

Abstract
Despite mounting evidence suggesting the involvement of the immune system in regulating brain function, the specific role of immune and inflammatory cells in neurodegenerative diseases remain poorly understood. Here we report that depletion of Natural Killer (NK) cells, a type of innate lymphocytes, alleviates neuroinflammation, stimulates neurogenesis, and improves cognitive function in a triple transgenic AD mouse model (3xTg-AD). NK cells in the brains of 3xTg-AD mice exhibited an enhanced pro-inflammatory profile. Depletion of NK cells by anti-NK1.1 antibodies drastically improved cognitive function of 3xTg-AD mice. NK cell depletion did not affect amyloid beta concentrations, but enhanced neurogenesis and reduced neuroinflammation. Notably, in 3xTg-AD mice depleted of NK cells, microglia demonstrated a homeostatic-like morphology, decreased proliferative response and reduced expression of neuro-destructive pro-inflammatory cytokines. Together, our results suggest a pro-inflammatory role for NK cells in 3xTg-AD mice, and indicate that targeting NK cells might unlock novel strategies to combat AD.
Introduction
Alzheimer’s disease (AD) is a devastating disease with unmet therapeutic needs. A hallmark of AD is chronic neuroinflammation mediated by dysregulated microglia (1–4). Microglia, the predominant brain-resident immune cells, appear to play dual wedged roles in the pathogenesis of Alzheimer’s disease (3, 5–7). While microglial phagocytosis is important in restraining the accumulation of amyloid beta and other toxic substances, deregulated microglia activation may lead to chronic neuroinflammation, synaptic loss, and impaired neurogenesis (3, 5–7). Chronic microglial activation in AD is demonstrated by multiple dynamic changes including altered morphology, abnormal proliferation, and increased secretion of neurotoxic pro-inflammatory mediators (8, 9). The cellular and molecular pathways that regulate microglia activity and brain inflammation in AD, however, remain largely unclear.
Increasing evidence indicates that no-glia immune cells might also play important roles in AD pathogenesis. The discovery that Aβ possesses anti-microbial activities has led to interesting infectious-disease hypotheses of Alzheimer’s disease (10–13). Peripheral innate immune cells, such as neutrophils, have been found to play important roles in promoting cognitive decline in mouse models of AD (14). Recent evident also suggests implication of lymphocytes in AD progression. Genetic deletion of lymphocytes in 5xFAD mice resulted in accelerated amyloid pathology and exacerbated neuroinflammation (15). In addition, amplification of regulatory T cells (Tregs) delayed progression of AD-like pathology in APPPS1 transgenic mice (16–18). Our recent work also indicates that activation of group-2 innate lymphoid cells can alleviate aging-associated cognitive decline (19). The precise roles of other adaptive and innate lymphocytes in AD development and exacerbation, however, remain poorly understood.
NK cells are innate cytotoxic lymphocytes that can be rapidly induced to secrete cytotoxic molecules and inflammatory cytokines such as granzymes, cathepsins, perforins, IFNγ and TNFα (20). NK cells are known for their capability to kill infected and malignant cells and to mediate antibody dependent cellular cytotoxicity. NK cells might also be involved in inflammatory disorders. Certain subsets of NK cells may produce multiple inflammatory cytokines and chemokines (21). The cytotoxic molecules secreted by NK cells, such as granzymes and cathepsins, may also possess proinflammatory functions when released into the extracellular micro-environment (22–28). However, the precise roles for NK cells in tissue homeostasis and inflammation remain largely unknown.
Here, we used a triple-transgenic AD mouse model (3xTg-AD) and anti-NK1.1 depleting antibodies to interrogate the role of NK cells in AD-associated cognitive decline. We found that NK cells exhibited an enhanced proinflammatory profile in a triple-transgenic mouse model of Alzheimer’s disease (3xTg-AD). Depletion of NK cells by anti-NK1.1 treatment significantly improved cognitive function of 3xTg-AD mice. Depletion of NK cells did not affect amyloid pathology, but repressed neuroinflammation and stimulated neurogenesis. Notably, microglia in NK-cell-depleted 3xTg-AD mice exhibited a homeostatic-like morphology, reduced proliferative responses, and decreased expression of multiple pro-inflammatory cytokines. Together, our results reveal a striking role for NK cells in promoting neuroinflammation and AD-associated cognitive decline, and indicate that targeting NK cells might unlock novel strategies to combat neurodegenerative diseases.
Materials and Methods
Mice
3xTg-AD and control B6129SF2/J were obtained from MMRRC JAX or the Jackson Laboratory, and bred in the animal facility of Albany Medical College. 7–8 months old female mice were used in this study. For anti-NK1.1 treatment, mice were treated with 25μg anti-NK1.1 (clone PK136, Bio X Cell) antibodies or isotype control (clone C1.18.4, Bio X Cell) every 4 days for 4 weeks. For anti-CD1d treatment, mice were treated with 500μg anti-CD1d (clone 19G11, Bio X Cell) antibodies or isotype control every other day for 4 weeks. Water Maze tests were performed on the day after the last treatment. Specifically, for anti-Nk1.1 treatment, mice were treated with anti-NK1.1 antibodies or isotype controls on day 1, 5, 9, 13, 17, 21, 25, 29; and Water Maze test was performed on day 30. For anti-CD1d treatment, mice were treated with anti-CD1d antibodies or isotype control on day 1, 3, 7, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27, 29; and Water Maze test was performed on day 30. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Albany Medical College.
Isolation of hematopoietic cells in the whole brain tissue
For isolation of hematopoietic cells in the whole brain tissue, mice were perfused with 50 ml of PBS. The whole brain tissue, including brain parenchyma, leptomeninges, choroid plexus, and perivascular space tissue, was harvested. The tissue was minced with scissors, and digested with 0.2 mg/ml of Liberase (Roche) and 0.1 mg/ml DNase I (Roche) for 30 mins at 37°C. Cells were filtered through a 70 μM strainer followed by gradient centrifugation with 40% Percoll (GE).
Flow Cytometry and Cell Sorting
Antibodies were purchased from Biolegend or Thermo. Antibodies for flow cytometry analysis included anti-B220 (RA3–6B2), anti-NK1.1 (PK136), anti-CD11b (M1/70), anti-CD3 (2C11), anti-CD45.2 (104), and anti-Ki67 (16A8). Mouse CD1d tetramers PBS57 were obtained from the NIH tetramer core (29). Intracellular staining of Ki67 was performed using the Foxp3 Fix/perm Kit (Thermo) according to the manufacturer’s instructions. EdU was detected using the Click-iT Plus EdU Flow Cytometry Assay Kit (Thermo) following the manufacturer’s instructions. Flow cytometric analysis was performed using a FACSCanto analyzer (BD). Cell sorting was performed using a FACSAria II sorter (BD).
Measurement of β-Amyloid concentration and in vivo β-Amyloid uptake
The concentrations of soluble and insoluble β-Amyloid were measured using the LEGEND MAX™ β-Amyloid x-42 ELISA kit (Biolegend) according to the manufacturer’s instructions. Specifically, brain samples were homogenized using a glass homogenizer with 6 passes on ice in TBS containing protease inhibitors. Samples were centrifuged at 350000xg for 20min. The supernatant containing soluble β-Amyloid was collected. The pellet was resuspended in TBS containing 1% triton and centrifuged again for collection of insoluble β-Amyloid. ELISA assays were performed immediately.
For measurement of in vivo β-Amyloid uptake, mice were injected i.p. with methoxy-X04 (4920, Tocris Bioscience) at the dose of 10mg/kg. Mice were euthanized at 3h after methoxy-X04 injection and flow cytometry analysis were performed to examine microglia uptake of methoxy-X04.
Gene Transcription Analysis, Microarray and RNA-sequencing
For QPCR, RNeasy Plus Mini Kit (Qiagen) was used to extract mRNA from samples. cDNA was synthesized using the Superscript II kit (Thermo). QPCR was performed with Taqman probes (Thermo).
RNA-seq was performed at the Center for Functional Genomics at the University at Albany. For bulk RNA-seq, cells were sorted by fluorescence activated cell sorting. cDNA was generated with the SMART-Seq v4 Ultra Low Input RNA kit (Takara) according to the manufacturer’s instructions. libraries were prepared using Nextera XT DNA Library Prep Kit (Illumina). Libraries were sequenced with a using NextSeq 500 (Illumina). Single-end 75 bp high throughput sequencing was performed. Data were aligned and normalized using STAR aligner. Differentially expressed genes were identified by DEseq2. Gene pathway analysis was performed with DAVID (30, 31).
For single-cell RNA-seq, cells were sorted by flow fluorescence activated cell sorting. Libraries were generated by 5’ gene expression kit (10Xgenomics) using the Chromium single-cell controller (10Xgenomics). Double-end 75 bp high throughout RNA-seq was performed using NextSeq 500 (Illumina). Initial data analysis was performed by Cellranger 3.1.1. Data were normalized, scaled, and mitochondria regression was performed using Seurat 3.1.1. Uniform Manifold Approximation and Projection (UMAP) was used for cell clustering. A wilcoxon rank-sum test was used to determine significance of differentially expressed genes.
scRNA-seq data for NK cells and microglia, and bulk RNA-seq data for microglia were deposit in the Gene Expression Omnibus under the accession number of GSE142853 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142853), GSE142858 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142858), and GSE142875 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142875), respectively.
Water Maze Test
Morris Water Maze was performed at 4 weeks after anti-NK1.1 treatment as we previously described (32). Specifically, mice for Water Maze Test were housed with 5 mice per Allentown cage. Mice were habituated to the testing facility for one hr before the behavior tests each day. A circular pool of opaque water with a diameter of 125 cm was used. Water temperature was kept at 21–22°C. The maze was conceptually divided into 4 quadrants, with visual cues on each side of the pool. On day 1 (visual trial), mice were trained to escape the maze by swimming to a clear plastic platform in the target quadrant. The platform was submerged by 1cm, and made visible by black tape around the platform and a 10 cm black cylinder on top of it. Mice were trained for 5 trials of up to 3 mins each, until the mouse found that platform and stayed on it for 10 secs. If a mouse failed to escape the maze within 3 mins, it was guided slowly to the platform by dragging its tail and stayed on the platform for 10 secs. On day 2 (Hidden trial), mice were trained to escape the maze with the same platform but the visual cues on the platform were removed. Mice were again trained for 5 trials for up to 3 mins each. On day 3 (Probe trial), the platform was removed, and the mice were assessed by one test of 3 mins. The behavior tests were recorded and analyzed by using the ANY-maze software (Stoelting Co.).
Immunofluorescence
For immunofluorescence, mice were perfused with 50 ml of PBS followed by 50 ml of 4% paraformaldehyde. The brains were harvested and fixed for 24 hrs in 4% paraformaldehyde. The samples were transferred to 30% sucrose in PBS, and frozen at OCT in −80°C until sectioning. at 40 μM sections were prepared using a Leica CM1950 cryostat. For staining of microglia, tissues were stained with goat anti-Iba-1 (Thermo), and Cy5-conjugated anti-goat secondary antibody (Jackson ImmunoResearch). Slides were imaged using a Zeiss Axio Observer fluorescence microscope with a 20X objective. The Zeiss Zen Blue software (Zeiss) was used to process the images.
For in vivo EdU labeling, mice were injected i.p. with 6 doses of EdU (100 mg/kg) over 2 days. EdU was detected by the Click-iT Plus EdU Imaging Kit (Invitrogen) according to the manufacturer’s instructions. After EdU detection, the sections were stained with anti-NeuN rabbit polyclonal ab (A60) (Millipore) and anti-rabbit Rhodamine secondary ab (Jackson). Slides were imaged using a Zeiss Axio Observer fluorescence microscope with a 10X objective. Images were processed by the Zeiss Zen Blue software (Zeiss).
Statistical analysis
Mann–Whitney U test was used to compare the differences in Water Maze Tests. Wilcoxon rank-sum test was used to compare gene expression difference between two groups for scRNA-seq data. Student’s t tests were used to calculate statistical significance for all other data. P<0.05 was considered significant.
Results
NK cells exhibit an enhanced proinflammatory profile in 3xTg-AD mice
Previous studies indicate that NK cell activity is altered in Alzheimer’s disease patients and mouse models of AD (33–41). However, the precise transcriptomal changes of NK cells in the inflamed microenvironment of AD remains unclear. Here we examined NK cells of middle-aged (7–8 months) triple transgenic 3xTg-AD and control wildtype mice. At this age, 3xTg-AD mice exhibit declined cognitive function, immunoreactivity to amyloid beta, and mild plaques (42–44). NK cells were identified as CD45+CD3−B220−NK1.1+DX5+ lymphocytes (Fig. 1A). The number of NK cells was moderately decreased in the whole brain tissue of 3xTg-AD mice, compared to wildtype control mice (Fig. 1B). Of note, in study, we examined NK cells in the whole brain tissue, including brain parenchyma, choroid plexus, leptomeninges, and perivascular space tissue. We noted that NK cells were enriched in the barrier tissues (choroid plexus, leptomeninges, and perivascular space tissues), whereas brain parenchyma was devoid of NK cells (Fig. S1A). We thus refer to these NK cells as “brain-associated NK cells”. To obtain sufficient numbers of brain-associated NK cells for in-depth transcriptomal analysis, we isolate NK cells from the whole brain tissue for single-cell RNA-seq (scRNA-seq). We sorted NK cells from the whole brain tissue by fluorescence activated cell sorting (FACS) and performed scRNA-seq. Recent work indicates that spleen and blood NK cells can be broadly divided into two subsets: Thy1−CD7−/low NK1 cells that express high amounts of cytotoxic molecules and Thy1+CD7hi NK2 cells that express Xcl1 and other effector molecules (45). The heterogeneity and single-cell transcriptomes of brain-associated NK cells remain unknown. Using Uniform Manifold Approximation and Projection (UMAP) analysis (46), our scRNA-seq results indicated that the majority of brain-associated NK cells were Thy1−CD7−/low NK1 cells that express relatively high amounts of cytotoxic molecules (Fig. 1C, 1D). Expression of Ifng was minimal among all brain-associated NK cell subsets (Fig. 1D); and expression of Tnf and Il17a was not detectable (not shown). Interestingly, UMAP also identified a subset of NK cells with a very distinct transcriptomal profile in 3xTg-AD mice, termed NK1AD here (Fig. 1C). NK1AD cells were marked by high expression of cytotoxic molecules Cstb and Ctsc (Fig. 1D). This subset was hardly distinguishable in control wildtype mice (Fig. 1C). Gene enrichment pathway analysis demonstrated that cytotoxic molecules and lymphocyte receptor signaling molecules were overrepresented in genes highly expressed in NK1AD cells (Fig. 1E). In particular, among all NK cell subsets, NK1AD cells expressed the highest amounts of the cytotoxic molecules Ctsc, Ctsd, pro-inflammatory chemokines Ccl3, Ccl4, and the adhesion molecule Icam1. The lymphocyte receptor signaling molecules highly expressed on NK1AD were predominantly NK cell activation molecules including Nfatc1, Tbx21, Nfkbia, Il12rb, and Klra9, suggesting that they are a hyperactivate proinflammatory subset (Fig. 1E–G). Other than NK1AD cells, the other NK1 cells in the brains of wildtype and 3xTg-AD mice can be further divided into two subsets, named NK1a and NK1b here (Fig. 1C). NK1a cells expressed lower amounts of cytotoxic molecules and activating genes than NK1b and NK1AD subset, suggesting that they might be at a relatively inactive state (Fig. 1D, 1G). Interestingly, compared to than those in control wildtype mice, NK1a and/or NK1b subsets in 3xTg-AD mice expressed higher amounts of cytotoxic and other effector molecules such Ctsc, Ctsd, Ccl3, and Ccl4, indicating enhanced activity (Fig. 1H). NK cells in 3xTg-AD mice also expressed higher amounts of the adhesion molecule ICAM1, which was verified by flow cytometric analysis (Fig. 1H, S1B, S1C). We were not able to obtain reliable antibodies to examine Ctsc and Ctsd protein levels by flow cytometric analysis, thus future efforts to verify changes in Ctsc and Ctsd protein expression would be worthwhile. Together, the brains in 3xTg-AD mice harbor a unique subset of NK1AD subset with a hyperactive proinflammatory profile and their other NK subsets also exhibit an enhanced pro-inflammatory profile than those in control wildtype mice.
Figure 1. NK cells in 3xTg-AD mice exhibited an enhanced pro-inflammatory profile.

A, Representative flow cytometry profiles of NK cells in 7–8 months 3xTg-AD and control mice. Plots were pre-gated on brain CD45+CD3−B220− lymphocytes. B, Numbers of NK cells in 7–8 months 3xTg-AD and control mice. C, UMAP analysis of sorted NK cells in 7 month old 3xTg-AD and control mice by single-cell RNA-seq (scRNA-seq). D, Expression of individual genes in sorted NK cells by scRNA-seq. E, Pathways of genes highly expressed in NK1AD subset. F, List of presentative genes highly expressed in NK1AD subset. G, Violin plots depicting expression of the indicated genes in each NK cell subsets. H, Expression of the indicated genes in NK1a and NK1b subsets in 3xTg-AD and control mice. Data are from 6 mice per group, pooled from two independent experiments (A and B), or are pooled from 6 mice per group (C-G). *p<0.05; **p<0.01.
Notably, all three brain-associated NK1 subsets, including NK1AD, express high amounts of the trafficking molecule S1pr5 (Fig. S1D). The expression of genes characteristic of tissue-resident lymphocytes, such as Itgae and CD69, was barely detectable in brain-associated NK Cells (Fig. S1D). Thus, NK1AD might be circulating NK cells that were recruited to brain barrier tissues.
Depletion of NK cells alleviates cognitive decline in 3xTg-AD mice.
We next sought to understand whether the presence of NK cells might influence the cognitive function of 3xTg-AD mice. 3xTg-AD mice exhibit progressive loss of cognitive function as early as 3–6 months, and impairment in cognitive function is very obvious in 7–8 months old (44). We depleted NK cells in middle-aged 3xTg-AD mice using anti-NK1.1 neutralizing antibody. Mice were treated with antibodies for 1 month, to allow sufficient time for potential effects of immune cells to take place. Flow cytometry analysis verified efficient depletion of NK cells (Fig. 2A). We used a Morris Water Maze test (47) to examine the cognitive function of 3xTg-AD mice that were treated with anti-NK1.1 antibody or isotype control (Fig. 2B). Mice were trained with a visible platform on day 1, followed by training with an invisible platform at the same place on day 2. Mice were then allowed to freely swim for 3 mins in the probe trial (no platform) on day 3. The activity and behavior of mice in the probe trial were recorded. We compared the mobility and cognitive function indexes between mice treated with isotype control and those treated with anti-Nk1.1 antibodies. NK cell depletion did not affect the general mobility of 3xTg-AD mice, demonstrated by comparable mean swimming speed between mice treated with anti-NK1.1 antibodies and those treated with isotype controls (Fig. 2C). Notably, the percentage of time spent in the target quadrant by isotype-treated 3xTg-AD mice was comparable to the predicted chance level (25%) in the probe trial, indicating a loss of hippocampal-dependent spatial recognition (Fig. 2D). Mice treated with anti-NK1.1 antibodies, however, spent significantly higher percentage of time in the target quadrant, suggesting improved spatial memory (Fig. 2D). Compared to control mice, mice treated with anti-NK1.1 antibodies also exhibited increased numbers of entries into target quadrant, reduced latency and increased path efficiency, verifying improved cognitive function (Fig. 2E–H). Together, multiplex indicators suggest that NK cell depletion by anti-NK1.1 treatment alleviated the cognitive decline of 3xTg-AD mice.
Figure 2. Depletion of NK cells improved cognitive function in 3xTg-AD mice.
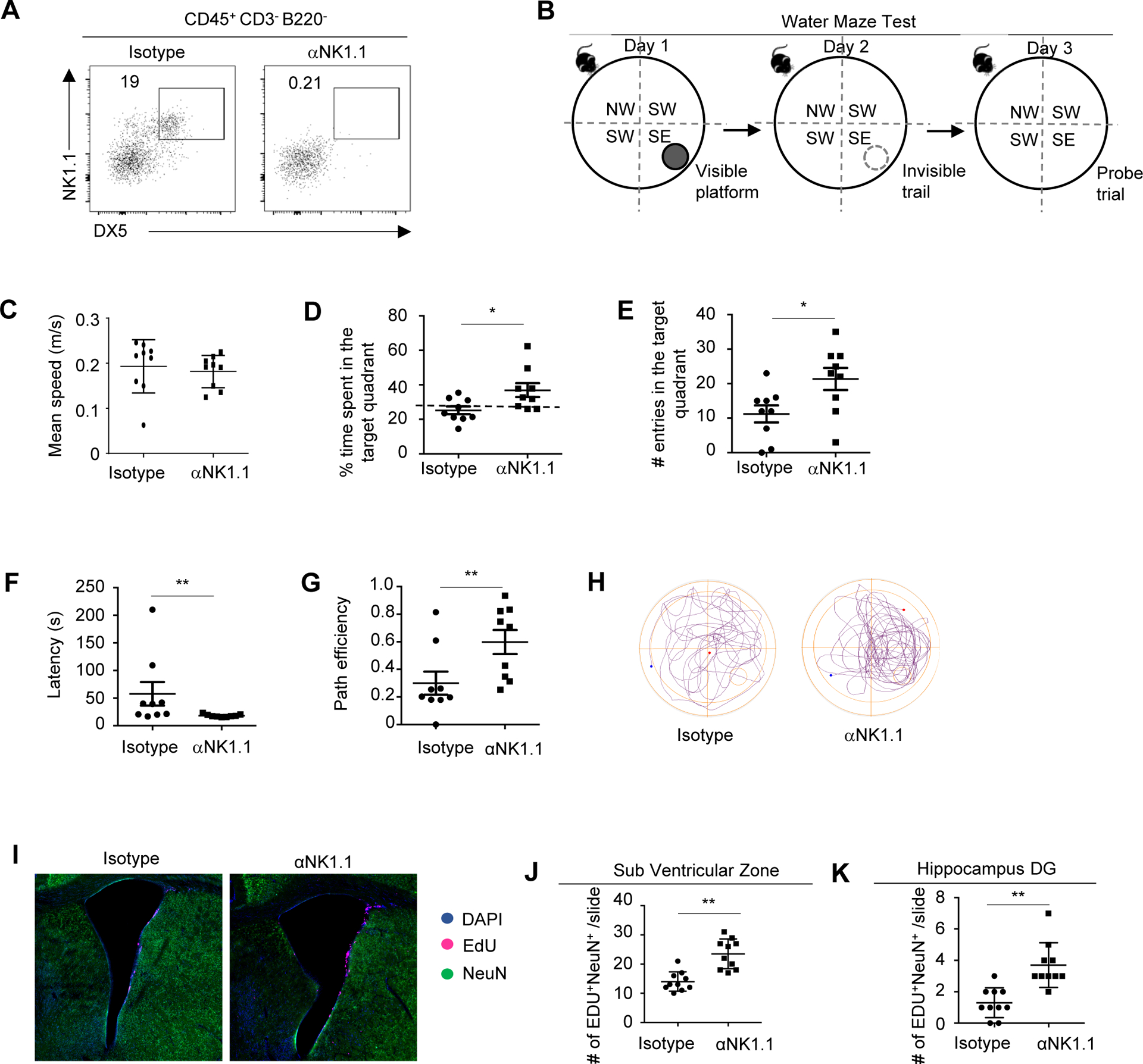
A, Representative flow cytometry profiles of NK cells in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. Plots were pre-gated on brain CD45+CD3−B220− lymphocytes. B, Experimental scheme of Water Maze Test. C, Mean swimming speed of 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls in the probe trial. D, Time spent in the target quadrant by 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls in the probe trial. E, Numbers of entries into the target quadrant in the probe trial. F, Latency to enter the target quadrant in the probe trial. G, Path efficiency of entering the target quadrant in the probe trial. H, Representative path profiles in the probe trial. I. Representative immunofluorescence imaging depicting Edu+ neurons in the SVZ region. J, Numbers of EdU+ neurons in SVZ regions. K. Numbers of EdU+ neurons in hippocampus dentate gyrus regions. DG, dentate gyrus. Data represents 3 independent experiments (A); or are from 9 mice per group and represent two independent experiments (B-H); or from 10 mice per group pooled from two independent experiments (I-K). *p<0.05; **p<0.01.
Of note, anti-NK1.1 depletion antibodies may also deplete NKT cells. We thus examined NKT cells in 3xTg-AD mice using CD1d tetramers. Notably, NKT cells were barely detectable in the whole brain tissue of 3xTg-AD mice (Fig. S2A). In addition, treatment with CD1d neutralizing antibodies did not significantly affect the cognitive function of 3xTg-AD mice (Fig. S2B). Thus, NK cells, but not NKT cells, might play a more predominant role in promoting cognitive decline in 3xTg-AD mice.
Depletion of NK cells stimulated neurogenesis, but did not affect amyloid beta concentrations in 3xTg-AD mice.
Declined neurogenesis is another notable feature of AD, which may contribute to declined cognitive function in AD (48). We thus examined neurogenesis in 3xTg-AD mice after 1 month of treatment with anti-NK1.1 depleting antibody or isotype control. We injected mice with six doses of 5-Ethynyl-2´-deoxyuridine (EdU) in 2 days to label proliferating cells and then co-labeled cells with NeuN to identify new neurons that had been born and survived to maturity. Compared to control mice, NK cell-depleted mice exhibited decreased number of EdU+ NeuN+ cells in the subventricular zone (SVZ) and also in the hippocampal dentate gyrus (DG) region, indicating enhanced neurogenesis (Fig. 2, I–K). Thus, depletion of NK cells stimulates neurogenesis in 3xTg-AD mice.
We next examined the effects of NK cell on β-amyloid pathologies. Results with ELISA assays indicated that the concentrations of soluble and insoluble β-amyloid in the brain of 3xTg-AD mice were not significantly altered by NK cell depletion (Fig. 3, A and B). To further examine whether anti-Nk1.1 depletion might influence microglia uptake of b-amyloid, we injected mice with methoxy-X04, a fluorescence probe for β-amyloid that can cross blood-brain barrier. Percentages of methoxy-X04+ Microglia were comparable in 3xTg-AD mice treated with anti-NK1.1 depleting antibodies and those treated with isotype controls, indicating that NK cell depletion did not affect β-amyloid uptake by microglia (Fig. 3, C and D). Together, NK cell depletion might improve cognitive function through mechanisms other than regulation of β-amyloid pathologies.
Figure 3. Depletion of NK cells did not affect amyloid beta concentrations.
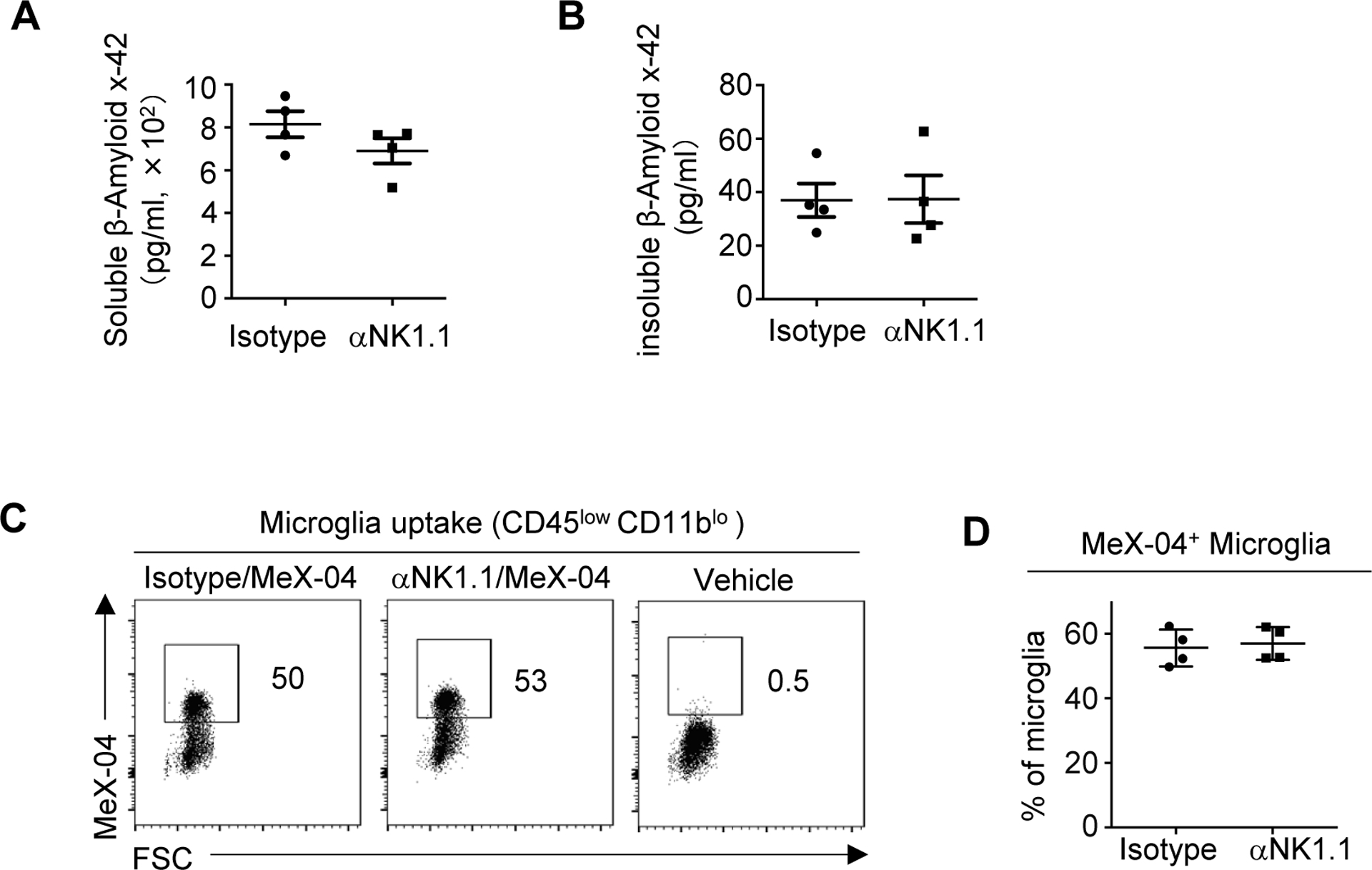
A, Concentrations of soluble b-amyloid x-42 measured by ELISA in 7–8 month old 3xTg-AD mice injected with MeX-04 and treated with anti-NK1.1 antibodies or isotype controls. B, Concentrations of insoluble b-amyloid x-42 measured by ELISA. C, Presentative flow cytometry profiles depicting uptake of b-amyloid by microglia in 7–8 month old 3xTg-AD mice injected with MeX-04 and treated with anti-NK1.1 antibodies or isotype controls. D, Percentage of MeX-04+ microglia in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. Data are from 4 mice per group and represent two independent experiments. *p<0.05; **p<0.01.
Depletion of NK cells ameliorated neuroinflammation in 3xTg-AD mice.
A hallmark of AD is neuroinflammation mediated by dysregulated microglia (4–9). We thus sought to determine whether NK cell depletion might affect neuroinflammation in 3xTg-AD mice. We first used flow cytometry analysis to examine microglia in 3xTg-AD mice treated with anti-NK1.1 antibody or isotype controls. The number of microglia was moderately reduced after NK cell depletion (Fig. 4A–B). Interestingly, microglia in NK cell-depleted 3xTg-AD mice demonstrated remarkedly reduced forward scatter (FSC) values than microglia in control 3xTg-AD mice, suggesting that NK cell depletion might alter the morphology of NK cells (Fig. 4C–D). Indeed, immunofluorescence assays indicated that microglia in NK cell-depleted 3xTg-AD mice exhibited a homeostatic-like morphology, in contrast to the more amoeboid-like morphology in control 3xTg-AD mice (Fig. 4E). We next performed bulk RNA-seq to examine the transcriptomal changes in microglia. Notably, RNA-seq suggested that anti-NK1.1 treatment reduced the expression of many proliferative genes such as Mik67 and Cdc42, as well as pro-inflammatory genes such as Tnf and Il1 (Fig. 4F–H). Consistent with decreased expression of proliferative genes, microglial proliferative responses in 3xTg-AD mice were significantly reduced by anti-NK1.1 treatment, demonstrated by increased proportion of microglia in the quiescent G0 phase and reduced percentage of cells in the activating cycling S/G2 phases (Fig4. I–J). QPCR verified that depletion of NK cells led to greatly reduced expression of Mki67 as well as pro-inflammatory cytokines Tnf, Il1a, Il1b and Il18, by microglia (Fig4. K). Together, these results indicate that depletion of NK cells lead to reduced microglial inflammation in 3xTg-AD mice.
Figure 4. Depletion of NK cells reduced neuroinflammation.
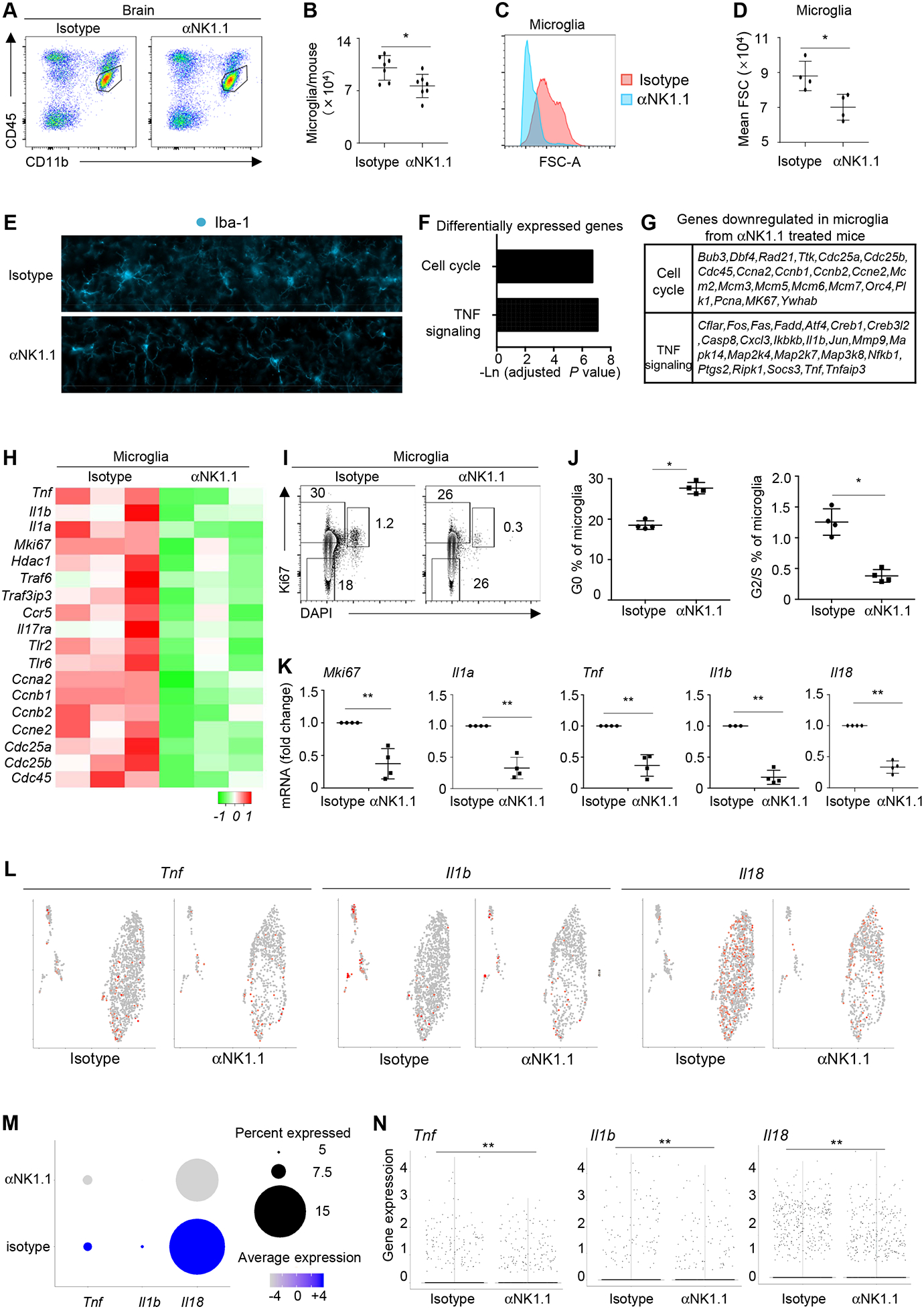
A, Representative flow cytometry profiles of microglia in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. B, Numbers of microglia in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. C, Histogram depicting forward scatter-A (FSC-A) of microglia in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. D, Mean FSC-A of microglia in 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. E, Representative immunofluorescence imaging of microglia from 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. F, RNA-seq was performed with sorted microglia from 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. Pathways of differentially expressed genes were identified. G, List of representative genes that were down-regulated in microglia from mice treated with anti-NK1.1 antibodies. H, Heatmap of representative genes whose expression is downregulated in microglia from 3xTg-AD mice treated with anti-NK1.1 antibodies. I, Flow cytometry profiles of Ki67 and DAPI staining in microglia from 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies and isotype controls. J, Percentages of microglia in G0 or S/G2 phases. K, Expression of the indicated genes by QPCR. L, scRNA-seq analysis of the indicated genes expressed by microglia from 7–8 month old 3xTg-AD mice treated with anti-NK1.1 antibodies or isotype controls. M, Percentages of microglia that express the indicated genes and the average gene expression by scRNA-seq. N, Violin plots depicting expression of the indicated genes by scRNA-seq. Data are from 7 mice per group, pooled from two independent experiments (A-B), or from 4 mice per group, representative of two independent experiments (C-E, I-J), or from four independent experiments (K), or from 3 mice per group (F-H), or pooled from 4 mice per group (J-M). *p<0.05; **p<0.01.
We next performed scRNA-seq analysis to better understand the effects of NK cell depletion on the transcriptomal changes of microglia in 3xTg-AD mice. Interestingly, UMAP analysis indicated that the expression of proinflammatory cytokines such as Il18 were broadly spread among all microglia clusters in 3xTg-AD mice, indicating that the capability to produce proinflammatory cytokines were not restricted to a specific microglia subset in 3xTg-AD mice (Fig. 4L). Depletion of NK cells by anti-NK1.1 treatment led to reduced frequency of microglia expressing proinflammatory cytokines Il18 and Il1b (Fig. 4L–N). More notably, the average expression of many proinflammatory cytokines including Il18, Tnf, and Il1b by cytokine-expressing microglia was markedly reduced in mice treated with anti-NK1.1 antibodies (Fig. 4L–N). Thus, depletion of NK cells may both reduce the frequency of microglia that express pro-inflammatory cytokines, and also decrease the cytokine production by microglia at per cell level.
Of note, anti-NK1.1 treatment did not affect the expression of Trem2 on microglia, indicating that depletion of NK cells might not affect the general fitness of microglia (Fig. S3A). Other innate immune cells, such as neutrophils, may also promote microglial inflammation in mouse models of Alzheimer’s disease (14). However, we did not detect significant changes in neutrophil numbers in the whole brain tissue of 3xTg-AD mice following anti-NK1.1 treatment (Fig. S3B, S3C). In addition, depletion of neutrophils reduces Aβ burdens in mouse models of Alzheimer’s disease (14); whereas depletion of NK cells improves cognitive function without affecting Aβ burdens (Fig. 3A, 3B). Thus, NK cells and neutrophils might promote neuroinflammation through distinctive mechanisms.
Discussion
Our work indicates a striking role for NK cells in promoting neuroinflammation and exacerbating cognitive decline in 3xTg-AD mice. Depletion of NK cells repressed neuroinflammation, stimulated neurogenesis and improved cognitive function in 3xTg-AD mice. Amyloid beta concentrations, however, were unaffected by NK cell depletion. This study thus establishes a novel role for NK cells in exacerbating AD-associated cognitive decline and suggests that targeting neuroinflammation may unlock new therapies to combat AD.
Several previous studies have indicated abnormal NK cell function in human patients of Alzheimer diseases (33–41). The results from these studies, however, were not entirely coherent (33–41). Some studies indicate increased functional capability of NK cells in Alzheimer’s disease; whereas other suggest decreased NK cell function (33–41). This might be partly because different in vitro assays have been employed to investigate NK cell function in different studies (33–41). In addition, the various stimulators used in in vitro assays could mask the homeostatic function of NK cells in physiologic conditions. Using scRNA-seq, our study provides an unbiased understanding of the physiologic biology of brain-associated NK cells in 3xTg-AD mice and control wildtype mice. Similar experiments in human patient samples would be highly worthwhile in future efforts.
Our results indicate that brain-associated NK cells are predominantly cytotoxic NK1 subsets with minimal cytokine expression. Ifng expression was barely detectable in brain-associated NK cells, and Tnf expression was not detected. Interestingly, such transcriptomal profile of brain-associated NK cells differs from previously published NK cell profile in the mouse spleen where equivalent percentages of NK1 and NK2 cells have been observed (45). Of note, the brain is a delicate organ in which excessive expression of stimulating cytokines might lead to significant detrimental effects. Thus, the nearly absence of cytokine-expressing NK cells might represent a protective mechanism that may help maintain brain homeostasis. Our results also indicate a hyper-active profile of NK cells in 3xTg-AD mice, which might be explained by several mechanisms. Aβ oligomers may directly bind to circulating lymphocytes through receptors such as RAGE and TLRs, the ligation of which may activate NK cells. The pro-inflammatory microenvironment in AD mice and humans, such as increased expression of IL-18, may also contribute to NK cell hyperactivation. Thus, a positive feedback loop might be formed between NK cell hyper-activation and microglial inflammation in AD, which might contribute to exacerbated neuroinflammation and cognitive decline.
Our data have revealed striking effects of NK cell depletion on improving cognitive function in 3xTg-AD mice. Of note, NK cells possess important beneficial function in healthy individuals, such as preventing tumor development and progression. Thus, long-term depletion of NK cells, particularly in humans, might incur unwanted side effects. As such, future exploration of more targeted therapies is warranted, which necessitates a more in-depth understanding of the specific molecular pathways by which NK cells promote neuroinflammation. Our results indicate that brain-associated NK cells predominantly express cytotoxic molecules such as granzymes and cathepsins. The roles of cytotoxic molecules and NK cells in immune and inflammatory disorders, beyond their cytotoxic function, remain poorly understood. Yet, increasing evidence indicates that, when secreted into the extracellular micro-environment, cytotoxic molecules such as cathepsins and granzymes can have important function in triggering myeloid cell activation and in processing pro-inflammatory cytokines (22, 23, 25–28). Granzymes have been found to promote the production and maturation of proinflammatory cytokines in myeloid cells and other innate cells (24, 25, 49–55). Cathepsins may also play important roles in processing pro-inflammatory cytokines and in modifying tissue microenvironment (22, 23, 56–60). NK cells are enriched in brain barrier tissues (choroid plexus, perivascular space, leptomeninges). In addition to their barrier function, these tissues also play essential roles in generating cerebrospinal fluid (CSF) that nitrifies the brain parenchyma. We thus speculate that, when released into the CSF, the cytotoxic molecules produced by NK cells might influence the behaviors of brain parenchymal cells such as microglia. And dysregulation of this process may underlie the pro-inflammatory property of brain-associated NK cells in AD.
Another characteristic feature of AD is sharply declined neurogenesis (48). Neurogenesis remains active in the SVZ, and to a less degree, in the hippocampus regions of healthy adult humans and mice (48, 61). Adult neurogenesis was diminished in the inflamed brains of AD patients and transgenic AD mouse models, which might contribute to declined cognitive function in AD (48, 62). We have found that NK cell depletion significantly enhanced neurogenesis in both the SVZ regions and in the hippocampus. The enhanced neurogenesis might be due to reduced neuroinflammation. Improved neurogenesis, as well as other beneficial effects conferred by reduced neuroinflammation, likely together contributes to the alleviated cognitive decline in NK cell-depleted 3xTg-AD mice.
Alzheimer’s disease remains a devastating disease with unmet therapeutic needs. The development of successfully treatments relies on a comprehensive understanding of the cellular and molecular mechanisms involved. Although amyloid beta pathologies are the defining features of AD, therapeutic efforts that target amyloid beta have been futile, indicating the importance of other mechanistic pathways (63). In this work, we show that NK cell depletion significantly improves cognitive function of 3xTg-AD mice without affecting amyloid beta concentrations. Strikingly, in NK cell depleted 3xTg-AD mice, microglia demonstrated homeostatic-like morphology, reduced proliferative response and decreased expression of proinflammatory cytokines. Interestingly, these microglia retained the capability to uptake amyloid-beta, indicating that NK cell depletion does not appear to affect the phagocyte capability of microglia. These microglia also exhibited intact expression of Trem2, a surface receptor that plays an essential role in promoting microglial metabolic fitness (64). Thus, depletion of NK cells might specifically reduce the proinflammatory property of microglia without affecting the general fitness of microglia. Together these results suggest that the presence of NK cells is pivotal in promoting neuroinflammation in 3xTg-AD mice. Our work thus indicates that targeting NK cells and neuroinflammation might provide new avenues to combat AD.
Supplementary Material
KEY POINTS.
NK cells exhibit enhanced proinflammatory phenotype in 3xTg-AD mice.
Depletion of NK cells enhances cognitive function of 3xTg-AD mice.
Depletion of NK cells alleviates neuroinflammation in 3xTg-AD mice.
Acknowledgments
This work was supported by the U.S. National Institutes of Health Grants R01HL137813 (to Q.Y.), R01AG057782(to Q.Y.), and R01NS110749 (to K.L.Z.)
Footnotes
Dr. Qi Yang reported a patent (U.S. Patent Application No.: 62/822,159). The authors declare no additional conflict of interest.
REFERENCES
- 1.Bradburn S, Murgatroyd C, and Ray N. 2019. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res Rev 50: 1–8. [DOI] [PubMed] [Google Scholar]
- 2.Calsolaro V, and Edison P. 2016. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 12: 719–732. [DOI] [PubMed] [Google Scholar]
- 3.Chaney A, Williams SR, and Boutin H. 2019. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J Neurochem 149: 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regen F, Hellmann-Regen J, Costantini E, and Reale M. 2017. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr Alzheimer Res 14: 1140–1148. [DOI] [PubMed] [Google Scholar]
- 5.Song WM, and Colonna M. 2018. The identity and function of microglia in neurodegeneration. Nat Immunol 19: 1048–1058. [DOI] [PubMed] [Google Scholar]
- 6.Hansen DV, Hanson JE, and Sheng M. 2018. Microglia in Alzheimer’s disease. J Cell Biol 217: 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McQuade A, and Blurton-Jones M. 2019. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J Mol Biol 431: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro V, Sanchez-Mejias E, Jimenez S, Munoz-Castro C, Sanchez-Varo R, Davila JC, Vizuete M, Gutierrez A, and Vitorica J. 2018. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front Aging Neurosci 10: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith JA, Das A, Ray SK, and Banik NL. 2012. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull 87: 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, and Moir RD. 2010. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, Frost EH, and Fulop T Jr. 2015. beta-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16: 85–98. [DOI] [PubMed] [Google Scholar]
- 12.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, and Moir RD. 2016. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: 340ra372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bode DC, Baker MD, and Viles JH. 2017. Ion Channel Formation by Amyloid-beta42 Oligomers but Not Amyloid-beta40 in Cellular Membranes. J Biol Chem 292: 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nani S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, and Constantin G. 2015. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 21: 880–886. [DOI] [PubMed] [Google Scholar]
- 15.Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, Fote GM, Lau L, Weinger JG, Lane TE, Inlay MA, Poon WW, and Blurton-Jones M. 2016. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 113: E1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, Jung J, Kim H, Lee S, Kim JS, Lee HJ, Shim I, Lee JH, and Bae H. 2016. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget 7: 69347–69357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciccocioppo F, Lanuti P, Pierdomenico L, Simeone P, Bologna G, Ercolino E, Buttari F, Fantozzi R, Thomas A, Onofrj M, Centonze D, Miscia S, and Marchisio M. 2019. The Characterization of Regulatory T-Cell Profiles in Alzheimer’s Disease and Multiple Sclerosis. Sci Rep 9: 8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, Cagnard N, Holzenberger M, Piaggio E, Aucouturier P, and Dorothee G. 2016. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139: 1237–1251. [DOI] [PubMed] [Google Scholar]
- 19.Fung ITH, Sankar P, Zhang Y, Robison LS, Zhao X, D’Souza SS, Salinero AE, Wang Y, Qian J, Kuentzel ML, Chittur SV, Temple S, Zuloaga KL, and Yang Q. 2020. Activation of group 2 innate lymphoid cells alleviates aging-associated cognitive decline. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abel AM, Yang C, Thakar MS, and Malarkannan S. 2018. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol 9: 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, and Zimmer J. 2016. Human CD56bright NK Cells: An Update. J Immunol 196: 2923–2931. [DOI] [PubMed] [Google Scholar]
- 22.Vidak E, Javorsek U, Vizovisek M, and Turk B. 2019. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zavasnik-Bergant T, and Turk B. 2006. Cysteine cathepsins in the immune response. Tissue Antigens 67: 349–355. [DOI] [PubMed] [Google Scholar]
- 24.Arias MA, Jimenez de Bagues MP, Aguilo N, Menao S, Hervas-Stubbs S, de Martino A, Alcaraz A, Simon MM, Froelich CJ, and Pardo J. 2014. Elucidating sources and roles of granzymes A and B during bacterial infection and sepsis. Cell Rep 8: 420–429. [DOI] [PubMed] [Google Scholar]
- 25.Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, and Granville DJ. 2011. Granzyme K activates protease-activated receptor-1. PLoS One 6: e21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, Kim S, Raja SM, Shi L, Simon MM, and Froelich CJ. 2008. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity 29: 720–733. [DOI] [PubMed] [Google Scholar]
- 27.Joeckel LT, Wallich R, Martin P, Sanchez-Martinez D, Weber FC, Martin SF, Borner C, Pardo J, Froelich C, and Simon MM. 2011. Mouse granzyme K has pro-inflammatory potential. Cell Death Differ 18: 1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Eck JA, Shan L, Meeldijk J, Hack CE, and Bovenschen N. 2017. A novel proinflammatory role for granzyme A. Cell Death Dis 8: e2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Goff RD, Zhou D, Mattner J, Sullivan BA, Khurana A, Cantu C 3rd, Ravkov EV, Ibegbu CC, Altman JD, Teyton L, Bendelac A, and Savage PB. 2006. A modified alpha-galactosyl ceramide for staining and stimulating natural killer T cells. J Immunol Methods 312: 34–39. [DOI] [PubMed] [Google Scholar]
- 30.Huang da W, Sherman BT, and Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 31.Huang da W, Sherman BT, and Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuloaga KL, Johnson LA, Roese NE, Marzulla T, Zhang W, Nie X, Alkayed FN, Hong C, Grafe MR, Pike MM, Raber J, and Alkayed NJ. 2016. High fat diet-induced diabetes in mice exacerbates cognitive deficit due to chronic hypoperfusion. J Cereb Blood Flow Metab 36: 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jadidi-Niaragh F, Shegarfi H, Naddafi F, and Mirshafiey A. 2012. The role of natural killer cells in Alzheimer’s disease. Scand J Immunol 76: 451–456. [DOI] [PubMed] [Google Scholar]
- 34.Martins LC, Rocha NP, Torres KC, Dos Santos RR, Franca GS, de Moraes EN, Mukhamedyarov MA, Zefirov AL, Rizvanov AA, Kiyasov AP, Vieira LB, Guimaraes MM, Yalvac ME, Teixeira AL, Bicalho MA, Janka Z, Romano-Silva MA, Palotas A, and Reis HJ. 2012. Disease-specific expression of the serotonin-receptor 5-HT(2C) in natural killer cells in Alzheimer’s dementia. J Neuroimmunol 251: 73–79. [DOI] [PubMed] [Google Scholar]
- 35.Masera RG, Prolo P, Sartori ML, Staurenghi A, Griot G, Ravizza L, Dovio A, Chiappelli F, and Angeli A. 2002. Mental deterioration correlates with response of natural killer (NK) cell activity to physiological modifiers in patients with short history of Alzheimer’s disease. Psychoneuroendocrinology 27: 447–461. [DOI] [PubMed] [Google Scholar]
- 36.Prolo P, Chiappelli F, Angeli A, Dovio A, Perotti P, Pautasso M, Sartori ML, Saba L, Mussino S, Fraccalini T, Fanto F, Mocellini C, Rosso MG, and Grasso E. 2007. Physiologic modulation of natural killer cell activity as an index of Alzheimer’s disease progression. Bioinformation 1: 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindowski K, Peters J, Gorriz C, Schramm U, Weinandi T, Leutner S, Maurer K, Frolich L, Muller WE, and Eckert A. 2006. Apoptosis of CD4+ T and natural killer cells in Alzheimer’s disease. Pharmacopsychiatry 39: 220–228. [DOI] [PubMed] [Google Scholar]
- 38.Solana C, Tarazona R, and Solana R. 2018. Immunosenescence of Natural Killer Cells, Inflammation, and Alzheimer’s Disease. Int J Alzheimers Dis 2018: 3128758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solerte SB, Cravello L, Ferrari E, and Fioravanti M. 2000. Overproduction of IFN-gamma and TNF-alpha from natural killer (NK) cells is associated with abnormal NK reactivity and cognitive derangement in Alzheimer’s disease. Ann N Y Acad Sci 917: 331–340. [DOI] [PubMed] [Google Scholar]
- 40.Solerte SB, Fioravanti M, Pascale A, Ferrari E, Govoni S, and Battaini F. 1998. Increased natural killer cell cytotoxicity in Alzheimer’s disease may involve protein kinase C dysregulation. Neurobiol Aging 19: 191–199. [DOI] [PubMed] [Google Scholar]
- 41.Solerte SB, Fioravanti M, Severgnini S, Locatelli M, Renzullo M, Pezza N, Cerutti N, and Ferrari E. 1996. Enhanced cytotoxic response of natural killer cells to interleukin-2 in Alzheimer’s disease. Dementia 7: 343–348. [DOI] [PubMed] [Google Scholar]
- 42.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, and LaFerla FM. 2003. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409–421. [DOI] [PubMed] [Google Scholar]
- 43.Billings LM, Oddo S, Green KN, McGaugh JL, and LaFerla FM. 2005. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45: 675–688. [DOI] [PubMed] [Google Scholar]
- 44.Stover KR, Campbell MA, Van Winssen CM, and Brown RE. 2015. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav Brain Res 289: 29–38. [DOI] [PubMed] [Google Scholar]
- 45.Crinier A, Milpied P, Escaliere B, Piperoglou C, Galluso J, Balsamo A, Spinelli L, Cervera-Marzal I, Ebbo M, Girard-Madoux M, Jaeger S, Bollon E, Hamed S, Hardwigsen J, Ugolini S, Vely F, Narni-Mancinelli E, and Vivier E. 2018. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 49: 971–986 e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McInnes LHJ; Melville J 2018. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 180203426. [Google Scholar]
- 47.Vorhees CV, and Williams MT. 2006. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1: 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, and Llorens-Martin M. 2019. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25: 554–560. [DOI] [PubMed] [Google Scholar]
- 49.Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, Reeves EP, McElvaney NG, Medema JP, Lavelle EC, and Martin SJ. 2011. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell 44: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irmler M, Hertig S, MacDonald HR, Sadoul R, Becherer JD, Proudfoot A, Solari R, and Tschopp J. 1995. Granzyme A is an interleukin 1 beta-converting enzyme. J Exp Med 181: 1917–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omoto Y, Yamanaka K, Tokime K, Kitano S, Kakeda M, Akeda T, Kurokawa I, Gabazza EC, Tsutsui H, Katayama N, Yamanishi K, Nakanishi K, and Mizutani H. 2010. Granzyme B is a novel interleukin-18 converting enzyme. J Dermatol Sci 59: 129–135. [DOI] [PubMed] [Google Scholar]
- 52.Sower LE, Froelich CJ, Allegretto N, Rose PM, Hanna WD, and Klimpel GR. 1996. Extracellular activities of human granzyme A. Monocyte activation by granzyme A versus alpha-thrombin. J Immunol 156: 2585–2590. [PubMed] [Google Scholar]
- 53.Sower LE, Klimpel GR, Hanna W, and Froelich CJ. 1996. Extracellular activities of human granzymes. I. Granzyme A induces IL6 and IL8 production in fibroblast and epithelial cell lines. Cell Immunol 171: 159–163. [DOI] [PubMed] [Google Scholar]
- 54.Spencer CT, Abate G, Sakala IG, Xia M, Truscott SM, Eickhoff CS, Linn R, Blazevic A, Metkar SS, Peng G, Froelich CJ, and Hoft DF. 2013. Granzyme A produced by gamma(9)delta(2) T cells induces human macrophages to inhibit growth of an intracellular pathogen. PLoS Pathog 9: e1003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wensink AC, Kemp V, Fermie J, Garcia Laorden MI, van der Poll T, Hack CE, and Bovenschen N. 2014. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc Natl Acad Sci U S A 111: 5974–5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aghdassi AA, John DS, Sendler M, Weiss FU, Reinheckel T, Mayerle J, and Lerch MM. 2018. Cathepsin D regulates cathepsin B activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J Biol Chem 293: 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao S, Zhu H, Zuo X, and Luo H. 2018. Cathepsin G and Its Role in Inflammation and Autoimmune Diseases. Arch Rheumatol 33: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hannaford J, Guo H, and Chen X. 2013. Involvement of cathepsins B and L in inflammation and cholesterol trafficking protein NPC2 secretion in macrophages. Obesity (Silver Spring) 21: 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakanishi H 2020. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen Res 15: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ortega-Gomez A, Salvermoser M, Rossaint J, Pick R, Brauner J, Lemnitzer P, Tilgner J, de Jong RJ, Megens RT, Jamasbi J, Doring Y, Pham CT, Scheiermann C, Siess W, Drechsler M, Weber C, Grommes J, Zarbock A, Walzog B, and Soehnlein O. 2016. Cathepsin G Controls Arterial But Not Venular Myeloid Cell Recruitment. Circulation 134: 1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, and Temple S. 2008. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell 3: 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, and Verkhratsky A. 2008. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One 3: e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panza F, Lozupone M, Logroscino G, and Imbimbo BP. 2019. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol 15: 73–88. [DOI] [PubMed] [Google Scholar]
- 64.Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, and Colonna M. 2017. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170: 649–663 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.