

Abstract
Emerging data from studies of pediatric-onset pulmonary arterial hypertension (PAH) indicate that the genomics of pediatric PAH is different than that of adults. There is a greater genetic burden in children, with rare genetic factors contributing to at least 35% of pediatric-onset idiopathic pulmonary arterial hypertension (IPAH) compared to ~11% of adult-onset IPAH. De novo variants are the most frequent genetic cause of PAH in children, likely contributing to ~15% of all cases. Rare deleterious variants in bone morphogenetic protein receptor 2 (BMPR2) contribute to pediatric-onset familial PAH and IPAH with similar frequency as adult-onset. While likely gene disrupting (LGD) variants in BMPR2 contribute across the lifespan, damaging missense variants are more frequent in early-onset PAH. Rare deleterious variants in T-box 4 containing protein (TBX4) are more common in pediatric-compared to adult-onset PAH, explaining ~8% of pediatric IPAH. PAH associated with congenital heart disease (APAH-CHD) and other developmental disorders account for a large proportion of pediatric PAH. SRY-related HMG box transcription factor (SOX17) was recently identified as an APAH-CHD risk gene, contributing less frequently to IPAH, with greater prevalence of rare deleterious variants in children compared to adults. The differences in genetic burden and genes underlying pediatric- vs adult-onset PAH indicate that genetic information relevant to pediatric PAH cannot be extrapolated from adult studies. Large cohorts of pediatric-onset PAH are necessary to identify the unique etiological differences of PAH in children, as well as the natural history and response to therapy.
Keywords: genomics, lung disease
Introduction
Pulmonary hypertension (PH) is a diverse group of pulmonary vascular diseases sharing a common pathophysiological endpoint. Endothelial dysfunction, aberrant cell proliferation and vasoconstriction give rise to increased pulmonary vascular pressures, increased vascular resistance, heart failure and premature death. The disease is caused by genetic, epigenetic and environmental factors, as well as gene x environment interactions wherein genetic contributions to disease risk are modified by environmental exposures. Most of our understanding of PH etiology is based upon studies in adults. Here, we highlight current knowledge of the genetic causes for World Symposium on Pulmonary Hypertension (WSPH) Group I PH, or pulmonary arterial hypertension (PAH), for pediatric-onset disease and highlight the need for dedicated studies of children with PAH since etiologies differ by age.
PAH usually manifests in early to mid-life with an estimated prevalence of 4.8–8.1 cases/million for pediatric-onset[1] and 15–50 cases/million for adult-onset disease[2]. Pediatric PAH differs from adult-onset disease in several important aspects including gender bias, disease etiology, clinical presentation, and response to therapy [3–5]. The 3–4-fold higher disease prevalence among females compared to males in adult-onset PAH is not observed in pediatric-onset disease, suggesting less dependence on sex-specific interacting factors in children[6–8]. Etiologically, pediatric-onset PAH has a higher proportion of idiopathic PAH (IPAH), PAH associated with congenital heart disease (APAH-CHD) and developmental lung diseases ((including persistent pulmonary hypertension of the newborn (PPHN)) compared to adult-onset disease[3, 4, 6, 8]. Data from the National Biological Sample and Data Repository for PAH (aka PAH Biobank, n=2572 cases) indicate that children present with slightly higher mean pulmonary arterial pressure, decreased cardiac output and increased pulmonary vascular resistance compared to adults at diagnosis[8] (Table 1). Due to the lack of pediatric clinical trial data, fewer therapeutic options are indicated for use in children with PAH. In practice, therapeutic regimens are based on experiences of individual centers [9] or recent statements of consensus guidelines [10, 11]. The management of pediatric PAH remains challenging due to the paucity of data regarding the natural history, mechanisms of disease and treatment response of PAH molecular subtypes in children.
Table 1. Clinical characteristics of child- vs adult-onset PAH cases at diagnosis.
Data from the National Biological Sample and Data Repository for PAH (n=2572 cases) (Zhu et al 2019 #8). Child-onset, <18 years of age at diagnosis. MPAP, mean pulmonary artery pressure; CO, cardiac output; PVR, pulmonary vascular resistance; dx, diagnosis.
| Group | Age at dx (y) | MPAP (mmHg) | CO, Fick (L/min) | PVR (Woods units) |
|---|---|---|---|---|
| Child (n=226) | 8 ± 6 (226) | 55 ± 18 (225) | 3.3 ± 1.6 (168) | 17.7 ± 11.6 (164) |
| Adult (n=2345) | 52 ± 19 (2345) | 50 ± 14 (2293) | 4.6 ± 1.7 (1630) | 10.0 ± 5.9 (1579) |
| P-value | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
Knowledge of genetic differences between pediatric- and adult-onset PAH are starting to emerge. Most of the data to date is from IPAH or APAH-CHD patients. As has been described for other pediatric developmental diseases including CHD [12–14] and congenital diaphragmatic hernia [15–17], de novo genetic variants contribute to a significant proportion of pediatric IPAH[7] (Figure 1). Furthermore, rare heritable variants in at least three developmental pathways and/or transcription factors have been implicated in pediatric PAH: BMPR2 (encoding bone morphogenetic protein receptor 2), TBX4 (T-box 4 containing protein) and SOX17 (SRY-related HMG box transcription factor) (Figures 1 and 2). Other known PAH risk genes such as ACVRL1, ENG, KCNK3 and SMAD9 rarely contribute to pediatric PAH [7] and in the aggregate account for ~3% of IPAH cases (Figure 1). No effect of sex on risk allele frequencies has been identified to date.
Figure 1.
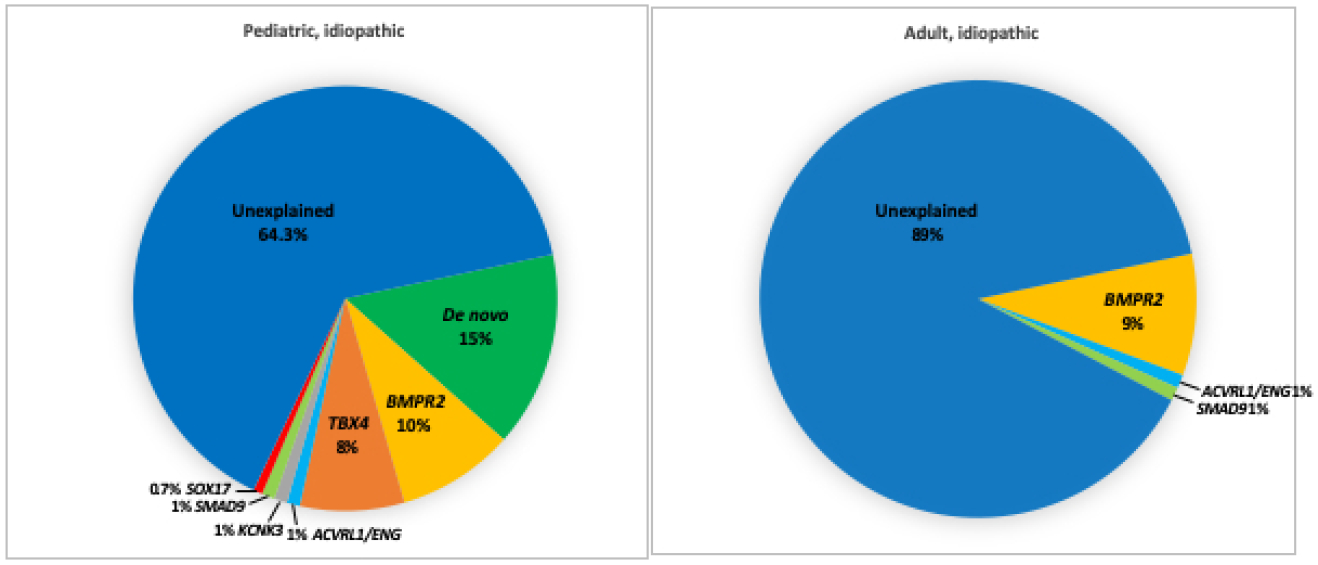
Relative contributions of de novo mutations and 11 established PAH risk genes in idiopathic pediatric- and adult-onset PAH in a cohort of 412 cases (130 pediatric IPAH, 178 adult IPAH). Risk genes included BMPR2, ACVRL1, BMPR1A, BMPR1B, CAV1, EIF2AK4, ENG, KCNK3, SMAD4, SMAD9, and TBX4.
Figure 2.
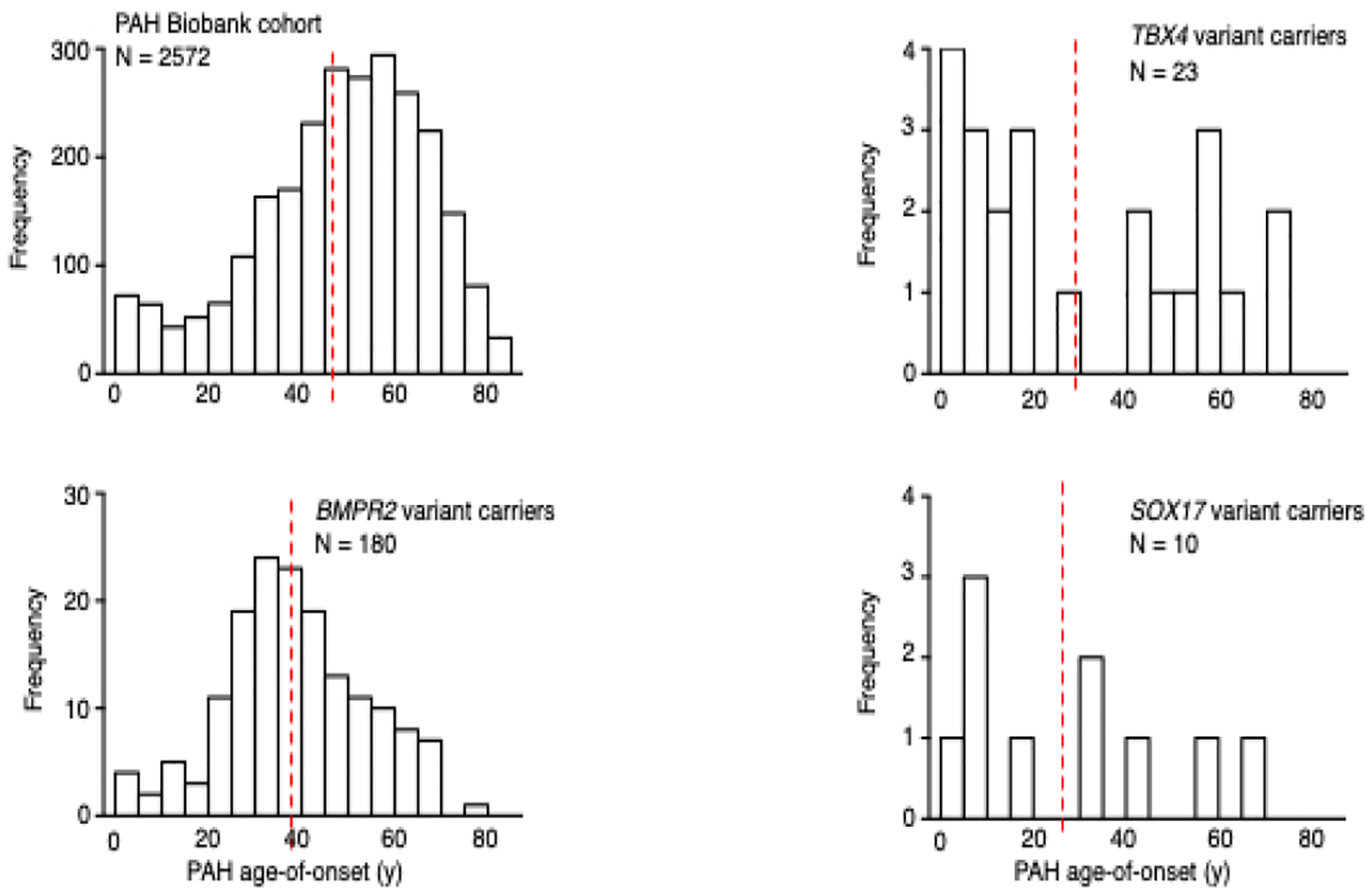
Age distributions for PAH cases from the National Biological Sample and Data Repository for PAH (n=2572). BMPR2, TBX4 and SOX17 variant carriers have younger mean age-of-onset compared to the whole cohort with significant enrichment of pediatric-onset cases among TBX4 variant carriers compared to the whole cohort (Zhu et al 2019 #8). Red vertical lines indicate the group means.
The PAH genetics community has introduced the term hereditary PAH (HPAH) to include familial PAH (PAH that occurs in two or more family members) as well as PAH without a family history of PAH when there is a clear genetic diagnosis. Thus, patients classified as IPAH at the time of diagnosis should be classified as HPAH when a causal genetic variant is identified.
Role of de novo variants
De novo variants have emerged as an important class of genetic factors underlying rare diseases, especially early-onset severe or lethal conditions[13, 17–19], due to strong negative selection decreasing reproductive fitness[20]. Using a cohort of 34 pediatric-onset idiopathic PAH (IPAH) cases for which we had samples from unaffected parents, we demonstrated a 2-fold enrichment of de novo variants in cases compared to an estimated background mutation rate [7]. The variants included both missense variants that change an amino acid with strong predictions of deleterious protein function (D-Mis) and likely gene damaging variants (LGD: frameshift, stop-gain, canonical splice site) often causing protein truncation and nonsense-mediated decay leading to a state of haploinsufficiency (loss of one functional copy of the gene). Among genes highly-expressed in developing heart and lung, the enrichment was increased 4-fold. All six of the LGD variants were identified in patients with an age-of-onset less than 5 years [7]. We now have additional data confirming the relative contribution of de novo variants in an expanded cohort of 124 trios with pediatric PAH probands, including both IPAH and APAH (primarily APAH-CHD) (manuscript in preparation). The majority of the cohort was recruited and consented at Columbia University Medical Center (CUMC) with whole exome sequencing and variant identification as described[7]. Whole genome sequencing data for ten trios was shared by the UK NIHR BioResource – Rare Diseases PAH Study[21] and analyzed together with the CUMC cohort data. The estimated fraction of pediatric PAH explained by de novo variants is ~15%. Some of the de novo variants occur in known risk genes (3 TBX4, 2 BMPR2, 1 ACVRL1, 1 ABCC8). However, the others occur in genes not previously implicated in PAH. Notably, some of the novel de novo variants occur in candidate genes with known or plausible roles in lung/vascular development. For example, AMOT (angiomotin) encodes an angiostatin-binding protein involved in embryonic endothelial cell migration and tube formation as well as endothelial cell tight junctions and angiogenesis [22–24]. KEAP1 (Kelch-like ECH associated protein 1) regulates oxidative stress and apoptosis through interactions with NRF2 in murine vascular cells[25], and endothelial-specific deletion of NRF2 reduces endothelial cell sprouting in vivo [26]. MAPK6 encodes ERK3 (extracellular signal-regulated kinase 3), and mice carrying null alleles exhibit intrauterine pulmonary hypoplasia and early neonatal death [27]. Due to the rarity of de novo mutations in the general population[28], statistical evidence of a candidate risk gene is effectively equivalent to multiplicity (≥2 occurrences) of rare deleterious variants among cases. While our data implicate a role for de novo variants in ~15% of pediatric PAH cases, larger trio cohorts will be required to confirm the role of individual genes with replication and develop a comprehensive list of PAH genes.
BMPR2
BMPR2 is a member of the TGFβ superfamily including TGFβ /BMP ligands, receptors, accessory proteins, activins, and downstream signaling mediators (including SMADs and NOTCH3).
Rare deleterious variants in BMPR2 underlie ~70% of FPAH and 10–20% of IPAH cases, with similar frequencies of BMPR2 variants in pediatric- vs adult-onset disease for both FPAH and IPAH [7, 8]. Overall, BMPR2 variant carriers have younger mean age-of-onset and more severe PAH compared to non-carrier patients, at least in part due to impaired response to oxidative stress [29]. However, BMPR2 variants are less frequent causes of APAH-CHD and have not been observed in PPHN. In a cohort of 258 APAH-CHD cases, only 7 (2.7%) cases carried rare deleterious BMPR2 variants [30]. In the PAH Biobank - comprised of 4% FPAH, 43% IPAH, 48% APAH and 5% other PAH - only 12/119 (10%) of all BMPR2 carriers had PAH diagnoses other than FPAH/IPAH [8]. Among 88 infants with PPHN, no rare deleterious variants in BMPR2 were identified [31]. The wide distribution of deleterious variant locations across the BMPR2 gene has limited genotype-phenotype analyses in the sample sizes studied to date due to small numbers of individuals carrying any one variant. However, an analysis of mutation type – truncating or missense – showed earlier age-of-onset and decreased survival among carriers of missense compared to truncating variants [32]. These data suggest that the presence of mutant/dysfunctional BMPR2 proteins arising from missense variants are more deleterious than haploinsufficiency of normal BMPR2 protein. Similarly, mice carrying a Bmpr2 extracellular domain missense mutation developed more severe PH in response to hypoxia or hypoxia with vascular endothelial growth factor inhibition than mice heterozygous for a Bmpr2 null allele [33]. In our cohort of 412 pediatric- and adult-onset PAH patients, there was a significant enrichment of deleterious missense variants in BMPR2 in patients with younger age-of-onset compared to LGD variant carriers[7] (Figure 3), providing independent confirmation of the importance of missense variants in early-onset disease. Thus, in pediatric PAH, rare deleterious BMPR2 variants contribute primarily to FPAH/IPAH with deleterious missense variants associated with increased disease severity.
Figure 3.

Age distributions of BMPR2 likely gene damaging (LGD) or predicted damaging missense (D-mis) variant carriers from a cohort of 412 pediatric- and adult-onset PAH patients. There was significant enrichment of D-mis variants among patients with younger age-of-onset compared to LGD variant carriers (n=83 total variant carriers)
TBX4
TBX4 is not part of the TGFB pathway. TBX4 encodes a transcription factor in the T-box gene family expressed in the developing atrium of the heart, limb buds, and mesenchyme of lung and trachea, with important roles in limb development as well as lung growth and branching [34]. Rare deleterious TBX4 variants have been associated with small patella syndrome [35] and PAH [7, 36]. TBX4 was first suggested as a candidate PAH risk gene because of its location on chromosome 17q23.1–23.2, where microdeletions were associated with severe neurodevelopmental delays and pulmonary hypertension [37, 38]. Kerstjens-Frederikse and colleagues [36] sequencedTBX2 and TBX4, both located within the 17q23.1–23.2 deletion, and identified three rare TBX4 variants among 49 adults with PAH and no rare variants in TBX2. In a small European cohort of 66 pediatric PAH cases, 3/40 FPAH/IPAH cases carried rare deleterious TBX4 variants[39] compared to 3/136 adult carriers in a Spanish PAH cohort [40]. In our larger cohort of 412 pediatric and adult onset FPAH/IPAH cases, we reported rare deleterious TBX4 variants in 13 cases with a significant enrichment of variants among pediatric (12/155) compared to adult onset (1/257) patients [7]. Furthermore, the mean age of onset was 20 years younger for TBX4 variant carriers compared to BMPR2 carriers. In the PAH Biobank, variants in TBX4 were the second most common genetic cause of PAH and accounted for ~1% of cases [8]. While 13/23 TBX4 variant carriers had a diagnosis of IPAH, the other diagnoses included APAH-CHD, PAH associated with connective tissue disease, FPAH and PAH due to dietary toxin exposure. Age-of-onset for the TBX4 carriers exhibited a bi-modal distribution with significant enrichment of variants among pediatric-onset cases (Figure 2). Interestingly, recent clinical and histological analysis of 19 children carrying rare deleterious TBX4 variants revealed a high frequency of severe developmental defects of the lung, skeleton and heart [41]. Ten of the infants presented with PPHN which resolved; however, the children were subsequently diagnosed with PAH later in infancy or childhood. This evidence indicates that TBX4 is especially important in pediatric-onset PAH, with rare deleterious variants causing a variety of disease subclasses and predicting disease recurrence in young patients initially diagnosed with PPHN.
SOX17
SOX17 also works outside of the TBFB pathway. SOX17 is a highly-constrained gene encoding a transcription factor involved in Wnt/β-catenin and Notch signaling during development [42]. Genetic studies in mice show that Sox17 is required for correct development and function of the pulmonary vascular tree. Endothelial-specific inactivation of Sox17 leads to impaired arterial specification and embryonic death or, with conditional postnatal inactivation, arterial-venous malformations [43]. Deletion in mesenchymal progenitor cells causes abnormal pulmonary vascular morphogenesis resulting in postnatal cardiopulmonary dysfunction and juvenile death [44]. Moreover, in an elegant endothelial lineage tracing study in mice, Liu and colleagues recently demonstrated that transcriptional activation of Sox17 via hypoxia-induced factor 1α, leads to upregulation of cyclin-E1 and endothelial regeneration in response to lung injury [45]. Thus, there are multiple mechanisms through which defective or deficient SOX17 could result in developmental cardiopulmonary defects or impaired response to hypoxic injury.
We identified SOX17 as a candidate risk gene for PAH using exome sequencing data in a cohort of 256 APAH-CHD patients [30]. Using a case-control gene-based association test, SOX17 was the only gene, out of ~18,000 genes, to reach genome-wide significance. Three of the top associated genes (BZW2, FTSJ3, BAZ1B) were putative SOX17 transcriptional targets[46], and enrichment analysis of a gene-set including 1947 putative SOX17 target genes revealed enrichment of rare missense variants in the patient cohort, suggesting that multiple genes in the SOX17 pathway way be important in APAH-CHD. The majority of these genes are expressed in pulmonary arterial endothelial cells or developing heart, and 28% (42/149) are expressed in the top quartile in both tissues/cell types. Pathway enrichment analysis showed that the SOX17 target genes with deleterious variants are overrepresented in developmental processes, transmembrane transport of small molecules, ion homeostasis and extracellular matrix interactions. The association signal for SOX17 was due to LGD and deleterious missense variants carried by 10 APAH-CHD patients, and 7/10 of these patients had pediatric-onset disease. Screening of our separate cohort of 412 FPAH/IPAH patients identified an additional three carriers (2 pediatric, 1 adult)[30]. Rare deleterious variants have been identified in two additional IPAH cohorts, comprised mostly of adults. Genome sequencing data from the UK NIHR BioResource – Rare Diseases PAH Study identified 9/1038 IPAH SOX17 variant carriers[21] and candidate gene analysis of a Japanese cohort found 4 (3 unrelated)/140 FPAH/IPAH SOX17 variant carriers[47]. In the PAH Biobank, rare deleterious SOX17 variants were identified in 10/2572 patients (6 IPAH, 2 APAH-CHD, 1 PAH associated with portopulmonary disease, and 1 PAH due to dietary toxin exposure)[8]. The mean age-of-onset for carriers in the PAH Biobank was 26 years, markedly younger than the overall cohort (48 years) or of BMPR2 variant carriers (38 years) (Figure 2). Notably, 15/18 rare missense variants carried by patients from all five cohorts are located in the high mobility group (HMG, DNA binding) domain of the SOX17 protein (Figure 4). The HMG domain is evolutionally conserved, down to unicellular yeast species, and is essential for target-specific transcriptional control[42, 48]. Based on these data, we estimate that rare deleterious variants in SOX17 contribute to ~7% (19/273) of pediatric-onset PAH, especially APAH-CHD, compared to ~0.4% (13/3455) of adult-onset PAH. In addition, common SNPs in a putative endothelial-acting enhancer region of SOX17 have been associated with PAH [49], suggesting that variation in SOX17 gene expression may increase risk for developing PAH or other vascular endothelium-related diseases more commonly.
Figure 4.
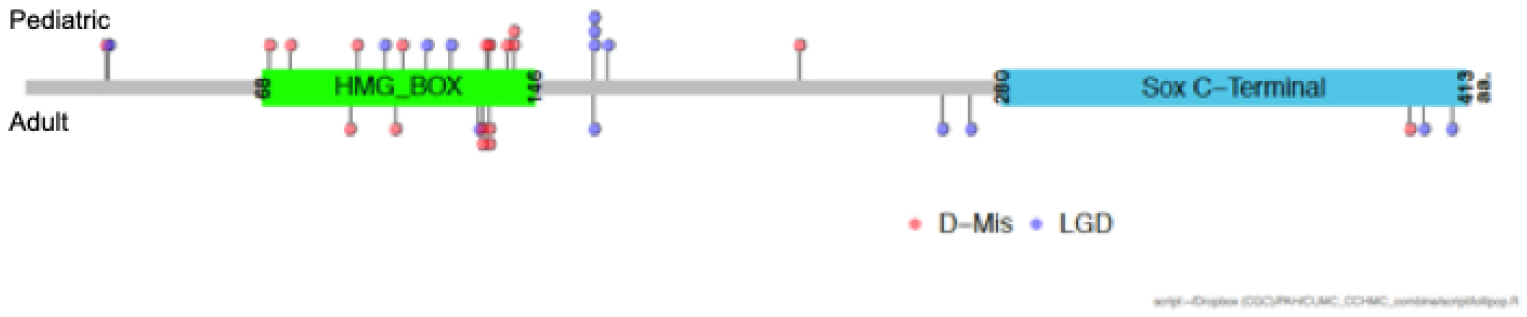
Locations of SOX17 likely gene damaging (LGD) and rare predicted deleterious (D-Mis) variants carried by PAH patients from five cohorts from the US, UK and Japan. Variants carried by pediatric patients (n=19) are shown above the protein schematic and variants carried by adult patients (n=13) below the schematic. The combined datasets include 273 pediatric and 3455 adult patients (Zhu et al 2018 #7; Zhu et al 2019 #8; Graf et al 2018 #21; Zhu et al 2018 #30; Hiraide et al 2018 #47).
Genetic ancestry
The contribution of individual genes in PAH is likely heterogeneous across different genetic ancestries. The results of genetic studies predominantly in individuals of European ancestry may not be generalizable to all other populations. A pediatric study of an Asian cohort revealed a higher carrier frequency of ALK1/ACVRLI variants (7/54, 12.9%) [50] compared to studies of predominantly Europeans. A recent association analysis involving 331 IPAH cases and 10,508 controls of Asian ancestry identified BMP9/GDF2 as a significant risk gene in this population, second in frequency to BMPR2 [51]. Among 22 carriers of rare deleterious GDF2 variants, three had pediatric-onset disease accounting for 5.2% of the 57 pediatric cases. In a case study of a five-year-old boy of Hispanic ancestry, a homozygous loss of function BMP9 variant, c.76C>T;p.Gln26Ter, was identified [52]. The gnomAD population database (gnomad.broadinstitute.org) contains only two heterozygous counts of this allele, both of Latino ancestry, suggesting that this might be an ancestry-specific allele. Clearly, larger studies of children with greater diversity are needed to define ancestral-specific genetic factors and their overall role in pediatric-onset PAH.
Genetic testing for PAH
Clinical genetic testing panels for PAH-associated genes are widely available. Typical panels include BMPR2, ACVRL1, CAV1, EIF2AK4, ENG, KCNK3, and SMAD9 with varied inclusion of additional genes. For children, targeted sequencing of BMPR2, TBX4 and SOX17 should be prioritized for sequence variants, as well as ACVRL1 and GDF2 for Asian patients. In addition, deletion/duplication analysis for BMPR2 should be performed. If the results of these tests are not diagnostic, then reflexive exome sequencing of family trios (affected child and unaffected biological parents) should be performed to identify de novo or inherited rare variants. Genetic diagnoses can inform management of PAH as well as risk stratification for relatives of patients. However, the low penetrance associated with PAH genes complicates risk prediction, and genetic counseling should be offered to families before genetic testing. Although there is no current means to prevent PAH, screening by echocardiogram to enable early diagnosis and treatment may improve outcomes.
Summary –
Genetic information relevant to pediatric PAH cannot be extrapolated from adults. Pediatric-onset PAH differs from adult-onset in many important aspects including the genetic burden and specific genes involved. Rare genetic factors contribute to ~35% of pediatric-onset IPAH compared to ~11% of adult-onset IPAH. De novo variants and rare deleterious variants in BMPR2, TBX4, and SOX17 currently explain most of the known genetic burden in pediatric PAH. However, ancestry-specific factors likely play a role as well, with ACVRL1 and GDF2 variants likely contributing in Asian patients. Large cohorts of pediatric-onset PAH are necessary to identify the unique etiological genes for PAH in children, as well as the natural history and response to therapy. Furthermore, genetic information is immediately clinically relevant and used by families with pediatric onset PAH to make reproductive decisions and screen family members. Identification of new causal genes will illuminate underlying disease pathophysiology and mechanisms that may identify therapeutic targets for adults and children. Given the increased burden of genetic etiologies in children, genomic studies in children with a trio design to identify inherited and de novo variants will yield more new targets than similarly powered studies in adults.
Acknowledgments
Funding support was provided by U01HL125218 and JPB Foundation (WKC) and R01 HL134802 (EDA).
References
- 1.Li L, Jick S, Breitenstein S, Hernandez G, Michel A, Vizcaya D: Pulmonary arterial hypertension in the USA: an epidemiological study in a large insured pediatric population. Pulm Circ 2017, 7:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, et al. : Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006, 173:1023–1030. [DOI] [PubMed] [Google Scholar]
- 3.Hansmann G: Pulmonary Hypertension in Infants, Children, and Young Adults. J Am Coll Cardiol 2017, 69:2551–2569. [DOI] [PubMed] [Google Scholar]
- 4.Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, Ivy DD, Berger RMF: Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J 2019, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saji T: Update on pediatric pulmonary arterial hypertension. Differences and similarities to adult disease. Circ J 2013, 77:2639–2650. [DOI] [PubMed] [Google Scholar]
- 6.Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD: Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012, 125:113–122. [DOI] [PubMed] [Google Scholar]
- 7.Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, et al. : Exome Sequencing in Children With Pulmonary Arterial Hypertension Demonstrates Differences Compared With Adults. Circ Genom Precis Med 2018, 11:e001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu N, Pauciulo MW, Welch CL, Lutz KA, Coleman AW, Gonzaga-Jauregui C, Wang J, Grimes JM, Martin LJ, He H, et al. : Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med 2019, 11:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humpl T, Berger RMF, Austin ED, Fasnacht Boillat MS, Bonnet D, Ivy DD, Zuk M, Beghetti M, Schulze-Neick I: Treatment initiation in paediatric pulmonary hypertension: insights from a multinational registry. Cardiol Young 2017, 27:1123–1132. [DOI] [PubMed] [Google Scholar]
- 10.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, et al. : Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015, 132:2037–2099. [DOI] [PubMed] [Google Scholar]
- 11.Hansmann G, Apitz C, Abdul-Khaliq H, Alastalo TP, Beerbaum P, Bonnet D, Dubowy KO, Gorenflo M, Hager A, Hilgendorff A, et al. : Executive summary. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102 Suppl 2:ii86–100. [DOI] [PubMed] [Google Scholar]
- 12.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. : De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. : De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350:1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. : Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 2017, 49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu L, Sawle AD, Wynn J, Aspelund G, Stolar CJ, Arkovitz MS, Potoka D, Azarow KS, Mychaliska GB, Shen Y, Chung WK: Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum Mol Genet 2015, 24:4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longoni M, High FA, Qi H, Joy MP, Hila R, Coletti CM, Wynn J, Loscertales M, Shan L, Bult CJ, et al. : Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum Genet 2017, 136:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, Zhu N, Kitaygorodsky A, Hernan R, Aspelund G, et al. : De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet 2018, 14:e1007822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epi KC, Epilepsy Phenome/Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, et al. : De novo mutations in epileptic encephalopathies. Nature 2013, 501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin ZB, Wu J, Huang XF, Feng CY, Cai XB, Mao JY, Xiang L, Wu KC, Xiao X, Kloss BA, et al. : Trio-based exome sequencing arrests de novo mutations in early-onset high myopia. Proc Natl Acad Sci U S A 2017, 114:4219–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veltman JA, Brunner HG: De novo mutations in human genetic disease. Nat Rev Genet 2012, 13:565–575. [DOI] [PubMed] [Google Scholar]
- 21.Graf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, Hodgson J, Liu B, Salmon RM, Southwood M, et al. : Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 2018, 9:1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L: Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol 2001, 152:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmgren L, Ambrosino E, Birot O, Tullus C, Veitonmaki N, Levchenko T, Carlson LM, Musiani P, Iezzi M, Curcio C, et al. : A DNA vaccine targeting angiomotin inhibits angiogenesis and suppresses tumor growth. Proc Natl Acad Sci U S A 2006, 103:9208–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Vertuani S, Nystrom S, Audebert S, Meijer I, Tegnebratt T, Borg JP, Uhlen P, Majumdar A, Holmgren L: Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circ Res 2009, 105:260–270. [DOI] [PubMed] [Google Scholar]
- 25.Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, de Cabo R, Csiszar A: Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol 2010, 299:H18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei Y, Gong J, Thimmulappa RK, Kosmider B, Biswal S, Duh EJ: Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc Natl Acad Sci U S A 2013, 110:E3910–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klinger S, Turgeon B, Levesque K, Wood GA, Aagaard-Tillery KM, Meloche S: Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc Natl Acad Sci U S A 2009, 106:16710–16715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. : A framework for the interpretation of de novo mutation in human disease. Nat Genet 2014, 46:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dyer LA, Pi X, Patterson C: The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol Metab 2014, 25:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu N, Welch CL, Wang J, Allen PM, Gonzaga-Jauregui C, Ma L, King AK, Krishnan U, Rosenzweig EB, Ivy DD, et al. : Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med 2018, 10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byers HM, Dagle JM, Klein JM, Ryckman KK, McDonald EL, Murray JC, Borowski KS: Variations in CRHR1 are associated with persistent pulmonary hypertension of the newborn. Pediatr Res 2012, 71:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Austin ED, Phillips JA, Cogan JD, Hamid R, Yu C, Stanton KC, Phillips CA, Wheeler LA, Robbins IM, Newman JH, Loyd JE: Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res 2009, 10:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frump AL, Datta A, Ghose S, West J, de Caestecker MP: Genotype-phenotype effects of Bmpr2 mutations on disease severity in mouse models of pulmonary hypertension. Pulm Circ 2016, 6:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arora R, Metzger RJ, Papaioannou VE: Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet 2012, 8:e1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bongers EM, Duijf PH, van Beersum SE, Schoots J, Van Kampen A, Burckhardt A, Hamel BC, Losan F, Hoefsloot LH, Yntema HG, et al. : Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet 2004, 74:1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, Leter EM, Douwes JM, Van Dijk A, Vonk-Noordegraaf A, Dijk-Bos KK, Hoefsloot LH, Hoendermis ES, et al. : TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. Journal of Medical Genetics 2013, 50:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, Thrush DL, Astbury C, Bartholomew D, McBride KL, Pyatt RE, et al. : Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet 2010, 86:454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nimmakayalu M, Major H, Sheffield V, Solomon DH, Smith RJ, Patil SR, Shchelochkov OA: Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am J Med Genet A 2011, 155A:418–423. [DOI] [PubMed] [Google Scholar]
- 39.Levy M, Eyries M, Szezepanski I, Ladouceur M, Nadaud S, Bonnet D, Soubrier F: Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J 2016, 48:1118–1126. [DOI] [PubMed] [Google Scholar]
- 40.Navas P, Tenorio J, Quezada CA, Barrios E, Gordo G, Arias P, Lopez Meseguer M, Santos-Lozano A, Palomino Doza J, Lapunzina P, Escribano Subias P: Molecular Analysis of BMPR2, TBX4, and KCNK3 and Genotype-Phenotype Correlations in Spanish Patients and Families With Idiopathic and Hereditary Pulmonary Arterial Hypertension. Rev Esp Cardiol (Engl Ed) 2016, 69:1011–1019. [DOI] [PubMed] [Google Scholar]
- 41.Galambos C, Mullen MP, Shieh JT, Schwerk N, Kielt MJ, Ullmann N, Boldrini R, Stucin-Gantar I, Haass C, Bansal M, et al. : Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and pediatric pulmonary hypertension. Eur Respir J 2019. [DOI] [PubMed] [Google Scholar]
- 42.Francois M, Koopman P, Beltrame M: SoxF genes: Key players in the development of the cardio-vascular system. Int J Biochem Cell Biol 2010, 42:445–448. [DOI] [PubMed] [Google Scholar]
- 43.Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, et al. : Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun 2013, 4:2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange AW, Haitchi HM, LeCras TD, Sridharan A, Xu Y, Wert SE, James J, Udell N, Thurner PJ, Whitsett JA: Sox17 is required for normal pulmonary vascular morphogenesis. Dev Biol 2014, 387:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, Benevolenskaya EV, Rehman J, Malik AB: Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun 2019, 10:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A: ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiraide T, Kataoka M, Suzuki H, Aimi Y, Chiba T, Kanekura K, Satoh T, Fukuda K, Gamou S, Kosaki K: SOX17 Mutations in Japanese Patients with Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2018, 198:1231–1233. [DOI] [PubMed] [Google Scholar]
- 48.Hou L, Srivastava Y, Jauch R: Molecular basis for the genome engagement by Sox proteins. Semin Cell Dev Biol 2017, 63:2–12. [DOI] [PubMed] [Google Scholar]
- 49.Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, Pauciulo MW, Hadinnapola C, Aman J, Girerd B, et al. : Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chida A, Shintani M, Yagi H, Fujiwara M, Kojima Y, Sato H, Imamura S, Yokozawa M, Onodera N, Horigome H, et al. : Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol 2012, 110:586–593. [DOI] [PubMed] [Google Scholar]
- 51.Wang XJ, Lian TY, Jiang X, Liu SF, Li SQ, Jiang R, Wu WH, Ye J, Cheng CY, Du Y, et al. : Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur Respir J 2019, 53. [DOI] [PubMed] [Google Scholar]
- 52.Wang G, Fan R, Ji R, Zou W, Penny DJ, Varghese NP, Fan Y: Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: a case report. BMC Pulm Med 2016, 16:17. [DOI] [PMC free article] [PubMed] [Google Scholar]