

Abstract
Cancer is a complex, dynamic disease that despite recent advances remains mostly incurable. Inter- and intratumoral heterogeneity are generally considered major drivers of therapy resistance, metastasis, and treatment failure. Recent advances in high-throughput experimentation have produced a wealth of data on tumor heterogeneity and researchers are increasingly turning to mathematical modeling to aid in the interpretation of these complex datasets. In this mini-review, we discuss three important classes of approaches for modeling cellular dynamics within heterogeneous tumors: agent-based models, population dynamics, and multiscale models. An important new focus, for which we provide an example, is the role of intratumoral cell-cell interactions.
Graphical Abstract
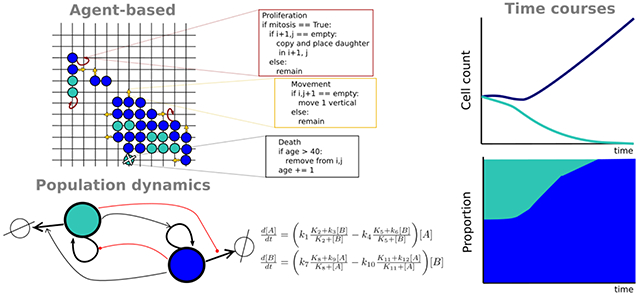
Introduction
Tumors are comprised of a heterogeneous mix of cell subpopulations with common traits and heritable phenotypes [1,2] (e.g., morphological, proliferative, tumorigenic, transcriptional) that compete for resources and interact with each other and with non-cancer cells in complex ways [3,4]. Tumor heterogeneity has been implicated as a source of therapeutic resistance and treatment failure and is seen both across tumors (among patients and between primary and metastatic sites within patients) and within individual tumors [1,2]. Factors contributing to intertumoral heterogeneity include genomic and epigenomic alterations, intrinsic differences between individuals, cell type of origin, and microenvironmental differences across anatomical (primary and secondary) sites. Intratumoral heterogeneity (Fig. 1) is driven by a complex mix of genomic instability, cellular plasticity, regional differences in the microenvironment (e.g., oxygen availability), cell-cell interactions, and intrinsic fluctuations in cell fate and gene expression.
Fig 1. Three sources of intratumoral heterogeneity.
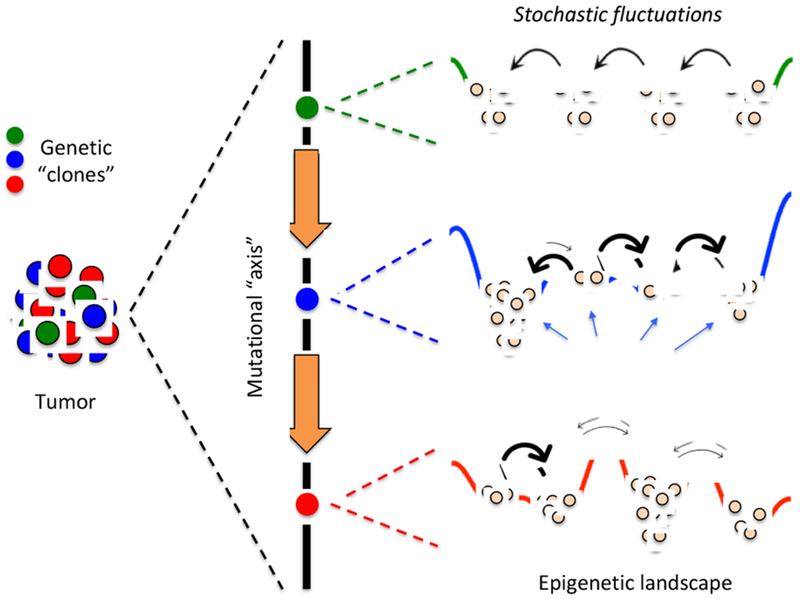
Genetic clones are cells with common origin but genetically diversified by mutations that either pre-exist or are acquired during the course of therapy [1,2]. Each genetic clone can be envisioned as existing along a “mutational axis” and having an associated “epigenetic landscape,” i.e., a quasi-potential energy surface where local minima, or “basins of attraction,” correspond to cellular subtypes [5]. From a molecular perspective, the epigenetic landscape is the consequence of the complex biochemical interaction networks that underlie cell fate decisions [6,7]. Thus, gene expression noise [8], and other sources of intrinsic (e.g., fluctuations in the production and contents of secreted factors) and extrinsic stochasticity can drive transitions between subtypes (thick arrows represent fast transitions, and vice versa). Altogether, at any point in time the subtype composition of a tumor will depend on the genetic clones present within the tumor, the topographies (depths of basins and heights of barriers) of the associated epigenetic landscapes, and the magnitudes of intracellular fluctuations within individual cells.
The complex nature of tumor heterogeneity can be understood within the framework of “epigenetic landscapes,” first proposed by Waddington [5] as a conceptual tool for understanding cellular differentiation during development. More recently, it has been applied to tumors, where “stem-like” cells have the capacity to differentiate into multiple phenotypes and seed tumor growth. Viewing tumor heterogeneity through the lens of epigenetic landscapes may serve as a means for better understanding tumor dynamics and therapy response [2]. Borrowing ideas from physical chemistry, an epigenetic landscape is a quasi-potential energy surface where local minima, or “basins of attraction,” correspond to cellular phenotypes [6,7]. The genetic state of a cell sets the topography of the landscape and genetic mutations can modify it, for example by changing the depths of the basins (Fig. 1). Cells can transition amongst these phenotypes at rates dependent on the heights of the barriers separating basins (Fig. 1). Fluctuations in intrinsic (e.g., gene expression [8]) and/or extrinsic (e.g., secreted factors [9], intracellular protein concentrations [10], oxygen production [11]) processes may serve as sources of noise that drive these transitions. This combination of epigenetic landscape heterogeneity and intrinsic/extrinsic stochasticity, together with genomic diversity (either acquired during normal tumor progression or over the course of therapy) results in a highly heterogeneous tumor (Fig. 1). This view of tumor heterogeneity is consistent with both known genetic clonality of tumors [12] as well as a growing body of literature on non-genetic (epigenetic) inheritance in cancer cells. The latter includes the cancer stem cell (CSC) hypothesis, which posits that phenotypically distinct tumorigenic subpopulations sit atop a hierarchy of non-tumorigenic progeny [13], as well as reports of stem-like cancer cells that are not organized in a classical hierarchy but are metastable and can reversibly shift phenotypes [14,15•]. Notably, stem cells are known to function within niches and interactions between stem-like and niche-like cells may govern tumor dynamics in an analogous manner to their tissue of origin. These cell-cell interactions may influence switching between epigenetic basins as well in response to tumor therapy.
In light of this confluence of genetic, epigenetic, and stochastic factors that underlie tumor heterogeneity, it is no surprise that durable anticancer therapies remain elusive. To tackle this complexity, a systems biology approach is required and has been steadily growing in popularity [16]. By combining high-throughput experimentation, ‘Big Data’ analysis, and mathematical modeling with in vitro and in vivo validation experiments, cancer systems biologists aim to disentangle the complex web of interactions, feedbacks, and dysregulated control mechanisms that has stymied clinical progress [1,2]. Mathematical models are essential to this endeavor because they can predict behaviors outside the range of the experimental conditions on which they are based and can explore the effects of drug dosing, scheduling, and changes in the microenvironment on tumor response [17]. Moreover, mathematical models can explicitly account for both inter- and intratumoral heterogeneity by varying model structure and parameter values. Intertumoral heterogeneity, for example, can be captured by creating variants of a model with different cellular and/or molecular species or by varying the values of rate parameters to account for genetic or microenvironmental differences across individuals or disease sites. Intratumoral heterogeneity can be accounted for by including multiple cell subtypes, interactions through physical contact and/or secreted factors, and by including spatial inhomogeneity of biological factors, such as oxygen, over the computational domain. Below, we provide a brief review of recent approaches for modeling tumor heterogeneity, followed by a short example from our own work.
Mathematical formalisms for modeling tumor growth
Agent-based models
At the resolution of single cells, agent-based models (ABMs) aim to capture complex and emergent behaviors of cell populations through simple and intuitive rules [18]. ABMs are particularly useful when detailed mechanisms of cell behavior are not known but there is a qualitative understanding of the conditions under which certain cellular behaviors are observed. The simplest type of ABM is the cellular automaton (CA) [19•], in which cells are organized in a regular grid and can take on one of a finite number of discrete states. Rules are defined that change the cell state based on its current value and those of its neighbors and updates can be performed synchronously or asynchronously (i.e., stochastically). Modern ABMs extend the basic CA by permitting cells to have multiple properties (e.g., age, phenotypic state, mutation), with discrete or continuous state values, and allowing rules to be either deterministic (occurring if a certain condition is met) or probabilistic (occurring with some probability if the condition is met) [20•]. An example of a rule governing cell death based on the age of a cell and its phenotypic state is (ccl.northwestern.edu/netlogo/models/Tumor)
if (not stem?) and (not metastatic?) and (age > 20)
[die]
if (not stem?) and metastatic? and (age > 4)
[die]
ABMs can also be on-lattice (i.e., grid-based) or off-lattice. On-lattice models are often used to model cell movement within a tumor microenvironment [21–26]. Off-lattice models can be beneficial for modeling cells as clusters or functional units, such as colon crypts [19•,27•–29]. ABMs can also be non-spatial, where the cell state is informed by its environment and the states of other cells but not by location or distance from other cells [30–32]. ABMs can be implemented in custom computer code or using one of numerous publicly available software packages (e.g., [33–36]).
Examples of ABMs applied in cancer include Enderling & Hahnfeldt [21], who constructed an on-lattice ABM of CSCs and non-CSCs, including crowding effects, and predicted that low proliferative capacity and a high rate of spontaneous cell death—which might be expected to keep tumor growth and metastasis in check—actually leads to a less densely seeded tumor where CSCs have the physical space to divide, produce progeny, and eventually escape and seed metastasis. Stichel et al. [27] developed an off-lattice ABM that includes displacement of cells in response to external forces from neighboring cells (e.g., repulsive forces at close cell-cell distances) and a vector-valued velocity term based on these forces. The model was able to reproduce diverse in vitro cell speed dynamics and migratory behaviors, such as straight cell fronts and the formation of cellular bridges.
Population dynamics models
In contrast to ABMs, population dynamics models do not track individual cells over time but rather the size of the cell population [37•]. The classic model of population growth is
which, if one assumes continuous and deterministic dynamics, gives the exponential growth equation
| (1) |
where N(t) is the cell count at time t and the growth rate constant k can be positive or negative. Since its inception, numerous extensions to the exponential growth model have been proposed, including adding an upper limit on the population size (“carrying capacity”) to account for nutrient consumption (e.g., logistic and Monod kinetics) [38] and reducing growth rates at small population sizes to account for the positive effects of cell-cell interactions on growth (e.g., the Allee effect) [39]. Models explicitly accounting for progression through the cell cycle, the apoptotic program, and entry into quiescence/senescence have also been developed [40,41]. Spatial heterogeneity can be accounted for by including cell diffusion and migration over either a continuous or discrete domain [42] and the inherent randomness of cell fate decisions can be modeled using stochastic methods, such as the chemical Master Equation or the Gillespie algorithm [43]. A variety of tools are available for constructing and simulating population dynamics models, including environments such as Matlab and Python and numerous general-purpose software packages (e.g., [44–50]).
In cancer, heterogeneous tumor growth and drug-response dynamics can be modeled by including multiple cell subpopulations that compete and interact [51]: clonal competition can be modeled by varying growth rates across subpopulations [52–54•]; genetic mutations can be included by allowing for spontaneous creation of new subpopulations with pre-specified growth rates [55•]; epigenetic states can be accounted for by allowing cells to reversibly transition between subpopulations [14,22,31,52,54•,56,57]; and cell-cell communication can be incorporated by including density-dependent growth rates [58–61] or by modeling (explicitly or implicitly) the secretion of factors that modify the growth dynamics of other cells [62–66]. As an example, Gupta et al. [14] constructed a population dynamics model describing phenotypic transitions among luminal, basal, and stem-like subpopulations in two breast cancer cell lines and recapitulated the experimental observation that isolated subpopulations recover the cell-state proportions of the parental cell line over time. Based on this, they predicted that after cessation of CSC-targeted therapies, surviving non-CSCs would transition back into CSCs and restart tumor growth, thereby reversing any therapeutic gains. In a subsequent study, Zhou et al. [52] extended this model to include subpopulation-specific growth rates and derived analytical expressions for the temporal dynamics of cell-state proportions as well as the conditions necessary for distinct subpopulations to coexist. Below, we present a similar model of subpopulation dynamics in tumors with the added feature of subtype interactions mediated by secreted factors (see “Example: modeling subtype interactions in small cell lung cancer”).
Multiscale models
Biological processes span multiple scales, from genes to proteins to cells to tissues to organisms [67•]. However, most mathematical models of biological processes operate at only a single scale and either ignore processes at other scales or try to incorporate their effects in some approximate way. Multiscale models attempt to address this shortcoming by explicitly modeling processes at different spatial and/or temporal scales and connecting these models through some type of information exchange interface [67•–71]. In multiscale tumor models, the “cell level” model can either be an ABM or a population dynamics model, which can be coupled to, e.g., a reaction-diffusion model governing background concentrations of bioactive species (e.g., oxygen, nutrients, extracellular matrix) [72–78] or a kinetic or Boolean model describing the intracellular biochemical networks driving cell fate (e.g., progression through the cell cycle, apoptosis) [19•,79]. Typically, simulations are performed by running a short simulation (possibly a single step) of a model at one scale, passing information generated from that simulation to another model at a different scale, running a short simulation of that model with updated initial and/or boundary conditions, and repeating. While in the majority of cases multiscale models are constructed by composing single-scale models in well-established modeling tools and linking them together with specialty purpose code [80], there is a rapidly growing list of general-purpose software packages for multiscale modeling [81–86]. These represent a community-wide effort to develop easy-to-use platforms that facilitate calibration, validation, reproducibility, and exchange of multiscale models using methods firmly rooted in formal mathematical theory [80].
As an example, Yan et al. [87•] used a multiscale hybrid discrete-continuum model to investigate vascular-tumor signaling in glioblastoma (GBM). The continuum (spatial population dynamics) model includes various GBM cells (stem, progenitor, terminally differentiated, dead), host tissue, and human primary endothelial cells, as well as nutrients and signaling molecules. Angiogenesis is modeled via a discrete, off-lattice ABM: blood vessels sprout, grow, and branch at rates dependent on tumor density and background concentrations of vascular endothelial growth factor (VEGF) and other signaling molecules. The vasculature also produces a factor that maintains the pool of GBM stem cells by increasing their proliferation rate and self-renewal capability. Central to the model is a positive feedback loop whereby nutrient-poor tumor cells produce VEGF, inducing vasculature growth, which increases GBM stem cell growth and leads to even more VEGF production. Using the model, the authors confirmed the experimental observation that anti-angiogenesis treatments have a negative effect of inducing the growth of invasive “fingers” that eventually develop into multifocal tumors. Blocking vasculature-secreted factors, however, disrupts growth and reduces tumor size without increasing invasiveness, as also seen experimentally [88,89].
Example: modeling subtype interactions in small cell lung cancer
SCLC is an aggressive and highly metastatic neuroendocrine (NE) carcinoma [90,91•] for which the standard of care (etoposide + cis-platinum and radiation), until very recently [92], had not changed in over 30 years. SCLC is characterized by loss-of-function mutations in TP53 and RBI and is often described as a homogeneous-looking cancer, with “small blue round cells” and minimal presence of non-tumor cells. Indeed, genomic analyses indicate that, on average, primary SCLC tumors are composed of ~84% tumor cells [93]. However, over time it has become clear that SCLC tumors are actually highly heterogeneous, with various cell subtypes coexisting, some of which may provide the trophic support that non-cancer cells often provide in other cancer types [94•,95].
Due to the challenges of acquiring human SCLC samples, heterogeneity in SCLC has been primarily studied in animal models [96]. A study using a genetically engineered mouse model of SCLC, with induced loss of p53, Rb, and p130 tumor suppressor genes in lung epithelial cells, demonstrated that in that model the most abundant tumor cells are NE self-renewing “tumor propagating cells” (TPCs), characterized by CD24high, CD44low, and EpCAMhigh surface expression [15•]. A non-NE population with high expression of Hes1 and lacking NE markers but active in the Notch signaling pathway was also identified [94•]. This subtype was shown to have increased resistance to chemotherapy in comparison to NE cells (TPCs), suggesting a possible role in chemoresistance. A third population, also non-NE, with high levels of CD44 and characterized by expression of mesenchymal markers, such as vimentin, has also been reported [97]. Furthermore, in vitro co-culture experiments of NE and Hes1+ non-NE cells show that NE cell growth is enhanced in the presence of non-NE cells [94•,97]. In vivo experiments have been reported showing that mice injected with an admixture of NE and CD44+ non-NE cells develop metastases but mice injected with only a single subpopulation do not [97]. Taken together, these data suggest that TPCs, Hes1+, and CD44+ cell subtypes form an SCLC tumor ecosystem that supports growth, metastasis, and treatment evasion.
Improving our understanding of the dynamics of SCLC tumors and the interactions between cell subtypes through mathematical modeling could lead to improved treatment options in the future [98]. Therefore, we present here a population dynamics model of the SCLC tumor ecosystem as an illustrative example of the model building and analysis process. We begin by enumerating a list of biological rules (Table 1) that encompass, to the best of our ability, all that is presently known about SCLC tumor heterogeneity and subtype interactions. Notably, these rules, which form the basis of our model, derive from the experimental results described above [15•,94•,97] and are primarily qualitative in nature. This is an important point as most biological data, especially in the early stages of a research project, are not quantitative [99]. As we shall show, it is nevertheless possible to construct a predictive mathematical model based on these data that can provide valuable insight and guide future, more quantitative experiments. These follow-up experiments can, in turn, provide the necessary data to perform more advanced modeling tasks, such as parameter estimation and identifiability analysis [100], which can significantly improve the scope and quality of the model predictions. This iterative process of model construction, experimentation, and refinement lies at the heart of the systems biology approach [16,17].
Table 1.
Biological rules used to construct the SCLC population dynamics model.
| Index | Description and Reference(s) |
|---|---|
| 1 | Tumor-propagating cells (TPCs) divide approximately once per day in culture [15•]. |
| 2 | TPCs have a 20-30% apoptosis rate in culture and a 0-5% apoptosis rate in vivo [15•]. |
| 3 | TPCs can differentiate into other cell types, e.g., Hes1+/Notch-active cells and CD44+ cells (and possibly other subtypes as well) [94•]. |
| 4 | Hes1+ cells have a lower division rate than TPCs [94•]. |
| 5 | Hes1+ cells support the growth of the TPC population by both enhancing cell division and reducing apoptosis [94•]. |
| 6 | There is evidence that TPCs inhibit proliferation of Hes1+ populations in vitro (30-50% reduction as assessed by BrdU incorporation) [94•]. |
| 7 | Role of CD44+ cells is unclear but they may be pro-metastatic and promote invasion by secretion of fibroblast growth factor, which activates ERK1/2 signaling in TPCs [93,97]. |
| 8 | In vivo tumors are comprised of approximately 50% TPCs, 25% Hes1+ cells, 5% CD44+ cells, and 20% unknown cell types [15•,94•]. |
| 9 | Interactions among cell types may involve secretion of soluble factors [94•,97]. |
With these biological rules in hand (Table 1), we next synthesize this knowledge into a simple diagram (Fig. 2A) that illustrates the basic biological processes we aim to model, namely, division, death, differentiation, and inhibition/enhancement of division and death via cell-cell interactions. We then translate this diagram into mathematical form by writing down eight kinetic equations representing division and death of each subtype and differentiation of TPCs into Hes1+ and CD44+ subtypes (Table 2). For five of these processes (Hes1+ death, CD44+ division/death, TPC differentiation), we currently have no information regarding any potential mediating factors (Table 1). Therefore, we make the simplest possible assumption, that the rates of these processes are directly proportional to the population sizes of the subtypes (known as the “law of mass action”). For the other three processes (Hes1+ division, TPC division/death), experimental data suggest that the rates are dependent on cell-cell interactions (Table 1). We assume these interactions to be mediated by secreted factors according to the following simple reaction motif,
where A and B are cell types, A secretes a factor x that interacts with B and converts it into an “active” form B* that has an increased or decreased rate of division or death, and ∅ represents cell death. Assuming that the concentration of x is stable, i.e., its rates of production and degradation are equal (known as the “quasi steady state assumption”), and that the rates of interconversion between B and B* are equal (known as the “partial equilibrium assumption”), we can derive an analytical expression for the rate of the cell fate (division or death) of cell type B as a function of the population size of cell type A,
| (2) |
where KD ≡ kr/kf, , and [B]T ≡ [B] + [B*] is the total amount of B. Note that Eq. (2) models an increase in the rate of division or death by the diffusible factor x if , reduction in the rate if , and reduces to vfate = kfate[B]T (mass-action kinetics) if (i.e., if x has no effect). Reactions 1–3 in Table 2 have rate expressions of this form, with the parameters kfate, , and written as numbered parameters ki or Ki, and [B]T = [TPC] or [Hes1+].
Fig. 2. Population dynamics model of subtype interactions in SCLC.
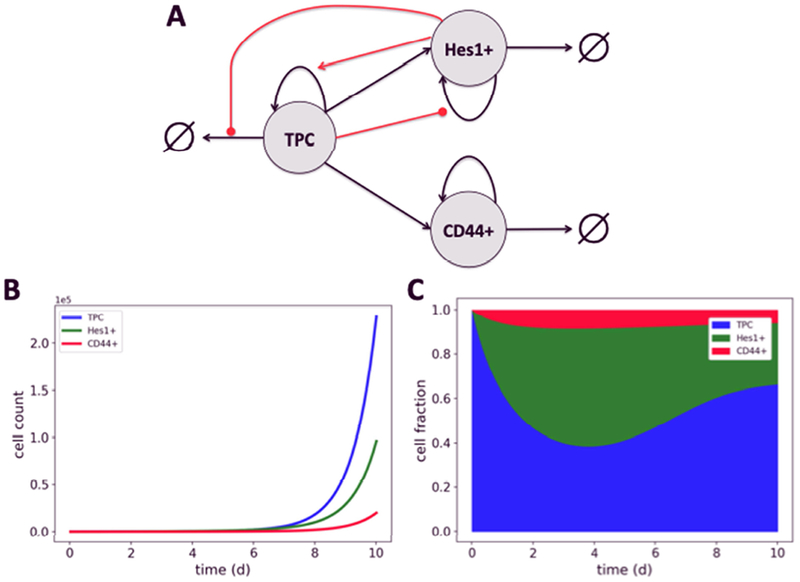
(A) Schematic representation. Black arrows represent cell fates (division, death, differentiation); red arrows/circles represent enhancement/inhibition of cell fate by secreted factors; ∅ represents cell death. (B,C) Simulated time courses of cell counts and fractions. ODE simulations were performed in PySB [49] with initial conditions [TPC]0 = 100, [Hes1+]0 = 0, [CD44+]0 = 0 and the following parameter values (see Table 2): k1 = 1; K2 = 1000; k3 = 2; k4 = 0.2; K5 =1000; k6 = 0.1; k7 = 0.5; K8 = 1000; k9 = 0.25; k10 = 0.2; k11 = 0.3; k12 = 0.2; k13 = 0.5; k14 = 0.1. These values were chosen to produce subtype proportions similar to those seen in vivo (Table 2, Rule 8; excluding the 20% unknown cell types).
Table 2. Kinetic formulation of the SCLC population dynamics model.
[X] is the population of cell type X; ∅ represents cell death.
| Index | Reaction | Rate expression | Description |
|---|---|---|---|
| 1 | TPC → 2 TPC | TPC cell division (k3 > k1) (Hes1+ promotes growth) | |
| 2 | TPC → ∅ | TPC cell death (k6 < k4) (Hes1+ inhibits apoptosis) | |
| 3 | Hes1+ → 2 Hes1+ | Hes1+ cell division (k9 < k7) (TPC inhibits growth) | |
| 4 | Hes1+ → ∅ | k10[Hes1+] | Hes1+ cell death |
| 5 | CD44+ → 2 CD44+ | k11[CD44+] | CD44+ cell division |
| 6 | CD44+ → ∅ | k12[CD44+] | CD44+ cell death |
| 7 | TPC → Hes1+ | k13[TPC] | TPC differentiation into Hes1+ |
| 8 | TPC → CD44+ | k14[TPC] | TPC differentiation into CD44+ |
It is important to emphasize that the model in Table 2 is certainly not the only possible mathematical interpretation of the diagram in Fig. 2A. It is simply our first attempt at codifying our knowledge of SCLC subtype interactions and dynamics in mathematical form. Over time, we expect to include additional interactions, cell types, and processes as experimental observations are made that cannot be captured by this preliminary model. A common approach when the details of a biological process are not well known is to construct numerous “candidate” models and then systematically eliminate unlikely candidates using various “model selection” metrics, such as Akaike or Bayesian information criteria [101]. Thus, rather than a single model, we expect to construct a compendium of SCLC tumor models that will grow and change over time. Numerous software tools and environments exist to facilitate such efforts (e.g., [49,102–105]). One such tool is PySB [49], a “rule-based” modeling platform [106,107] written in Python that includes an easy-to-use modeling language and a variety of deterministic and stochastic simulators [46,50,108,109] and analysis tools [110]. We constructed the model in Table 2 in PySB (available at github.com/lh64/CurrOpinSysBiol_2019) and used it to automatically generate the following set of coupled ordinary differential equations,
| (3) |
| (4) |
| (5) |
These equations were then numerically integrated using algorithms available in the Python package SciPy [108].
Since our experimental data is qualitative (Table 1), for our preliminary simulation analysis (Fig. 2B,C) we decided to manually choose parameter values using the experimental observations as a guide (see caption to Fig. 2 for values). For example, the division rate for TPCs unaffected by secreted factors (k1 in Table 2) was set to 1/day, consistent with experimental observations (Table 1, Rule 1). The division rate for unaffected Hes1+ cells (k7 in Table 2) was then set to 0.5/day based on the observation that they divide at a lower rate than TPCs (Table 1, Rule 4). Initial values for all other rate parameters were chosen based on similar reasoning. All parameter values were then manually varied until values were found that produced steady-state subtype proportions similar to those seen in in vivo mouse tumors (Table 1, Rule 8; Fig. 2C). Of note is that the rate of differentiation from TPCs to Hes1+ cells (k13 in Table 2) had to be set to a value five times larger than that for TPCs to CD44+ cells (k14 in Table 2) because modulating the division and death rates of the subtypes alone could not achieve the desired subtype proportions.
This result is a simple illustration of the non-trivial relationships that often exist between parameter values and model outputs in systems biology models [111]. An obvious next step in the model analysis process would be to quantify these relationships through a “sensitivity analysis” [112] that would identify parameters (or sets of parameters) that act as “control points” for modulating model behavior. These could then be tested experimentally to either validate or refute the current version of the model and suggest possible refinements. As mentioned above, these validation experiments may be quantitative. In such cases, one could then move beyond sensitivity analysis and perform a formal “parameter estimation” [113], which would allow for quantitative model predictions that could be further tested experimentally. Modern parameter estimation algorithms utilize Monte Carlo (or similar) techniques to generate “ensembles” of parameter sets that provide similar fits to experimental data [110,111,114]. Model predictions are then generated by sampling over the ensemble, producing a predictive “envelope” (e.g., [54]) that hopefully is not excessively broad (if it is, further experiments could be designed to reduce the breadth of the envelope [115]). We do not go into this level of sophistication here as our goal is simply to demonstrate how a preliminary model can be built and analyzed but we will employ these techniques in the future as the model grows in complexity and additional experimental data is collected.
Finally, even though our model is based on qualitative observations and static measurements of the compositions of in vivo mouse tumors, the population dynamics formalism that we use allows us to explore and speculate on the temporal dynamics of tumor growth. In Fig. 2B,C, we show time courses for a 10 day in silico experiment starting with a pure population of 100 TPCs. Interestingly, we see that while the total cell count grows continuously throughout the simulation (because the current model lacks a carrying capacity; Fig. 2B), the cell-state proportions stabilize around Day 10 (Fig. 2C). This is consistent with prior theoretical work [14,52], which was experimentally validated [14], showing that phenotypic state transitions can lead to stable steady-state proportions in heterogeneous cell populations. However, our simulation also shows pronounced non-linear dynamics prior to Day 10, with the Hes1+ proportion peaking around Day 4 (Fig. 2C). This is a direct result of modeling cell-cell interactions (which were not considered in [14,52]) between the TPC and Hes1+ subtypes (Fig. 2A and Table 2). Thus, these non-linear dynamics represent a prediction of the model that could be tested experimentally: if observed it would provide support for the underlying assumptions of our model and verify the importance of cell-cell interactions in SCLC tumor dynamics.
Conclusion
Significant progress has been made in recent years in cancer therapy, including the development of new chemotherapies, targeted agents, and immunotherapies. However, cancer morbidity remains stubbornly high, in large part due to inter- and intratumoral heterogeneity. To overcome this challenge, an integrative systems approach involving experimentation, bioinformatics, and mathematical modeling is beginning to take root and produce results [16]. ABMs, population dynamics, and multiscale models are central to this effort as they provide a window onto the underlying mechanisms of therapy resistance [17]. Going forward, a major goal in the field will be to develop patient-specific tumor models, parameterized by clinical data and spanning genetic, cellular, and tissue scales, which can act as in silico platforms for testing and designing personalized anticancer therapies. The promise of this approach is a significant improvement in clinical outcomes and quality of life for millions worldwide suffering from this deadly disease.
Highlights.
Tumors are highly heterogenous, composed of multiple interacting cell types
Cell-cell interactions can be modeled at the single cell and cell population levels
Modeling approaches include agent-based, population dynamics, and multiscale models
Small cell lung cancer is a highly heterogeneous and aggressive tumor
A small cell lung cancer cell-cell interaction model predicts non-linear dynamics
Acknowledgements
We thank Drs. Vito Quaranta, Darren Tyson, and Julien Sage for useful comments regarding this manuscript. This work was supported by NIH grants U54CA217450, F32CA217064, and T32GM007347.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marusyk A, Polyak K: Tumor heterogeneity: Causes and consequences. Biochim Biophys Acta - Rev Cancer 2010, 1805:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marusyk A, Almendro V, Polyak K: Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer 2012, 12:323–334. [DOI] [PubMed] [Google Scholar]
- 3.Nowell PC: The clonal evolution of tumor cell populations. Science 1976, 194:23–28. [DOI] [PubMed] [Google Scholar]
- 4.Greaves M, Maley CC: Clonal evolution in cancer. Nature 2012, 481:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waddington CH: Canalization of development and the inheritance of acquired characters. Nature 1942, 150:563–565. [DOI] [PubMed] [Google Scholar]
- 6.Huang S: The molecular and mathematical basis of Waddington’s epigenetic landscape: a framework for post-Darwinian biology? BioEssays 2012, 34:149–157. [DOI] [PubMed] [Google Scholar]
- 7.Huang S: Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev 2013, 32:423–448. [DOI] [PubMed] [Google Scholar]
- 8.Raj A, van Oudenaarden A: Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 2008, 135:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batsilas L, Berezhkovskii AM, Shvartsman SY: Stochastic Model of Autocrine and Paracrine Signals in Cell Culture Assays. Biophys J 2003, 85:3659–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK: Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anastasiou D: Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer 2017, 116:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, et al. : A Big Bang model of human colorectal tumor growth. Nat Genet 2015, 47:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meacham CE, Morrison SJ: Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES: Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146:633–644. [DOI] [PubMed] [Google Scholar]
- 15.•.Jahchan NS, Lim JS, Bola B, Morris K, Seitz G, Tran KQ, Xu L, Trapani F, Morrow CJ, Cristea S, et al. : Identification and targeting of long-term tumor-propagating cells in small cell lung cancer. Cell Rep 2016, 16:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Werner HMJ, Mills GB, Ram PT: Cancer Systems Biology: a peek into the future of patient care? Nat Rev Clin Oncol 2014, 11:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson ARA, Quaranta V: Integrative mathematical oncology. Nat Rev Cancer 2008, 8:227–234. [DOI] [PubMed] [Google Scholar]
- 18.An G, Mi Q, Dutta-Moscato J, Vodovotz Y: Agent-based models in translational systems biology. WIRES Syst Biol Med 2009, 1:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.•.Metzcar J, Wang Y, Heiland R, Macklin P: A review of cell-based computational modeling in cancer biology. JCO Clin Cancer Inform 2019, 3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; A thorough review of agent-based (i.e., cell-based) models in cancer, with examples dedicated to important processes in tumors, including hypoxia, angiogenesis, and metastasis.
- 20.•.Poleszczuk J, Macklin P, Enderling H: Agent-based modeling of cancer stem cell driven solid tumor growth. Methods Mol Biol 2016, 1516:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]; A useful step-by-step protocol for developing ABMs of cancer stem cell tumors, including instructions for setting up the lattice, defining cell characteristics, and performing simulations.
- 21.Enderling H, Hahnfeldt P: Cancer stem cells in solid tumors: Is “evading apoptosis” a hallmark of cancer? Prog Biophys Mol Biol 2011, 106:391–399. [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Quan Y, Fu Q, Liu Y, Liang Y, Wu J, Yang G, Luo C, Ouyang Q, Wang Y: Dynamics between cancer cell subpopulations reveals a model coordinating with both hierarchical and stochastic concepts. PLoS One 2014, 9:e84654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norton K-A, Wallace T, Pandey NB, Popel AS: An agent-based model of triple-negative breast cancer: the interplay between chemokine receptor CCR5 expression, cancer stem cells, and hypoxia. BMC Syst Biol 2017, 11:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poleszczuk J, Enderling H: Cancer stem cell plasticity as tumor growth promoter and catalyst of population collapse. Stem Cells Int 2016, 2016:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kather JN, Poleszczuk J, Suarez-Carmona M, Krisam J, Charoentong P, Valous NA, Weis CA, Tavernar L, Leiss F, Herpel E, et al. : In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res 2017, 77:6442–6452. [DOI] [PubMed] [Google Scholar]
- 26.Sabzpoushan SH, Pourhasanzade F: A new method for shrinking tumor based on microenvironmental factors: Introducing a stochastic agent-based model of avascular tumor growth. Physica A 2018, 508:771–787. [Google Scholar]
- 27.•.Stichel D, Middleton AM, Müller BF, Depner S, Klingmüller U, Breuhahn K, Matthäus F: An individual-based model for collective cancer cell migration explains speed dynamics and phenotype variability in response to growth factors. NPJ Syst Biol Appl 2017, 3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors used a two-dimensional computational cell migration model to capture experimentally observed behaviors of highly motile non-small cell lung cancer cells, including straight cell fronts and cellular bridges.
- 28.Hoehme S, Bertaux F, Weens W, Grasl-Kraupp B, Hengstler JG, Drasdo D: Model prediction and validation of an order mechanism controlling the spatiotemporal phenotype of early hepatocellular carcinoma. Bull Math Biol 2018, 80:1134–1171. [DOI] [PubMed] [Google Scholar]
- 29.Van Liedekerke P, Neitsch J, Johann T, Alessandri K, Nassoy P, Drasdo D: Quantitative agent-based modeling reveals mechanical stress response of growing tumor spheroids is predictable over various growth conditions and cell lines. PLOS Comput Biol 2019, 15:e1006273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Brenes IA, Wodarz D: Preventing clonal evolutionary processes in cancer: Insights from mathematical models. Proc Natl Acad Sci U S A 2015, 112:8843–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palm MM, Elemans M, Beltman JB: Heritable tumor cell division rate heterogeneity induces clonal dominance. PLoS Comput Biol 2018, 14:e1005954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobile MS, Vlachou T, Spolaor S, Bossi D, Cazzaniga P, Lanfrancone L, Mauri G, Pelicci PG, Besozzi D: Modeling cell proliferation in human acute myeloid leukemia xenografts. Bioinformatics 2019, doi: 10.1093/bioinformatics/btz063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minar N, Burkhart R, Langton C, Askenazi M: The Swarm simulation system: a toolkit for building multi-agent simulations. 1996. [Google Scholar]
- 34.Tisue S, Wilensky U: NetLogo: A simple environment for modeling complexity. In International Conference on Complex Systems 2004:16–21. [Google Scholar]
- 35.Solovyev A, Mikheev M, Zhou L, Dutta-Moscato J, Ziraldo C, An G, Vodovotz Y, Mi Q: SPARK. Int J Agent Technol Syst 2010, 2:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collier N, North M: Parallel agent-based simulation with Repast for High Performance Computing. Simulation 2013, 89:1215–1235. [Google Scholar]
- 37.•.Charlebois DA, Balázsi G: Modeling cell population dynamics. In Silico Biol 2018, 13:21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]; An excellent review of the theory and application of computational models of the dynamics of cell populations, including various population growth models, Markov chain models, agent-based models, and evolutionary models.
- 38.Kargi F: Re-interpretation of the logistic equation for batch microbial growth in relation to Monod kinetics. Lett Appl Microbiol 2009, 48:398–401. [DOI] [PubMed] [Google Scholar]
- 39.Courchamp F, Berec L, Gascoigne J: Allee effects in ecology and conservation. Oxford University Press; 2008. [Google Scholar]
- 40.Fuentes-Garí M, Misener R, García-Munzer D, Velliou E, Georgiadis MC, Kostoglou M, Pistikopoulos EN, Panoskaltsis N, Mantalaris A: A mathematical model of subpopulation kinetics for the deconvolution of leukaemia heterogeneity. J R Soc Interface 2015, 12:20150276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greene JM, Levy D, Fung KL, Souza PS, Gottesman MM, Lavi O: Modeling intrinsic heterogeneity and growth of cancer cells. J Theor Biol 2015, 367:262–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith S, Grima R: Spatial stochastic intracellular kinetics: a review of modelling approaches. Bull Math Biol 2018, 81:2960–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillespie DT: Stochastic simulation of chemical kinetics. Annu Rev Phys Chem 2007, 58:35–55. [DOI] [PubMed] [Google Scholar]
- 44.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U: COPASI--a COmplex PAthway SImulator. Bioinformatics 2006, 22:3067–3074. [DOI] [PubMed] [Google Scholar]
- 45.Resasco DC, Gao F, Morgan F, Novak IL, Schaff JC, Slepchenko BM: Virtual Cell: computational tools for modeling in cell biology. WIRES Syst Biol Med 2012, 4:129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanft KR, Wu S, Roh M, Fu J, Lim RK, Petzold LR: StochKit2: software for discrete stochastic simulation of biochemical systems with events. Bioinformatics 2011, 27:2457–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fange D, Mahmutovic A, Elf J: MesoRD 1.0: Stochastic reaction-diffusion simulations in the microscopic limit. Bioinformatics 2012, 28:3155–3157. [DOI] [PubMed] [Google Scholar]
- 48.Drawert B, Engblom S, Hellander A: URDME: a modular framework for stochastic simulation of reaction-transport processes in complex geometries. BMC Syst Biol 2012, 6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lopez CF, Muhlich JL, Bachman JA, Sorger PK: Programming biological models in Python using PySB. Mol Syst Biol 2013, 9:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris LA, Hogg JS, Tapia J-J, Sekar JAP, Gupta S, Korsunsky I, Arora A, Barua D, Sheehan RP, Faeder JR: BioNetGen 2.2: Advances in rule-based modeling. Bioinformatics 2016, 32:3366–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tabassum DP, Polyak K: Tumorigenesis: It takes a village. Nat Rev Cancer 2015, 15:473–483. [DOI] [PubMed] [Google Scholar]
- 52.Zhou JJX, Pisco AAO, Qian H, Huang S, Yu M, Huang S, Kauffman S, Wang R-S, Saadatpour A, Albert R, et al. : Nonequilibrium population dynamics of phenotype conversion of cancer cells. PLoS One 2014, 9:e110714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piretto E, Delitala M, Ferraro M: Combination therapies and intra-tumoral competition: Insights from mathematical modeling. J Theor Biol 2018, 446:149–159. [DOI] [PubMed] [Google Scholar]
- 54.•.Paudel BB, Harris LA, Hardeman KN, Abugable AA, Hayford CE, Tyson DR, Quaranta V: A nonquiescent “idling” population state in drug-treated, BRAF-mutated melanoma. Biophys J 2018, 114:1499–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]; Combined experimental/modeling study describing a state of near-zero net growth that melanoma cell populations enter under continued drug treatment, with subsequent model-based inference of cell line-specific epigenetic landscapes.
- 55.•.Chowell D, Napier J, Gupta R, Anderson KS, Maley CC, Wilson Sayres MA: Modeling the subclonal evolution of cancer cell populations. Cancer Res 2018, 78:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors use an evolutionary model of subclonal heterogeneity that includes genetic drift and driver mutation selection to quantify fitness effects over 88,000 simulated tumor initiation events, a scale not reachable by experimentation.
- 56.Kaveh K, Kohandel M, Sivaloganathan S: Replicator dynamics of cancer stem cell: Selection in the presence of differentiation and plasticity. Math Biosci 2016, 272:64–75. [DOI] [PubMed] [Google Scholar]
- 57.Zhong H, Brown S, Devpura S, Li XA, Chetty IJ: Kinetic modeling of tumor regression incorporating the concept of cancer stem-like cells for patients with locally advanced lung cancer. Theor Biol Med Model 2018, 15:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korolev KS, Xavier JB, Gore J: Turning ecology and evolution against cancer. Nat Rev Cancer 2014, 14:371–380. [DOI] [PubMed] [Google Scholar]
- 59.Konstorum A, Hillen T, Lowengrub J: Feedback regulation in a cancer stem cell model can cause an Allee effect. Bull Math Biol 2016, 78:754–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown JS, Cunningham JJ, Gatenby RA: Aggregation effects and population-based dynamics as a source of therapy resistance in cancer. IEEE Trans Biomed Eng 2017, 64:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neufeld Z, von Witt W, Lakatos D, Wang J, Hegedus B, Czirok A: The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Comput Biol 2017, 13:e1005818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirouac DC, Ito C, Csaszar E, Roch A, Yu M, Sykes EA, Bader GD, Zandstra PW: Dynamic interaction networks in a hierarchically organized tissue. Mol Syst Biol 2010, 6:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu M, Huang B, Hanash SM, Onuchic JN, Ben-Jacob E: Modeling putative therapeutic implications of exosome exchange between tumor and immune cells. Proc Natl Acad Sci U S A 2014, 111:E4165–E4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sehl ME, Shimada M, Landeros A, Lange K, Wicha MS: Modeling of cancer stem cell state transitions predicts therapeutic response. PLoS One 2015, 10:e0135797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun X, Bao J, Shao Y: Mathematical modeling of therapy-induced cancer drug resistance: connecting cancer mechanisms to population survival rates. Sci Rep 2016, 6:22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baar M, Coquille L, Mayer H, Hölzel M, Rogava M, Tüting T, Bovier A: A stochastic model for immunotherapy of cancer. Sci Rep 2016, 6:24169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.•.Deisboeck TS, Wang Z, Macklin P, Cristini V: Multiscale cancer modeling. Annu Rev Biomed Eng 2011, 13:127–155. [DOI] [PMC free article] [PubMed] [Google Scholar]; A review with an excellent section on the concept of “hierarchy integration” and an extensive presentation of case studies of multiscale models combining submodels over various scales.
- 68.Meier-Schellersheim M, Fraser IDC, Klauschen F: Multiscale modeling for biologists. WIRES Syst Biol Med 2010, 1:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dada JO, Mendes P: Multi-scale modelling and simulation in systems biology. Integr Biol 2011,3:86–96. [DOI] [PubMed] [Google Scholar]
- 70.Walpole J, Papin JA, Peirce SM: Multiscale computational models of complex biological systems. Annu Rev Biomed Eng 2013, 15:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu JS, Bagheri N: Multi-class and multi-scale models of complex biological phenomena. Curr Opin Biotechnol 2016, 39:167–173. [DOI] [PubMed] [Google Scholar]
- 72.Anderson ARA, Weaver AM, Cummings PT, Quaranta V: Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006, 127:905–915. [DOI] [PubMed] [Google Scholar]
- 73.Anderson ARA, Hassanein M, Branch KM, Lu J, Lobdell NA, Maier J, Basanta D, Weidow B, Narasanna A, Arteaga CL, et al. : Microenvironmental independence associated with tumor progression. Cancer Res 2009, 69:8797–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qiao M, Wu D, Carey M, Zhou X, Zhang L: Multi-scale agent-based multiple myeloma cancer modeling and the related study of the balance between osteoclasts and osteoblasts. PLoS One 2015, 10:e0143206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ibrahim-Hashim A, Robertson-Tessi M, Enriquez-Navas PM, Damaghi M, Balagurunathan Y, Wojtkowiak JW, Russell S, Yoonseok K, Lloyd MC, Bui MM, et al. : Defining cancer subpopulations by adaptive strategies rather than molecular properties provides novel insights into intratumoral evolution. Cancer Res 2017, 77:2242–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghadiri M, Heidari M, Marashi SA, Mousavi SH: A multiscale agent-based framework integrated with a constraint-based metabolic network model of cancer for simulating avascular tumor growth. Mol Biosyst 2017, 13:1888–1897. [DOI] [PubMed] [Google Scholar]
- 77.Jagiella N, Müller B, Müller M, Vignon-Clementel IE, Drasdo D: Inferring growth control mechanisms in growing multi-cellular spheroids of NSCLC cells from spatial-temporal image data. PLoS Comput Biol 2016, 12:e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butner JD, Cristini V, Wang Z: Development of a three dimensional, multiscale agent-based model of ductal carcinoma in situ. In 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC) IEEE; 2017:86–89. [DOI] [PubMed] [Google Scholar]
- 79.Kim E, Kim JY, Smith MA, Haura EB, Anderson ARA: Cell signaling heterogeneity is modulated by both cell-intrinsic and -extrinsic mechanisms: An integrated approach to understanding targeted therapy. PLoS Biol 2018, 16:e2002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoekstra A, Chopard B, Coveney P: Multiscale modelling and simulation: a position paper. Philos Trans R Soc A Math Phys Eng Sci 2014, 372:20130377. [DOI] [PubMed] [Google Scholar]
- 81.Letort G, Montagud A, Stoll G, Heiland R, Barillot E, MacKlin P, Zinovyev A, Calzone L: PhysiBoSS: A multi-scale agent-based modelling framework integrating physical dimension and cell signalling. Bioinformatics 2019, 35:1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swat MH, Thomas GL, Belmonte JM, Shirinifard A, Hmeljak D, Glazier JA: Multi-scale modeling of tissues using CompuCell3D. Methods Cell Biol 2012, 110:325–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghaffarizadeh A, Heiland R, Friedman SH, Mumenthaler SM, Macklin P: PhysiCell: an open source physics-based cell simulator for 3-D multicellular systems. PLOS Comput Biol 2018, 14:e1005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mirams GR, Arthurs CJ, Bernabeu MO, Bordas R, Cooper J, Corrias A, Davit Y, Dunn S-J, Fletcher AG, Harvey DG, et al. : Chaste: an open source C++ library for computational physiology and biology. PLoS Comput Biol 2013, 9:e1002970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bravo R, Baratchart E, West J, Schenck RO, Miller AK, Gallaher J, Gatenbee CD, Basanta D, Robertson-Tessi M, Anderson ARA: Hybrid Automata Library: a modular platform for efficient hybrid modeling with real-time visualization. bioRxiv 2018, doi: 10.1101/411538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Starruß J, de Back W, Brusch L, Deutsch A: Morpheus: a user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30:1331–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.•.Yan H, Romero-Lopez M, Frieboes HB, Hughes CCW, Lowengrub JS: Multiscale modeling of glioblastoma suggests that the partial disruption of vessel/cancer stem cell crosstalk can promote tumor regression without increasing invasiveness. IEEE Trans Biomed Eng 2016, 64:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors use a multiscale mathematical model to reproduce observations from several experimental studies of glioblastoma tumor-vasculature interactions and draw novel conclusions regarding different treatment strategies.
- 88.Galan-Moya EM, Le Guelte A, Fernandes EL, Thirant C, Dwyer J, Bidere N, Couraud P-O, Scott MGH, Junier M-P, Chneiweiss H, et al. : Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep 2011, 12:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yan G-N, Yang L, Lv Y-F, Shi Y, Shen L-L, Yao X-H, Guo Q-N, Zhang P, Cui Y-H, Zhang X, et al. : Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol 20l4, 234:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gazdar AF, Bunn PA, Minna JD: Small-cell lung cancer: What we know, what we need to know and the path forward. Nat Rev Cancer 2017, 17:725–737. [DOI] [PubMed] [Google Scholar]
- 91.•.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV., Johnson JE, Lehman JM, MacPherson D, et al. : Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019, 19:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important review, with contributions from many of the most active researchers in the SCLC field, of the various SCLC subtypes that have been described in the literature to date along with characterizations of subtype behavior and suggestions for directions the field should take going forward.
- 92.First-Line Atezolizumab OK’d for SCLC. Cancer Discov 2019, 9:568–569. [DOI] [PubMed] [Google Scholar]
- 93.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. : Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.•.Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, Luca VC, Yang Di, Jahchan NS, Hamard C, et al. : Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important report detailing cell-cell interactions between TPCs and supportive cells in SCLC tumors, as well as signaling alterations and phenotypic transitions that result from these interactions.
- 95.Shamai Y, Alperovich DC, Yakhini Z, Skorecki K, Tzukerman M: Reciprocal reprogramming of cancer cells and associated mesenchymal stem cells in gastric cancer. Stem Cells 2019, 37:176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shue YT, Lim JS, Sage J: Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl Lung Cancer Res 2018, 7:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, Berns A: A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011, 19:244–256. [DOI] [PubMed] [Google Scholar]
- 98.Salgia R, Mambetsariev I, Hewelt B, Achuthan S, Li H, Poroyko V, Wang Y, Sattler M, Salgia R, Mambetsariev I, et al. : Modeling small cell lung cancer (SCLC) biology through deterministic and stochastic mathematical models. Oncotarget 2018, 9:26226–26242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitra ED, Dias R, Posner RG, Hlavacek WS: Using both qualitative and quantitative data in parameter identification for systems biology models. Nat Commun 2018, 9:3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Villaverde AF, Barreiro A, Papachristodoulou A: Structural identifiability of dynamic systems biology models. PLOS Comput Biol 2016, 12:e1005153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kirk P, Thorne T, Stumpf MP: Model selection in systems and synthetic biology. Curr Opin Biotechnol 2013, 24:767–774. [DOI] [PubMed] [Google Scholar]
- 102.Blischak JD, Davenport ER, Wilson G: A quick introduction to version control with Git and GitHub. PLoS Comput Biol 2016, 12:e1004668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chelliah V, Laibe C, Le Novère N: BioModels Database: a repository of mathematical models of biological processes. Methods Mol Biol 2013, 1021:189–199. [DOI] [PubMed] [Google Scholar]
- 104.Xu W, Smith AM, Faeder JR, Marai GE: RuleBender: a visual interface for rule-based modeling. Bioinformatics 2011, 27:1721–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wenskovitch JE, Harris LA, Tapia J-J, Faeder JR, Marai GE: MOSBIE: a tool for comparison and analysis of rule-based biochemical models. BMC Bioinformatics 2014, 15:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chylek LA, Harris LA, Tung C-S, Faeder JR, Lopez CF, Hlavacek WS: Rule-based modeling: a computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip Rev Syst Biol Med 2014, 6:13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chylek LA, Harris LA, Faeder JR, Hlavacek WS: Modeling for (physical) biologists: an introduction to the rule-based approach. Phys Biol 2015, 12:045007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McKinney W: Data structures for statistical computing in Python. In Proceedings of the 9th Python in science conference (SCIPY 2010) Edited by van der Walt S, Millman J. 2010:51–56. [Google Scholar]
- 109.Harris LA, Nobile MS, Pino JC, Lubbock ALR, Besozzi D, Mauri G, Cazzaniga P, Lopez CF: GPU-powered model analysis with PySB/cupSODA. Bioinformatics 2017, 33:3492–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shockley EM, Vrugt JA, Lopez CF: PyDREAM: high-dimensional parameter inference for biological models in Python. Bioinformatics 2018, 34:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP: Universally sloppy parameter sensitivities in systems biology models. PLoS Comput Biol 2007, 3:1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zi Z: Sensitivity analysis approaches applied to systems biology models. IET Syst Biol 2011,5:336–346. [DOI] [PubMed] [Google Scholar]
- 113.Ashyraliyev M, Fomekong-Nanfack Y, Kaandorp JA, Blom JG: Systems biology: parameter estimation for biochemical models. FEBS J 2009, 276:886–902. [DOI] [PubMed] [Google Scholar]
- 114.Eydgahi H, Chen WW, Muhlich JL, Vitkup D, Tsitsiklis JN, Sorger PK: Properties of cell death models calibrated and compared using Bayesian approaches. Mol Syst Biol 2013, 9:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tönsing C, Timmer J, Kreutz C: Cause and cure of sloppiness in ordinary differential equation models. Phys Rev E 2014, 90:023303. [DOI] [PubMed] [Google Scholar]