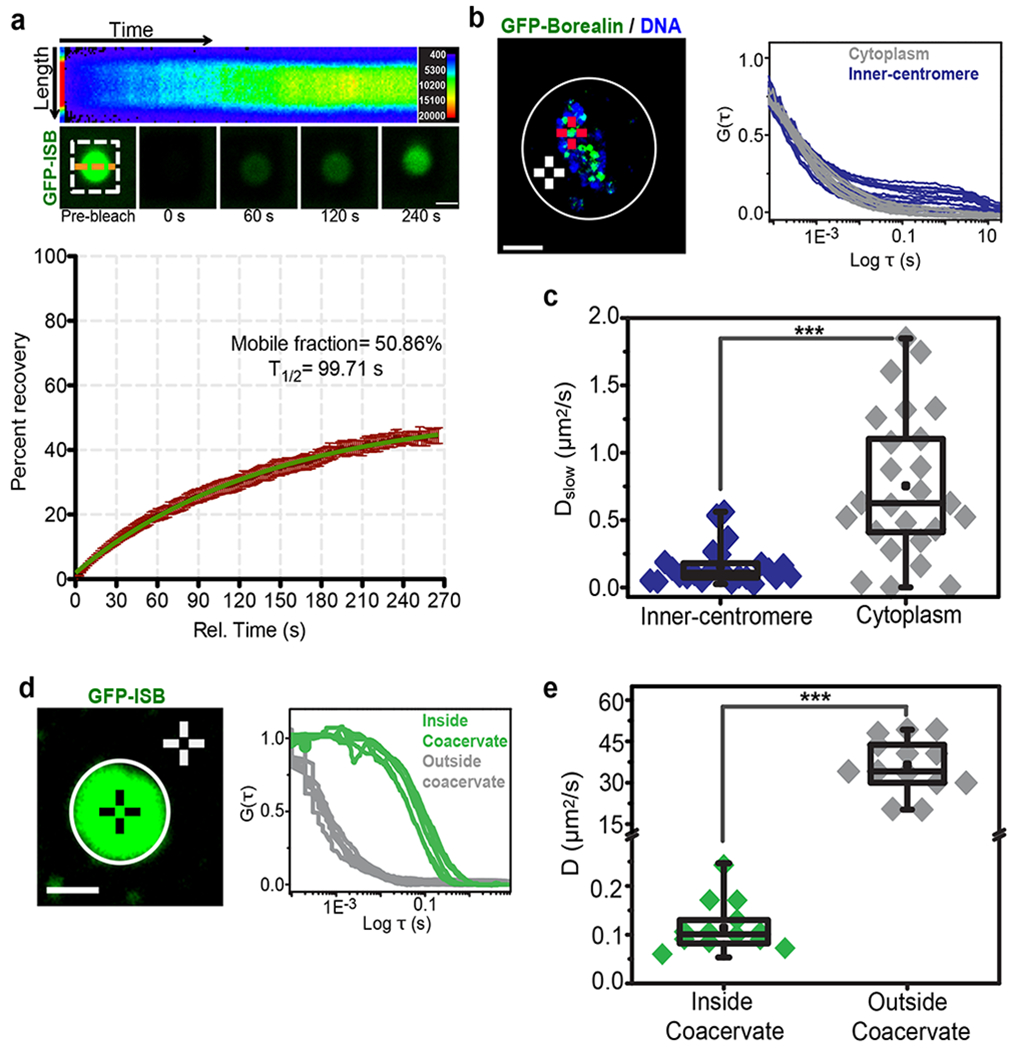
(a) FRAP analysis of GFP-ISB coacervates. GFP- INCENP1-58 was photo-bleached in the ISB coacervates and fluorescence recovery was monitored (white dotted box indicates the bleached area). (Top) Pseudo-colored kymograph of the fluorescent intensity at the orange dotted line of the FRAP experiment. Pseudo-color in the kymograph corresponds to the fluorescent intensity as shown in the key on top right (3 independent repeats). (Bottom) Time-lapse images from the FRAP experiment (bottom) Graph of fluorescence recovery after photo bleaching inside the droplet over time showing mean and SEM at each time point (n=14 coacervates, combined data from 3 independent repeats). Green line indicates curve fitted with one phase association kinetics equation. Mobile fraction is 50.86% with 95% confidence interval ranging from 48.89% to 52.82%. T1/2 is 99.71 s with 95% confidence interval ranging from 90.13 to 111.6 s. (b) (Left) Image of a mitotic cell expressing GFP-Borealin treated with 3.3 μM nocodazole. Red crosshair and white crosshair are examples of the inner-centromeric and cytoplasmic region selected for FCS measurement respectively (3 independent repeats). Scale bar 10 μm. (Right) Normalized FCS curves of GFP-Borealin measurements at inner-centromere (Blue) or in the cytoplasm (Grey). (c) Box and whisker graph of GFP-Borealin’s diffusion rate as part of CPC (Dslow) measured at inner-centromere (n=25 inner-centromeres) or cytoplasm (n=25 spots in cytoplasm) calculated form graph shown in b, combined data from 3 independent repeats, P=3.34E-06. (d) (Left) Image of GFP-ISB coacervate, black crosshair and white crosshair are examples of region selected for FCS measurements inside coacervates or outside coacervates respectively (3 independent repeats). Scale bar is 1 μm. (Right) Normalized FCS curves of GFP-ISB measurements inside the coacervates (Green) or outside the coacervates (Grey). (e) Box and whisker graph of GFP-ISB diffusion rate inside the coacervates (n=13 coacervates) or outside the coacervates (n=14 spots outside coacervates), combined data from 3 independent repeats. For statistical analysis two-tailed T-test was applied, P= 1.54E-08. ***P<0.001, **P<0.01, *P<0.05 and ns P>0.05. All box and whisker graphs represent the median (central line), 25th-75th percentile (bounds of the box), and 5th-95th percentile (whiskers). Source data for a-e are shown in Supplementary Table 2.
ISB coacervates enrich inner-centromere components
The CPC is recruited to inner-centromeres by multivalent interactions with at least three proteins: Histone H3pT3, Sgo1 and HP1α. The interactions that drive ISB coacervation may block the binding surfaces for other inner-centromere proteins. To test whether Survivin in coacervates still bind histone tails we mixed fluorescent H3T3ph peptides with preformed ISB coacervates and observed the partition of these peptides. H3T3ph peptides were enriched in coacervates more than unphosphorylated peptides. Coacervates generated with ISB containing a Survivin H3pT3 binding mutation (H80A), did not enrich H3pT3 over H3 peptides (Fig.3a, Supplementary Fig.4d). Thus, Survivin retains the ability to “read” the histone tails within coacervates. A key heterochromatin regulator, HP1α, controls CPC activity and localization in mitosis30,31,32. We mixed CF-555 labeled HP1α with preformed ISB coacervates and observed robust enrichment of HP1α into ISB coacervates (Fig.3b). We employed conditions where HP1α is unable to phase-separate on its own. Sgo1 interacts with the Borealin subunit of the CPC through its N-terminus and drives its localization to the inner-centromere 16. We observed strong enrichment of CF-555-hSgo11-112 in the ISB coacervates (Fig.3c). In contrast, molecules that either don’t interact with the CPC or are not inner-centromere components such as GFP, GFP-Mad2, or Cy3-azide showed marginal or no enrichment in the ISB phase (Fig.3g, Supplementary Fig.4b, c). Thus, a number of inner-centromere regulators robustly interact with ISB in the droplet phase.
Figure 3: Inner-centromere components are enriched in ISB coacervates.
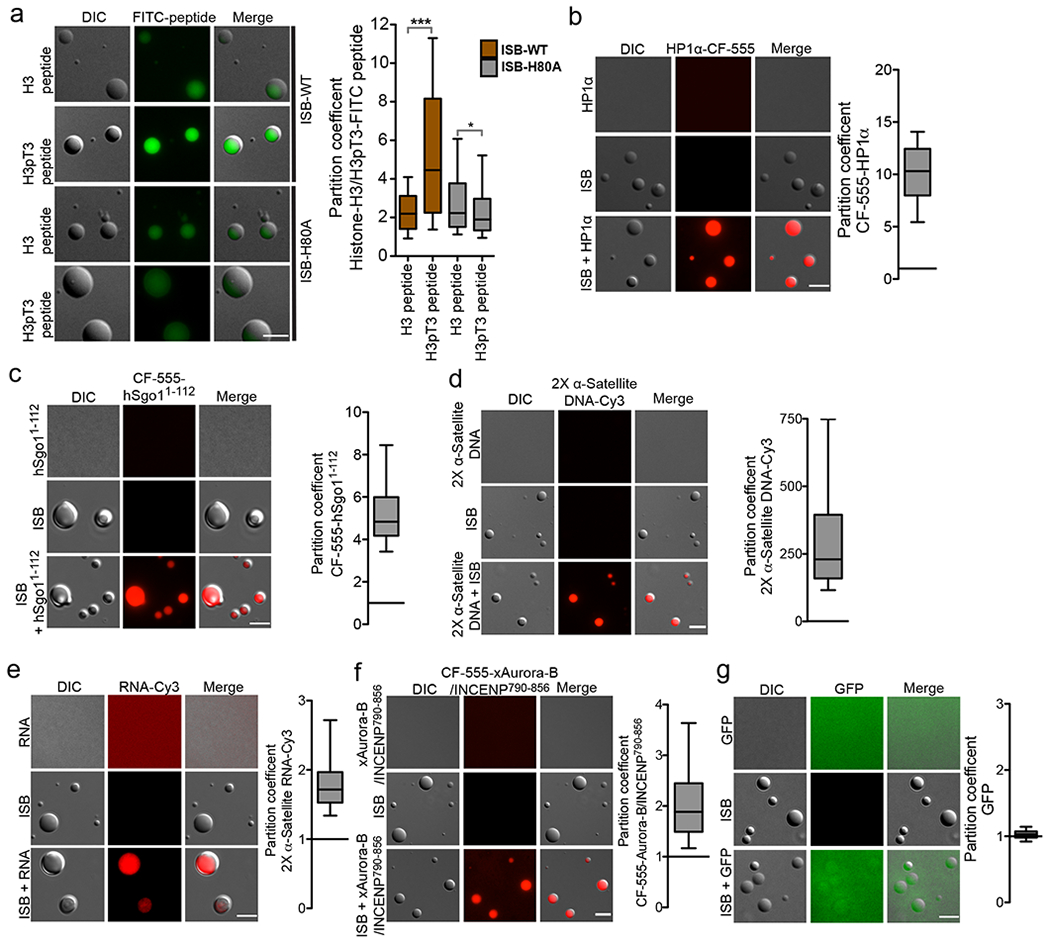
a-g ISB coacervates were mixed with the indicated fluorescent inner-centromere constituents and images for enrichment. (a) Representative micrographs showing enrichment of FITC-histone H3 peptide or FITC-histone H3pT3 peptide in ISB-WT or ISB-H80A mutant coacervates. Control micrographs for FITC peptide alone and ISB-WT or ISB-H80A mutant coacervates alone are shown in Supplementary fig.4d. Graph shows partition coefficient of the H3 and H3pT3 peptide in ISB-WT or ISB-H80A mutant coacervates (82, 63, 76 and 95 coacervates were analyzed for ISB-WT+H3, ISB-WT+H3pT3, ISB-H80A+H3 and ISB-H80A+H3pT3 respectively). For statistical analysis two tailed Mann-Whitney test was applied, ***P<0.0001 and * indicates P=0.0498. (b) Purified HP1 protein was labeled with CF555 (CF555-HP1α), incubated with ISB coacervates and imaged. Representative micrograph and graph of partition coefficient (n=171 coacervates), (c) The partition coefficient of CF555-hSgo11-112 was measured as in B (n=129 coacervates), (d) 2X α-satellite DNA-Cy3 (n=193 coacervates), (e) 2X α-satellite RNA-Cy3 (n=129 coacervates), (f) CF555-xAurora-B/INCENP790-856 (n=200 coacervates), and (g) GFP (n=133 coacervates) in ISB phase. For a-g, combined data from 3 independent experiments. Scale bar is 5 μm. All box and whisker graphs represent the median (central line), 25th-75th percentile (bounds of the box), and 5th-95th percentile (whiskers). Source data for c-g are shown in Supplementary Table 2.
Another subassembly of the CPC, xAurora-B/xINCENP790-847 also partitioned into the ISB coacervates (Fig.3f), confirming previous results33,34. This observation provides evidence that the current models of autoactivation are compatible with phase-separation, since the kinase was not excluded from the droplet. Nucleic acids (α-satellite RNA and DNA) were enriched in ISB coacervates (Fig.3 d, e; Supplementary Fig.4a). Surprisingly, coacervates also bound mononucleosomes independent of T3 phosphorylation, demonstrating that this simple assay can be used to identify new CPC activities.
α/β-tubulin dimers were enriched in ISB coacervates (Supplementary Fig.3e). Surprisingly, ISB coacervates nucleated microtubules at concentrations of α/β-tubulin dimers that are 20-fold below the critical concentration (Supplementary Fig.3f), similar to what has been observed for C. elegans centrosome proteins26. No microtubule nucleation was seen in absence of ISB or GTP (Supplementary Fig.3f). Interestingly, the CPC regulates microtubule formation near chromosomes, although this has been attributed to its ability to inhibit the microtubule depolymerase MCAK35.
Inner-centromere components enhance ISB phase-separation
ISB spontaneously phase-separates in physiological conditions at ~6 μM concentrations, which is lower than that estimated to be present at the inner-centromere (10 μM) (Fig.1g). However, the presence of other inner-centromere components or mitotic post-translational modifications might promote or inhibit phase-separation. We observed that α-satellite DNA, histone H3.3 polynucleosomes, Histone H3.1 mononucleosomes, H3T3ph mononucleosomes and microtubule bundles all induce phase-separation under conditions where ISB exists in a homogeneous phase on its own (Fig.4a–c). Thus, a number of inner-centromere components promote the ISB phase-separation.
Figure 4: Inner-centromere components can induce ISB phase-separation.
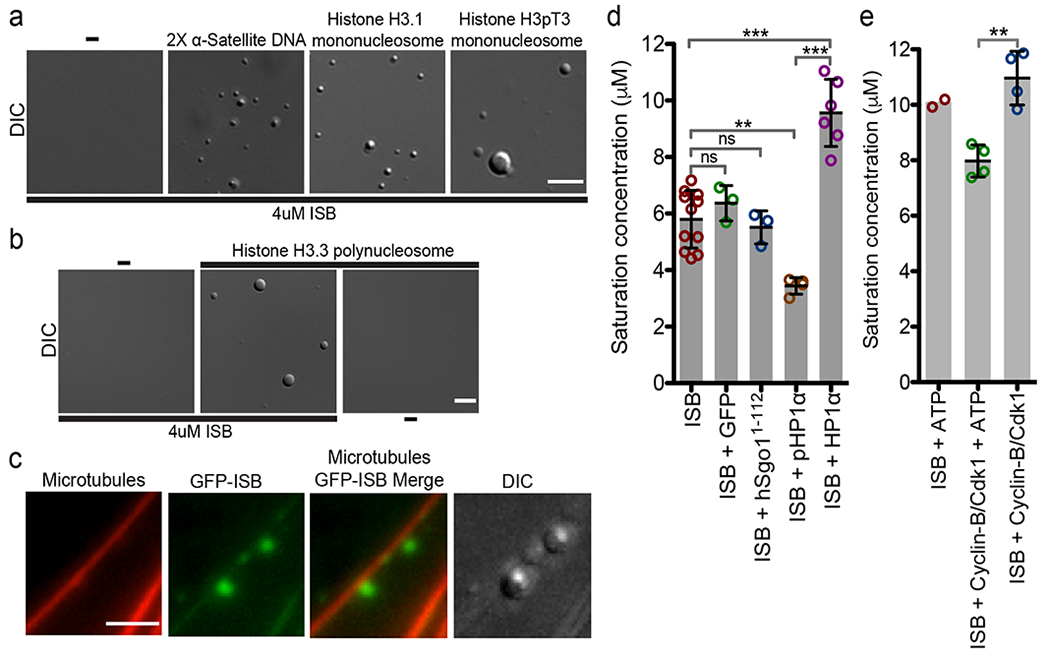
(a) Micrographs showing phase-separation of 4 μM ISBWT induced by 13 ng/ul of 2X α-satellite, Histone H3.1 and H3pT3 mono-nucleosomes in buffer containing 150 mM NaCl. Scale bar is 5 μm. (b) Micrographs showing phase-separation of 4uM ISBWT induced by 80 ng/ul Histone H3.3 poly-nucleosomes in buffer containing 150 mM NaCl. Scale bar is 5 μm. (c) Micrographs showing phase-separation of GFP-ISB (5 μM) induced on paclitaxel-stabilized rhodamine labeled microtubules (1μM) in buffer containing 150 mM NaCl. Scale bar is 3 μm. For a-c the experiment was repeat independently 3 times. (d) Mitotically phosphorylated HP1α lowers the saturation concentration of ISB. Saturation concentrations of ISB (n=11) or when its mixed with equimolar amounts of GFP (n=3), hSgo11-112 (n=3), pHP1α (n=3), and HP1α (n=6) in buffer containing 150 mM NaCl measured by spin-down method. (e) CDK phosphorylation lowers saturation concentration of ISB. Saturation concentration of ISB upon incubation with ATP (n=2), Cyclin-B/Cdk1 and ATP (n=4), and Cyclin-B/Cdk1 (n=4) in kinase buffer containing 150 mM NaCl measure by spin-down method. For d and e, “n” stands for the number of independent experiments. For statistical analysis in d and e, One-way Analysis of Variance followed by Bonferroni’s Multiple Comparison Test was used. ***P<0.001, **P<0.01, *P<0.05 and ns P>0.05. All Bar graphs show mean (central dark line) and error bars are ±s.d.; Open circles show the individual data points. Source data for d and e are shown in Supplementary Table 2.
HP1α can undergo phase-separation and is a regulator of the CPC in the inner-centromere. HP1α is phosphorylated on both its N-terminus and hinge region during mitosis36. We measured the saturation concentration of the ISB in the presence and absence of HP1a by measuring the concentration of the ISB present in the homogenous phase after centrifugation. The saturation concentration of ISB was ~6 μM at physiological ionic strength, consistent with the phase diagram. Interestingly, phosphorylated HP1α promoted phase-separation of ISB (reducing the saturation concentration of the ISB to ~3.5 μM), whereas the unphosphorylated HP1α inhibited phase-separation (increasing the saturation concentration to ~9 μM) (Fig.4d). In contrast, neither GFP nor Sgo11-112 affected the ISB saturation concentration (Fig.4d).
Borealin is phosphorylated at multiple sites by CDK1 during mitosis to promote inner-centromere localization. We measured the saturation concentration of ISB after phosphorylation by Cyclin-B/CDK1. Pre-incubation of ISB with Cyclin-B/CDK1 and ATP reduced the saturation concentration from 10 μM to ~8 μM (Fig.4e). This effect was not seen in absence of ATP (Fig.4e). The effect of CDK phosphorylation is probably an underestimate since it is unlikely that the ISB was fully phosphorylated in vitro. These in vitro data strongly argue for a model where the initial local concentration of the CPC on chromatin by the phospho-histone marks nucleates phase-separation at the inner-centromere and other factors such as the presence of pHP1α and CDK1 activity further enhances phase-separation of the CPC.
Borealin phase-separates in vivo
We used the Cry2 opto-droplet system to test whether Borealin can phase-separate in vivo37. A phase-separating protein fused to the light inducible dimerizing protein Cry2 will form droplets/puncta in vivo after exposure to blue light. We fused Cry2 to the Borealin subunit because it had the highest likelihood to participate in phase-separation amongst CPC subunits by CatGranule, which is a machine learning algorithm that predicts the propensity of a protein to undergo phase-separation (Supplementary Fig.5g). ~95% of cells expressing mCherry-Cry2-Borealin formed distinct puncta in interphase nuclei after exposure to blue light (Fig.5a; Supplementary Fig.5a). In contrast, none of the cells expressing mCherry-Cry2 formed foci (Fig.5a).
Figure 5: Borealin phase-separates in vivo and inner-centromeric CPC is sensitive to inhibitors of phase-separation.
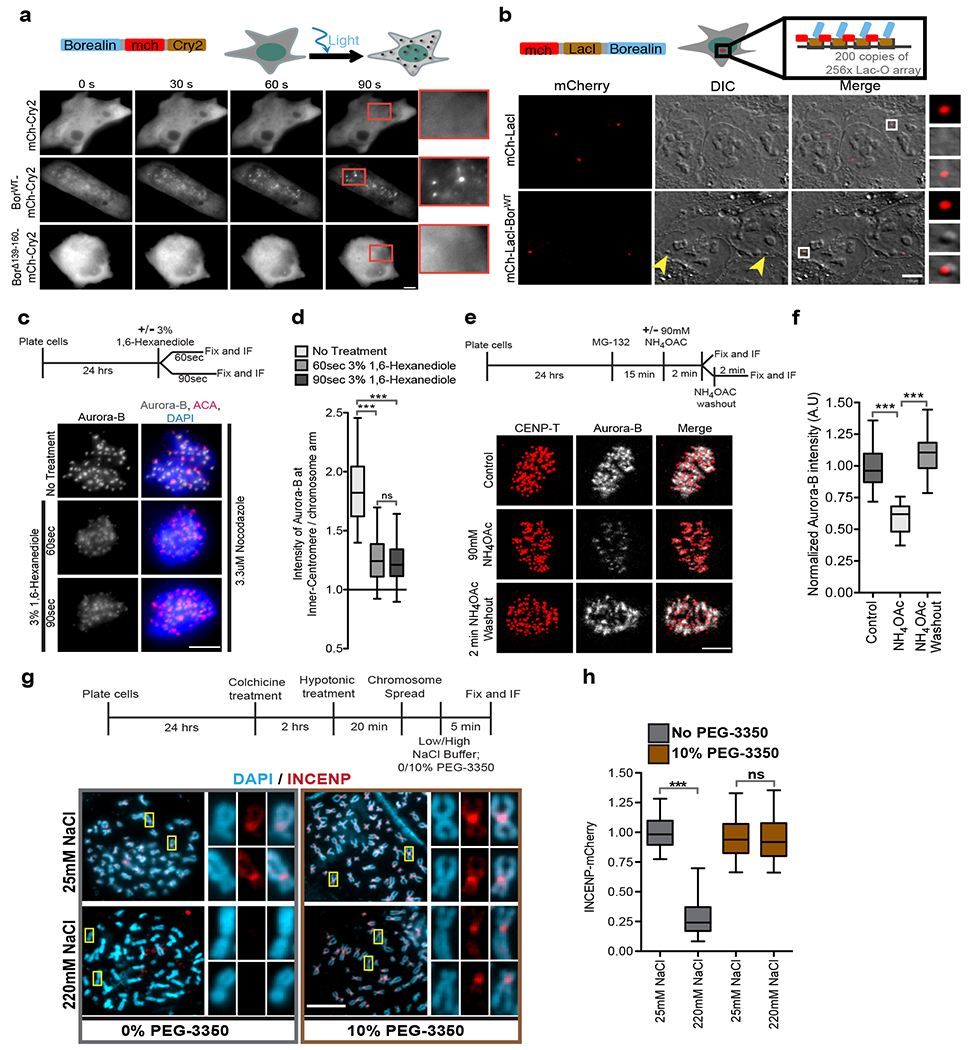
(a) Optodroplet assay. Cells expressing Borealin-mCherry- Cry2 protein were exposed to blue light and followed by time lapse imaging. Quantitation in Supplementary Fig.5a. (b) (Top) Schematic of system to target LacI-Borealin to a LacO array. (Bottom) DIC and mCherry images of cells expressing mCherry-LacI or mCherry-LacI-BorealinWT showing the formation of DIC spot (yellow arrowheads) at the lacO array upon targeting. White box marks the region in the insets. Quantitation in Supplementary Fig.5b. (c) (Top) Experimental setup for c and d. (Bottom) Images (c) and quantification (d) of cells arrested in mitosis with 3.3 μM nocodazole and treated with vehicle control for 90 s or 3% 1,6-Hexanediol for 60 s or 90 s and stained for Aurora-B (grey), ACA (red), and DAPI (blue)(n=158 from 11 cells, n=149 from 10 cells, and n=190 from 12 cells for no-treatment, 60 s and 90 s 1,6-Hexanediol treatment respectively). (e) (Top) Experimental setup for e and f. (Bottom) Images showing Aurora-B (grey) and CENP-T (red) in cells treated with vehicle control or 90 mM NH4OAc or 2 min after NH4OAc washout. (f) Aurora-B fluorescent intensity normalized to control under indicated conditions from e (n=131 from 11 cells for control and n=76 from 9 cells each for NH4OAC treatment and washout). One-Way ANOVA followed by Dunn’s multiple comparison test was applied for d and f. (g) (Top) Experimental setup for g and h. (bottom) Chromosome spread showing DAPI (blue) and INCENP-mCherry (red) under indicated conditions, white boxes mark the regions shown in the insets. (h) Normalized to intensity of INCENP-mCherry at the inner-centromeres under indicated conditions from e. (n=318, 430, 412, and 892 for 25 mM NaCl, 220 mM NaCl, 25 mM NaCl + 10% PEG-3350, and 220 mM + 10% PEG-3350 respectively). Two-tailed T-test was applied (ns indicated P=0.6688). “n” is the number of inner-centromeres measured and *** indicates P<0.0001 and ns indicates P>0.05. Scale bar=5 μm except b=7μm. Two independently experimental repeats for a-h. Box and whisker graphs: median (central line), 25th-75th percentile (box), and 5th-95th percentile (whiskers). Source data for d,f and h are shown in Supplementary Table 2.
Ectopic targeting of phase-separating low complexity protein regions to a specific genomic locus has been shown to induce foci in interphase cells that can be identified by Differential Interference Contrast (DIC) imaging38. Borealin fused to LacI-mCherry was targeted to a locus on chromosome 1 containing 200 copies of 256×LacO/96×tetracycline-responsive element (TRE) arrays. The targeting of Borealin generated a distinct DIC foci compared to chromatin next to it (Fig.5b; Supplementary Fig.5b). Targeting mCherry-LacI did not generate DIC foci (Fig.5b; Supplementary Fig.5b). The change in DIC signal was difficult to distinguish on highly condensed mitotic chromosomes restricting our analysis to interphase nuclei. We conclude that Borealin/CPC can undergoes phase-separation in vivo either upon artificial targeting to a chromosomal locus or upon increase in valency.
The CPC displays condensate-like properties at the inner-centromere.
Inhibiting weak hydrophobic interactions by using aliphatic alcohol 1,6-Hexanediol disrupts many phase-separated organelles in cells39. 1,6-Hexanediol treatment reduced the CPC enrichment in the inner-centromere and the CPC redistributed to the chromosome arms (Fig.5c, d). 1,6-Hexanediol also disrupted ISB coacervates in vitro (Supplementary Fig.5c).
Our in vitro analysis suggested that the CPC at inner-centromeres should be sensitive to increases in ionic strength. Treatment with the cell-permeable salt ammonium acetate has been used to disrupt the phase-separation and gelation of repeat-containing RNA in cells40 and it also disrupted ISB coacervates in vitro (Supplementary Fig.5d). Two minutes of ammonium acetate treatment displaced the CPC from the inner-centromeres of mitotic cells (Fig.5e, f). Inner-centromeric Aurora-B completely recovered two minutes after ammonium acetate washout (Fig.5e, f).
PEG-3350 drastically reduces saturation concentration of ISB allowing phase-separation to occur in the presence of high NaCl concentrations, (Fig.1 b–d; Supplementary Fig.5e), providing an opportunity to investigate whether the reduction of CPC levels was due to disruption of phase-separation or other effects of increasing ionic concentrations. High salt significantly reduced the levels of endogenously tagged Aurora-B and INCENP at inner-centromeres of isolated mitotic chromosomes. In contrast, the CPC was maintained at high levels in presence of high salt and PEG-3350 (Fig.5g, h; Supplementary Fig.5f). Thus, treatments that disrupt membraneless organelles disrupt the CPC at inner centromeres.
Borealin dependent phase-separation drives inner-centromere localization of the CPC.
We identified mutants of Borealin that are defective in phase-separation. We deleted two regions in the central unstructured region of Borealin that were predicted to have a high propensity to phase-separate by CatGranule41,42 (Fig.6a, Supplementary Fig.5g–i). ISB lacking 139-160 amino acids of Borealin was deficient in both spontaneous and DNA induced phase-separation (Fig.6b–d). In contrast, the ISBΔ165-180 behaves similar to ISBWT in these assays (Fig.6b–d). These deletions did not compromise ISB complex formation or hydrodynamic properties (Supplementary Fig.1a, b). BorealinΔ139-160 was also deficient in the Optodroplet assay (Fig.5a; Supplementary Fig.5a). The deleted region in BorealinΔ139-160 is adjacent to a CDK phosphorylation region that has been postulated to interact with the Sgo1 protein. We did not affect any CDK phosphorylation sites in any of our mutants but we still checked whether the interaction between ISB and Sgo1 was affected by the deletion. ISBWT and ISBΔ139-160 interacted equally well with hSgo11-112, inside droplets formed in 10% PEG3350, which lowers the saturation concentration of ISB and allows formation of coacervates even with the ISBΔ139-160 protein (Supplementary Fig.6l,m). CDK phosphorylation was not essential for interaction between ISBWT and Sgo11-112 within coacervates (Fig.3c), rather, we found that CDK phosphorylation lowered the saturation concentration of ISB (Fig.4e), suggesting an alternative explanation to the requirement of CDK in inner-centromere formation. We cannot completely rule out the possibility that the region that drives phase-separation also has an unappreciated second function that controls CPC localization.
Figure 6: The phase-separation property of the CPC is crucial for its localization to inner-centromeres and midzones.
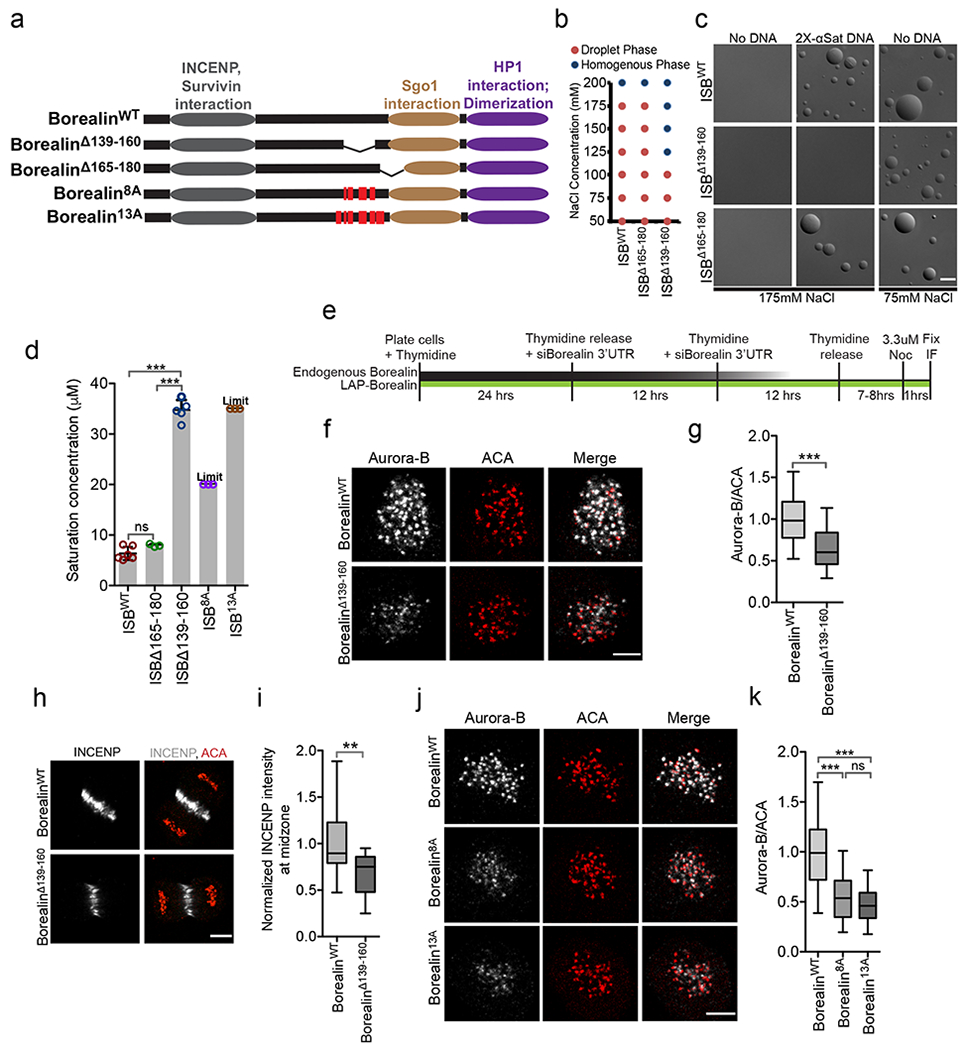
(a) Borealin domains and mutants tested in b-k, red vertical lines indicate the mutated residues. (b) Phase of 20 μM ISBWT, ISBΔ165-180 or ISBΔ139-160 at indicated NaCl concentration. (c) Micrographs of ISBWT or mutant at indicated condition in presence 8 μM ISB and +/−58 ng/ul 2X α-satellite DNA, repeated twice. (d) Saturation concentration of ISBWT (n=6), ISBΔ165-180 (n=3), ISBΔ139-160 (n=5), ISB8A (n=3), and ISB13A (n=3) in buffer containing 150 mM NaCl. “n” is number of independent experiments. “Limit” indicates that phase-separation was not observed at the maximum concentration that could be tested. Bar graphs: mean, error bars ±s.d.; open circles= individual data points. One-way Analysis of Variance followed by Bonferroni’s Multiple Comparison Test. ***P<0.001, **P<0.01, *P<0.05 and ns P>0.05. (e) Experimental design for knockdown and replacement of endogenous Borealin for f, g, j and k. (f) Aurora-B and ACA in cells rescued with either LAP-BorealinWT or LAP-BorealinΔ139-160 and treated with 3.3 μM nocodazole. (g) Normalized intensity of Aurora-B/ACA from experiment shown in f (n=235 from 13 cells for LAP-BorealinWT, n=195 from 14 cells for LAP-BorealinΔ139-160, repeated twice). Two-tailed unpaired T-test with Welch correction was applied. (h) Staining of INCENP and ACA in anaphase cells rescued with either LAP-BorealinWT or LAP-BorealinΔ139-160. (i) Normalized INCENP intensity (to the mean intensity of LAP-BorealinWT) at the midzone form experiment shown in h (n=16 and n=15 midzones for LAP-BorealinWT and LAP-BorealinΔ139-160). Two-tailed T-test was applied; **P=0.0045. (j) Aurora-B and ACA in cells rescued with LAP-BorealinWT (n=154 from 9 cells), LAP-Borealin8A (n=123 from 8 cells) or LAP-Borealin13A (n=97 from 9 cells) and treated with 3.3 μM nocodazole; repeated twice. (k) Normalized intensity of Aurora-B/ACA from experiment shown in j. “n” indicates number of inner-centromeres measured for g and k. 2 independent repeats for f-k. For statistical analysis One-way analysis of variance followed by Bonferroni’s Multiple comparison test was applied. For the whole figure *** indicates P<0.0001 and ns indicates P>0.05. Scale bar is 5 μm. Box and whisker plots: median (line), 25th-75th percentile (box), and 5th-95th percentile (whiskers). Source data for b, d, g, i and k are shown in Supplementary Table 2.
The strong regulation of ISB phase-separation by ionic concentration in vivo and in vitro suggested that charge is important for phase-separation. We mutated 8 positively charged residues in the 139-160 region of Borealin to alanine (ISB8A). We also mutated 5 additional positively charged residues surrounding 139-160aa region to alanine (ISB13A) (Fig.6a; Supplementary Fig.5g). Both of these mutants were defective in phase-separation (Fig.6d).
To uncover the effect of these mutations on localization of the CPC during mitosis, we depleted the endogenous Borealin by siRNA and complemented with either LAP (GFP and S-peptide) tagged wild type Borealin (BorealinWT) or lacking the amino acids 139-160 (BorealinΔ139-160) (Supplementary Fig.6a, c). The BorealinΔ139-160 had reduced CPC in the inner-centromere and midzones compared to the WT (Fig.6e–i, Supplementary Fig.6d). H3T3ph, H2AT120ph and hSgo1 levels at the inner-centromere were similar in cells complemented with BorealinWT and BorealinΔ139-160 (Supplementary Fig.6f–k). Cells complemented with Borealin8A or Borealin13A also had lower amount of Aurora-B in the inner-centromere (Fig.6j, k; Supplementary Fig.6b). We conclude that CPC exists in a phase-separated state at the inner-centromere.
Phase-separation property of the CPC is important for its mitotic function.
We depleted the endogenous Borealin and complemented it with either BorealinWT or BorealinΔ139-160 and imaged cells going through mitosis. Cells expressing BorealinΔ139-160 were deficient in correcting improper kinetochore-microtubule attachments compared to BorealinWT expressing cells as measured by the frequency of anaphases with lagging chromosomes (Fig.7a–c). A modest increase in duration of mitosis was observed in cells complemented with BorealinΔ139-160 (Supplementary Fig.7a–c). In addition, the paclitaxel induced spindle assembly checkpoint arrest was deficient in cells complemented with BorealinΔ139-160 compared to BorealinWT (Fig.7d–f, Supplementary Fig.7 d). We conclude that the phase-separation property of the CPC is important for correcting kinetochore-microtubule attachments and maintaining the spindle checkpoint.
Figure 7: The phase-separation property of the CPC is important for its mitotic functions.
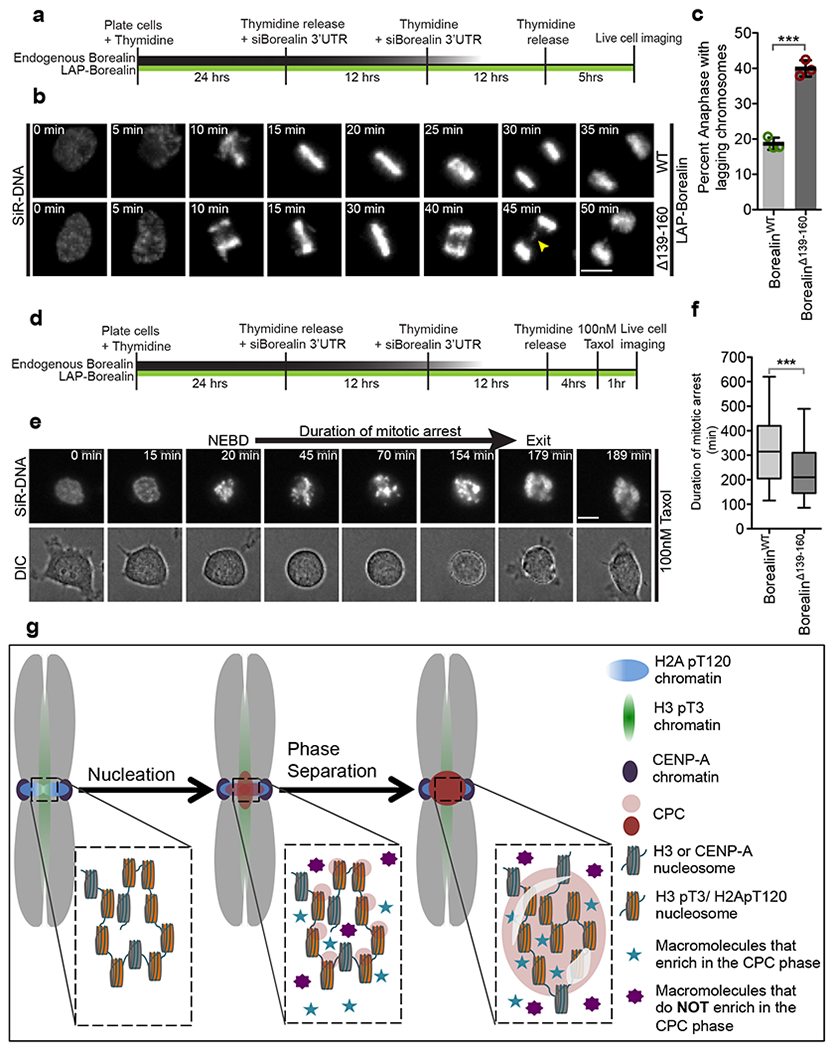
(a) Schematic of the experimental setup for B used to analyze mitotic progression in first mitosis after depletion of endogenous Borealin and complementation with either LAP-BorealinWT or LAP-BorealinΔ139-160. (b) Representative frames from the time-lapse images of cells complemented with either LAP-BorealinWT or LAP-BorealinΔ139-160 undergoing mitosis. Yellow arrow points to the lagging chromosomes in anaphase. 3 independent repeats. (c) Graph showing percent of anaphases with lagging chromosomes in cells rescued with LAP-BorealinWT or LAP-BorealinΔ139-160 (data from n=3 independent experiment (121,107, and 125 cells were analyzed for LAP-BorealinWT; 106, 103, and 86 cells were analyzed for LAP-BorealinΔ139-160). Open circles show individual data points. Error bars are s.d. and central line is mean. For statistical analysis two-tailed unpaired T-test was applied. *** Indicates P=0.0002. Cumulative frequency graph of duration of taxol arrest and duration of various phases of mitosis is shown in Supplementary Fig.7. (d) Schematic of the experimental setup for e used to analyze duration of taxol induced mitotic arrest in first mitosis replacement of endogenous Borealin with either LAP-BorealinWT or LAP-BorealinΔ139-160. (e) Representative frames from the time-lapse images of cells undergoing mitosis in presence of 100nM paclitaxel. “NEBD” refers to the time of nuclear envelope break down and “exit” refers to the time of mitotic exit. Duration from NEBD to exit/death is defined as duration of mitotic arrest. 2 independent repeats. (f) Graph showing duration of mitotic arrest in cells rescued with LAP-BorealinWT or LAP-BorealinΔ139-160 (n=220 cells were analyzed per condition, combined data from 2 independent experiment). Box and whisker plots: median (line), 25th-75th percentile (box), and 5th-95th percentile (whiskers). For statistical analysis two tailed Mann-Whitney test was applied. *** Indicates P<0.0001. For b and e SiR-DNA was used to visualize DNA. Scale bar is 5 μm. (g) Model for the localization of the CPC to the inner-centromere driven by initial nucleation by phospho-histone marks followed by phase-separation of the CPC. Furthermore, some components are enriched in the CPC coacervates (blue stars), while others are excluded (Purple 8-pointed star). Source data for c and f are shown in Supplementary Table 2.
Discussion
We demonstrate that phase-separation is a central biochemical function of the CPC that works in concert with Aurora kinase activity to regulate chromosome segregation. The centromere-targeting region of the CPC undergoes phase-separation driven by an unstructured region on Borealin that is critical for the CPC’s mitotic functions. Phase-separated CPC retained its ability to interact with many inner-centromere components and some stimulated coacervate formation. We were able to provide six lines of evidence that the CPC exists in a phase-separated state at the inner-centromeres. First, the CPC centromere targeting subunits form coacervates at concentrations below those measured at the inner-centromere. Second, the turnover and diffusion rates of the CPC subunits in coacervates and in the inner-centromere strongly correlated. Third, 1,6-Hexanediol or ammonium acetate, which disrupt phase-separation, reduced inner-centromeric CPC. Fourth, ectopic targeting of the Borealin subunit to a LacO array imparted droplet-like optical properties to this chromatin region. Fifth, Borealin subunits formed foci in the Optodroplet assay. Sixth, Borealin mutants that reduced the capacity to phase-separate in vitro were deficient at localizing to inner-centromeres and midzones and at regulating mitotic events. Thus, we suggest phase-separation underlies the unique properties of the inner-centromere and it may become more appropriate to refer to this chromosome area as the inner-centromere body to reflect this insight.
Although the ISB can phase-separate on its own in vitro, the fact that nucleosomes, HP1α and CDK phosphorylation all lowered the saturation concentration suggests they form a multivalent interaction network that drives inner centromeres body formation. This multivalent interaction network may also explain how interactions that either are sub-stoichiometric (Sgo1) or are more dynamic (Survivin) can still anchor CPC stably at the inner-centromere.
CPC signals from inner-centromeres are chromosome autonomous1. Phase-separation may provide a mechanism to restrict signaling events to a single chromosome, although we don’t think that kinase activity can be absolutely contained, as it must escape droplets to generate gradients of soluble activity on chromatin and from the center of the anaphase spindle10,11. The CPC distinguishes merotelic from amphitelic attachments by interaction with microtubules that lie near inner-centromeric chromatin43. The propensity of the CPC to phase-separate on either chromatin or microtubules suggests the exciting possibility that merotelic K-fiber bundles act as conduits for phase-separated CPC to flow from the inner-centromeres onto merotelic microtubules as a mechanism to correct attachments.
Biomolecular condensates provide a mechanism to concentrate components to control biochemical reactions within cells4. We have shown that inner-centromere components preferentially partition to the ISB condensate, suggesting that functions in the inner-centromere such as cohesion protection emerge from the concentration of key proteins in coacervates (Fig.7g). Our in vitro data are performed with a subassembly that contains all the activities required to localize the CPC to inner-centromeres, however, we have not been able to purify the full-length CPC and therefore it is possible that the rest of the CPC may alter its propensity to form coacervates or interact with other proteins.
Our data suggests pericentric heterochromatin undergoes a switch from HP1α as the primary liquid-demixing agent in interphase to the CPC during mitosis. HP1α initiates the activation of Aurora-B kinase activity before mitosis30. The CPC disrupts HP1α binding to H3K9me3 in mitosis44,45 and then directly recruits HP1α to the inner-centromere30,31,46. We have shown that mitotically phosphorylated HP1α36 increased ISB phase-separation, suggesting that the liquid-demixing properties of HP1α and the CPC cooperate to control pericentric heterochromatin.
How cells assemble and disassemble phase-separated bodies is an area of intense research. We suggest that the inner-centromere body is an excellent system to dissect these activities as their assembly and disassembly is spatiotemporally defined. We propose that the initial localization of the CPC through phospho-histone marks (nucleation) concentrates the CPC on mitotic chromatin until it reaches a critical concentration after which it phase-separates (Fig.7g).
Methods:
Protein purification:
BL21-pLysS (DE3) cells were transformed with a tri-cistronic pET28a vector containing 6XHis-INCENP1-58, Survivin, Borealin (WT, Δ139-169, or Δ165-180) sequence; for GFP-ISB, GFP was cloned between 6His and INCENP1-58 to yield 6His-GFP-ISB construct. Cells were then grown in presence of 30 ug/ml Kanamycin to OD 0.8 and protein expression was induced with 0.45 mM IPTG for 16-18hrs at 18°C. The media was also supplemented with 60mg/L ZnCl2 and 0.2% glucose. Cells were then pelleted and lysed in buffer containing 500mM NaCl; 50mM Tris pH7.5, 0.5mM TCEP, 5% glycerol, 5mM Imidazole and protease inhibitor cocktail (Roche) using EmulsiFlex-C3 Homogenizer. The lysate was then cleared with centrifugation and incubated with Ni-NTA beads (Qiagen) for 4 hours at 4°C. After washing Ni-NTA beads (Qiagen) with 200ml buffer containing 500mM NaCl, 50mM Tris pH7.5, 0.5mM TCEP, 25mM Imidazole, and 5% glycerol. The protein was eluted with buffer containing 500mM NaCl, 50mM Tris pH7.5, 0.5mM TCEP, 250mM Imidazole, and 5% glycerol. The eluted protein was then gel filtered on Superdex-200 column 10/300 GL size-exclusion column (GE Life Sciences) in buffer containing 500mM NaCl, 50mM Tris pH7.5, 0.5mM TCEP and 5% glycerol. The desired fractions were collected and concentrated with Amicon Ultra-4 Centrifugal Filter Unit with 3KDa cutoff. HP1α, pHP1α5 and xAurora-B/INCENP790-856 47 were expressed and purified by following the previously published purification scheme. GFP was expressed from a pET28a-GFP vector in BL21-pLysS (DE3) cells. Cells were lysed in PBS containing 0.5mM TCEP and 5mM Imidazole and purified using Ni-NTA beads (Qiagen). After washing with PBS supplemented with 0.5mM TCEP and 25mM Imidazole, GFP was eluted in PBS supplemented with 0.5mM TCEP and 250mM Imidazole. GFP was then dialyzed in PBS supplemented with 0.5mM TCEP. GFP-xMad2 was generated by cloning the gene encoding 6His-eGFP onto he N-terminus of Xenopus Mad2 in pET28a vector and the protein was then purified from E. coli on Ni2+-Agarose.
hSgo11-112 was cloned in pMCSG17 containing N-terminus 6His+MBP-tag to obtain pMCSG13-6His-MBP-hSgo11-112 construct with TEV cleavage site between MBP and hSgo11-112. BL21-pLysS (DE3) cells containing pMCSG13-6His-MBP-hSgo11-112 was grown to 0.8 O.D. and recombinant protein expression was induced with 0.3M IPTG at 18°C for 16-18 hours. The cells were then harvested and lysed in in buffer containing 150mM NaCl; 20mM Tris pH7.5, 0.5mM TCEP, 5% glycerol, 5mM Imidazole and protease inhibitor cocktail (Roche) using EmulsiFlex-C3 Homogenizer. The lysate was incubated with Ni-NTA beads (Qiagen) for 4 hours at 4°C. The beads were then washed with buffer containing 150mM NaCl; 20mM Tris pH7.5, 0.5mM TCEP, 5% glycerol, 25mM Imidazole. The protein was then eluted with buffer containing 150mM NaCl; 20mM Tris pH7.5, 0.5mM TCEP, 5% glycerol, 250mM Imidazole. The 6His-MBP tag was then cleaved from the protein during dialysis with TEV protease in buffer containing 150mM NaCl; 20mM Tris pH7.8, 0.5mM TCEP, 5% glycerol. The resulting cleaved protein was then subjected to gel filtration on Superdex-200 column 10/300 GL size-exclusion column (GE Life Sciences) in buffer containing 150mM NaCl, 20mM Tris pH7.5, 0.5mM TCEP and 5% glycerol. The fraction containing hSgo11-112 were then further purified by ion-exchange chromatography using HiTrap SP HP column (GE lifesciences) with 75mM – 500mM KCl gradient. The pure hSgo11-112 was collected and stored in −80°C.
Phase-separation assay:
Phase-separation was induced by diluting the indicated amount of ISB in the low salt buffer (50mM Tris pH7.5, 1mM DTT) to achieve the indicated final concentration of protein and NaCl. To induce phase-separation in presence of a molecular crowding agent the ISB at the indicated concentration was incubated in the buffer containing 5% PEG-3350, 50mM Tris pH7.5, 1mM DTT and indicated amount of NaCl. Indicated concentration of 2X-α-satellite DNA, histone H3.3 poly-nucleosome (containing 12 nucleosomes) (Active motif), 2X-α-satellite RNA, or paclitaxel stabilized microtubules was incubated with 8uM ISB in buffer containing 150mM NaCl, 50mM Tris pH7.5 and 1mM DTT. Phase-separation was observed by adding a drop of the reaction on the coverslip and imaged on 63X objective in Zeiss Observer Z1 wide-field microscope by fluorescence and DIC imaging or on JEOL 1230 for transmission electron microscopy. For time lapse imaging of ISB droplet fusion, ISB dorplets were formed in indicated condition and immediately images under DIC every second.
Partitioning of constituents into ISB coacervates:
ISB coacervates were made by incubating 5-6 μM ISB in buffer containing 150 mM NaCl, 50 mM Tris pH7.5, 1 mM DTT and 5% PEG-3350. 400 nM (unless otherwise stated) of the indicated agent molecules were incubated with ISB coacervates for 2 mins at room temperature and imaged immediately. For calculating partition coefficients with histone peptides, the ISB coacervates were generated by incubating 20 μM of ISB in buffer containing 150 mM NaCl, 50 mM Tris pH7.5, 1 mM DTT. Fluorescence signal was calculated by using Volocity (V6.3, PerkinElmer). Partition coefficient was calculated by dividing fluorescence signal per unit area inside the coacervates by the fluorescence signal per unit area outside the coacervates after subtraction of background fluorescence. Background fluorescence was calculated by imaging the coacervates in absence of fluorescent agent molecules.
Unmodified histone H3 or H3 pT3 synthetic peptides (ARTKQTARKSTGGKAPRKQLY-fluorescein (note the additional tyrosine, to allow concentration measurement at 280 nm, and C-terminal fluorescein) (GenScript) were previously described. 400 nM rhodamine labeled α/β-Tubulin dimers (Cytoskeleton Inc.), 400 nM HP1α-CF-555, 1 μM hSgo11-112-CF555, 100 nM xAurora-B/xINCENP790-856-CF-555, 400nM Cy3-2X-α-satellite DNA, 400nM 2X-α-satellite RNA, 400nM Cy3-azide, 400nM GFP, 400nM GFP-xMad2 were incubated with phase separated ISB coacervates. 2X-α-satellite RNA was made by in vitro transcription of 2X-α-satellite-DNA PCR product, which was amplified from vector containing 2X-α-satellite-DNA sequence (a kind gift from Dan Foltz) using primer containing the T7 transcription initiation sequence (Fw: TAATACGACTCACTATAGGGAGAAAGTGGATATACAGACCCC; Rv: TCCACTTGCAGACTTTACAAACAG). The in vitro transcription was carried out by using Megascript T7 Transcription Kit (Thermo Fisher) spiked with Cy3-UTP, RNA was then treated with DNAase and isolated by using RNA Clean and Concentrator Kit (Zymo Research). Cy3-2X-α-satellite-DNA was made by PCR, spiked with Cy3-dCTP; using primers listed above and purified using gel extraction. HP1α, hSgo11-112 and xAurora-B/xINCENP790-856 were labeled with Mix-n-Stain™ CF-555 labeling kit (Sigma-Aldrich) according to manufacturer’s protocol followed by dialysis to remove unreacted dye molecules.
Measurement of saturation concentration by spin-down method:
ISB alone or mixed with other indicated protein was incubated in buffer containing 150 mM NaCl, 50 mM Tris pH7.5, and 0.5 μM TCEP to induce phase-separation in a 15 μl reaction. The reaction was allowed to stand for 5 min and then spun at 16100 g for 10 min in order to separate the soluble phase from the droplet phase. 5 μl from the top phase was removed and ran on an SDS-PAGE gel to determine the saturation concentration. Same concentration of ISB that was used in the reaction was also added to buffer with high salt to prevent phase-separation and a serial dilution of this solution was loaded on the same gel along with the top phase of spun down phase-separated solution to create a standard curve (often with R2>0.99), which was used to accurately determine saturation concentration of ISB. The SDS-PAGE gel was stained with GelCode Blue (Biorad) according to manufacturer’s protocol and subjected to densitometry using ImageJ. For experiments shown in fig.4D 15 μM ISB was incubated with equimolar concentration of indicated proteins in buffer containing 150 mM NaCl, 50 mM Tris pH7.5, and 0.5 μM TCEP. For experiment shown in Fig.4E 40 μM ISB was incubates with 10 units of CDK1/Cyclin-B (Enzo, Cat.no. BML-SE128-0100) in buffer containing 5 mM Tris7.5, 300 mM NaCl, 0.1 mM DTT, 5% Glycerol, 50 mM HEPES, 10 mM MgCl2, 1.1 mM EGTA, 0.011% Brij-35, 0.5 μM TCEP and 1mM ATP (ATP or CDK1/Cyclin-B was left out for indicated condition and replaced with their respective dissolving buffer) for 1.5 hours at room temperature. Phase-separation was then induced by lowering the NaCl concentration to 150 mM by diluting the reaction mixture 2-fold with buffer containing 50 mM Tris pH7.5, 0.5 μM TCEP followed by centrifugation and gel analysis as described above.
Microtubule preparation and microtubule dependent phase-separation:
Paclitaxel-stabilized microtubules were prepared by polymerizing bovine brain tubulin dimers spiked with rhodamine labeled α/β-Tubulin dimers in BRB80 (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8 with NaOH), 1 mM DTT and 1 mM GTP with increasing concentration of paclitaxel, paclitaxel stabilized MTs were then separated from the un-polymerized tubulin dimers by centrifuging through a 40% glycerol cushion at 137, 000 x g. 6 μM GFP-ISB was incubated with 1 μM paclitaxel stabilized rhodamine-MTs in buffer containing 50 mM Tris pH7.5, 150 mM NaCl, 1 mM DTT and 20 μM paclitaxel for 15min at room temperature. A drop of the reaction was squished between coverslips and images at 63X on Zeiss Observer Z1 wide-field microscope.
Cell culture and stable cell line generation:
HeLa T-REx cell (ThermoFisher Scientific) and HeLa Kyoto cells with endogenously tagged INCENP-mCherry and Aurora-B-GFP (a gift from Jan Ellenberg)29 were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (Gibco). U2OS-LacO-TRE cells were grown in DMEM-Glutamax medium (Gibco). All the cells were grown in presence of 5% CO2 in a humidified incubator at 37°C.
In order to generate HeLa T-REx cells stably expressing LAP-BorealinWT and LAP-BorealinΔ139-160, the BorealinWT transgene fragments were sub-cloned in to pCDNA5/FRT vector (Invitrogen) containing N-terminal LAP (GFP and S-peptide) tag sequence. LAP- BorealinΔ139-160 was generated by deleting region coding for Borealin 139-160aa by site directed mutagenesis. LAP-Borealin8A and LAP-Borealin13A were created by sub-cloning gblocks (IDT) containing sequences of Borealin8A and Borealin13A in pCDNA5/FRT vector (Invitrogen) containing N-terminal LAP (GFP and S-peptide) tag sequence. The resulting LAP-BorealinWT, LAP-Borealin8A, LAP-Borealin13A and LAP-BorealinΔ139-160 plasmids were co-transfected with the pOG44 plasmid (Invitrogen) using Lipofectamine 2000 (Invitrogen). Cells were subjected to selection for 15 days in presence of 200ug/ml Hygromycin B (Invitrogen). To get cells with homogenous expression of the transgenes, selected colonies were pooled and FACS sorted for GFP (for LAP-Borealin transgenes) expression.
Plasmid and siRNA transfection:
Plasmid transfection was done using Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol.
For knockdown and replacement experiments Borealin 3’UTR siRNA (AGGUAGAGCUGUCUGUUCAdTdT)25 was transfected by using RNAiMAX (Invitrogen) according to manufacturer’s protocol. To analyze mitotic phenotypes in the first mitosis after complementation Borealin stable cell lines were plated in presence of 2mM thymidine, 24hrs after plating cells were release in fresh media and siRNA was transfected. Another round of siRNA transfection was done after 12 hours of the 1st siRNA treatment and 2mM Thymidine was added. After 12-14 hours of 2nd siRNA treatment cells were released from thymidine in fresh media. For, immunofluorescence analysis cells were fixed after 8-10 hours and for live cell imaging cell were treated as indicated.
Live cell imaging:
For live cell imaging, cells were plated in the 4 well imaging chamber (Labtek) in presence 2 mM thymidine followed by siRNA treatment. 3-4 hours after 2nd thymidine release 200 nM SiR-DNA (Cytoskeleton Inc.) dye was added to the cells. 1.5 hours post SiR-DNA treatment time-lapse images were taken 16 hours at 5 min interval on a Zeiss Observer-Z1 in a humidified environmental chamber maintained at 37°C in presence of 5%CO2. Movies were analyzed using Volocity software (V6.3, PerkinElmer).
Fluorescence recovery after photo bleaching (FRAP):
GFP-ISB coacervates were generated by incubating 6uM GFP-ISB in buffer containing 150 mM NaCl, 50m M Tris pH7.5, 1m M DTT and 5% PEG-3350 supplemented with oxygen scavenging solution containing 40 mM Glucose, 130 mg/ml Glucose oxidase, and 24 mg/ml Catalase. GFP-ISB coacervates were then placed in a flow chamber constructed by placing two strips of double-sided tape on a coverslip and placing a second coverslip on top of it to form a grove. FRAP experiment was performed on a Zeiss 880 confocal microscope by acquiring 2 time points before bleaching and then bleaching with 20 cycles of 488 laser at 100% power and imaging every 1.5 sec for 265 sec. Fluorescence intensities were measured using Volocity (V6.3, PerkinElmer). Recovered intensity corrected for photo bleaching that occurred due to imaging was termed as Percent-corrected recovered intensity (Nt) and was calculated by using following equation at each time point.
Where,
Nt = Percent-corrected recovered intensity.
Ft = Fluorescence intensity at time (t).
F0postbleach = Fluorescence intensity just after bleaching.
Ftunbleached = Fluorescence intensity at time (t) of unbleached coacervate.
F0prebleach = Fluorescence intensity before bleaching.
F0unbleached = Fluorescence intensity of unbleached coacervate at the same time as F0prebleach.
Graph of Nt vs time was fitted with one-phase association equation by least square fitting method using Prism software (GraphPad) (shown in Green in Fig.2a). Mobile fraction and T1/2 with 95% confidence interval were extracted from this curve using Prism software (GraphPad).
Fluorescence Correlation Spectroscopy (FCS)
All in vivo FCS measurements were performed on a Zeiss LSM 880 microscope equipped with the quasar spectral array of 32 GaAsP detector, an Argon laser (Melles-Griot) set 488 nm excitation and a Zeiss Plan-Aapochromat 63x/1.2 NA DIC M27 Oil objective. The acquisition modality was set with 0.2 μs sampling time, the laser power was set 0.1% and the pin-hole of 90 a.u. was used. Correlations curves were obtained acquiring intensity fluctuations for 30 s, and then the correlation curves were computed by ZEN Black Zeiss software. An image of the FCS spot was taken before and after recording the FCS trace in order to evaluate if the cell underwent any movement or conformational change.
For live cell FCS, HeLa-TReX cells stably expressing GFP-Borealin were plated in 35 mm imaging petri dish and allowed to growth overnight. Cells were then incubated for 1 h with 3.3 μM of Nocodazole and stained with 20 μl of NucBlue (Thermofisher Scientific, Waltham, MA, USA) diluted in 2 ml of cell culture medium. Cells were incubated during the imaging at 37 C and 5.1% of CO2. Many cells in mitotic and interphase were chosen. For mitotic cells, FCS was carried out in the same cells at the inner-centromere region and in the cytoplasm. For cells in interphase FCS were acquired only inside the nucleus.
All FCS measurements of the in vitro GFP-ISB coacervates were carried out on a Zeiss LSM 510 microscope equipped with ConfoCor3 Single Photocounting Photodiodes (SAPD), an Argon laser for 488 nm excitation and 40x water immersion C-Apochormat NA 1.2 Zeiss objective. GFP-ISB coacervates were generated incubating 5 μM of GFP-ISB with 15 μM of ISB in buffer that finally contains 150 mM NaCl, 50 mM Tris pH 7.5, 0.5 μM DTT. GFP-ISB coacervates solutions were then placed in a flow chamber constructed by two coverslips. The acquisition modality was set with 0.2 μs per time point, and pin-hole of 100 a.u. GFP-ISB FCS are recorded centrally for many droplets and in solution in order to measure the diffusion coefficients of the proteins not phase separated (5 μM in 200 mM NaCl, 50 mM Tris pH 7.5, 0.5 μM DTT).
All the correlations curves were obtained acquiring fluorescence intensity fluctuations over 30 s, and then computed by Zeiss LSM software. Correlation plots were fitted using two populations (in live cells) and one population models (in vitro) using SimFCS software developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu), as described in previously published papers48,49. FCS fitting parameters were obtained with the same settings of the experiment measuring FCS of EGFP as reported previously50.
Optodroplet assay:
Plasmids containing mCherry-Cry2 were obtained from Brangwynne lab37, mCherry-Cry2 was PCR amplified and cloned into pCDNA5-Borealin vectors. pCDNA5 containing mCherry-Cry2, BorealinWT-mCherry-Cry2, and BorealinΔ139-160-mCherry-Cry2 were transfected in HeLa TREx cells 24 hours after plating into 4 well imaging chamber (Labtek). Constructs were allowed to express for 9-10 hours. The cells were then imaged on Zeiss Observer-Z1 in a humidified environmental chamber maintained at 37°C in presence of 5%CO2. Cells with similar expression levels of exogenous proteins were selected based on the mCherry intensity. To induce phase separation cells were exposed to 488nm wavelength light every 5 s and single Z-plane images were taken in the 568nm channel to observe foci-formation. Quantification was performed in Volocity software (V6.3, Perkin Elmer) by manually encircling foci at the 95 s timepoint to measure intensity per unit area in the foci. Enrichment of mCherry in the foci was quantitated by calculating the ratio of intensity per unit area of the foci to that of an area near but not overlapping a foci (Supplementary Fig.5a). Statistical analysis was performed using Prism software (GraphPad).
LacO array experiment:
U2OS-LacO-TRE cells containing 200 copies of 256×LacO/96×tetracycline-responsive element (TRE) arrays integrated in the Chromosome 151 were plated in a 4-well imaging chamber (Labtek). Cells were imaged live with DIC and mCherry channel 24 hours post transfection with either pDF287 Plasmid containing LacI-mCherry alone or LacI-mCherry-Borealin. The DIC signal at the region with differential refractive index is characterized by the presence of a dark area (low intensity region) and a bright area (high intensity region) adjacent to each other yielding a characteristic horizontal “S” shaped profile which lies over the mCherry signal in our experiments. The DIC and mCherry signal was quantitated by measuring intensities along a line (Cyan arrow in Supplementary Fig.5b) using ZEN Black Zeiss software. The direction of the line was chosen to go from the darkest to brightest area of the DIC foci. When no DIC foci was observed the direction was chosen based on the DIC signal of the nearby nuclear bodies. The line-profiles were normalized to the intensities of the first pixel which lied outside a foci to control for variation in background intensities between images.
Mitotic chromosome spreads:
100 ng/ml KaryoMAX Colcemid (Gibco) was added to HeLa Kyoto cells expressing endogenously tagged INCENP-mCherry and Aurora-B-eGFP for 2 hours. Mitotic cells were collected by mitotic shake off spun down and resuspended in a hypotonic buffer (25 mM KCl, 0.27% Na-Citrate in distilled water) for 20 min at 37°C. Swollen cells were then broken open and chromosomes were spread on coverslips using a Cytospin 4 (ThermoShandon). The coverslip was then transferred to the indicated buffer (20 mM HEPES, 5 mM MgCl2, 25 mM NaCl or 220 mM NaCl, 0 or 10% PEG-3350, 1 mM DTT, 20 mM β-glycerophosphate, 1X protease inhibitor cocktail (Roche)) for 5 min and washed once more with the same buffer. The chromosomes were then fixed with 2% PFA in PBS for 15 min. The chromosomes were then stained with DAPI and mounted on a slide for imaging. Chromosomes were imaged at 63X on a Zeiss Observer-Z1. The intensity of INCENP-mCherry and DAPI were quantitated at the inner-centromere by using Volocity (V6.3, PerkinElmer). These values were plotted using Prism software (GraphPad) and indicated statistical tests where applied.
Immunofluoresence microscopy:
Cells were seeded on poly-L-Lysine (Sigma) coated coverslips and treated as indicated. Cells were then fixed with 4% paraformaldehyde in PHEM buffer (25 mM HEPES, 60 mM Pipes, 10 mM EGTA, and 4 mM MgCl2, pH 6.9) containing 0.5% Triton-X 100 for 20 minutes at room temperature. After 3X Tris buffered saline (TBS) wash, cells were blocked for 1hour with 3% BSA in TBS-T at room temperature. Cells were then incubated with primary antibodies in 3% BSA in TBS-T (TBS + 0.1% Tween20) for 1 hour at room temperature. Cells were then washed 3X with TBS-T (10 min each), and incubated with fluorescent secondary antibodies (1:2000) (Jackson Immuno-Research). Cells were then washed 4 times with TBS-T and stained with 0.5 μg/ml DAPI for 5 minutes before mounting on the slides using ProlongGold antifade (Invitrogen) and sealed with nail polish. Image acquisition was performed as described previously52. Image processing and analysis was done using Volocity (V6.3, PerkinElmer). To quantify fluorescence levels at centromeres, we used an intensity thresholding algorithm to mark all centromeres on the basis of ACA intensity. To eliminate changes in fluorescence intensity due to differences in centromere size, the total fluorescence intensity was divided by the total volume of the selected area. Background intensity was subtracted and the intensity/volume of the desired channel were normalized against the corresponding ACA intensity/volume. For quantitating Aurora-B intensity at the midzones, area in the furrow showing Aurora-B intensity was selected manually on a maximum intensity projection image of an anaphase cells and sum of the intensity inside the selected area was measured. The sum of the intensity was divided by area to normalize difference in area of selected regions. A region away from the midzone was selected as background and intensity/area of this region was subtracted from that of the midzones. “Normalized intensity” throughout the paper refers to normalized intensity with respect to the intensity of control cells or of cells expressing LAP-BorealinWT. The final normalized intensity values were graphed and analyzed using Prism software (GraphPad) and indicated statistical tests were applied. For Fig.5c, d Aurora-B intensity at inner-centromere was measure by encircling region between ACA lobes and Aurora-B intensity on the chromosome arm was measured by selecting a region of similar dimension adjacent but not between to ACA lobes on the chromosome arms. The sum of the intensity was divided by the volume of the selected region and the background intensity/volume was subtracted from it. Background intensity was determined by selecting the region away from the chromatin. The ratio of Aurora-B intensity/volume at inner-centromere to that on the chromosome arm was plotted. For Fig.5e, f, Aurora-B intensity/volume at the inner-centromere was measured by drawing region of interest between two CENP-T spots encircling the inner-centromere. Background intensity/volume was determined by selecting a region of interest away from the inner-centromere and subtracted from the intensity/volume measured at the inner-centromere. Most primary antibodies used in this study are from commercial sources and are listed in supplementary table 1with the exception of Borealin52 and CENP-T53 antibodies which have been previously characterized.
Statistics and Reproducibility:
All key experiments were repeated multiple times as indicated in the figure legends. For example, Figure 1 b–f, Figure 2 a–e, Figure 3 a–g, Figure 4 a–e, Figure 6d, Figure 7b,c were all performed independently at least 3 times with similar results. Figure 5 a–h and Figure 6 c, f–k, Figure 7 e,f were performed twice independently. Indicated statistical analysis was done using Prism software (V5, Graphpad). Were box and whisker graphs are used the middle line indicates median; the box indicates 25-75th percentile and the whiskers represent data from 5-95th percentile.
Data Availability:
Source data for Fig. 1–7 and Supplementary Fig. 1–7 have been provided as Supplementary Table 2. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments:
The authors thank Jan Ellenberg, Clifford Brangwynne, Geeta Narlikar and Dan Foltz for reagents and Dan Burke and Sathyan Mattada for helpful discussions. P.T.S. and P.T. were funded by R01GM124042 and R24OD023697. In addition, F.P. and M.A.D. were supported in part by a grant from NSF MCB-1615701. M.A.D. and E.G were funded by NIH P41-GM103540.
Footnotes
Conflict of interest statement:
The authors declare no conflict of interest.
References:
- 1.Trivedi P & Stukenberg PT A Centromere-Signaling Network Underlies the Coordination among Mitotic Events. Trends in Biochemical Sciences (2016). doi: 10.1016/j.tibs.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom KS Centromeric Heterochromatin: The Primordial Segregation Machine. Annu. Rev. Genet (2014). doi: 10.1146/annurev-genet-120213-092033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaqaman K et al. Kinetochore alignment within the metaphase plate is regulated by centromere stiffness and microtubule depolymerases. J. Cell Biol. (2010). doi: 10.1083/jcb.200909005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: Organizers of cellular biochemistry. Nature Reviews Molecular Cell Biology (2017). doi: 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson AG et al. Liquid droplet formation by HP1α suggests a role for phase-separation in heterochromatin. Nature (2017). doi: 10.1038/nature22822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strom AR et al. Phase-separation drives heterochromatin domain formation. Nature (2017). doi: 10.1038/nature22989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabari BR et al. Coactivator condensation at super-enhancers links phase-separation and gene control. Science (80-. ). (2018). doi: 10.1126/science.aap9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahen R et al. Comparative assessment of fluorescent transgene methods for quantitative imaging in human cells. Mol. Biol. Cell (2014). doi: 10.1091/mbc.E14-06-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sessa F et al. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol. Cell (2005). doi: 10.1016/j.molcel.2005.03.031 [DOI] [PubMed] [Google Scholar]
- 10.Fuller BG et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature (2008). doi: 10.1038/nature06923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang E, Ballister ER & Lampson MA Aurora B dynamics at centromeres create a diffusion-based phosphorylation gradient. J. Cell Biol. (2011). doi: 10.1083/jcb.201103044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamagishi Y, Honda T, Tanno Y & Watanabe Y Two Histone Marks Establish the. (2009). [DOI] [PubMed]
- 13.Wang F et al. Histone H3 Thr-3 phosphorylation by haspin positions Aurora B at centromeres in mitosis. Science (80-. ). (2010). doi: 10.1126/science.1189435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly AE et al. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science (80-. ). (2010). doi: 10.1126/science.1189505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamagishi Y, Honda T, Tanno Y & Watanabe Y Two histone marks establish the inner centromere and chromosome bi-orientation. Science (80-. ). (2010). doi: 10.1126/science.1194498 [DOI] [PubMed] [Google Scholar]
- 16.Tsukahara T, Tanno Y & Watanabe Y Phosphorylation of the CPC by Cdk1 promotes chromosome bi-orientation. Nature (2010). doi: 10.1038/nature09390 [DOI] [PubMed] [Google Scholar]
- 17.Niedzialkowska E et al. Molecular basis for phosphospecific recognition of histone H3 tails by Survivin paralogues at inner centromeres. Mol. Biol. Cell (2012). doi: 10.1091/mbc.E11-11-0904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du J, Kelly AE, Funabiki H & Patel DJ Structural basis for recognition of H3T3ph and Smac/DIABLO N-terminal peptides by human survivin. Structure (2012). doi: 10.1016/j.str.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delacour-Larose M, Molla A, Skoufias DA, Margolis RL & Dimitrov S Distinct dynamics of aurora b and survivin during mitosis. Cell Cycle (2004). doi: 10.4161/cc.3.11.1203 [DOI] [PubMed] [Google Scholar]
- 20.Beardmore VA Survivin dynamics increases at centromeres during G2/M phase transition and is regulated by microtubule-attachment and Aurora B kinase activity. J. Cell Sci. (2004). doi: 10.1242/jcs.01242 [DOI] [PubMed] [Google Scholar]
- 21.Wühr M et al. Deep proteomics of the xenopus laevis egg using an mRNA-derived reference database. Curr. Biol (2014). doi: 10.1016/j.cub.2014.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hindriksen S, Lens SMA & Hadders MA The Ins and Outs of Aurora B Inner Centromere Localization. Front. Cell Dev. Biol. (2017). doi: 10.3389/fcell.2017.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hauf S et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. (2003). doi: 10.1083/jcb.200208092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hengeveld RCC, Vromans MJM, Vleugel M, Hadders MA & Lens SM A. Inner centromere localization of the CPC maintains centromere cohesion and allows mitotic checkpoint silencing. Nat. Commun (2017). doi: 10.1038/ncomms15542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein UR, Nigg EA & Gruneberg U Centromere Targeting of the Chromosomal Passenger Complex Requires a Ternary Subcomplex of Borealin , Survivin , and the N-Terminal Domain of INCENP. 17, 2547–2558 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodruff JB et al. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell (2017). doi: 10.1016/j.cell.2017.05.028 [DOI] [PubMed] [Google Scholar]
- 27.Wheelock MS, Wynne DJ, Tseng BS & Funabiki H Dual recognition of chromatin and microtubules by INC ENP is important for mitotic progression. J. Cell Biol. (2017). doi: 10.1083/jcb.201609061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanley ML, Yoo TY, Sonnett M, Needleman DJ & Mitchison TJ Chromosomal passenger complex hydrodynamics suggests chaperoning of the inactive state by nucleoplasmin/nucleophosmin. Mol. Biol. Cell (2017). doi: 10.1091/mbc.E16-12-0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wachsmuth M et al. High-throughput fluorescence correlation spectroscopy enables analysis of proteome dynamics in living cells. Nat. Biotechnol (2015). doi: 10.1038/nbt.3146 [DOI] [PubMed] [Google Scholar]
- 30.Ruppert JG et al. HP1α targets the chromosomal passenger complex for activation at heterochromatin before mitotic entry. EMBO J. (2018). doi: 10.15252/embj.201797677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X et al. Chromatin protein HP1α interacts with the mitotic regulator borealin protein and specifies the centromere localization of the chromosomal passenger complex. J. Biol. Chem (2014). doi: 10.1074/jbc.M114.572842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abe Y et al. HP1-Assisted Aurora B Kinase Activity Prevents Chromosome Segregation Errors. Dev. Cell (2016). doi: 10.1016/j.devcel.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J et al. Survivin enhances aurora-B kinase activity and localizes aurora-B in human cells. J. Biol. Chem (2003). doi: 10.1074/jbc.M211119200 [DOI] [PubMed] [Google Scholar]
- 34.Wheatley SP, Carvalho A, Vagnarelli P & Earnshaw WC INCENP is required for proper targeting of Survivin to the centromeres and the anaphase spindle during mitosis. Curr. Biol (2001). doi: 10.1016/S0960-9822(01)00238-X [DOI] [PubMed] [Google Scholar]
- 35.Sampath SC et al. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell (2004). doi: 10.1016/j.cell.2004.06.026 [DOI] [PubMed] [Google Scholar]
- 36.Chakraborty A, Prasanth KV & Prasanth SG Dynamic phosphorylation of HP1α regulates mitotic progression in human cells. Nat. Commun (2014). doi: 10.1038/ncomms4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin Y et al. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell (2017). doi: 10.1016/j.cell.2016.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chong S et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science (80-. ). (2018). doi: 10.1126/science.aar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kroschwald S, Maharana S & Simon A Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters (2017). doi: 10.19185/matters.201702000010 [DOI] [Google Scholar]
- 40.Jain A & Vale RD RNA phase transitions in repeat expansion disorders. Nature (2017). doi: 10.1038/nature22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ambadipudi S, Biernat J, Riedel D, Mandelkow E & Zweckstetter M Liquid-liquid phase-separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun (2017). doi: 10.1038/s41467-017-00480-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolognesi B et al. A concentration-dependent liquid phase-separation can cause toxicity upon increased protein expression. Cell Rep. (2016). doi: 10.1016/j.celrep.2016.05.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trivedi P et al. The binding of Borealin to microtubules underlies a tension independent kinetochore-microtubule error correction pathway. Nat. Commun (2019). doi: 10.1038/s41467-019-08418-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirota T, Lipp JJ, Toh BH & Peters JM Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature (2005). doi: 10.1038/nature04254 [DOI] [PubMed] [Google Scholar]
- 45.Fischle W et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature (2005). doi: 10.1038/nature04219 [DOI] [PubMed] [Google Scholar]
- 46.Ainsztein AM, Kandels-Lewis SE, Mackay AM & Earnshaw WC INCENP centromere and spindle targeting: Identification of essential conserved motifs and involvement of heterochromatin protein HP1. J. Cell Biol. (1998). doi: 10.1083/jcb.143.7.1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosasco-Nitcher SE, Lan W, Khorasanizadeh S & Stukenberg PT Centromeric Aurora-B activation requires TD-60, microtubules, and substrate priming phosphorylation. Science (80-. ). (2008). doi: 10.1126/science.1148980 [DOI] [PubMed] [Google Scholar]
- 48.Cardarelli F, Lanzano L & Gratton E Fluorescence Correlation Spectroscopy of Intact Nuclear Pore Complexes. Biophys. J (2011). doi: 10.1016/j.bpj.2011.04.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moens PDJ, Gratton E & Salvemini IL Fluorescence correlation spectroscopy, raster image correlation spectroscopy, and number and brightness on a commercial confocal laser scanning microscope with analog detectors (Nikon C1). Microsc. Res. Tech (2011). doi: 10.1002/jemt.20919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scipioni L, Lanzanó L, Diaspro A & Gratton E Comprehensive correlation analysis for super-resolution dynamic fingerprinting of cellular compartments using the Zeiss Airyscan detector. Nat. Commun (2018). doi: 10.1038/s41467-018-07513-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janicki SM et al. From silencing to gene expression: real-time analysis in single cells. Cell (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banerjee B, Kestner CA & Stukenberg PT EB1 enables spindle microtubules to regulate centromeric recruitment of Aurora B. J. Cell Biol. (2014). doi: 10.1083/jcb.201307119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sathyan KM, Fachinetti D & Foltz DR α-amino trimethylation of CENP-A by NRMT is required for full recruitment of the centromere. Nat. Commun (2017). doi: 10.1038/ncomms14678 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Fig. 1–7 and Supplementary Fig. 1–7 have been provided as Supplementary Table 2. The data that support the findings of this study are available from the corresponding author upon reasonable request.