

Abstract
The inflammatory response to SARS-CoV-2 infection has a direct impact on the clinical outcomes of COVID-19 patients. Of the many innate immune pathways that are engaged by SARS-CoV-2, we highlight the importance of the inflammasome pathway. We discuss available pharmaceutical agents that target a critical component of inflammasome activation, signaling leading to cellular pyroptosis, and the downstream cytokines as a promising target for the treatment of severe COVID-19-associated diseases.
While the race for a vaccine against the severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) is in full swing, current treatments available for the coronavirus disease 2019 (COVID-19) are limited to supportive care. One of the greatest enigmas surrounding COVID-19 is the diverse disease trajectories of the COVID-19 patients. Some displayed little to no symptoms, while others develop severe fever and pneumonia, leading to acute respiratory distress syndrome (ARDS) and ultimately to death. It is becoming increasingly clear that the innate immune system is a major player in patients’ response to the virus infection. Serum levels of both pro- and anti-inflammatory cytokines are markedly higher in severe cases than in moderate cases of COVID-19, suggesting that a cytokine storm, also known as cytokine release syndrome, is associated with increasing disease severity (1). Additionally, leukocytosis and lymphocytopenia are hallmark clinical features of severe cases of COVID-19 (1). These observations allude to an overdrive in inflammation as a mismanaged antiviral response against SARS-CoV-2 that lead to poor clinical outcomes.
The SARS-CoV-2 is a positive sense RNA virus. As such, its pathogen associated molecular patterns will be recognized by RNA sensing pattern recognition receptors, including TLR3, TLR7, TLR8 in the endosome, as well as retinoic acid-inducible gene I (RIG-I)-like receptors in the cytosol (2). Suggestion of SARS-CoV-2 activating the inflammasomes and pyroptosis being at the core of pathogenesis comes from the fact that lactate dehydrogenase (LDH) levels are highly elevated in patients that go on to develop severe disease (3). LDH is a cytosolic enzyme that is released to the extracellular environment upon membrane rupture. In fact, LDH release is used to monitor pyroptosis (4). Second, cytokine released as a result of inflammasome activation, IL-1β, as well as its response gene product, IL-1R, are found to be elevated in the sera of COVID-19 patients (5).
The key to overcoming excessive inflammatory activity is to target a crucial regulator of cellular inflammation while leaving the antiviral pathways intact. Pathogen- or alarmin-induced activation of NOD-like receptors (NLRs), leads to inflammasome assembly into a colossal molecular scaffold which generates a platform for the mass recruitment and activation of caspase-1 with the help of a ‘bridge’ filament protein, the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) (Figure 1). Proteolytic activation of caspase-1 subsequently catalyzes the maturation and secretion of pro-inflammatory cytokines, specifically IL-1β and IL-18 (6). The most well-characterized of the inflammasomes is the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, which has been implicated in a plethora of diseases ranging from autoinflammatory diseases to neurological disorders. Importantly, the NLRP3 inflammasome is also involved in antiviral responses and virus-associated illnesses.
Figure 1. Activation of the NLRP3 inflammasome by SARS coronavirus.
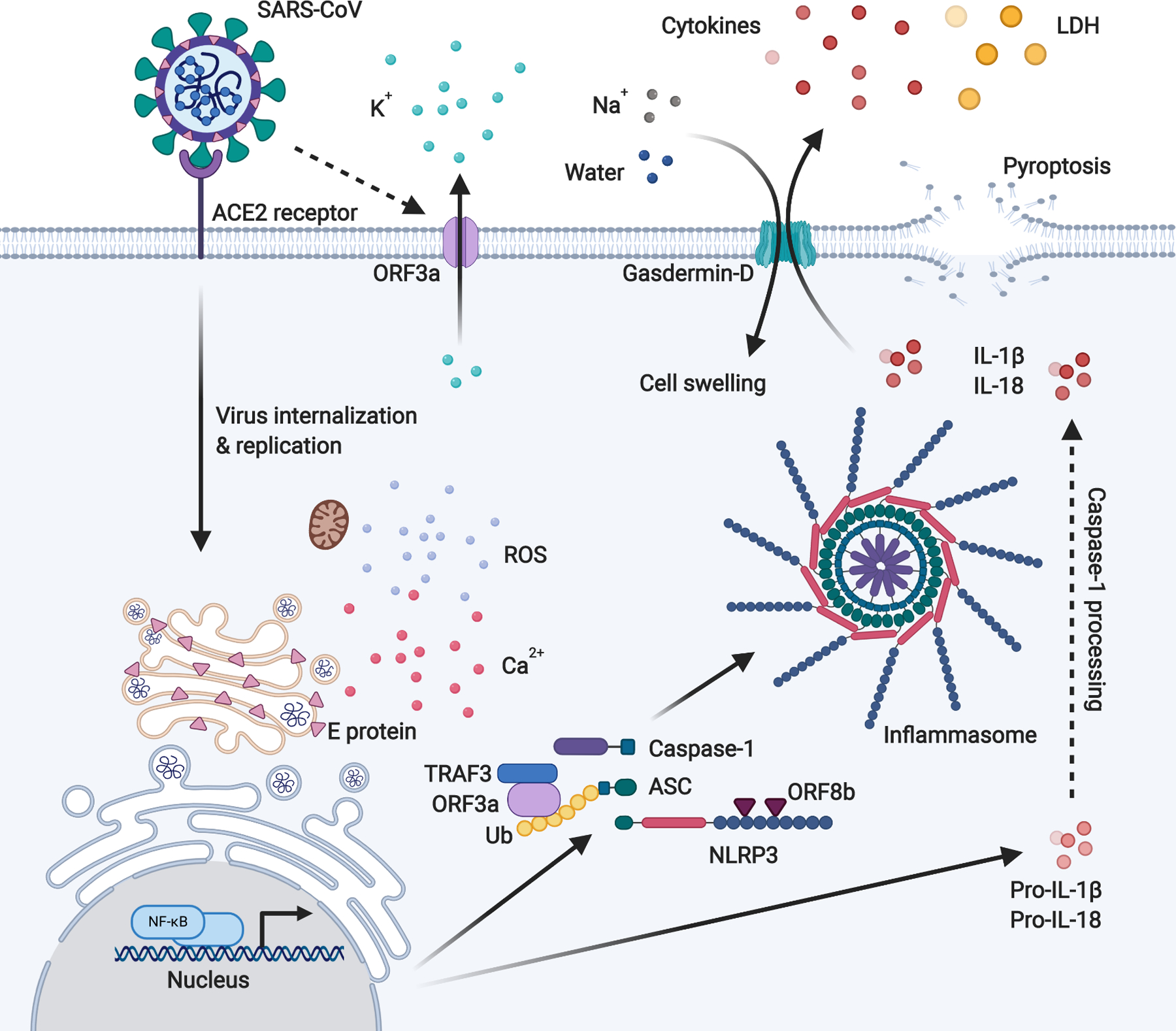
SARS-CoV E protein induces Ca2+ leakage to the cytosol from Golgi storage, while ORF3a induces K+ efflux at the plasma membrane to the extracellular spaces. These imbalance in the ionic concentration within the cells, and the resultant ROS generated by damaged mitochondria, triggers NLRP3 inflammasome activation. In addition to inducing K+ efflux, ORF3a promotes inflammasome assembly through TRAF3-mediated ubiquitination of ASC. ORF8b interacts directly with LRR of NLRP3 to stimulate its activation independent of ion channel activity. Inflammasome activation induces the formation of gasdermin-D pores on the cell membrane, causing IL-1β and IL-18 secretion, and the influx of water molecules leading to cell swelling and subsequent rupture (pyroptosis).
It is presently unclear if SARS-CoV-2 activates the NLRP3 inflammasome. However, taking lessons from its predecessor, the severe acute respiratory syndrome-related coronavirus (SARS-CoV) which caused the SARS global epidemic between 2002 and 2003, was shown to express at least 3 proteins which activate the NLRP3 inflammasome: Envelop (E), ORF3a and ORF8b. E protein localizes at the membrane enfolding the Golgi complex and the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) and function as an ion channel (viroporin) that facilitate Ca2+ leakage to the cytosol (7). On the other hand, ORF3a localizes at the Golgi complex and plasma membrane, acting as a K+ channel (8). As NLRP3 is sensitive to high cytosolic Ca2+ but is instead inhibited by high K+ concentration, the viroporin activity of SARS-CoV presumably induce inflammasome activation via E protein-mediated Ca2+ leakage from intracellular storage and ORF3a-mediated cellular K+ efflux at the plasma membrane to the extracellular spaces (8, 9). The resultant disruption of intracellular ionic balance also promotes mitochondrial damage and generation of reactive oxygen species (ROS) which co-activates NLRP3 (8). SARS-CoV could also activate inflammasomes independent of its viroporin activities. E protein and ORF3a are able to stimulate NF-κB signaling to drive the transcription of inflammatory cytokines and chemokines including IL-1β, IL-18 and IL-8, and to prime NLRP3 expression to its functional level (10–13). ORF3a also activates NLRP3 inflammasome by promoting TNF receptor-associated factor 3 (TRAF3)-mediated ubiquitination of ASC (13). Finally, ORF8b activates NLRP3 through direct interaction of the leucine-rich repeat (LRR) domain of NLRP3 (14). Given that the SARS-CoV-2 share approximately 79% overall genetic similarity with SARS-CoV, and the amino acid sequences of SARS-CoV-2 and SARS-CoV E protein are 94.7% conserved, it is likely that SARS-CoV-2 could similarly activate the NLRP3 inflammasome (15, 16). Interestingly however, a study on SARS-CoV-2 consensus sequence HKU-SZ-005b showed a remarkable distinction in ORF8 from that of SARS-CoV, and lack the aggregation motif found in SARS-CoV ORF8b that trigger NLRP3 activation (15). ORF3a share 72% amino acid sequence identity between the two viruses and of note, ORF3b is another region with distant sequences at only 32% identity (15). It would be interesting to determine whether SARS-CoV-2 ORF3a and ORF8 can likewise interact with NLRP3 or at least function as ion channels that would indirectly induce inflammasome activation. While it is unknown for SARS-CoV2 infection, various innate immune receptors are proposed as the upstream contributors of RNA virus-induced NLRP3 inflammasome activation. These include Z-DNA binding protein 1 (ZBP1) - receptor interacting protein kinase 1 (RIPK1) - RIPK3 signaling (17, 18) and 2’, 5’-oligoadenylate synthetase (OAS)/RNaseL pathway (19). Moreover, RIG-I is proposed to interact with ASC and induce IL-1β secretion after vesicular stomatitis virus (VSV) infection independently of NLRP3 (20). In parallel with the viral protein-mediated inflammasome activation, it is possible that the RNA sensing pathways trigger inflammasome activation upon SARS-CoV-2 infection.
There are various other NLRs beyond NLRP3 and the inflammasomes that may be just as consequential in host immune response against viral infections like SARS-CoV-2. These include NLRs that intensify inflammatory processes, such as nucleotide-binding oligomerization domain 1 (NOD1) and NOD2 that similarly form multiprotein complexes known as NODosomes. NOD1 and NOD2 are expressed in leukocytes and epithelial cells, and the assembled NODosomes drives NF-κB signaling and type I interferon production (21). Conversely, there exist a unique subgroup of NLRs which function as negative regulators of inflammation, including nucleotide-binding oligomerization domain-like receptor X1 (NLRX1), NLRP12 and NLR family CARD domain containing 3 (NLRC3). These NLRs attenuate inflammation by modulating NF-κB signaling, type I interferon response and ROS production, among other processes (21). Interestingly, it was reported that SARS-CoV-2 ORF9c protein can activate negative regulators of host inflammatory responses, including NLRX1, to block mitochondrial antiviral-signaling protein (MAVS) to hinder NF-κB-mediated cytokine production (22).
As a mechanism to drastically intensify disease pathogenesis, inflammasome activation can trigger cellular pyroptosis, a type of programmed cell death characterized by gasdermin D- mediated influx of sodium ions and water, causing the cells to swell excessively and rupture the membrane, and spontaneous release of cytosolic contents into the extracellular spaces. Upon inflammasome activation, caspase-1 and other non-canonical inflammasome caspases such as caspase-4, caspase-5 and caspase-11, activates gasdemin-D which subsequently form pores on the cell membrane (23). These gasdermin-D pores facilitate the secretion of IL-1β and IL-18, and importantly, they also enable simultaneous influx of Na+ and water molecules, causing excessive cell swelling to the point of membrane rupture (23, 24). Pyroptosis of macrophages which have phagocytosed viruses rapidly release a myriad of alarmins including viral particles, cytokines, chemokines, LDH, ATP and ROS, prompting an immediate reaction from surrounding immune cells and thus induces a pyroptotic chain reaction. Moreover, pyroptosis would allow viral antigens and RNA to be disseminated in the circulation and possibly generating immune complex and deposition in target organs such as kidney to initiate severe inflammatory cascade.
SARS-CoV-2-induced inflammasome activation and pyroptosis in alveolar macrophages and recruited monocyte-derived macrophages could drastically aggravate symptoms of pneumonia including ARDS and fever. It was established that the route of SARS-CoV-2 entry into cells through the angiotensin-converting enzyme 2 (ACE2) receptor, and these are indeed expressed by cells in the lungs, including alveolar type 2 cells, respiratory epithelial cells and macrophages, making them suitable targets for viral infection and potential inflammasome induction leading to pyroptosis (25, 26). The epithelial cells lining the airways are particularly vulnerable to pathogenic insults owing to its large area of exposure to external environment. Against influenza A virus infection, the RIG-I receptor is essential in regulating NLRP3 inflammasome activation in response to elevated type I interferon production to induce pyroptosis of lung epithelial cells (27, 28). Pyroptosis of lung epithelial cells may confer protection against pathogens, as demonstrated in mice models of melioidosis (29). However, Inflammasome signaling in lung epithelial cells is significantly enhanced in asthmatic patients, which aggravates tissue inflammation and worsen viral pathogenesis (30). It is predicted that pyroptosis in lung epithelial cells is likewise detrimental given the severe pneumonia experienced by COVID-19 patients. On the other hand, pyroptosis in alveolar macrophage induces acute lung injury and exacerbates lung inflammation by promoting neutrophil infiltration into the lungs and augmented alveolar concentrations of cytokines IL-6, TNFα, and IL-1β (31). The combination effects between leukocytosis and pyroptosis may be a major contributor to cytokine storms observed in COVID-19 patients. Another unsettling observation that is especially relevant to severe COVID-19 patients is that mechanical stretch of the lungs further amplify lung inflammation via NLRP3 activation in alveolar macrophages and mitogen-activated protein kinase kinase 6-mediated high-mobility group box 1 (HMGB1) protein expression in alveolar epithelial cells (32, 33). Therefore, the use of NLRP3 suppressors in patients requiring the use of ventilators might be helpful in mitigating excessive lung tissue damage.
Widespread pyroptosis might lead to excessive tissue inflammation, organ failure and death within minutes (34). Uncontrolled pyroptosis is especially detrimental in the elderly who are already experiencing an age-related chronic inflammatory condition known as ‘inflammaging’ (35). Moreover, ageing individuals have impaired capacity to produce type I and type III interferons due to TRAF3 degradation (36). These interferons are not only key antiviral resistance factors, but also are potent regulator of the inflammasomes (37). When tested in a mouse model of influenza A virus disease, the absence of innate resistance (due to deficiencies in TLR7 and RIG-I like receptor signaling) led to a lethal disease only in the presence of caspase-1/caspase-11 activation (38). In this setting, recruitment of neutrophils to the lung and activation of NETosis led to the pathological and lethal disease. Treatment with DNase (to break up the DNA released by the NETs) as well as IL-1R antagonist (Anakinra) was able to reduce the severity of the disease. Thus, the impairment in antiviral resistance and unregulated inflammasome activation may underlie the perfect storm for the severe disease we observe in the COVID-19 patients.
While there are indeed ample evidences advocating for inflammasome inhibition as a viable solution to hyper-inflammatory responses in virus infections, there have been conflicting results in the roles of immune receptors in host immune defense against virus infections. For example, it was shown that mice lacking NLRP3 and caspase-1 exhibit much greater mortality to influenza A virus infection due to compromised immune response, including a reduction in neutrophil and monocyte migration, as well as decreased secretion of cytokines and chemokines (39, 40). This imply the significant temporal, cell type, and disease-specific functions that would alter their therapeutic potential in a particular context, and it is imperative that these factors should be taken into consideration in the design and use of inflammasome inhibitors.
Inflammasome activation and pyroptosis could be underappreciated events that is central to COVID-19 pathogenesis. It was reported that abnormalities in blood coagulation leading to thrombotic complications, including pulmonary embolism are associated with poor prognosis in COVID-19 patients (41, 42). The suppression of inflammasome-mediated pyroptosis in macrophages might mitigate anomalous blood clotting by preventing the release of tissue factor, which is an initiator of blood coagulation cascades (43). Inhibition of complement-induced pyroptosis was able to reduce local inflammation at the lungs and spleen of mice infected with the Middle East respiratory syndrome-related coronavirus (MERS-CoV) (44). Yet another potential benefit of NLRP3 inhibition is the possibility of ameliorating comorbidities associated with COVID-19, including hypertension, chronic obstructive pulmonary disease, type 2 diabetes and cardiovascular disease, as NLRP3 inflammasome activation are implicated in these diseases as well (45–48). These comorbidities strongly influence COVID-19 severity and mitigating them might improve COVID-19 prognosis and significantly decrease the risk of death.
Several repurposed compounds with regulatory effects on inflammasome activity are currently being appraised in clinical trials as treatment for COVID-19. An example is tranilast, a tryptophan analogue which has a direct inhibitory action against NLRP3 (49), which is currently undergoing a randomized control trial in COVID-19 patients (Registration number: ChiCTR2000030002 on the Chinese Clinical Trial Registry). Tranilast is initially approved for the treatment of allergic and inflammatory diseases such as bronchial asthma, atypical dermatitis, allergic conjunctivitis, keloids and hypertrophic scars (50). Now, its effectiveness has also been recognized in the treatment of fibrosis, proliferative disorders, cancer, cardiovascular problems, autoimmune disorders, ocular diseases, diabetes and renal diseases (50). Tranilast might be an attractive intervention for COVID-19 patients with co-morbidities, given its wide array of therapeutic effects with minimal side effects. However, tranilast should not be used together with the anticoagulant drug warfarin, in the event that the latter is used clinically to control blood clotting in COVID-19 patients, as the two drugs are known to interact with each other synergistically to produce serious side effects (51). In an exciting recent development, a study shows that disulfiram, an FDA-approved drug used to treat alcohol addiction, is a potent inhibitor of pyroptosis and gasdermin D-dependent cytokine release (52) and holds promise for COVID-19 therapy. Preventing gasdermin D pore formation without disrupting inflammasome activation represents a promising approach, as one can restrict viral replication within cells by eliciting inflammasome-mediated apoptotic cell death instead of pyroptosis and cytokine release, thus limiting widespread tissue inflammation (53). Efforts are also on the way to block the cytokines downstream of inflammasomes, including IL-1β using Anakinra which are currently being tested at Phase 3 clinical trials (ClinicalTrials.gov identifier: NCT04330638 and NCT04324021).
Various dedicated inhibitors of NLRP3 have existed mostly in the form of experimental drugs and small molecules, as reviewed by Zahid et al. (54). These compounds could either inhibit NLRP3 indirectly, or directly target the NLRP3 core protein or its constituents such as ASC and caspase-1. Pharmacological compounds that disrupt the signaling pathways upstream of inflammasome activation also holds promise. For example, in addition to their selective inhibition of NLRP3 function itself, the anti-inflammatory natural compound parthenolide and the synthetic IκB kinase-β inhibitor Bay 11–7082 both inhibit the NF-κB pathway, thereby preventing the priming step of NLRP3 activation and the transcription of inflammatory cytokines (55). Inhibition of NF-κB-mediated inflammation was shown to improve survivability of SARS-CoV-infected mice (10).
Clinically approved drugs such as non-steroidal anti-inflammatory drugs (NSAIDs) can also be repurposed to selectively inhibit NLRP3. NSAIDs of the fenemate type such as flufenamic acid and mefenamic acid were shown to inhibit NLRP3 inflammasome by reversibly blocking volume-regulated anion channels (VRAC) which regulates Cl− transport across plasma membrane (56). Additionally, it was suggested that NSAIDs also contribute in limiting the secretion of pro-inflammatory cytokines through their cyclooxygenase-1 (COX-1)-independent activity. At present, there are no evidence for or against the use of NSAIDs as COVID-19 treatment. Nevertheless, it is recommended that NSAIDs should be prescribed cautiously to COVID-19 patients, including when used as analgesic (57).
Finally, as discussed above, type I and type III interferons can be used to suppress transcription of both IL-1β as well as inflammasome components. Type I interferons have demonstrated efficacy against SARS-CoV infection in in vitro experiments, but generally failed in human trials (58). However, the use of type I interferon in COVID-19 might still be effective for COVID-19 as SARS-CoV-2 is unusually sensitive to type I interferon pretreatment compared to SARS-CoV (59). On the other hand, an advantage that type III interferons have in COVID-19 treatment over type I interferons is that the former do not induce pro-inflammatory effects in the lungs (60). Tests in pre-clinical models have also supported the effectiveness of type III interferons in reducing the disease severity (61). Nonetheless, a careful clinical trial is warranted for the use of interferon treatments as, to date, the precise pathogenesis of COVID-19 is still unclear at this stage.
Conclusions
In this review, we propose that the benefits of inhibiting inflammasomes and/or pyroptosis are multifaceted and the search for inhibitor drugs in COVID-19 therapy would prove to be a worthwhile effort. However, NLRP3 inhibition may prove to be a promising intervention, a functional and balanced immune activity is still paramount for infection control and pathogen clearance. The importance of NLRP3 in repressing SARS-CoV-2 virulence is emphasized in a study which demonstrated that significantly dampened NLRP3-mediated inflammation in bats conferred disease tolerance in these hosts, providing an ideal reservoir for a range of zoonotic viruses, including SARS-CoV, MERS-CoV, and likely SARS-CoV-2 (62). In fact, some viruses such as the influenza virus, measles virus, Sendai virus and Nipah virus have evolved mechanisms to suppress the NLRP3 inflammasome (63). Although shown to interact with anti-inflammatory immune receptors, whether SARS-CoV-2 also suppresses inflammasomes in infected cells is unknown. Considering the gravity of the present situation, it is worth expanding the screen for available NLRP3 inhibitors to evaluate their effectiveness in mitigating aberrant inflammatory responses in COVID-19 patients. It would also be beneficial to determine the safety and the most suitable dosage of such inhibitor drugs through clinical trials as early as possible.
Acknowledgements
BioRender was used to make the figure.
Research in our laboratory is supported by NIH grants 1R01AI127429, 75N93019C00051, 1R01NS111242, 2U19AI089992 (to A.I.). COVID-19 research in the laboratory is supported by Women’s Health Research at Yale Pilot Project Program, Fast Grant from Emergent Ventures at the Mercatus Center (George Mason University), Mathers Foundation, and the Ludwig Family Foundation. A.I. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations used in this article:
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- COVID-19
coronavirus disease 2019
- LDH
lactate dehydrogenase
- NLRP3
nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain-containing 3
- NSAIDs
non-steroidal anti-inflammatory drugs
- RIG-I
retinoic acid-inducible gene I
- ROS
reactive oxygen species
- SARS-CoV
severe acute respiratory syndrome-related coronavirus
- SARS-CoV-2
severe acute respiratory syndrome-related coronavirus 2
References
- 1.Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, and Ning Q. 2020. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Invest 130: 2620–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwasaki A 2012. A Virological View of Innate Immune Recognition. Annu. Rev. Microbiol 66: 177–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han Y, Zhang H, Mu S, Wei W, Jin C, Xue Y, Tong C, Zha Y, Song Z, and Gu G. 2020. Lactate dehydrogenase, a Risk Factor of Severe COVID-19 Patients. medRxiv. doi: 10.1101/2020.03.24.20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rayamajhi M, Zhang Y, and Miao EA. 2013. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol. Biol 1040: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, and Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broz P, and Dixit VM. 2016. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16: 407–420. [DOI] [PubMed] [Google Scholar]
- 7.Nieto-Torres JL, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, Torres J, Aguilella VM, and Enjuanes L. 2015. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 485: 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen IY, Moriyama M, Chang MF, and Ichinohe T. 2019. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol 10: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, and Horng T. 2012. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U. S. A 109: 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeDiego ML, Nieto-Torres JL, Regla-Nava JA, Jimenez-Guardeno JM, Fernandez-Delgado R, Fett C, Castano-Rodriguez C, Perlman S, and Enjuanes L. 2014. Inhibition of NF-B-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol 88: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanzawa N, Nishigaki K, Hayashi T, Ishii Y, Furukawa S, Niiro A, Yasui F, Kohara M, Morita K, Matsushima K, Le MQ, Masuda T, and Kannagi M. 2006. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 580: 6807–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley N, Jeltema D, Duan Y, and He Y. 2019. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci 20: E3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan CP, Yuen KY, Enjuanes L, and Jin DY. 2019. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 33: 8865–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi CS, Nabar NR, Huang NN, and Kehrl JH. 2019. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 5: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan JFW, Kok KH, Zhu Z, Chu H, To KKW, Yuan S, and Yuen KY. 2020. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect 9: 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, and Tan W. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 395: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1: aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, and Zhou R. 2014. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol 15: 1126–1133. [DOI] [PubMed] [Google Scholar]
- 19.Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M, Núñez G, and Silverman RH. 2015. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe. 17: 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, and Ruland J. 2010. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1B production. Nat. Immunol 11: 63–69. [DOI] [PubMed] [Google Scholar]
- 21.Coutermarsh-Ott S, Eden K, and Allen IC. 2016. Beyond the inflammasome: Regulatory nod-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol 97: 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huettenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang X-P, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu D, Wang H-Y, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, and Krogan NJ. 2020. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lieberman J, Wu H, and Kagan JC. 2019. Gasdermin D activity in inflammation and host defense. Sci. Immunol 4: eaav1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovacs SB, and Miao EA. 2017. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou X, Chen K, Zou J, Han P, Hao J, and Han Z. 2020. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med 14:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi F, Qian S, Zhang S, and Zhang Z. 2020. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun 526: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pothlichet J, Meunier I, Davis BK, Ting JPY, Skamene E, von Messling V, and Vidal SM. 2013. Type I IFN Triggers RIG-I/TLR3/NLRP3-dependent Inflammasome Activation in Influenza A Virus Infected Cells. PLoS Pathog. 9: e1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, Hirohama M, Noguchi M, Nagata K, and Kawaguchi A. 2018. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J. Virol 92: e00396–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, and Re F. 2018. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 14: e1007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer RN, Brighton LE, Mueller L, Xiang Z, Rager JE, Fry RC, Peden DB, and Jaspers I. 2012. Influenza enhances caspase-1 in bronchial epithelial cells from asthmatic volunteers and is associated with pathogenesis. J. Allergy Clin. Immunol 130: 958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, Billiar TR, Wilson MA, Shi X, and Fan J. 2016. TLR4-Upregulated IL-1β and IL-1RI Promote Alveolar Macrophage Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci. Rep 6: 31663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, and Hu G. 2013. Activation of NLRP3 Inflammasome in Alveolar Macrophages Contributes to Mechanical Stretch-Induced Lung Inflammation and Injury. J. Immunol 190: 3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding N, Wang F, Han Y, Xiao H, Xu L, and She S. 2012. Mitogen-activated protein kinase kinase 6 mediates mechanical stretch-induced high-mobility group box 1 protein expression in pulmonary alveolar epithelial cells. J Trauma Acute Care Surg. 72: 162–168. [DOI] [PubMed] [Google Scholar]
- 34.Von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, Van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, and Vance RE. 2012. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 490: 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franceschi C, Garagnani P, Parini P, Giuliani C, and Santoro A. 2018. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 14: 576–590. [DOI] [PubMed] [Google Scholar]
- 36.Molony RD, Nguyen JT, Kong Y, Montgomery RR, Shaw AC, and Iwasaki A. 2017. Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci. Signal 10: eaan2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, Farlik M, Decker T, Du Pasquier RA, Romero P, and Tschopp J. 2011. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity. 34: 213–223. [DOI] [PubMed] [Google Scholar]
- 38.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, Solis AG, Bielecki P, Mohanty S, Trentalange M, Homer RJ, Flavell RA, Wagner DD, Montgomery RR, Shaw AC, Staeheli P, and Iwasaki A. 2016. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 352: 463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, and Kanneganti TD. 2009. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity. 30: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, and Ting JPY. 2009. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity. 30: 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang N, Li D, Wang X, and Sun Z. 2020. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost 18: 844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, and Endeman H. 2020. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, Shi J, Li XA, Daugherty A, Smyth SS, Kirchhofer D, Shiroishi T, Shao F, Mackman N, Wei Y, and Li Z. 2019. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity. 50: 1401–1411.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang Y, Li J, Teng Y, Sun H, Tian G, He L, Li P, Chen Y, Guo Y, Li J, Zhao G, Zhou Y, and Sun S. 2019. Complement receptor c5ar1 inhibition reduces pyroptosis in hdpp4-transgenic mice infected with mers-cov. Viruses. 11: E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, Diep H, Kett MM, Samuel CS, Kemp-Harper BK, Robertson AAB, Cooper MA, Peter K, Latz E, Mansell AS, Sobey CG, Drummond GR, and Vinh A. 2019. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res 115: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Colarusso C, Terlizzi M, Molino A, Pinto A, and Sorrentino R. 2017. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget. 8: 81813–81824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, and Jo EK. 2013. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 62: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.An N, Gao Y, Si Z, Zhang H, Wang L, Tian C, Yuan M, Yang X, Li X, Shang H, Xiong X, and Xing Y. 2019. Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front. Immunol 10: 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, Deng X, Liang G, Zhang H, Jiang W, and Zhou R. 2018. Tranilast directly targets NLRP 3 to treat inflammasome‐driven diseases. EMBO Mol. Med 10: e8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darakhshan S, and Pour AB. 2015. Tranilast: A review of its therapeutic applications. Pharmacol. Res 91: 15–28. [DOI] [PubMed] [Google Scholar]
- 51.Matsumura T, Matsui M, Iwata Y, Asakura M, Saito T, Fujimura H, and Sakoda S. 2018. A pilot study of tranilast for cardiomyopathy of muscular dystrophy. Intern. Med 57: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, Wang J, Hollingsworth LR, Magupalli VG, Zhao L, Lou HR, Kim J, Lieberman J, and Wu H. 2020. Disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, Kinoshita T, Kushiyama H, Sakurai M, Shiroishi T, and Suda T. 2019. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun 10: 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zahid A, Li B, Kombe AJK, Jin T, and Tao J. 2019. Pharmacological inhibitors of the nlrp3 inflammasome. Front. Immunol 10: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP, and Alnemri ES. 2010. Anti-inflammatory compounds parthenolide and bay 11–7082 are direct inhibitors of the inflammasome. J. Biol. Chem 285: 9792–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daniels MJD, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S, Wong R, Latta C, Yu S, Jackson J, Fischer N, Koziel V, Pillot T, Bagnall J, Allan SM, Paszek P, Galea J, Harte MK, Eder C, Lawrence CB, and Brough D. 2016. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun 7: 12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russell B, Moss C, Rigg A, and Van Hemelrijck M. 2020. COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience 14: 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, and Peiffer-Smadja N. 2020. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 178: 104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, and Menachery VD. 2020. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv. doi: 10.1101/2020.03.07.982264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prokunina-Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R, Kotenko SV, Lazear HM, O’Brien TR, Odendall C, Onabajo OO, Piontkivska H, Santer DM, Reich NC, Wack A, and Zanoni I. 2020. COVID-19 and emerging viral infections: The case for interferon lambda. J. Exp. Med 217: e20200653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Brien TR, Thomas DL, Jackson SS, Prokunina-Olsson L, Donnelly RP, and Hartmann R. 2020. Weak Induction of Interferon Expression by SARS-CoV-2 Supports Clinical Trials of Interferon Lambda to Treat Early COVID-19. Clin. Infect. Dis ciaa453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, Wen M, Chia WN, Mani S, Wang LC, Ng JHJ, Sobota RM, Dutertre CA, Ginhoux F, Shi ZL, Irving AT, and Wang LF. 2019. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat. Microbiol 4: 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao C, and Zhao W. 2020. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front. Immunol 11: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]