

Summary
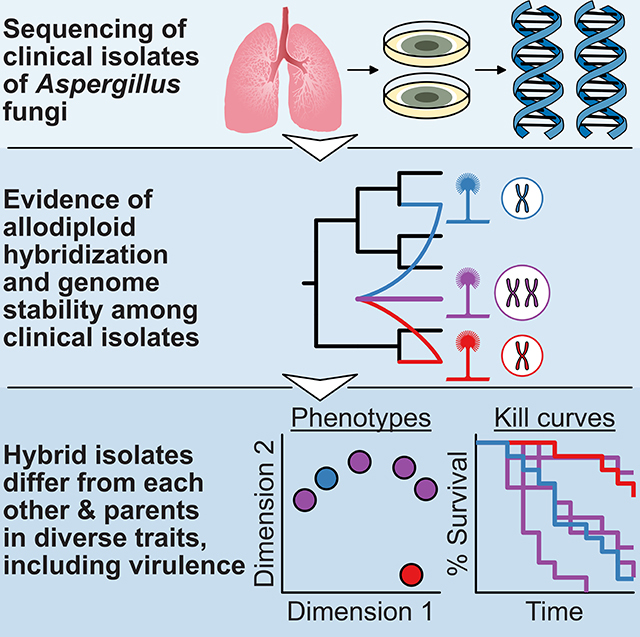
Interspecific hybridization substantially alters genotypes and phenotypes and can give rise to new lineages. Hybrid isolates that differ from their parental species in infection-relevant traits have been observed in several human-pathogenic yeasts and plant-pathogenic filamentous fungi, but have yet to be found in human-pathogenic filamentous fungi. We discovered 6 clinical isolates from patients with aspergillosis originally identified as Aspergillus nidulans (section Nidulantes) that are actually allodiploid hybrids formed by the fusion of Aspergillus spinulosporus with an unknown close relative of Aspergillus quadrilineatus, both in section Nidulantes. Evolutionary genomic analyses revealed that these isolates belong to Aspergillus latus, an allodiploid hybrid species. Characterization of diverse infection-relevant traits further showed that A. latus hybrid isolates are genomically and phenotypically heterogeneous but also differ from A. nidulans, A. spinulosporus, and A. quadrilineatus. These results suggest that allodiploid hybridization contributes to the genomic and phenotypic diversity of filamentous fungal pathogens of humans.
Keywords: hybridization, allopolyploidy, pathogenicity, Eurotiomycetes, aspergillosis, cryptic species, fungal pathogen evolution
eTOC Blurb
Steenwyk, Lind et al. identify clinical isolates of Aspergillus latus, an allodiploid species of hybrid origin. These isolates exhibit phenotypic heterogeneity in infection-relevant traits and are distinct from closely related species. The results suggest that allodiploid hybridization contributes to the evolution of filamentous fungal pathogens.
Graphical Abstract
Introduction
Interspecific hybridization can result in the formation of new species that substantially differ in their genomic and phenotypic characteristics from either parental species. One common mechanism by which interspecific hybrids can originate is allopolyploidy, which merges and multiplies the parental species’ chromosomes [1,2]. Allopolyploid hybrids may be more similar to one parent in some traits, reflect both parents in others, or may differ from both in the rest. Hybrids’ distinct phenotypic profiles means that they can potentially colonize new habitats [3,4], whereas their polyploidy means that they can quickly become reproductively isolated from both parental species, forming a new species in the process [2]. Allopolyploids are relatively common in plants, but are also found in several other lineages, including in animals [5] and fungi [6,7].
Among fungi, the most well-known example of allopolyploidy is the whole genome duplication in an ancestor of the baker’s yeast Saccharomyces cerevisiae [8–10]. Importantly, allopolyploidy is known from both fungal pathogens of plants [6,11] and of animals [12,13]. For example, the crucifer crop pathogens Verticillium longisporum and Verticillium dahliae are allopolyploid hybrids, as is the onion pathogen Botrytis allii [6,14,15]. Among fungal pathogens that infect humans, allopolyploidy has been reported in the ascomycete budding yeasts Candida metapsilosis [16] and Candida orthopsilosis [17] and in the basidiomycete yeast Cryptococcus neoformans X Cryptococcus deneoformans [18]. To our knowledge, allopolyploidy has never been reported in human pathogenic filamentous fungi.
Aspergillus-related diseases, collectively known as aspergillosis, are caused by various species in the Aspergillus genus of filamentous fungi [19]. Although the saprophytic and ubiquitous airborne species Aspergillus fumigatus (section Fumigati) is responsible for most infections, several other species are also pathogenic [20–23]. One such species is A. nidulans (section Nidulantes); even though rarely pathogenic, A. nidulans is of interest because it is a major cause of invasive aspergillosis infections in chronic granulomatous disease (CGD) patients [24]. CGD is a genetic disorder that compromises the ability of phagocytes to produce reactive oxygen species, which act as broad range antimicrobial chemicals [24,25]. Strikingly, among CGD patients, A. nidulans is harder to treat than the more common pathogen A. fumigatus [24,26].
To gain insights into A. nidulans pathogenicity, we sequenced 9 clinical isolates that were originally identified as A. nidulans from patients with various pulmonary diseases, including 2 isolates from CGD patients. Two of these isolates belong to A. nidulans and have been described in detail elsewhere [27]. However, our phenotypic and genomic analyses showed that 6 of the remaining 7 isolates are not A. nidulans but rather allodiploid hybrids of Aspergillus latus, a species that arose from allodiploid hybridization between Aspergillus spinulosporus and an unknown second parental species closely related to Aspergillus quadrilineatus (both from section Nidulantes). Our analyses also revealed that the seventh isolate belongs to A. spinulosporus.
Phenotypic characterization of A. latus isolates, their parental species, and A. nidulans for diverse infection-relevant traits revealed two key findings. The first finding is that A. latus isolates exhibit strain heterogeneity for several infection-relevant traits. For example, we observed wide variation between A. latus isolates in their virulence in a disease model as well as in their interactions with human immune cells. The second finding is that A. latus isolates are phenotypically distinct from A. spinulosporus, A. quadrilineatus, and A. nidulans. For example, we found that A. latus hybrid spores are better at evading macrophage engulfment as well as evading hyphal killing and inhibition of germination by neutrophils than A. nidulans or A. spinulosporus and are more resistant to antifungals and oxidative stressors than A. nidulans and A. quadrilineatus. From a clinical perspective, our discovery of allodiploid fungal pathogens of humans suggests that accurate taxonomic identification of Aspergillus clinical isolates is a key first step to disease management. From an evolutionary perspective, our discovery suggests that allodiploid hybridization is a general mechanism of genomic and phenotypic diversification among human fungal pathogens.
Results
Six clinical isolates previously characterized as A. nidulans are diploid
To gain insights into the genetic diversity of clinical isolates of A. nidulans, we analyzed 7 isolates from patients with different pulmonary diseases and compared them to haploid (A4) and the laboratory-induced diploid (R21/R153) reference strains of A. nidulans (Table 1). Using microscopy-based and/or molecular biology methods, all 7 isolates had previously been identified as A. nidulans, all are similar in appearance when grown in standard laboratory conditions (Figure S1), and two were analyzed as A. nidulans isolates in previous experimental studies [28]. Examination of DNA content revealed that 6 / 7 isolates were more similar to the diploid A. nidulans R21/ R153 strain than to the haploid A. nidulans A4 strain, suggesting that these 6 isolates are diploid (Figure 1A). The volume of asexual spores (conidia) is frequently proportional to the DNA content of the nucleus [29] and examination of their size showed that the same 6 isolates and the diploid A. nidulans strain have significantly larger spores than isolates with haploid genomes (p < 0.001, respectively; Dunn’s test with Benjamini-Hochberg method of multi-test correction for both tests) (Figure 1B).
Table 1.
Isolates used in this study
| Strain / Isolate | Isolation source | Reference/Source |
|---|---|---|
| A. spinulosporus NRRL 2395T | Soil, Buenos Aires, Argentina | [37] |
| A. nidulans A4 | E. Yuill, Birmingham, United Kingdom | Fungal Genetics Stock Center |
| A. quadrilineatus NRRL 201T | Soil, New Jersey, USA | USDA |
| 4060 | Clinical isolate from patient with chronic granulomatous disease | [28] |
| A. latus NRRL 200T | Unknown origin | USDA |
| MM151978 | Clinical isolate from patient with chronic obstructive pulmonary disease (COPD) | Laboratory of Dr. Donald Sheppard, McGill University, Canada. Patient sex: Unknown. Year isolated unknown. |
| NIH | Clinical isolate from patient with chronic granulomatous disease | [28] |
| ASFU1710 | Clinical isolate from patient with primary immunodeficiency | Sputum material, Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven, Leuven, Belgium. Patient sex: Female. Isolated in year 2011 |
| ASFU1908 | Clinical isolate from patient with asthma, Allergic Bronchopulmonary Aspergillosis (ABPA), and IgG3 deficiency | Sputum material, Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven, Leuven, Belgium. Patient sex: Female. Isolated in year 2011. Isolated from the same patient as ASFU2033 |
| ASFU2033 | Clinical isolate from patient with asthma, Allergic Bronchopulmonary Aspergillosis (ABPA), and IgG3 deficiency | Sputum material, Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven, Leuven, Belgium. Patient sex: Female. Isolated from the same patient as ASFU1908 in year 2012 |
| MO46149 | Clinical isolate from patient with myelodysplastic syndrome with pneumonia | Sputum material, Clinical Laboratory of Dr. Catarina Lameiras, Department of Microbiology, Portuguese Oncology Institute of Porto, Porto, Portugal. Patient sex: Male. Isolated in year 2011 |
Figure 1. Six clinical isolates previously characterized as Aspergillus nidulans and the type strain of Aspergillus latus are diploids.

(A) Fluorescence-assisted cell sorting analysis suggests that the type strain of Aspergillus latus NRRL 200T and 6 clinical isolates (MM151978, NIH, ASFU1710, ASFU1908, ASFU2033, and MO46149) previously identified as Aspergillus nidulans have diploid genomes. In contrast, Aspergillus spinulosporus NRRL2395 and clinical isolate 4060 have haploid genomes. The haploid A. nidulans strain A4 and the laboratory-induced diploid A. nidulans strain R21/R23 were used as references of haploid and diploid genomes, respectively. (B) Asexual spore diameter is significantly different between the 6 diploid clinical isolates, the haploid A. quadrilineatus, A. spinulosporus, and A. nidulans, and the laboratory-induced diploid A. nidulans (χ2 = 399.54, df = 2, p < 0.001; Kruskal-Wallis rank sum test). Additional pairwise comparisons are shown by brackets; all comparisons used Dunn’s test with Benjamini-Hochberg method of multi-test correction. *** p-value ≤ 0.001. (C) The 6 diploid clinical isolates and the A. latus NRRL 200T strain have substantially larger genome sizes, gene numbers, and percent duplicated BUSCO genes compared to haploid genomes of representative Aspergillus species (A. clavatus NRRL 1, A. flavus NRRL 3357, A. fumigatus Af293, A. nidulans A4, A. niger CBS 513.88, A. sydowii CBS 593.65, and A. versicolor CBS 583.65). Genus and species names are abbreviated using the following scheme: A. latus (Alat), A. spinulosporus (Aspi), A. quadrilineatus (Aqua), and A. nidulans (Anid). CI represents clinical isolates. Dark grey represents A. nidulans; red represents A. quadrilineatus; blue represents A. spinulosporus and CI 4060; purple represents A. latus and diploid isolates. See also Table 1, S1, S2, S3, and Figure S1.
To gain further insight into the genomes of the 6 diploid isolates and 1 haploid isolate, we sequenced them and compared their genome size and gene number with those of representative Aspergillus species known to be haploid (A. clavatus NRRL 1, A. flavus NRRL 3357, A. fumigatus Af293, A. nidulans A4, A. niger CBS 513.88, A. sydowii CBS 593.65, and A. versicolor CBS 583.65) [30–35]. We found that the genomes and gene numbers of the diploids were significantly larger (average genome size = 69.09 ± 5.68 Mb, average gene number = 21,321.57 ± 2,342.13) than those of the haploid representative Aspergillus species (average genome size = 32.62 ± 3.05 Mb, average gene number = 11,330.75 ± 1,838.70) (Figure 1C; figshare image 1, 10.6084/m9.figshare.8114114; p < 0.001; Wilcoxon rank sum test for both tests; Table S1). Similarly, examination of gene content completeness revealed a significantly higher number of duplicated near-universally single copy (BUSCO) genes in the diploids relative to representative Aspergillus species (Figure 1C; p = 0.001; Wilcoxon rank sum test). Thus, we concluded that 6 / 7 clinical isolates are diploids.
Diploid clinical isolates are Aspergillus latus, a species of hybrid origin
To examine the evolutionary origin of the clinical isolates, we retrieved their calmodulin and β-tubulin sequences and performed molecular phylogenetic analysis in the context of sequences of the two genes from all available taxa in the section Nidulantes phylogeny [36]. We found that the haploid clinical isolate 4060 had nearly identical calmodulin and β-tubulin sequences to other strains of A. spinulosporus and formed a monophyletic group with them, suggesting that it belongs to A. spinulosporus (Figure 2A; figshare: 10.6084/m9.figshare.8114114). Notably, we found that all 6 diploid clinical isolates contained two different copies of the calmodulin and β-tubulin genes; one copy was nearly identical to A. spinulosporus sequences whereas the other was nearly identical to A. latus ones (Figure 2A; figshare: 10.6084/m9.figshare.8114114), raising the hypothesis that the diploids originated from interspecific hybridization between A. spinulosporus and A. latus.
Figure 2. The 6 clinical diploids belong to A. latus, an allodiploid species formed via hybridization of A. spinulosporus and a close relative of A. quadrilineatus.

(A) The type strain of A. latus NRRL 200T and the 6 diploid clinical isolates have each two copies of the taxonomic markers β-tubulin and calmodulin. Phylogenetic analysis of their β-tubulin and calmodulin sequences together with sequences from representative taxa in section Nidulantes [36] suggests that clinical isolate 4060 belongs to A. spinulosporus whereas A. latus NRRL 200T and the 6 diploid clinical isolates are derived from two parental genomes. Interestingly, neither of the parental genomes is A. nidulans; rather one is A. spinulosporus and the other is a species closely related to Aspergillus quadrilineatus. Newly sequenced isolates are shown in red and blue. (B) Examination of sequence divergence (Ks; x-axis) between each gene in an allodiploid and its best blast hit in A. spinulosporus confirms that the 6 diploid clinical isolates and the type strain of A. latus are allodiploid hybrids. In contrast, the 7th clinical isolate (4060) is a haploid A. spinulosporus. Similarly, we found no evidence of A. quadrilineatus NRRL 201T forming via allodiploid hybridization. (Bi) Examination of the haploid, non-hybrid genome of A. fumigatus strain Af293 (negative control) shows a unimodal distribution, whereas examination of the diploid, hybrid genome Zygosaccharomyces parabailii strain NBRC1047/ATCC56075 (Zpar) shows a bimodal distribution (positive control; grey represents genes from one parent; black represents genes from the other parent). Red represents genes assigned to the A. quadrilineatus-like parental genome; blue represents genes assigned to the A. spinulosporus parental genome. See also Figure S2, S3, S4, and Data S1.
To test this hypothesis, we analyzed the genome of A. spinulosporus strain NRRL 2395T [37] and sequenced the type strain NRRL 200T of A. latus. Examination of the DNA content and asexual spore size of these two species’ genomes showed that A. spinulosporus NRRL 2395T had similar values as clinical isolate 4060 (Figure 1) and was also placed in the same phylogenetic clade (Figure 2A); these findings confirm that clinical isolate 4060 belongs to A. spinulosporus and that A. spinulosporus is one of the parental species involved in the interspecific hybridization event that gave rise to the 6 clinical isolates.
In contrast, the DNA content and spore size of the genome of the type strain of A. latus NRRL 200T were similar to those of the 6 diploid clinical isolates (Figure 1). Furthermore, like the 6 clinical isolates, A. latus NRRL 200T also had two copies of the calmodulin and β-tubulin gene sequences; one copy was nearly identical to A. spinulosporus sequences and the other copy was closely related to, but distinct from, A. quadrilineatus sequences (Figure 2A; figshare: 10.6084/m9.figshare.8114114). These results suggest that the 6 diploid clinical isolates belong to A. latus, and that A. latus is an allodiploid hybrid species that originated via interspecific hybridization between A. spinulosporus and a species closely related to A. quadrilineatus.
We tested this hypothesis by performing two different sets of analyses. In the first set of analyses, we sequenced, assembled, and annotated the genome of the type strain (NRRL 201T) of A. quadrilineatus. Consistent with our hypothesis that A. latus is an allodiploid hybrid, we found that the A. quadrilineatus genome contains a single copy of the calmodulin and β-tubulin gene sequences, that these sequences form a monophyletic group with their orthologous sequences retrieved from the genome of a different A. quadrilineatus strain (strain CBS 853.96; https://www.ncbi.nlm.nih.gov/sra/SRX5010607), and that the A. quadrilineatus sequences form a sister group with one of the two sets of the A. latus sequences (Figure 2A).
In the second set of analyses, we estimated the sequence divergence of each gene in the genomes of the 7 clinical isolates as well as of A. latus NRRL 200T and A. quadrilineatus NRRL 201T from A. spinulosporus NRRL 2395T. Under this analysis, the genomes of non-hybrids are expected to show a unimodal distribution (e.g., see control non-hybrid, A. fumigatus; Figure 2Bi left), whereas the genomes of hybrids are expected to show a bimodal distribution whose two modes correspond to the distributions of gene sequence divergence values from each parental genome (e.g., see control hybrid, Zygosaccharomyces parabailii; Figure 2Bi right). We found that the haploid A. spinulosporus 4060 clinical isolate and A. quadrilineatus NRRL 201T had unimodal distributions reflecting a history devoid of hybridization while the 6 diploid clinical isolates and A. latus NRRL 200T had bimodal distributions consistent with allodiploidy (Figure 2Bii). Furthermore, all 6 diploid isolates and A. latus NRRL 200T contained nearly equal percentages of A. spinulosporus and A. quadrilineatus-like genes (51.43 ± 0.74% A. spinulosporus : 48.57 ± 0.74% A. quadrilineatus-like; Figure 2B, pie charts), including nearly the full sets of A. spinulosporus and A. quadrilineatus-like secondary metabolic gene clusters (Table S2; figshare: 10.6084/m9.figshare.8114114). Putative homeologs exhibited an average nucleotide sequence divergence of 7.15 ± 0.03%, a value very similar to the average divergence of 7.14% observed between the 8,523 orthologs of A. spinulosporus NRRL 2395T and A. quadrilineatus NRRL 201T (Figure S2). These two sets of analyses confirm that the 6 diploid clinical isolates belong to A. latus, and that A. latus is an allodiploid hybrid species that originated via interspecific hybridization between A. spinulosporus and a species closely related to A. quadrilineatus.
We next assessed whether the allodiploid hybrid species A. latus stems from a single hybridization event by comparing the genome-scale phylogenies constructed from the A. spinulosporus and the A. quadrilineatus-like parental genomes of the A. latus isolates (Figure S3). We found that the relationships of the A. latus isolates differed between the two phylogenies (Figure S3). This incongruence may stem from biological reasons (e.g., multiple hybridization events or recombination between the two parental genomes). However, the low level of support for relationships among isolates, especially in the phylogeny from the A. spinulosporus parental genome (Figure S3A), means that we cannot exclude the possibility that the two phylogenies are not statistically significantly different. To test this, we evaluated whether the two topologies were statistically different using the approximately unbiased topology constraint test [38]. Using the A. spinulosporus data matrix, we found that we could not reject the topology inferred based on the A. quadrilineatus-like data matrix as statistically inferior; similarly, we could not reject the A. spinulosporus topology when we using the A. quadrilineatus-like data matrix (p-value = 0.50 for both tests). These results are consistent with the hypothesis that the two parental genomes of A. latus share the same evolutionary history.
To provide more insight on whether the two parental genomes A. latus hybrids undergo recombination, we first examined whether A. latus hybrids undergo the sexual cycle to produce sexual spores (ascospores). We found that all A. latus hybrids produce sexual spores and that the viability of these spores is similar to that of the sexual spores of their parental species (figshare image 2, 10.6084/m9.figshare.8114114). We next examined if any contigs in the genomes of A. latus isolates had evidence of recombination events. Examination of long (≥ 100 kb) contigs revealed that most genes in most contigs contained genes from one or the other parent and that very few contigs contained substantial percentages of genes from both parents (Figure S4). For example, only an average of 2.67 ± 0.71% contigs per A. latus hybrid genome contained substantial percentages of genes from both parental species (Figure S4). However, interpretation of these data is challenging for two reasons. First, the high sequence similarity of the two parental genomes means that identification of parent of origin for highly-conserved genes is difficult and likely explains the sporadic presence of one or a handful of genes from one parent in contigs comprised mostly of genes from the other parent. Second, alignment of several of the contigs that contain large numbers of genes from both parents to the A. nidulans A4 reference genome suggests that they are often patchworks of A. nidulans contigs; for example, a long stretch of an A. latus contig that matches one parent is homologous to A. nidulans chromosome 5 and the rest of the contig, which matches the other parent, is homologous to A. nidulans chromosome 7. The absence of A. latus contigs that contain genes from both parental species and map to a single A. nidulans chromosome suggests that A. latus contigs that contain genes from both parental species may stem from assembly artifacts. These results suggest that A. latus hybrids likely undergo little to no recombination between the two parental genomes.
The genomes of the A. latus allodiploid hybrid isolates are stable
To assess the genome stability of the A. latus isolates, we begun by examining the gene content completeness of each parental genome. We found that each parental genome contained nearly all of the 1,315 near-universally single-copy orthologous (BUSCO) genes from the fungal phylum Ascomycota (93.50 ± 1.88% A. spinulosporus and 94.30 ± 0.40% A. quadrilineatus-like) (figshare image 3, 10.6084/m9.figshare.8114114). Considering that gene content completeness from each parent is only slightly below that from haploid representative species (average = 96.33 ± 0.78%; min = 95.70%, A. spinulosporus; max = 97.3%, A. nidulans A4), these results suggest little loss of each parental genome by either aneuploidy or loss of heterozygosity events.
To further test this observation genome-wide, we examined the fraction of orthologous genes shared between the A. spinulosporus NRRL 2395T strain and the parental genomes of A. latus isolates that stem from A. spinulosporus. We found that the A. spinulosporus parental genomes from A. latus hybrids shared a minimum of 9,227 / 9,611 orthologous genes with A. spinulosporus NRRL 2395T; the sole exception was A. latus NRRL 200T, which shared 8,749 orthologs (figshare: 10.6084/m9.figshare.8114114). Interestingly, the A. spinulosporus parental genome of A. latus NRRL 200T shows by far the highest evolutionary rate in our phylogenomic analyses (Figure S3), suggesting that the A. spinulosporus parental genome of this strain may be more genetically unstable than those of the clinical isolates.
Examination of loss of heterozygosity and aneuploidy events in A. latus genomes revealed relatively little evidence for either. Two isolates contained loss of heterozygosity regions. The A. latus NRRL 200T strain contained a ~1.2 Mb region homologous to the end of A. nidulans chromosome VIII that contained two copies of the A. quadrilineatus-like parental genome and lacked a copy of the A. spinulosporus genome. This region contains several BUSCO genes, which explains why this strain has a higher proportion of missing BUSCO genes from the A. spinulosporus parental genome compared to the 6 clinical isolates (figshare image 3, 10.6084/m9.figshare.8114114). The clinical isolate MO46149 contained a ~1 Mb region homologous to the beginning of A. nidulans chromosome V with two copies of the A. spinulosporus genome and lacked a copy of the A. quadrilineatus-like genome (Data S1). We did not find evidence for chromosome-scale aneuploidies (Data S1).
Lastly, by comparing the gene lengths of homeolog pairs as a signature of pseudogenization, we found evidence of pseudogenization in at least one gene among an average of 11.67 ± 0.004% homeologs (Figure S2). These results suggest that the genomes of the A. latus allodiploid hybrids are generally stable, that loss of heterozygosity is rare, that major aneuploidies have not occurred, and that both genes in ~88% of homeolog pairs are intact.
Hybrids exhibit wide variation for infection-relevant traits
To examine variation in infection-relevant traits between the hybrid isolates, one of their known parental species (A. spinulosporus), the closest known relative of their other parental species (A. quadrilineatus), and the species they were originally identified as (A. nidulans), we tested the virulence of all isolates in an invertebrate disease model and phenotypically characterized them across a wide variety of infection-relevant conditions, including interactions with host immune cells, drug susceptibility, oxidative stress, iron starvation, and temperature stress (Figure 3 and S5). Principal component analysis (PCA) and examination of the traits with the greatest contributions to the observed variance among isolates revealed two major findings. First, the 7 A. latus hybrids exhibit substantial heterogeneity in their phenotypic profiles (Figure 3A). Second, the A. latus hybrids are phenotypically distinct from A. nidulans and their parental species but are more similar to A. spinulosporus than to A. quadrilineatus (Figure 3A). Among the traits tested, those with the largest contributions to the observed variation among isolates and species were interactions with host immune cells, antifungal drug susceptibility, and oxidative stress resistance (figshare image 3A and 4, 10.6084/m9.figshare.8114114). Here, we discuss exemplary phenotypic traits that highlight these two major findings (see Figure S5 for other phenotypes).
Figure 3. A. latus hybrids exhibit strain heterogeneity and differ from parental species, A. quadrilineatus, and A. nidulans in infection-relevant phenotypes.

(A) Principal component analysis of diverse infection-relevant phenotypes reveals strain heterogeneity among A. latus hybrids and that they differ from the closest relative of the unknown parent, A. quadrilineatus, and the A. nidulans A4 strain. Data for each phenotype was scaled prior to principal component analysis. (B, C) Wide phenotypic variation among strains of A. latus hybrids as well as the various species tested was observed for virulence in Galleria moth model of disease and NETosis. (D) Examination of the percentage of hyphal viability between the various species revealed significant differences (F(3) = 24.514, p < 0.001; Multi-factor ANOVA). (E) Examination of the caspofungin drug susceptibility profiles among A. nidulans, A. spinulosporus, and the A. latus revealed differences between the three species (F(3) = 56.01, p < 0.001; Multi-factor ANOVA). At 1 μg / mL caspofungin treatment, A. spinulosporus and A. latus hybrids grew more than A. nidulans and A. quadrilineatus (p < 0.001 for both comparisons; Tukey honest significant differences test). We found no statistically significant differences between the various species at 8 μg / mL of caspofungin but observed a qualitative difference similar to growth in 1 μg / mL of caspofungin. (F) Examination of growth in the presence of the oxidative stress agent paraquat revealed differences among the various species (F(3) = 30.25, p < 0.001; Multi-factor ANOVA). Genus and species names are abbreviated using the following scheme: A. latus (Alat), A. spinulosporus (Aspi), A. quadrilineatus (Aqua), and A. nidulans (Anid). Dark grey represents A. nidulans; red represents A. quadrilineatus; blue represents A. spinulosporus; purple represents A. latus. All pairwise comparisons shown by brackets were examined using a Tukey honest significant differences test. * 0.01 ≤ p-value ≤ 0.05; ** 0.001 ≤ p-value ≤ 0.01; *** p-value ≤ 0.001. See also Figure S5.
Phenotypic variation or strain heterogeneity among A. latus hybrids was observed for nearly every trait measured (Figure 3 and S5). For example, examination of virulence in the invertebrate greater wax moth (Galleria mellonella) model revealed substantial variation among isolates (p < 0.001; log-rank test; Figure 3B). Specifically, we observed that A. latus isolate ASFU1710 was the most virulent and A. latus isolate MO46149 was the least virulent. Similarly, we found substantial strain heterogeneity in how much lytic and non-lytic NETosis (a process where neutrophils release neutrophil extracellular traps, or NETs, to kill microbes; [39]) was stimulated by A. latus hybrid isolates (Figure 3C). For example, A. latus NIH did not substantially stimulate NETosis while A. latus ASFU1710 did. Strain heterogeneity was less pronounced for other traits, such as hyphal viability, drug susceptibility, and oxidative stress, yet all exhibited variation across isolates (Figure 3D, E, and F). One A. latus isolate that was consistently different from the rest is MO46149; for example, this isolate was twice as susceptible to hyphal killing by neutrophils compared to the other A. latus isolates, it was the isolate most sensitive to the antifungal caspofungin, as well as the isolate most tolerant to the oxidative stressor paraquat.
Phenotypic variation was also pronounced when we compared infection-relevant traits between A. latus, its two parental species, and A. nidulans (Figure 3A). For example, we found that A. latus hybrids (and A. quadrilineatus) were less susceptible to killing by neutrophils compared to A. spinulosporus (Figure 3D). In contrast, we found A. latus isolates (and A. spinulosporus) differed in their susceptibility to low doses of caspofungin (Figure 3E) or to high doses of oxidative stress (Figure 3F) from A. quadrilineatus and A. nidulans.
In summary, we found substantial heterogeneity among A. latus hybrid isolates as well as between A. latus and closely related or parental species for diverse infection-relevant traits. Generally, A. latus hybrids are more similar to their known parent, A. spinulosporus, compared to the closest known relative of their other parent, A. quadrilineatus. Importantly, A. latus hybrids are also phenotypically distinct from A. nidulans, the species they were originally misdiagnosed as.
Discussion
Infections by filamentous fungal pathogens affect hundreds of thousands of humans and exhibit very high mortality rates [40], so understanding the evolutionary mechanisms underlying their pathogenicity is of great interest. We have discovered several clinical isolates previously identified as A. nidulans, an important pathogen of CGD patients, which in reality are allodiploid hybrids that arose via interspecific hybridization between A. spinulosporus and a close relative of A. quadrilineatus and belong to A. latus. In line with clinical misidentification of these species, A. nidulans, A. spinulosporus, A. quadrilineatus, and A. latus are known to be nearly indistinguishable with the exception of aspects of their ascospore micromorphology and their secondary metabolic profiles [36]. The allodiploid hybrids show strain heterogeneity in their phenotypic profiles and differ from their parental species and A. nidulans with respect to several infection-relevant traits. Below, we discuss the implications of these results for disease management and the evolution of fungal pathogenicity.
Application of molecular typing, and more recently genomic, approaches to delineate fungal species and pathogens has revealed the existence of multiple, closely related species that are morphologically indistinguishable but genomically distinct from each other [41]. This “hidden” or “cryptic” genomic diversity is found in species from many genera that harbor major fungal pathogens, including Aspergillus [42–44]. Alarmingly, application of molecular methods on fungal clinical isolates has too begun to reveal that a significant portion of fungal infections are caused by these cryptic species. In the case of Aspergillus, studies in both the USA and Spain report that 10 to 15% of aspergillosis infections stem from cryptic species [45,46]. Understanding the biology of these cryptic species is essential for guidance in therapy, as many show high levels of antifungal drug resistance [45–47]. Several A. fumigatus-related cryptic species exhibit decreased susceptibility (relative to A. fumigatus) to antifungal drugs [48]; similarly, we found notable differences in certain phenotypic traits, including drug susceptibility, between the allodiploid hybrids and A. nidulans (Figure 3E and S5).
A growing body of literature suggests that phenotypic heterogeneity among isolates of the same species is an under-appreciated factor in understanding fungal pathogenicity [49]. For example, several recent studies have identified phenotypic and genomic differences between A. fumigatus strains that are associated with virulence [50–52]; strain heterogeneity is also observed among A. nidulans strains [27]. In line with these studies, our work reveals considerable heterogeneity in infection-relevant traits among A. latus hybrids (Figure 3 and S5), further highlighting the importance of strain heterogeneity in understanding Aspergillus pathogenicity [49].
Allopolyploid hybrids typically have unstable genomes [12]. Evolutionary paths to achieve stability after hybridization include whole-genome duplication, total or partial chromosome loss, gene loss, and loss of heterozygosity [12]. For example, an ancient allodiploid hybridization event in the budding yeast lineage that includes the baker’s yeast Saccharomyces cerevisiae [9] was quickly followed by rapid gene loss in the parental genomes [53], with estimates suggesting that 10% of genes were lost in the first 10 million years following hybridization [53]. Similar rates of gene loss have been reported in plants [54,55] and animals [56], suggesting that rapid gene loss is a common outcome of hybridization. In contrast to these studies, we found that most A. latus hybrids (with the possible exception of A. latus NRRL 200T) contain both copies of most homeolog gene pairs and have relatively stable genomes (Figure S2 and S4). Consistent with our genomic analyses, A. latus isolates exhibit minimal sectoring when grown in culture (Figure S1) and have similar ascospore viability compared to their closest relatives, A. spinulosporus and A. quadrilineatus (figshare image 2, 10.6084/m9.figshare.8114114). Furthermore, laboratory studies that have created synthetic Aspergillus hybrids—including those that created interspecies hybrids from distinct species in section Nidulantes—reported that some of these hybrids are relatively stable [57–60]. Taken together, these results suggest that Aspergillus hybrids may be more stable than other hybrid allopolyploids.
Although several examples of interspecies hybridization are known in fungi [6,8,18,9–12,14–17], most of them are ancient. Consequently, the steps that led to hybrid formation and maintenance are harder to elucidate. As the A. latus allodiploid hybrids originated much more recently, and the mechanisms that underlie synthetic hybrid formation in Aspergillus have been extensively studied [57–60], we can propose a model to explain the origin and lifecycle of A. latus (Figure 4). Under our model, the first step in the formation of the A. latus allodiploid hybrid was the cellular fusion (or plasmogamy) of an A. spinulosporus parental isolate and an A. quadrilineatus-like parental isolate through a parasexual or a sexual cycle. In the next step, the distinct nuclei contributed by the two parental isolates underwent nuclear fusion (or karyogamy) to create a single nucleus with a diploid genome comprised from the A. spinulosporus and A. quadrilineatus-like genomes. Once formed, the allodiploid hybrid species A. latus has been capable of undergoing both asexual and sexual reproduction and forms viable asexual spores (conidia; Figure 1) and sexual spores (ascospores; figshare image 2, 10.6084/m9.figshare.8114114S5).
Figure 4. Proposed model for the evolution of A. latus via allodiploid hybridization.

Under the model, haploid nuclei of an A. spinulosporus isolate and of an isolate from an A. spinulosporus-like species underwent cellular fusion (plasmogamy) forming a heterokaryotic mycelium (i.e., a mycelium where cells contain two distinct nuclei). Next, nuclear fusion (karyogamy) resulted in the merging of the two genetically distinct nuclei and their genomes into a single one, giving rise to the allodiploid species A. latus which, is capable of undergoing asexual and sexual reproduction to produce asexual spores (conidia) or sexual spores (ascospores).
Hybrids have been observed in several fungal pathogens of animals and plants [6,12,16,18], suggesting that hybridization of pathogenic fungi poses threats to plant and animal health. Hybridization can result in the acquisition of new traits, such as host expansion or increased virulence. For example, two powdery mildew species that specialize in infecting different species of plants have been shown to hybridize and infect a plant species that neither parent can [61]. Similarly, hybridization is thought to contribute to virulence among human yeast pathogens but clear examples are currently lacking [12]. To our knowledge, our results are the first report of hybrid clinical isolates in a filamentous fungal pathogen of humans (Figure 2). Like previous studies, we observe that the hybrids exhibit infection-relevant traits that differentiate them from their parental relatives and may provide a fitness advantage inside human hosts (Figure 3). However, it should be noted that the potential selection pressure for hybrids in patients is unknown; although we isolated the hybrids from patients with diverse pulmonary diseases, we do not know if the hybrids’ primary lifestyle is that of a pathogen or whether they originated inside a host. Importantly, the type strain of A. latus (NRRL 200T) is not a clinical isolate, suggesting that the species is also found in the environment. More broadly, the existence of hybrids in a diverse set of pathogenic fungi infecting a diverse set of animal and plant hosts raise the hypothesis that allodiploid hybridization contributes to the evolution and diversity of all kinds of fungal pathogens, perhaps to a greater extent than currently realized.
In summary, viewed from a medical mycology perspective, our discovery of allodiploid hybrid clinical isolates reveals the importance of accurate isolate identification and strain heterogeneity. Viewed from an evolutionary perspective, our and previous results suggest that hybridization contributes to the genomic and phenotypic diversification of filamentous fungal pathogens of humans and argue that hybridization represents a general mechanism that can be potentially employed by all fungal pathogens to adapt to all kinds of hosts.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Antonis Rokas (antonis.rokas@vanderbilt.edu).
Materials Availability
This study did not generate new unique reagents. Aspergillus strains are available upon request.
Data and Code Availability
All data are publicly available through NCBI or a figshare repository. Genome assemblies are available through BioProject IDs PRJNA542678 and PRJNA542141; raw reads are available through BioProject IDs PRJNA542395, PRJNA542181, and PRJNA542141. Genome assembly and raw reads for A. quadrilineatus NRRL 201T are available through BioProject ID PRJNA623402. Strain specific BioProject or BioSample IDs can be found in Table S3. The figshare repository (doi: 10.6084/m9.figshare.8114114) contains phenotypic measurements, parent of origin analysis, homeologs per hybrid isolate, fluorescence-assisted cell sorting files, phylogenetic and phylogenomic data matrices and tree files, biosynthetic gene cluster prediction results, genome assemblies and gene predictions. Additional scripts and commands used throughout computational analyses are available through GitHub https://github.com/JLSteenwyk/Pathogenic_allodiploid_hybrids_of_Aspergillus_fungi.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
All Aspergillus spp. strains were obtained from public collections, in laboratories, or hospitals. Upon receipt, these strains were grown on YAG medium and after 5 days growth at 30°C, and asexual spores (conidia) were harvested and stored either at 4°C in water for further usage or at −80°C in a 30% glycerol solution in the laboratory collection. In experiments for DNA extraction and genome sequencing, the conidia were inoculated into Erlenmeyer-flasks on an orbital shaker (150 rpm) at 30°C for 24 hours. A. quadrilineatus NRRL 201T was acquired separately from the NRRL culture collection. Upon receipt of the cryopreserved vial, the contents of the frozen material were aseptically removed from the vial and transferred to 50 ml of YESD (Yeast Extract, Soy peptone, Dextrose) media. After, 3–4 days of shaking on an orbital shaker (100 rpm) at room temperature, the resulting mycelium was aseptically transferred on potato dextrose agar for all subsequent experiments, including DNA extraction and genome sequencing. Cultures of A. quadrilineatus NRRL201T are currently preserved at room temperature on potato dextrose agar (Difco).
Primary cell cultures
To obtain macrophages for a phagocytosis assay of Aspergillus asexual spores (conidia), we used bone marrow-derived macrophages that were isolated as described previously [62]. Briefly, macrophages were recovered from femurs of male C57BL/6 (wild-type, WT, Jax 000664) at 6–8 weeks old and were incubated in an RPMI medium (Gibco) supplemented with 30% (volume / volume) L929 growth conditioning media, 20% inactivated fetal bovine serum (Gibco), 2 millimolar glutamine and 100 units / milliliter of penicillin-streptomycin (Life Technologies). Fresh media was added after 4 days of cultivation and macrophages were collected after 7 days and used for subsequent experiments. The animals were housed in the animal facility of the Ribeirao Preto Medical School, University of Sao Paulo, under optimized hygienic conditions.
Human polymorphonuclear cells (PMNs) were isolated from 8 mL of peripheral blood of adult male healthy volunteers by density centrifugation using Mono-Poly™ Resolving Medium (MP Biomedicals LLC, Irvine, CA, USA) according to the manufacturer’s instruction. The PMNs (5×106 / mL) were resuspended in Hank’s Balanced Salt Solution, without calcium or magnesium, containing 5% fetal bovine serum (Gibco®, South American, Brazil).
Animals
Galleria mellonella larvae are cultivated in a greenhouse at 27°C. The larvae are kept in glass jars, in which they are separated by stages, moths, eggs, larvae and pupa. The larvae are fed once a week, with a feed composed of (yeast extract 1.5%; soy 1%, powdered milk 1%, honey 1% and beeswax 2%).
In the sixth week of the larval stage, the larvae are adult and larger, facilitating infection. We separated and assembled the groups with the larvae (n = 10) in petri dishes. The groups are composed of larvae that are approximately 300 milligrams in weight and 2 centimetres long. Moth sex was not accounted for due to the impossibility of visually determining sex at the sixth week of larval development.
Ethics statement
The principles that guide our studies are based on the Declaration of Animal Rights ratified by the UNESCO on the 27th January 1978 in its 8th and 14th articles. All protocols used in this study were approved by the local ethics committee for animal experiments from Universidade de São Paulo, Campus Ribeirão Preto (permit number 08.1.1277.53.6). All animals were cared for in strict accordance with the principles outlined by the Brazilian College of Animal Experimentation (Princípios Éticos na Experimentação Animal—Colégio Brasileiro de Experimentação Animal [COBEA]) and the Guiding Principles for Research Involving Animals and Human Beings of the American Physiological Society.
METHOD DETAILS
Fluorescence-assisted cell sorting for DNA content determination
Asexual spores (conidia) were collected, centrifuged (13,000 rounds per minute for 3 minutes) and washed with sterile 1 x phosphate-buffered saline (8 grams sodium chloride, 0.2 grams potassium chloride, 1.44 grams disodium phosphate, 0.24 grams monopotassium phosphate per liter of sterilized water). For cell staining, the protocol described by Almeida et al. [63] was followed with modifications. Following overnight fixation with 70% ethanol (volume / volume) at 4°C, spores were harvested, washed and suspended in 850 microliter of sodium citrate buffer (50 millimolar sodium citrate; pH=7.5). Spores were subsequently sonicated using four ultrasound pulses at 40W for 2 seconds with an interval of 1 to 2 seconds between pulses. Sonicated spores were treated for 1 hour at 50°C with RNase A (0.50 milligrams / milliliter; Invitrogen, Waltham, Massachusetts, USA) and for 2 hours at 50°C with proteinase K (1 mg/mL; Sigma-Aldrich, St. Louis, Missouri, USA). Spores were stained overnight with SYBR Green 10,000x (Invitrogen™, Carlsbad, CA, USA) diluted 10-fold in Trisethylenediaminetetraacetic acid (pH 8.0), at a concentration of 2% (volume / volume) at 4°C. Finally, Triton® X-100 (Sigma-Aldrich) was added to samples at a final concentration of 0.25% (volume / volume). Stained spores were analyzed in a Fluorescence-assisted cell sorting LSRII flow cytometer (Becton Dickinson, NJ, USA) with a 488 nanometer excitation laser. Signals from a minimum of 30,000 cells per sample were captured in fluorescein isothiocyanate channel (530 nanometers ± 30 nanometers) at low flow rate of ~1,000 cells / second and an acquisition protocol was defined to measure forward scatter and side scatter on a four-decade logarithmic scale and green fluorescence on a linear scale. Fluorescence-assisted cell sorting DIVA was used as the acquisition software. Results were analyzed with the R package FLOWVIZ, version 1.46.1 [64].
Asexual spore size measurements
The diameters of 100 spores for each isolate were measured under a Carl Zeiss (Jena, Germany) AxioObserver.Z1 fluorescent microscope equipped with a 100-W HBO mercury lamp using the 100 x magnification oil immersion objective and the AXIOVISION, software v.3.1.
DNA extraction and sequencing
Frozen mycelia of all isolates were ground in liquid nitrogen and genomic DNA was extracted as previously described [65,66]. Standard techniques for manipulation of DNA were used [67]. The genomes of all clinical isolates and the type strain of A. latus (9 in total) were sequenced at the Genomic Services Lab of Hudson Alpha (Huntsville, Alabama, USA) on an Illumina HiSeq 2500 sequencer; the sole exception was A. quadrilineatus NRRL 201T, which was sequenced using on a NovaSeq S4 at the Vanderbilt Technologies for Advanced Genomes facility (Nashville, Tennessee, USA). All isolates were sequenced using paired-end sequencing (150 bp) with the Illumina TruSeq library kit. Additionally, the type strain of A. latus and the clinical isolates MM151978 and NIH were also sequenced using mate-pair sequencing (150 bp) using the Illumina Nextera Mate Pair Library kit with an insert size of 4 kilobases. The genome coverage of each isolate was greater than 150X. Both the raw short-read sequence data and the genome assemblies are publicly available (see Table S3 for accession numbers).
Genome assembly and annotation
All genomes were assembled with the iWGS pipeline [68] using DIPSPADES, version 1.0 [69] or SPADES, version 3.6.2 [70]. Optimal k-mer lengths were selected using KMERGENIE, version 1.6982 [71], and assembly quality was evaluated using QUAST, version 3.2 [72]. Protein-coding genes were predicted using AUGUSTUS, version 3.3 [73], using Aspergillus nidulans gene annotation parameters. Predicted genes in each A. latus hybrid genome were annotated by reciprocal-best-BLAST against a database of all A. nidulans and A. spinulosporus proteins.
Prediction of secondary metabolic gene clusters
Secondary metabolic gene clusters (SMGCs) were predicted in all assembled genomes using ANTISMASH, version 3.0.5.1 [74]. In addition, we used literature-curated SMGC annotations from the well-characterized A. nidulans A4 genome [35] to identify SMGCs not captured by the ANTISMASH software.
Assigning genes in hybrid genomes to parents of origin
To determine the most likely parent-of-origin for each gene in a hybrid genome, we measured the sequence divergence between every gene in a hybrid genome and its ortholog in the A. spinulosporus NRRL 2395T parent [75]. Specifically, for each gene in a hybrid genome, we used BLASTP, version 2.3.0 from NCBI’s BLAST+ package [76], to identify the putative A. spinulosporus ortholog. The resulting pair was then aligned using MAFFT, version 7.294b [77], with the BLOSUM 62 matrix of substitutions [78], a gap penalty of 1.0, 1,000 maximum iterations, a single guide tree, and the ‘genafpair’ parameter. The associated DNA sequences were then forced onto the protein alignment using PAL2NAL, version 14 [79], and the synonymous substitution rate Ks was calculated according to LWL85m method using YN00 module in PAML, version 4.9 [80]. By examining the bimodal distribution of Ks values, genes with relatively low Ks values (0 ≤ Ks < 0.05) were inferred to be from the A. spinulosporus parent genome and genes with high Ks values (0.05 ≤ Ks <10) were inferred to stem from the A. quadrilineatus-like parent genome. We performed the same analysis on the known, non-hybrid haploid genome of Aspergillus fumigatus strain A1163 compared to A. fumigatus strain Af293 [32] and on the known hybrid diploid genome of Zygosaccharomyces parabailii strain NBRC1047/ATCC56075 compared to their closest homologs in the known parent Zygosaccharomyces bailii strain CLIB213 [75] as controls.
To determine the completeness of each parental genome and evaluate the efficacy of assigning genes in hybrid genomes to parent-of-origin, we created separate proteome FASTA files for genes from the A. spinulosporus and A. quadrilineatus-like regions of the genome and evaluated how many near-universally single copy orthologous genes were present in each parental genome using the BUSCO pipeline [81] with the ASCOMYCOTA database (creation date: 2016–02-13, number of species: 75, number of BUSCOs: 1315) from ORTHODB, version 9 [82].
To determine homeolog pairs between each parental genome, we used a reciprocal best blast hit approach. Specifically, we used the FASTA files of genes created in the previous step and conducted a reciprocal best blast hit analysis between the two parental genomes using an e-value cutoff of 10−3. To determine if one gene was putatively pseudogenized, we employed a previously established approach, which compares gene lengths between the two homeologs; genes whose length is substantially shorter (we used an upper threshold of 80%) than their homeolog pair are inferred to be pseudogenes [75].
Maximum likelihood phylogenetic and phylogenomic analyses
To establish the taxonomic identity of the sequenced isolates, we retrieved their β-tubulin and calmodulin sequences by performing a nucleotide BLAST of the Aspergillus nidulans A4 β-tubulin and calmodulin sequences against each assembled genome. In addition, β-tubulin and calmodulin sequences from two strains of each of Aspergillus foveolatus, Aspergillus latus, Aspergillus nidulans, Aspergillus pachycristatus, Aspergillus rugulosus, Aspergillus spinulosporus, and Aspergillus striatus and from one strain of Aspergillus corrugatus were retrieved from GenBank. We also retrieved the same sequences from one strain of Aspergillus sydowii, which served as an outgroup for the phylogeny [36]. Sequences were aligned with MAFFT, version 7.310 [77], gaps were removed with TRIMAL, version 1.2.rev59 [83], using the ‘gappyout’ parameter, and the β-tubulin and calmodulin sequences for each individual strain were concatenated. A phylogeny was constructed from the concatenated sequences with RAxML, version 8.2.11 [84], with the GTRGAMMAX model and 1,000 rapid bootstrap replicates. Branches with bootstrap support less than 80 were collapsed.
To determine the number of hybridization events that gave rise to A. latus isolates, we conducted phylogenomic analyses to reconstruct the evolutionary history of the A. spinulosporus parental genome in the A. latus hybrids and of the genes of A. spinulosporus strains NRRL2395 and 4060. We first identified single-copy orthologous genes across the 9 strains using ORTHOFINDER, version 2.3.8 [85] with default parameters, which employs a Markov clustering algorithm [86] on gene sequence similarity information derived from an ‘all-vs-all’ approach using NCBI’s BLAST+, version 2.3.0. Out of the inferred 12,596 groups of orthologous genes, 5,894 were single-copy (i.e., each of the 9 isolates were represented by a single sequence). All 5,894 sets of corresponding nucleotide sequences were aligned using MAFFT, version 7.402 [77], with the ‘genafpair’ parameter and 1,000 maximum iterative sequence alignment refinements. Alignments were trimmed using TRIMAL, version 1.2rev59 [83], using the ‘gappyout’ parameter. The resulting sequences were concatenated into a single nucleotide data matrix (8,405,004 sites). The strain phylogeny was inferred using IQTREE, version 1.6.1 [87], with the nbest parameter set to 10 to increase the number of best trees used during the search. Bipartition support was evaluated using 5,000 ultrafast bootstrap approximation replicates [88]. The evolutionary history of the A. quadrilineatus-like parental genome among the A. latus hybrids and two A. quadrilineatus strains (NRRL201T and CBS 853.96) was inferred using the same approach; in this case, the nucleotide data matrix was comprised of 7,385,465 sites (from 5,079 single-copy orthologous genes out of a total of 11,814 groups of orthologous genes).
Lastly, we conducted approximately unbiased topology tests [38] using the data matrices from each parental genome with the ‘au’ parameter in IQTREE, version 1.6.1 [87]. Specifically, we examined if the two topologies among the 7 A. latus hybrids inferred using the A. spinulosporus or A. quadrilineatus-like parental genomes were significantly different for either genome-scale data matrix inferred from the parental genomes. During constrained tree search, we used a substitution model of a general time-reversible model with empirical base frequencies, a discrete Gamma model with 4 rate categories, and allowed for a proportion of invariable sites (GTR+I+F+G4) [89–92].
Examination of loss of heterozygosity using copy number variation analysis
To conservatively and accurately instances of loss of heterozygosity, we measured copy number variation using CONTROL-FREEC, version 9.1 [93] and CNVNATOR, version 0.3.2 [94]. More specifically, we evaluated false discovery rate (FDR) and false positive rate (FPR) using reads from A. spinulosporus that were aligned to a “concatenated” genome of the A. spinulosporus and A. nidulans FGSC-A4 using 10 different window sizes (500, 750, 1000, 1250, 1500, 1750, 2000, 3000, 4000, and 5000 base pairs). Reads were aligned with BWA-MEM, version 0.7.12 [95], and duplicates reads were removed with PICARDTOOLS, version 2.1.1 (http://broadinstitute.github.io/picard/). In this way, we inferred the window size parameter that minimizes the FDR and FPR CN variable regions identified in the hybrid genomes. Specific parameters used for CONTROL-FREEC include a minimum and maximum expected GC-content of 0.3 and 0.5, respectively, and a telocentrometric parameter of 10,000. Default parameters were used for CNVNATOR. To identify statistically significant CN variable loci, we implemented a Wilcoxon Rank Sum test [96] and a Kolmogorov-Smirnov test [97], in the case of CONTROL-FREEC and a T-test in the case of CNVNATOR. Using the resulting set of significant CN variants per window size for each program, FDR and FPR were calculated using the following formulas:
where TP represents true positives, FP represents false positives, and TN represents true negatives. Across the 10 different window sizes, we found that CONTROL-FREEC often, but not always, slightly outperformed CNVNATOR (figshare image 5, 10.6084/m9.figshare.8114114S5S10). More importantly, we found that CONTROL-FREEC had an FDR and FPR of 0 (i.e., had no false negatives or false positives) when using a window size of 1000 and 1250. Therefore, we used CONTROL-FREEC with a window size of 1000 to identify CN variable loci in all other isolates.
Macrophage isolation
To obtain macrophages for a phagocytosis assay of Aspergillus asexual spores (conidia), we used bone marrow-derived macrophages that were isolated as described previously [62]. Briefly, macrophages were recovered from femurs of C57BL/6 wild-type mice (6-weeks old) and were incubated in an RPMI medium (Gibco) supplemented with 30% (volume / volume) L929 growth conditioning media, 20% inactivated fetal bovine serum (Gibco), 2 millimolar glutamine and 100 units / milliliter of penicillin-streptomycin (Life Technologies). Fresh media was added after 4 days of cultivation and macrophages were collected after 7 days and used for subsequent experiments.
In vitro phagocytosis by macrophages
Phagocytosis of asexual spores (conidia) by wild-type macrophages were carried out according as previously described with modifications [98]. 24-well plates containing a 15-mm-diameter coverslip in each well (phagocytosis assay) and 2×105 macrophages per well were incubated with 1 ml of RPMI-FBS [(RPMI medium (Gibco) supplemented with 10% inactivated fetal bovine serum (Gibco), 2 millimolar glutamine and 100 units / milliliter of penicillin-streptomycin (Life Technologies)] at 37°C, 5% carbon dioxide for 24 hours. Wells were washed with 1 milliliter of phosphate-buffered saline before the same volume of RPMI-FBS medium supplemented with 1×106 spores (1:5 macrophage / spore ratio) was added in the same conditions.
To determine phagocytosis, macrophages were incubated with spores for 1.5 hours before supernatant was removed and 500 μl of phosphate-buffered saline containing 3.7% formaldehyde was added for 15 minutes at room temperature. Sample coverslips were washed with 1 milliliter of ultrapure water and incubated for 20 minutes with 500 microliters of 0.1 milligrams / milliliter calcofluor white to stain the cell wall of non-phagocytosed spores. Samples were washed and coverslips were viewed under a Zeiss Observer Z1 fluorescence microscope. In total, 100 spores were counted per sample and the phagocytosis index was calculated. Experiments were performed in biological triplicates.
Viability of Aspergillus hyphae
Human neutrophils from fresh venous blood of healthy adult volunteers were isolated according to a previous study with slight modifications [99], through centrifugation over isotonic Percoll, lysed, and resuspended in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-buffered saline solution. Since we did not observe any neutrophil-mediated killing of Aspergillus asexual spores, as also previously described [100], we followed the protocol previously reported for neutrophil-mediated inhibition of germination. Aspergillus asexual spores were incubated with neutrophils (0.5, 1.0, or 2.5×105 cells / milliliter; effector : target cell ratios of 1:1000, 1:500, or 1:200, respectively) in a 96-well plate overnight at 37°C in RPMI 1640 medium containing glutamine and 10% fetal calf serum (Life). The neutrophils were lysed in water / sodium hydroxide (pH 11.0) and spore germination was determined using an MTT (thiazolyl blue; Sigma-Aldrich) assay as previously reported [101]. Each isolate’s viability was calculated relative to incubation without neutrophils, which was set at 100% for each isolate and evaluated separately. To evaluate the viability of Aspergillus hyphae in the presence of neutrophils, we used a previously described protocol [100]. Aspergillus asexual spores were incubated overnight at 37°C in RPMI 1640 medium containing glutamine and 10% fetal calf serum (Life) upon formation of a monolayer, as verified by microscope. Freshly isolated neutrophils (0, 1.0, 2.0, or 3.0 × 105 cells / milliliter) were cultured for 1 hour on the Aspergillus monolayer at 37°C. Subsequently, the cells were lysed in water / sodium hydroxide (pH 11.0) and the MTT assay was performed. Each isolate’s hyphal viability was calculated as a percentage of its viability after incubation without neutrophils. The experiments were repeated three times, each performed in triplicate.
NETosis assays
Human polymorphonuclear cells (PMN) were isolated from 8 mL of peripheral blood of adult male healthy volunteers by density centrifugation using Mono-Poly™ Resolving Medium (MP Biomedicals LLC, Irvine, CA, USA) according to the manufacturer’s instruction. PMN (5×106 / mL) were resuspended in Hank’s Balanced Salt Solution, without calcium or magnesium, containing 5% FBS (Gibco®, South American, Brazil). For flow cytometry analysis, 100 μL (5×105 / tube) of PMN were seeded in sterile round-bottom polystyrene tubes, stimulated with 10 nM of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO, USA) or fungi samples (5×105 / tube) and then incubated for 3 hours at 37°C and 5% CO2. In the last 30 minutes, 1000x diluted LIVE/DEAD™ (Invitrogen, Eugene, OR, USA) was added. After that, PMN were made to react with SYTOX™ Green Nucleic Acid Stain (1 μM) (Invitrogen) for 10 minutes at room temperature. Data on cells were acquired by flow cytometry using a BD FACSCanto II instrument (BD Bioscience, Franklin Lakes, NJ, USA). One hundred thousand events per sample were collected, doublet discrimination was performed using Forward Scatter Area (FSC-A) versus Forward Scatter Height (FSC-H) parameters, and the PMN were gated according to size (FSC-A) and granularity (Side Scatter Area, SSC-A). LIVE/DEAD™ and SYTOX™ positive cells were analyzed with FlowJo software (TreeStar, Ashland, OR, USA). For fluorescence microscopy, 100 μL (5×105 / well) of PMN were seeded on 13 mm glass coverslips in 24-wells plate and pre-incubated for 30 minutes at 37°C and 5% CO2. After adherence, PMN were stimulated with PMA (10 nM) or fungi samples (5×105 / tube) and then incubated for 3 hours at 37°C and 5% CO2. PMN were made to react with SYTOX™ Green (1 μM) (Life Technologies) for 10 minutes at room temperature. The glass coverslips were removed and the slides were fixed with ProLong Gold Antifade Mountant with DAPI (Invitrogen). The images were obtained using a Leica DMI6000 Fluorescence Microscope (Leica Microsystems, Wetzlar, Germany). Details on the flow cytometry gating strategy used and microscopy images are available in the figshare repository (doi: 10.6084/m9.figshare.8114114).
Growth in the presence of different stresses
To study variation in infection-relevant phenotypes between the hybrid isolates, we compared the radial growth of A. nidulans, A. spinulosporus, A. latus, and of all the clinical isolates in different temperatures (30°C, 37°C and 44°C), in the presence of increasing concentrations of oxidative stress-inducing compounds (paraquat and menadione), and on iron starvation. Although the importance of oxidative stress susceptibility is contentious for Aspergillus virulence [102,103], we chose to examine these phenotypes because it is well established that hosts produce reactive oxygen species in response to infection [104]. To test for the effects of iron starvation on fungal growth, MM was prepared without any iron source and supplemented or not with 200 μM of the iron chelators Bathophenanthrolinedisulfonic acid (4,7-diphenyl-1,10-phenanthrolinedisulfonic acid [BPS]) (Sigma-Aldrich) and 300 μM of 3-(2-pyridyl)-5,6-bis(4-phenylsulfonic acid)-1,2,4-triazine (ferrozine) (Sigma-Aldrich). For radial growth, isolates were grown in triplicate from 105 spores and incubated at 37°C (except for the temperature test) for 5 days. Growth results in the presence of oxidative stress were expressed as ratio, dividing colony radial diameter (cm) of growth in the stress condition by colony radial diameter in the control (no stress condition).
Hydrogen peroxide tolerance
To test the viability of Aspergillus hyphae after exposure to hydrogen peroxide, we performed the XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)2H-tetrazolium- 5-carboxanilide sodium salt]) (Sigma-Aldrich) assay as described by Henriet et al. (2011), but with modifications. We obtained hyphae from each strain by incubating 1×105 asexual spores/well in 96-well plates containing MM. After 16 hours at 37°C, the medium was removed and the wells washed twice with PBS. Subsequently, 100 microliters of minimal media supplemented or not (control) with different concentrations of hydrogen peroxide (1, 3 and 5 millimolar) were added to each well. Hyphal viability was assayed after 90 minutes of incubation (37°C) to avoid overgrowth of hyphae. One hundred microliters of PBS and 100 microliters of XTT-menadione solution was added to each well, obtaining a final concentration of 200 micrograms / milliliter XTT and 4.3 micrograms / milliliter menadione and the plates were incubated for 2 hours in the dark. After centrifugation (3,000 × g for 10 min), the supernatants were transferred to another plate and read at 450 nm in a spectrophotometer. Fungal damage was defined as the percent reduction in metabolic activity compared to that of controls without hydrogen peroxide (viability = 100%).
Antifungal susceptibility assays
Antifungal susceptibility testing for voriconazole (Sigma-Aldrich), posaconazole (Sigma-Aldrich), itraconazole (Sigma-Aldrich) and amphotericin B was performed by determining the minimal inhibitory concentration (MIC) according to the protocol established by the Clinical and Laboratory Standards Institute (CLSI, 2017). For caspofungin (Sigma-Aldrich) susceptibility, the radial growth in MM supplemented with different concentration of the drug was carried out similarly as described before (see Growth in the presence of different stresses). The results were presented as a ratio: growth in caspofungin(cm)/growth in the control/without caspofungin (cm).
Lastly, to determine the extent of phenotypic differences across all strains tested and all traits measured, we conducted principal component analysis. To do so, we first scaled (i.e., standardized) the data to account for variables that are measured in different scales (e.g., MIC, radial growth, virulence). We then conducted principal component analysis and examined each variable’s contribution to the variance along principal components using the R, version 3.5.2, packages FACTOEXTRA, version 1.0.5 [105], and FACTOMINER, version 1.41 [106].
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in R, version 3.5.2 (https://www.r-project.org/). Throughout the manuscript, comparisons were made between A. spinulosporus, A. quadrilineatus, A. latus, and/or A. nidulans (e.g., Figure 1, 3, and S5). For these comparisons, we used a multi-factor ANOVA. If the null hypothesis was rejected, we used a Tukey’s honest significant differences test to examine pairwise comparisons. To visualize these comparisons, we used violin plots with embedded boxplots (Figure 1B) or bar graphs (Figures 3 and S5). Violin plots depict the distribution of the underlying data; boxplot hinges depict the first, second, and third quartiles and whiskers extend to the largest value no greater than 1.5 times the interquartile range from the first or third quartile hinge; bar graphs depict the mean value measured across replicates and error bars depict the standard deviation across replicates. To conduct the principal component analysis (PCA) shown in Figure 1A, we first scaled all the data. To examine heterogeneity in Galleria mellonella killing curves, we employed a log-rank test. To compare Galleria mellonella killing curves solely across Aspergillus strains, we removed the negative control (PBS) prior to conducting statistical analysis. Other statistical analyses were automatically performed by publicly available software (e.g., substitution model testing for phylogenomic analyses was automatically performed by IQ-TREE).
Supplementary Material
Data S1. Genomic regions with signatures of aneuploidy and loss of heterozygosity. Related to Figure 2, S3, and S4.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| A. spinulosporus NRRL 2395T | [37]; USDA ARS Culture Collection Database | Strain # NRRL 2395T |
| A. nidulans A4 | [35]; Fungal Genetics Stock Center | Strain # A4 |
| A. quadrilineatus NRRL 201T | This study; USDA ARS Culture Collection Database | Strain # NRRL 201T |
| A. spinulosporus 4060 | [28]; Laboratory of Dr. Donald Sheppard, McGill University | Strain # 4060 |
| A. latus NRRL 200T | This study; USDA ARS Culture Collection Database | Strain # NRRL 200T |
| A. latus MM151978 | This study; Laboratory of Dr. Donald Sheppard, McGill University | Strain # MM151978 |
| A. latus NIH | This study; Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven | Strain # NIH |
| A. latus ASFU1710 | This study; Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven | Strain # ASFU1710 |
| A. latus ASFU1908 | This study | |
| A. latus ASFU2033 | This study; Clinical Laboratory of Dr. Katrien Lagrou, Department of Microbiology, Immunology and Transplantation, KU Leuven | Strain # ASFU2033 |
| A. latus MO46149 | This study; Clinical Laboratory of Dr. Catarina Lameiras, Department of Microbiology, Portuguese Oncology Institute of Porto | Strain # MO46149 |
| Macrophages | This study | N/A |
| Neutrophils | This study | N/A |
| Human polymorphonuclear cells | This study | N/A |
| Human blood samples | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Caspofungin | Sigma-Aldrich | Cat#SML0425 |
| Amphotericin B | Sigma-Aldrich | Cat#A2942 |
| Itraconazole | Sigma-Aldrich | Cat#I6657 |
| Voriconazole | Sigma-Aldrich | Cat#PZ0005 |
| Posaconazole | Sigma-Aldrich | Cat#P-103 |
| Menadione | Sigma-Aldrich | Cat#M5625 |
| Hydrogen peroxide | Sigma-Aldrich | Cat#H3410 |
| Paraquat | Sigma-Aldrich | Cat#36541 |
| Bathophenanthrolinedisulfonic acid (4,7-diphenyl-1,10-phenanthrolinedisulfonic acid [BPS]) | Sigma-Aldrich | Cat#B1375 |
| 3-(2-pyridyl)-5,6-bis(4-phenylsulfonic acid)-1,2,4-triazine (ferrozine) | Sigma-Aldrich | Cat#P9762 |
| XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)2H-tetrazolium- 5-carboxanilide sodium salt]) | Sigma-Aldrich | Cat#X4626 |
| MTT | Sigma-Aldrich | Cat#298-93-1 |
| PMA (phorbol 12-myristate 13-acetate) | Sigma-Aldrich | Cat#16561-29-8 |
| LIVE/DEAD™ | Invitrogen | Cat#R37601 |
| SYTOX™ Green Nucleic Acid Stain | Invitrogen | Cat#S7020 |
| Percoll | GE Healthcare | Cat#17-5445-02 |
| Proteinase K | Sigma-Aldrich | Cat#39450-01-6 |
| SYBR Green 10,000x | Thermo Fisher Scientific Inc. | Cat#S7567 |
| Triton® X-100 | Sigma-Aldrich | Cat#9002-93-1 |
| Deposited Data | ||
| Raw reads used for genomics; See Table S3 | This study | N/A |
| Genome assemblies; See Table S3 | This study | N/A |
| Predicted gene boundaries | This study | figshare: 10.6084/m9.figshare.8114114 |
| Phenotypic measurements | This study | figshare: 10.6084/m9.figshare.8114114 |
| FACS data | This study | figshare: 10.6084/m9.figshare.8114114 |
| Phylogenetic and phylogenomic data matrices | This study | figshare: 10.6084/m9.figshare.8114114 |
| Parent of origin analysis | This study | figshare: 10.6084/m9.figshare.8114114 |
| Homeologs per hybrid isolates | This study | figshare: 10.6084/m9.figshare.8114114 |
| Biosynthetic gene cluster predictions | This study | figshare: 10.6084/m9.figshare.8114114 |
| Experimental Models: Organisms/Strains | ||
| The greater wax moth (Galleria mellonella) | Goldman lab | N/A |
| Software and Algorithms | ||
| In Silico Whole Genome Sequencer and Analyzer (iWGS), version 1.1 | [68] | https://github.com/zhouxiaofan1983/iWGS |
| AUGUSTUS, version 3.3 | [73] | http://bioinf.uni-greifswald.de/augustus/ |
| Blast+, version 2.3.0 | [107] | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+ |
| PAML, version 4.9 | [80] | http://abacus.gene.ucl.ac.uk/software/paml.html |
| Mafft, version 7.402 | [77] | https://mafft.cbrcjp/alignment/software/ |
| trimAl, version 1.2rev59 | [83] | http://trimal.cgenomics.org/downloads |
| RAxML, version 8.2.11 | [84] | https://cme.h-its.org/exelixis/web/software/raxml/ |
| IQTREE, version 1.6.1 | [87] | http://www.iqtree.org |
| Control-FREEC, version 9.1 | [93] | https://github.com/BoevaLab/FREEC |
Highlights.
Aspergillus latus clinical isolates were previously misidentified as A. nidulans
A. latus is an allodiploid that arose via hybridization of two Aspergillus species
A. latus hybrid isolates exhibit heterogeneity for several infection-relevant traits
A. latus is phenotypically distinct from parental and closely related species
Acknowledgements
We thank members of the Rokas and Goldman laboratories, in particular Xing-Xing Shen, Matthew E. Mead, and Abigail L. Labella, for their helpful suggestions and discussions. We thank Drs. Catarina Lameiras and Donald C. Sheppard for kindly sharing Aspergillus clinical isolates. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University (http://www.accre.vanderbilt.edu/). AR was supported in part by the National Science Foundation (DEB-1442113), the Vanderbilt Discovery Grant Program, the Burroughs Wellcome Fund, and the Guggenheim Foundation, JLS and AR were supported by the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program, ALL by a U.S. National Library of Medicine training grant (grant number 2T15LM007450), GHG by the Brazilian São Paulo Research Foundation (FAPESP) (grant number 2016/07870–9) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), FA by FAPESP (grant number 2016/03322–7), LNAR by FAPESP (grant number 2017/14159–2), and FR by the Northern Portugal Regional Operational Programme (NORTE 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (FEDER) (NORTE-01–0145-FEDER-000013).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, Boughman J,Brelsford A, Buerkle CA, Buggs R, et al. (2013). Hybridization and speciation. J. Evol. Biol. 26, 229–246. [DOI] [PubMed] [Google Scholar]
- 2.Baack EJ, and Rieseberg LH (2007). A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 17, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rieseberg LH (2003). Major Ecological Transitions in Wild Sunflowers Facilitated by Hybridization. Science (80-. ). 301, 1211–1216. [DOI] [PubMed] [Google Scholar]
- 4.Rieseberg LH, Kim S-C, Randell RA, Whitney KD, Gross BL, Lexer C, and Clay K (2007). Hybridization and the colonization of novel habitats by annual sunflowers. Genetica 129, 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mable BK, Alexandrou MA, and Taylor MI (2011). Genome duplication in amphibians and fish: an extended synthesis. J. Zool. 284, 151–182. [Google Scholar]
- 6.Depotter JR, Seidl MF, Wood TA, and Thomma BP (2016). Interspecific hybridization impacts host range and pathogenicity of filamentous microbes. Curr. Opin. Microbiol. 32, 7–13. [DOI] [PubMed] [Google Scholar]
- 7.Dunn B, and Sherlock G (2008). Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 18, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolfe KH (2015). Origin of the Yeast Whole-Genome Duplication. PLOS Biol. 13, e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcet-Houben M, and Gabaldón T (2015). Beyond the Whole-Genome Duplication: Phylogenetic Evidence for an Ancient Interspecies Hybridization in the Baker’s Yeast Lineage. PLOS Biol. 13, e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfe KH, and Shields DC (1997). Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387, 708–713. [DOI] [PubMed] [Google Scholar]
- 11.Stukenbrock EH (2016). The Role of Hybridization in the Evolution and Emergence of New Fungal Plant Pathogens. Phytopathology 106, 104–112. [DOI] [PubMed] [Google Scholar]
- 12.Mixão V, and Gabaldón T (2018). Hybridization and emergence of virulence in opportunistic human yeast pathogens. Yeast 35, 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin X, Patel S, Litvintseva AP, Floyd A, Mitchell TG, and Heitman J (2009). Diploids in the Cryptococcus neoformans Serotype A Population Homozygous for the α Mating Type Originate via Unisexual Mating. PLoS Pathog. 5, e1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inderbitzin P, Davis RM, Bostock RM, and Subbarao KV (2011). The Ascomycete Verticillium longisporum Is a Hybrid and a Plant Pathogen with an Expanded Host Range. PLoS One 6, e18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen K, and Yohalem DS (2001). Origin of a Polyploid Botrytis Pathogen through Interspecific Hybridization between Botrytis aclada and B. byssoidea. Mycologia 93, 1064. [Google Scholar]
- 16.Pryszcz LP, Németh T, Saus E, Ksiezopolska E, Hegedűsová E, Nosek J, Wolfe KH, Gacser A, and Gabaldón T (2015). The Genomic Aftermath of Hybridization in the Opportunistic Pathogen Candida metapsilosis. PLOS Genet. 11, e1005626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schröder MS, Martinez de San Vicente K, Prandini THR, Hammel S, Higgins DG, Bagagli E, Wolfe KH, and Butler G (2016). Multiple Origins of the Pathogenic Yeast Candida orthopsilosis by Separate Hybridizations between Two Parental Species. PLOS Genet. 12, e1006404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhodes J, Desjardins CA, Sykes SM, Beale MA, Vanhove M, Sakthikumar S, Chen Y, Gujja S, Saif S, Chowdhary A, et al. (2017). Tracing Genetic Exchange and Biogeography of Cryptococcus neoformans var. grubii at the Global Population Level. Genetics 207, 327–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes PD, and Marr KA (2006). Aspergillosis: Spectrum of Disease, Diagnosis, and Treatment. Infect. Dis. Clin. North Am. 20, 545–561. [DOI] [PubMed] [Google Scholar]
- 20.van de Veerdonk FL, Gresnigt MS, Romani L, Netea MG, and Latgé J-P (2017). Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 15, 661–674. [DOI] [PubMed] [Google Scholar]
- 21.Brown NA, and Goldman GH (2016). The contribution of Aspergillus fumigatus stress responses to virulence and antifungal resistance. J. Microbiol. 54, 243–253. [DOI] [PubMed] [Google Scholar]
- 22.Paulussen C, Hallsworth JE, Álvarez-Pérez S, Nierman WC, Hamill PG, Blain D, Rediers H, and Lievens B (2017). Ecology of aspergillosis: insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb. Biotechnol. 10, 296–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rokas A, Mead ME, Steenwyk JL, Oberlies NH, and Goldman GH (2020). Evolving moldy murderers: Aspergillus section Fumigati as a model for studying the repeated evolution of fungal pathogenicity. PLOS Pathog. 16, e1008315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henriet SSV, Verweij PE, and Warris A (2012). Aspergillus nidulans and Chronic Granulomatous Disease: A Unique Host–Pathogen Interaction. J. Infect. Dis. 206, 1128–1137. [DOI] [PubMed] [Google Scholar]
- 25.Fang FC (2011). Antimicrobial Actions of Reactive Oxygen Species. MBio 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dotis J, and Roilides E (2004). Osteomyelitis due to Aspergillus spp. in patients with chronic granulomatous disease: comparison of Aspergillus nidulans and Aspergillus fumigatus. Int. J. Infect. Dis. 8, 103–110. [DOI] [PubMed] [Google Scholar]
- 27.Bastos RW, Valero C, Silva LP, Schoen T, Drott M, Brauer V, Silva-Rocha R, Lind A, Steenwyk JL, Rokas A, et al. (2020). Functional Characterization of Clinical Isolates of the Opportunistic Fungal Pathogen Aspergillus nidulans. mSphere 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MJ, Liu H, Barker BM, Snarr BD, Gravelat FN, Al Abdallah Q, Gavino C, Baistrocchi SR, Ostapska H, Xiao T, et al. (2015). The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLOS Pathog. 11, e1005187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heagy FC, and Roper JA (1952). Deoxyribonucleic Acid Content of Haploid and Diploid Aspergillus Conidia. Nature 170, 713–714. [DOI] [PubMed] [Google Scholar]
- 30.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, et al. (2008). Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nierman WC, Yu J, Fedorova-Abrams ND, Losada L, Cleveland TE, Bhatnagar D, Bennett JW, Dean R, and Payne GA (2015). Genome Sequence of Aspergillus flavus NRRL 3357, a Strain That Causes Aflatoxin Contamination of Food and Feed. Genome Announc. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, et al. (2005). Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438, 1151–1156. [DOI] [PubMed] [Google Scholar]
- 33.Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, et al. (2007). Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat. Biotechnol. 25, 221–231. [DOI] [PubMed] [Google Scholar]
- 34.de Vries RP, Riley R, Wiebenga A, Aguilar-Osorio G, Amillis S, Uchima CA, Anderluh G, Asadollahi M, Askin M, Barry K, et al. (2017). Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 18, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galagan JE, Calvo SE, Cuomo C, Ma L-J, Wortman JR, Batzoglou S, Lee S-I, Baştürkmen M, Spevak CC, Clutterbuck J, et al. (2005). Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438, 1105–1115. [DOI] [PubMed] [Google Scholar]
- 36.Chen AJ, Frisvad JC, Sun BD, Varga J, Kocsubé S, Dijksterhuis J, Kim DH, Hong S-B, Houbraken J, and Samson RA (2016). Aspergillus section Nidulantes (formerly Emericella): Polyphasic taxonomy, chemistry and biology. Stud. Mycol. 84, 1–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steenwyk JL, Shen X-X, Lind AL, Goldman GH, and Rokas A (2019). A Robust Phylogenomic Time Tree for Biotechnologically and Medically Important Fungi in the Genera Aspergillus and Penicillium. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimodaira H (2002). An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 51, 492–508. [DOI] [PubMed] [Google Scholar]
- 39.Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, and Papayannopoulos V (2014). Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 15, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, and White TC (2012). Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 4, 165rv13–165rv13. [DOI] [PubMed] [Google Scholar]
- 41.Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, and Fisher MC (2000). Phylogenetic Species Recognition and Species Concepts in Fungi. Fungal Genet. Biol. 31, 21–32. [DOI] [PubMed] [Google Scholar]
- 42.Balajee SA, Gribskov JL, Hanley E, Nickle D, and Marr KA (2005). Aspergillus lentulus sp. nov., a New Sibling Species of A. fumigatus. Eukaryot. Cell 4, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geiser DM, Pitt JI, and Taylor JW (1998). Cryptic speciation and recombination in the aflatoxin-producing fungus Aspergillus flavus. Proc. Natl. Acad. Sci. 95, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pringle A, Baker DM, Platt JL, Wares JP, Latgé JP, and Taylor JW (2005). Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus. Evolution 59, 1886–99. [PubMed] [Google Scholar]
- 45.Perlin DS, Rautemaa-Richardson R, and Alastruey-Izquierdo A (2017). The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect. Dis. 17, e383–e392. [DOI] [PubMed] [Google Scholar]
- 46.Alastruey-Izquierdo A, Alcazar-Fuoli L, and Cuenca-Estrella M (2014). Antifungal Susceptibility Profile of Cryptic Species of Aspergillus. Mycopathologia 178, 427–433. [DOI] [PubMed] [Google Scholar]
- 47.Verweij PE, Ananda-Rajah M, Andes D, Arendrup MC, Brüggemann RJ, Chowdhary A, Cornely OA, Denning DW, Groll AH, Izumikawa K, et al. (2015). International expert opinion on the management of infection caused by azole-resistant Aspergillus fumigatus. Drug Resist. Updat. 21–22, 30–40. [DOI] [PubMed] [Google Scholar]
- 48.Van Der Linden JWM, Warris A, and Verweij PE (2011). Aspergillus species intrinsically resistant to antifungal agents. Med. Mycol. 49, S82–S89. [DOI] [PubMed] [Google Scholar]
- 49.Keller NP (2017). Heterogeneity confounds establishment of “a” model microbial strain. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kowalski CH, Beattie SR, Fuller KK, McGurk EA, Tang Y-W, Hohl TM, Obar JJ, and Cramer RA (2016). Heterogeneity among Isolates Reveals that Fitness in Low Oxygen Correlates with Aspergillus fumigatus Virulence. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ries LNA, Steenwyk JL, de Castro PA, de Lima PBA, Almeida F, de Assis LJ, Manfiolli AO, Takahashi-Nakaguchi A, Kusuya Y, Hagiwara D, et al. (2019). Nutritional Heterogeneity Among Aspergillus fumigatus Strains Has Consequences for Virulence in a Strain- and Host-Dependent Manner. Front. Microbiol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kowalski CH, Kerkaert JD, Liu K-W, Bond MC, Hartmann R, Nadell CD, Stajich JE, and Cramer RA (2019). Fungal biofilm morphology impacts hypoxia fitness and disease progression. Nat. Microbiol. 4, 2430–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scannell DR, Frank AC, Conant GC, Byrne KP, Woolfit M, and Wolfe KH (2007). Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc. Natl. Acad. Sci. 104, 8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowers JE, Chapman BA, Rong J, and Paterson AH (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438. [DOI] [PubMed] [Google Scholar]
- 55.Paterson AH, Bowers JE, and Chapman BA (2004). Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. 101, 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brunet FG, Crollius HR, Paris M, Aury J-M, Gibert P, Jaillon O, Laudet V, and Robinson-Rechavi M (2006). Gene Loss and Evolutionary Rates Following Whole-Genome Duplication in Teleost Fishes. Mol. Biol. Evol. 23, 1808–1816. [DOI] [PubMed] [Google Scholar]
- 57.Macdonald D, Thomson DD, Johns A, Contreras Valenzuela A, Gilsenan JM, Lord KM, Bowyer P, Denning DW, Read ND, and Bromley MJ (2018). Inducible Cell Fusion Permits Use of Competitive Fitness Profiling in the Human Pathogenic Fungus Aspergillus fumigatus. Antimicrob. Agents Chemother. 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olarte RA, Worthington CJ, Horn BW, Moore GG, Singh R, Monacell JT, Dorner JW, Stone EA, Xie D-Y, and Carbone I (2015). Enhanced diversity and aflatoxigenicity in interspecific hybrids of Aspergillus flavus and Aspergillus parasiticus. Mol. Ecol. 24, 1889–1909. [DOI] [PubMed] [Google Scholar]
- 59.Kevei F, and Perberdy JF (1984). Further Studies on Protoplast Fusion and Interspecific Hybridization within the Aspergillus nidulans Group. Microbiology 130, 2229–2236. [DOI] [PubMed] [Google Scholar]
- 60.Kevei F, and Peberdy JF (1979). Induced segregation in interspecific hybrids of Aspergillus nidulans and Aspergillus rugulosus obtained by protoplast fusion. MGG Mol. Gen. Genet. [DOI] [PubMed] [Google Scholar]
- 61.Menardo F, Praz CR, Wyder S, Ben-David R, Bourras S, Matsumae H, McNally KE, Parlange F, Riba A, Roffler S, et al. (2016). Hybridization of powdery mildew strains gives rise to pathogens on novel agricultural crop species. Nat. Genet. 48, 201–205. [DOI] [PubMed] [Google Scholar]
- 62.Weischenfeldt J, and Porse B (2008). Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. Cold Spring Harb. Protoc. 2008, pdb.prot5080-pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 63.Almeida AJ, Matute DR, Carmona JA, Martins M, Torres I, McEwen JG, Restrepo A, Leão C, Ludovico P, and Rodrigues F (2007). Genome size and ploidy of Paracoccidioides brasiliensis reveals a haploid DNA content: Flow cytometry and GP43 sequence analysis. Fungal Genet. Biol. [DOI] [PubMed] [Google Scholar]
- 64.Sarkar D, Le meur N, and Gentleman R (2008). Using flowViz to visualize flow cytometry data. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malavazi I, and Goldman GH (2012). Gene Disruption in Aspergillus fumigatus Using a PCR-Based Strategy and In Vivo Recombination in Yeast. In, pp. 99–118. [DOI] [PubMed] [Google Scholar]
- 66.Mead ME, Raja HA, Steenwyk JL, Knowles SL, Oberlies NH, and Rokas A (2019). Draft Genome Sequence of the Griseofulvin-Producing Fungus Xylaria flabelliformis Strain G536. Microbiol. Resour. Announc. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sambrook J, Russell DW (2001). Molecular cloning : a laboratory manual, 3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- 68.Zhou X, Peris D, Kominek J, Kurtzman CP, Hittinger CT, and Rokas A (2016). in silico Whole Genome Sequencer & Analyzer (iWGS): A Computational Pipeline to Guide the Design and Analysis of de novo Genome Sequencing Studies. G3 Genes|Genomes|Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Safonova Y, Bankevich A, and Pevzner PA (2015). dipSPAdes: Assembler for Highly Polymorphic Diploid Genomes. J. Comput. Biol. 22, 528–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chikhi R, and Medvedev P (2014). Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37. [DOI] [PubMed] [Google Scholar]
- 72.Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stanke M, and Waack S (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225. [DOI] [PubMed] [Google Scholar]
- 74.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, et al. (2015). antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ortiz-Merino RA, Kuanyshev N, Braun-Galleani S, Byrne KP, Porro D, Branduardi P, and Wolfe KH (2017). Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madden T (2013). The BLAST sequence analysis tool. BLAST Seq. Anal. Tool, 1–17. [Google Scholar]
- 77.Katoh K, and Standley DM (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mount DW (2008). Using BLOSUM in Sequence Alignments. Cold Spring Harb. Protoc. 2008, pdb.top39-pdb.top39. [DOI] [PubMed] [Google Scholar]
- 79.Suyama M, Torrents D, and Bork P (2006). PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang Z (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- 81.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, and Zdobnov EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- 82.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, and Kriventseva EV (2013). OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 41, D358–D365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Capella-Gutierrez S, Silla-Martinez JM, and Gabaldon T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li L, Stoeckert CJ, and Roos DS (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Dongen S (2000). Graph clustering by flow simulation. Graph Stimul. by flow Clust. PhD thesis, University of Utrecht. [Google Scholar]
- 87.Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, and Vinh LS (2018). UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Z (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 39, 306–314. [DOI] [PubMed] [Google Scholar]
- 90.Yang Z (1996). Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 11, 367–372. [DOI] [PubMed] [Google Scholar]
- 91.Tavaré S (1986). Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. life Sci. 17, 57–86. [Google Scholar]
- 92.Vinet L, and Zhedanov A (2011). A ‘missing’ family of classical orthogonal polynomials. J. Phys. A Math. Theor. 44, 085201. [Google Scholar]
- 93.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, and Barillot E (2012). Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22155870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abyzov A, Urban AE, Snyder M, and Gerstein M (2011). CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM.
- 96.Wallace D (2004). The Mann- Whitney Test. J. Am. Soc. Inf. …, 1–5. [Google Scholar]
- 97.Panchenko P (2006). Kolmogorov-Smirnov Test. Stat. Appl, 83–90. [Google Scholar]
- 98.Bom VLP, de Castro PA, Winkelströter LK, Marine M, Hori JI, Ramalho LNZ, dos Reis TF, Goldman MHS, Brown NA, Rajendran R, et al. (2015). The Aspergillus fumigatus sitA Phosphatase Homologue Is Important for Adhesion, Cell Wall Integrity, Biofilm Formation, and Virulence. Eukaryot. Cell 14, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Drewniak A, Gazendam RP, Tool ATJ, van Houdt M, Jansen MH, van Hamme JL, van Leeuwen EMM, Roos D, Scalais E, de Beaufort C, et al. (2013). Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121, 2385–2392. [DOI] [PubMed] [Google Scholar]
- 100.Gazendam RP, van Hamme JL, Tool ATJ, Hoogenboezem M, van den Berg JM, Prins JM, Vitkov L, van de Veerdonk FL, van den Berg TK, Roos D, et al. (2016). Human Neutrophils Use Different Mechanisms To Kill Aspergillus fumigatus Conidia and Hyphae: Evidence from Phagocyte Defects. J. Immunol. 196, 1272–1283. [DOI] [PubMed] [Google Scholar]
- 101.Dos Reis Almeida FB, Carvalho FC, Mariano VS, Alegre ACP, Silva R. do N., Hanna ES, and Roque-Barreira MC(2011). Influence of N-Glycosylation on the Morphogenesis and Growth of Paracoccidioides brasiliensis and on the Biological Activities of Yeast Proteins. PLoS One 6, e29216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lessing F, Kniemeyer O, Wozniok I, Loeffler J, Kurzai O, Haertl A, and Brakhage AA (2007). The Aspergillus fumigatus Transcriptional Regulator AfYap1 Represents the Major Regulator for Defense against Reactive Oxygen Intermediates but Is Dispensable for Pathogenicity in an Intranasal Mouse Infection Model. Eukaryot. Cell 6, 2290–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lambou K, Lamarre C, Beau R, Dufour N, and Latge J-P (2010). Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 75, 910–923. [DOI] [PubMed] [Google Scholar]
- 104.Warris A, and Ballou ER (2019). Oxidative responses and fungal infection biology. Semin. Cell Dev. Biol. 89, 34–46. [DOI] [PubMed] [Google Scholar]
- 105.Kassambara A, and Mundt F (2017). factoextra. R Packag. v. 1.0.5. [Google Scholar]
- 106.Lê S, Josse J, and Husson F (2008). FactoMineR : An R Package for Multivariate Analysis. J. Stat. Softw. 25, 1–18. [Google Scholar]
- 107.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Genomic regions with signatures of aneuploidy and loss of heterozygosity. Related to Figure 2, S3, and S4.
Data Availability Statement
All data are publicly available through NCBI or a figshare repository. Genome assemblies are available through BioProject IDs PRJNA542678 and PRJNA542141; raw reads are available through BioProject IDs PRJNA542395, PRJNA542181, and PRJNA542141. Genome assembly and raw reads for A. quadrilineatus NRRL 201T are available through BioProject ID PRJNA623402. Strain specific BioProject or BioSample IDs can be found in Table S3. The figshare repository (doi: 10.6084/m9.figshare.8114114) contains phenotypic measurements, parent of origin analysis, homeologs per hybrid isolate, fluorescence-assisted cell sorting files, phylogenetic and phylogenomic data matrices and tree files, biosynthetic gene cluster prediction results, genome assemblies and gene predictions. Additional scripts and commands used throughout computational analyses are available through GitHub https://github.com/JLSteenwyk/Pathogenic_allodiploid_hybrids_of_Aspergillus_fungi.