

Abstract
Purpose of Review
This review summarizes recent progress in our understanding how environmental adjuvants promote the development of asthma.
Recent Findings
Asthma is a heterogeneous set of lung pathologies with overlapping features. Human studies and animal models suggest that exposure to different environmental adjuvants activate distinct immune pathways, which in turn give rise to distinct forms, or endotypes, of allergic asthma. Depending on their concentrations, inhaled TLR ligands can activate either type 2 inflammation, or Th17 differentiation, along with regulatory responses that function to attenuate inflammation. By contrast, a different category of environmental adjuvants, proteases, activate distinct immune pathways and prime predominantly type 2 immune responses.
Summary
Asthma is not a single disease, but rather a group of pathologies with overlapping features. Different endotypes of asthma likely arise from perturbations of distinct immunologic pathways during allergic sensitization.
Keywords: asthma, endotypes, allergic sensitization, TLR ligands, proteases
Introduction
A wealth of evidence links environmental exposures to the development of asthma and to exacerbations of that disease1. In particular, exposures in early life have a profound and often life-long impact on asthma development2. However, in most cases, the molecular and cellular pathways that link specific exposures to disease processes remain poorly understood. One reason for that is that asthma is no longer regarded as a single disease, but rather as a spectrum of lung pathologies that display overlapping features. The relatively new concept of asthma endotypes includes the idea that perturbations of different signaling pathways give rise to distinct forms of disease. It seems likely that events during allergic sensitization, the presumptive first step in asthma development, can affect the nature, severity and longevity of the disease. Accordingly, recent investigations have utilized animal models to uncover the relationship between different environmental adjuvants and distinct forms of allergic airway disease. Toll-like receptor (TLR) ligands, such as lipopolysaccharide (LPS) and bacterial flagellin (FLA), can be readily found in samples of house dust, and inhalation of each of these products act as potent adjuvants for allergic sensitization through the airway. Similarly, protease activity is also found in house dust, and many common allergens from dust mites and fungi possess protease activity.
Asthma and the environment
Allergic asthma is a chronic inflammatory disease of the airways characterized by intermittent shortness of breath secondary to airway obstruction, airway hyperresponsiveness (AHR) and inflammation3. The increasing prevalence of this disease over the last several decades suggests that a change in the environment is driving the development of this disease, as the time period during that increase is too short for genetic drift to have had a significant effect. Furthermore, individuals moving from countries with low asthma prevalence to countries with high asthma prevalence display increased asthma prevalence in the new country4. A wealth of epidemiologic evidence links the prevalence of allergic asthma with exposure to specific types of environmental exposures, including various components of air pollution, such as ozone and particulate matter, as well as organic products derived from bacteria and fungi. However, dissecting the mechanisms through which these agents affect asthma is not straightforward because they could conceivably act either during allergic sensitization to increase the incidence of asthma, or alternatively during allergen re-exposures to increase the severity of exacerbations. The timing of exposures might also influence the magnitude, or even the direction, of the effect, and simultaneous exposures to multiple environmental agents could have unforeseen consequences. An improved understanding of how the environment impacts asthma will be critical for not only revealing novel therapeutic pathways, but also for establishing public health policies that reduce the incidence and severity of this disease. Although factors such as diet, exercise, stress, and the microbiota of the gut and lung are often regarded as environmental factors, their impacts on asthma have been recently reviewed elsewhere5–10, and will not be discussed here. Instead, I summarize here recent progress in our understanding of how inhalation of airborne products in the environment can function as adjuvants to promote allergic sensitization and asthma.
Asthma subtypes
Asthma has been traditionally regarded as a disease of the airways in which relatively large numbers of eosinophils are present in the sputum, airway and/or blood. In general, individuals with this form of asthma respond well to glucocorticoid treatment, the gold standard asthma therapy11. However, approximately half of patients with asthma have a non-eosinophilic form of this disease, often with predominantly neutrophilic inflammation of the airway12, 13. Analysis of induced sputum has helped to classify asthma as being eosinophilic, neutrophilic, mixed inflammation, and paucigranulocytic (relatively few granulocytes)11, 14. These and other studies have revealed that patients with non-eosinophilic forms of asthma are poorly responsive to inhaled corticosteroids11. Indeed, steroids might actually exacerbate disease in these patients by inhibiting neutrophil apoptosis15. Other patients display both neutrophilic and eosinophilic inflammation, and this form of asthma is associated with particularly low lung function and high health care costs16.
The terms eosinophilic asthma and non-eosinophilic asthma are phenotypic designations because they refer to observable characteristics. A different term, ‘endotype’, has been proposed17 to additionally include the idea that different forms of asthma are caused by perturbations of different cellular and molecular pathways11, 12. This is an important distinction because an improved mechanistic understanding of how different pathways give rise to different forms of asthma should in principle lead to novel therapies that selectively target different types of asthma without compromising protective immune responses.
Eosinophilic asthma
It has long been recognized that in ‘allergic asthma’, allergen-specific T helper (Th)2 cells produce the type 2 cytokines, IL-4, −5, and −13, which act synergistically to drive airway pathology. IL-4 promotes immunoglobulin (Ig) class switching to IgE antibodies, which bind to high affinity IgE receptors (FcεR1) present on the surface of mast cells and basophils (see below). IL-5 acts as an eosinophil growth factor in the bone marrow and also induces eosinophil chemoattractants such as eotaxin-1, −2 and −3 (CCL11, CCL24, and CCL26, respectively), which directly recruit eosinophils to the lung. These cells contain granules that can release pro-inflammatory mediators, including major basic protein, eosinophil peroxidase, and eicosanoids into the extracellular space. Finally, IL-13 promotes goblet cell hyperplasia and airway hyperresponsiveness (AHR)18, 19. This general form of asthma is sometimes referred to as type 2 (T2)-high to contrast it with T2-low forms of the disease that tend to have fewer eosinophils and lower levels of type 2 cytokines.
Neutrophilic asthma
The origin of neutrophilic asthma remains uncertain, but likely includes a strong innate immune component. However, several lines of evidence suggest that IL-17-producing Th17 cells20 contribute to asthma by driving recruitment of neutrophils to the airway21–25. In support of this, numbers of CD4+ Th17 cells in the peripheral blood positively correlate with asthma severity26, 27. Moreover, Th17 cells are resistant to glucocorticoids, which might at least partly explain the observation that neutrophilic asthma tends to be steroid-resistant. The degree to which infections drive neutrophilic asthma remains unclear, but Chlamydia and Haemophilus infections increase molecules that drive Th17 differentiation, including NLRP3, caspase-1, and IL-1β. Furthermore, neutrophilic airway inflammation, disease severity, and steroid resistance in human asthma correlate with NLRP3 and IL-1β expression28. Together, these observations suggest that some bacteria or their products might promote neutrophilic asthma by driving Th17 responses to inhaled bystander allergens. It is critical to develop a more comprehensive understanding of these pathways so that effective therapies can be developed that selectively target specific asthma endotypes.
Allergic sensitization
Classically, allergic sensitization refers to the development of allergen-specific immune responses that include production of allergen-specific IgE. When allergens bind the exposed, variable region of IgE molecules on cell surfaces, the intracellular regions of these antibodies undergo cross-linking, thereby triggering the release of secretory granules that contain multiple pro-inflammatory mediators, including histamine, cytokines, and proteases. This leads to an allergen-specific inflammatory response that is the basis for a positive response in skin prick tests for allergic sensitization to individual allergens. An improved understanding of the cellular and molecular basis of allergic sensitization is important because it is likely one of the first events in the development of allergic asthma. Children that display both wheeze and sensitization to perennial allergens during the first 5 years of their lives are much more likely to develop severe asthma than children with wheeze that do not display sensitivity to allergens29. Moreover, the earlier in life that allergic sensitization occurs, the more likely it is that this sensitization will be predictive of asthma30. Identifying mechanisms that drive allergic sensitization might identify novel molecules and pathways that can be targeted therapeutically to prevent asthma or reduce its incidence.
Environmental pathogens and their products
It has long been thought that exposure to various components of the environment can contribute to the development of asthma. However, some environmental exposures can also protect against developing this disease. For example, the hygiene hypothesis postulates that increased hygiene, smaller families, and consequent decreased exposure to pathogens and their products is at least partially responsible for the recent increase in allergic diseases such as asthma31. Indeed, some lower respiratory tract infections have been shown to be associated with protection from asthma32. However, this does not hold true for all pathogens, as early life infections with RSV and rhinovirus are associated with an increased risk of asthma. It remains unclear whether the infections associated with asthma predispose for the development of this disease, or, conversely, whether children predisposed to develop asthma are more likely to become infected with these viruses33. Some bacterial products have also been linked to either protection from, or susceptibility to, asthma. The most widely studied of these is LPS, a major component of the outer cell wall of Gram-negative bacteria. LPS is ubiquitous in the environment and is active at extremely low concentrations. Some studies have indicated that high levels of LPS in the environment, particularly on farms, can protect against developing asthma34, 35. In agreement with this finding, extracts of house dust from Amish households (which have relatively high levels of endotoxin) are protective in an animal model of asthma, whereas extracts from Hutterite homes (which have lower amounts of endotoxin) are not protective36. Paradoxically, however, other studies have demonstrated a positive association between asthma and high household levels of bacteria or endotoxin37. While bacterial products might themselves have direct effects on promoting, or protecting from, asthma, it is also possible that these products simply serve as surrogate markers for the bacteria that produce them. A wealth of evidence in recent years has underscored the profound impact of microbiota on health. Accordingly, the ‘old friends’ hypothesis postulates that allergic diseases stem in part from the loss of symbiotic relationship between humans and various microorganisms38. In particular, exposure to a wide spectrum of organisms appears to protect against developing atopic diseases. Conversely, frequent use of antibiotics and ‘cleaner’ living might lead to a less diverse microbiota that carries an increased risk of asthma. It seems highly likely that the keen interest in how the microbiota affects human health will lead to a rapid improvement in our understanding of which microorganisms and their products impact the development and progression of asthma, and the mechanisms by which they do so.
LPS-mediated allergic sensitization
The positive association seen in some studies between asthma and high household levels of bacteria or endotoxin39 is consistent with the ability of bacterial products to act as adjuvants to promote adaptive immune responses40, including allergic responses to innocuous inhaled allergens41–44. Furthermore, almost all commercially available allergens, including house dust mite allergens are ‘contaminated’ by LPS, which contribute to their allergenicity45. Thus, it is critical to understand how LPS impacts allergic sensitization.
The experimental allergen, ovalbumin (OVA), is frequently used in animal models of asthma. On its own, OVA is not allergenic, but Eisenbarth and colleagues reported several years ago that large amounts of inhaled LPS (10 μg) promote Th1 responses to co-administered OVA, whereas more moderate amounts of this product (100 ng) promote Th2 responses. Still lower amounts, which might be more similar to those encountered in natural environments, were not tested, nor were the effects of LPS on Th17 and Treg responses. Our laboratory showed that the moderate dose of 100 ng LPS strongly promotes the development of OVA-specific Th17 cells, which are critical for airway neutrophilia and airway hyperresponsiveness (AHR)25. Subsequent studies showed that the nature of allergen-specific immune responses is exquisitely sensitive to the dose of LPS used during allergic sensitization46. In cultures of cells from lung-draining lymph nodes (LNs), type 2 cytokines generally increased in concert with the dose of LPS used during the sensitization, except for highest dose of LPS (10 μg), where almost no IL-4 or IL-5 was detected in LNs41. By contrast, the concentrations of IFN-γ and IL-17 in LN cultures continued to increase up to and including the highest dose of 10 μg LPS.
A more complicated picture emerged when mice sensitized using different amounts of LPS were evaluated for their biologic responses to subsequent OVA challenge(s). Remarkably, animals that had been sensitized to OVA using the extremely low dose of 0.1 pg LPS as the adjuvant developed airway eosinophilia after a single allergen challenge, and additional challenges led to stronger eosinophilia. Using a moderately higher dose of LPS (10 pg) as the adjuvant during sensitization led to stronger eosinophilic responses to subsequent allergen challenge, but using still larger amounts of LPS during the sensitization phase led to fewer eosinophils and lower pulmonary levels of IL-5 (Figure 1). This result contrasts with the very high amounts of type 2 cytokines in the regional LNs of these animals. Moreover, when the dose of LPS used during sensitization is increased to 100 ng LPS, mice continue to display relatively strong responses to a single allergen challenge, but the intensity of those responses is suppressed by additional daily challenges. Thus, although increasing doses of LPS leads to progressively higher amounts of type 2 cytokines in regional LNs, those responses are not predictive of either the intensity or the duration of responses to allergen challenge. In contrast to these findings with type 2 cytokines and eosinophilia, increasing doses of LPS during allergic sensitization led to more IL-17 and neutrophils in the airway, consistent with the increased amounts of IL-17 in regional LNs following allergic sensitization.
Figure 1.
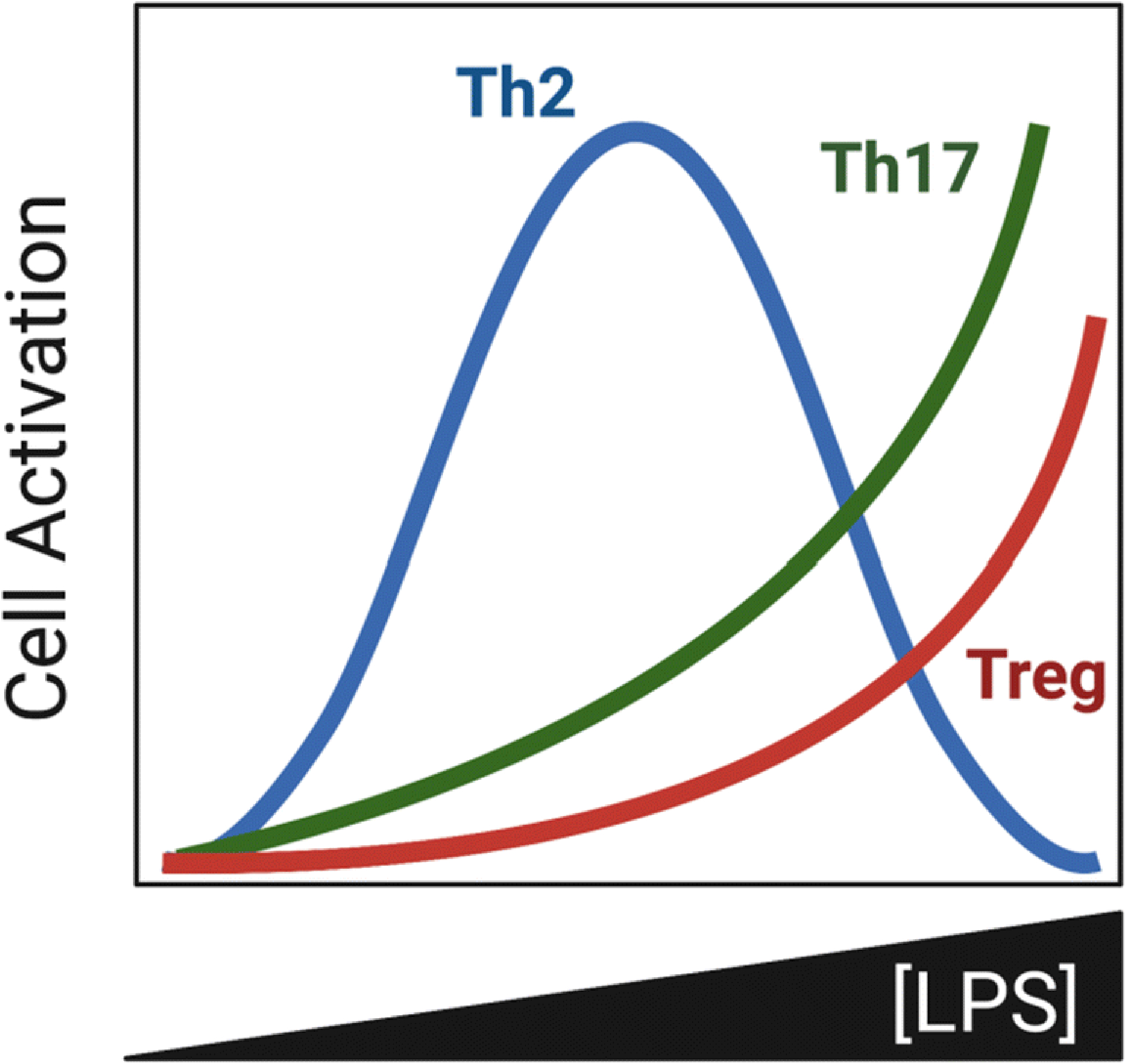
Impact of LPS concentration during sensitization on abundance of T cell subsets in the lung following allergen challenge.
It is unlikely that the reduction in asthma-like features seen when higher doses of LPS are used as the adjuvant is due to a switch from Th2 to Th1 responses because it was not associated with increased pulmonary levels of IFN-γ. However, higher amounts of LPS during sensitization does lead to higher numbers of regulatory T cells (Tregs), especially ICOS+ Foxp3+ Tregs46. This observation is in agreement with the finding that suppression of airway inflammation and AHR is associated with high levels of ICOS expression on CD4+ Tregs47 and that adoptive transfer of ICOS+ CD4+ T cells, but not ICOS− cells, suppresses established AHR in mice48.
FLA-mediated allergic sensitization
Given the potency of LPS in the development and maintenance of asthma, we asked if other TLR ligands in the environment might display similar properties. We found that the TLR9 agonist, CpG oligonucleotides, and the TLR2/6 agonist, FSL-1, promote adaptive immune responses leading to airway neutrophilia upon OVA challenge, whereas the bacterial protein, FLA, promoted responses leading to eosinophilic inflammation49. The adaptive immune responses promoted by FLA also include production of type 2 cytokines in regional LNs and the airway, as well as increased IgE synthesis. With potential relevance to human asthma, goblet cell hyperplasia and AHR are also seen in OVA-challenged mice previously sensitized to that protein using FLA as the adjuvant. FLA also primes Th17 responses, as IL-17-containing CD4+ T cells can be found in lungs of mice sensitized to OVA using FLA as an adjuvant.
HDE
Humans are routinely exposed to indoor house dust, although its composition can vary widely from household to household. Our laboratory and others have explored the use of house dust extracts (HDE) as environmentally-relevant sources of adjuvant activity in animal models of asthma. We found that although most HDEs promote adaptive responses to inhaled OVA, they differ widely in potency. Moreover, whereas some HDEs prime responses that give rise to predominantly eosinophilic inflammation upon subsequent exposure to OVA, others prime neutrophilic responses (Figure 2). Thus, an important avenue for future research will be identification of the components in HDEs that drive eosinophilic and neutrophilic responses, respectively. As might be anticipated, very small amounts of HDE generally prime relatively weak initial responses to OVA challenge, and those responses are sustained following multiple OVA challenges. By contrast, higher doses of HDE promote stronger initial eosinophilic responses to OVA, but they are not sustained in the face of multiple daily OVA challenges. These observations are reminiscent of our findings with LPS and suggest that the latter represents a major source of adjuvant activity in HDE, but can also suppress allergic responses. In support of this, adding moderate amounts of LPS to an HDE with low endogenous endotoxin content increased initial responses to OVA, but blunted responses to prolonged allergen challenge. These results are consistent with findings showing that indoor dust from Amish homes, which have relatively high endotoxin concentrations, suppress allergic responses in a mouse model of asthma, whereas dust from homes of the genetically-similar Hutterite population has much less endotoxin and fails to suppress allergic responses36. Collectively, these findings might help to explain why endotoxin has been found to be associated with increased asthma in some studies, but reduced asthma in other studies.
Figure 2. Heterogeneity in adjuvant activity associated with HDE.
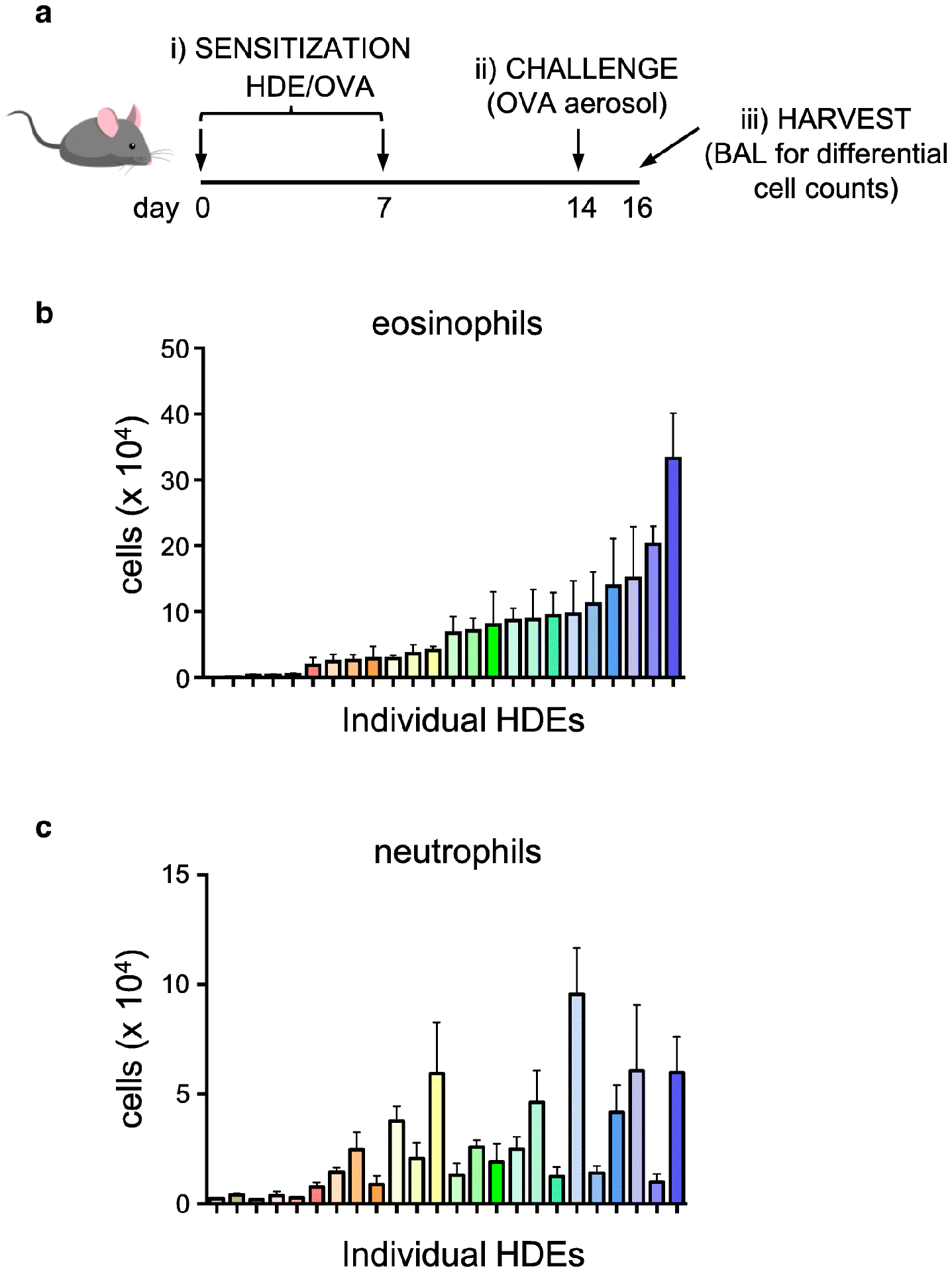
A) Timeline of asthma model, including allergic sensitization to OVA using different HDEs as the adjuvant, followed by challenge with OVA alone. B, C) Eosinophils (B) and neutrophils (C) in BALF following OVA challenge of mice previously sensitized using different samples of HDE. Note that each HDE is color-coded to allow comparisons in their ability to prime responses leading to eosinophilic and neutrophilic inflammation, respectively.
Having found that purified FLA and LPS can both function as strong adjuvants in the airway, we studied the individual requirement for each of these bacterial products in HDE-mediated allergic sensitization. Tlr4−/− mice, which do not respond to LPS, had fewer airway neutrophils and less IL-17 than did wild-type (WT) mice after HDE-mediated allergic sensitization followed by allergen challenge. By contrast, numbers of airway eosinophils and the concentration of IL-5 in BALF were as high, or higher, in Tlr4−/− mice compared with WT mice49. This suggests that LPS is required for neutrophilic airway inflammation and contributes to AHR, but is dispensable for HDE-mediated type 2 responses and consequent eosinophilic airway inflammation. Conversely, Tlr5−/− mice had fewer airway eosinophils and had lower concentrations of IL-5 than did WT or Tlr4−/− mice. WT and Tlr4−/− mice sensitized with HDE plus OVA developed AHR after OVA challenge, whereas this response was markedly reduced in Tlr5−/− mice. Although these results are likely dependent, at least in part, on the specific sample of HDE tested, they suggest that in at least some dust samples, FLA provides adjuvant activity leading to asthma-like responses to inhaled antigens.
HDE as both allergen and adjuvant
OVA is widely used in animal models of allergic pulmonary inflammation, but it is not a clinically relevant allergen for asthma. By contrast, household dust typically contains many allergens, including those derived from dust mites, cockroaches and animal dander, and multiple instillations of HDE alone (without OVA) is sufficient to trigger allergic responses in mice50. In these experiments, neutrophils can be recruited to the airway by innate immune responses to the LPS component in HDE, as well as by allergen-specific Th17 responses. Both responses are dependent on TLR4. However, WT and Tlr4−/− mice have similar numbers of airway eosinophils in these experiments, indicating that the LPS component of HDE is dispensable for type 2 responses and eosinophilia. Tlr5−/− mice had significantly fewer eosinophils in the airway than did WT mice, suggesting that at least for some extracts, FLA is a more critical driver of HDE-mediated type 2 responses than is LPS.
A caveat to experiments with Tlr5−/− mice is that TLR5 can sense and respond to FLA in gut microbiota and affect its composition from a very early point in life51, 52. Moreover, these changes can affect immune responses to pulmonary pathogens53. It remains to be determined whether this microbiota sensing function of TLR5 – or its ability to sense the FLA in dust samples – contributes more to the development of allergic asthma. However, Tlr5−/− mice display dysbiosis and metabolic syndrome, which might be expected to exacerbate disease, not protect its development. Thus, animal studies suggest that it is the direct sensing of inhaled FLA by TLR5 that contributes to asthma. Of more direct relevance to human asthma, individuals carrying a dominant negative form of TLR5, and therefore unable to respond to FLA, appear to be partially protected from asthma exacerbations54. A more extensive analysis of the microbiota of these individuals, and of the amounts of FLA in dust samples from their homes, should help to disentangle the direct effects of TLR5 in the lung and the indirect effects of this receptor on gut microbiota.
Mechanisms of TLR ligand-meditated allergic sensitization
The TLR ligands, LPS and FLA, both rapidly induce production of the cytokines, TNF, IL-1α, IL-1β, and granulocyte macrophage colony-stimulating factor (GM-CSF)55. HDE, which contains both LPS and protease activity, also induces those same cytokines, whereas proteases do not. The induction of TNF by TLR ligands is interesting because this cytokine is also associated with human asthma56. In mice, soluble TNF is sufficient to promote allergic sensitization to OVA when the two proteins are co-administered to the airways, and upon OVA challenge these mice develop allergic airway disease41. Furthermore, compared with WT mice, animals lacking both TNF receptors, TNFR1 and TNFR2 (and therefore unable to respond to either soluble or membrane-bound TNF) display marked attenuation of multiple asthma-like features in the LPS/OVA model, including eosinophils, neutrophils, and AHR55. In particular, there is a requirement for TNFR1, which is found on most cells, and can respond to either soluble or membrane-bound TNF. By contrast, TNFR2, which signals in response to membrane-bound TNF, is dispensable for allergic sensitization in this model. Compared to WT mice, TNFR1-deficient animals also had reduced amounts of type 2 cytokines in regional LNs shortly after LPS/OVA-mediated allergic sensitization, whereas TNFR2 mice had as high or higher amounts of these cytokines. No differences between these genotypes were seen for IL-17, indicating that although LPS can prime both Th2 and Th17 responses, the TNFR1 signaling pathway is required for the former, but not the latter.
Macrophages are the primary source of TNF, and studies of bone marrow chimeric mice generated using WT and Tnfr1–/– bone marrow show that TNF responsiveness in radio-resistant cells, not hematopoietic cells, is necessary for airway inflammation in TLR ligand-dependent models of asthma55. This suggests that macrophages communicate with epithelial cells (or another stromal cell) during allergic sensitization to drive allergic sensitization. Future studies should identify which specific TNFR1-dependent changes in these stromal cells are required for TLR ligand-mediated asthma.
Data from clinical trials suggest that although antagonizing TNF is not helpful to all patients, it is beneficial to a subgroup of asthmatics, including those that have severe disease57, 58. It will be important to identify individuals whose known environmental exposures and genetic predisposition are predictive of a positive response to TNF pathway blockade. Furthermore, because blockade of TNF itself is associated with increased infections59, an improved understanding of how other molecules in the TNF pathway contribute to asthma is necessary for design of novel therapies that ameliorate asthma exacerbations without this complication.
An important aspect of understanding how LPS acts as an adjuvant for promoting asthma is the identification of the cell types(s) in which responsiveness to LPS is critical for sensitization. The preponderance of data suggest that the answer to this question depends on whether type 2 responses or Th17/neutrophilic responses are being studied. Several years ago, Hammad and colleagues generated bone marrow chimeric mice using WT and Tlr4–/– animals, and found that radio-resistant cells, which they assumed to be epithelial cells, are critical for the induction of type 2 responses to inhaled house dust mite (HDM) allergens. Similar findings were found for bone marrow chimeric mice when LPS was used as the adjuvant for allergic sensitization60, 61. Experiments of this type are confounded by the persistence of large numbers of tissue resident macrophages and dendritic cells (DCs) after irradiation 62 and by radiation-induced transcriptional changes in epithelial cells63. However, the requirement of Tlr4 in stromal cells for type 2 responses was confirmed genetically following the generation of conditionally mutant (floxed) Tlr4 mice in which the Vav1 promoter drives cre recombinase expression in hematopoietic cells and the Ssh promoter drives cre expression in stromal cells60. This same study further showed that Th17 responses, and consequent neutrophilic inflammation of the airway, is dependent on Tlr4 expression in hematopoietic cells.
A recent study used a more selective approach to target myeloid differentiation primary response 88 (Myd88) deletion to either lung epithelial cells using Surfactant protein C (Sftpc)-cre, or to antigen presenting cells using Cd11c-cre64. This study showed, consistent with previous findings, that type 2 responses to both FLA-mediated allergic sensitization to OVA, and allergic sensitization to HDM, are dependent on Myd88 expression in lung epithelial cells, whereas Th17 responses and neutrophilic inflammation are dependent on Myd88 expression in antigen presenting cells. Interestingly, this study also revealed that Myd88 expression in lung epithelial cells can affect the chromatin accessibility and transcriptional profiles of lung dendritic cells and alveolar macrophages. This exciting finding raises several important questions, including which type of epithelial cells are communicating with antigen presenting cells, what molecules convey this cell-to-cell communication, which type of dendritic cells are responding, and what changes in dendritic cells allow them to drive Th2 differentiation. The answers to these questions should greatly enhance our understanding of how TLR ligands promote allergic sensitization.
Proteases and allergic sensitization
One of the highest risk factors for the development and exacerbation of asthma is exposure to fungi and their associated products65, 66. Again, the impact of these exposures is particularly potent during early life67, 68, and children exposed to high levels of mold are much more likely to develop asthma than are children raised with very little mold67. Interestingly, exposure to fungi during early childhood is associated with the development of asthma even in individuals that are not sensitized to the fungal products themselves69. This suggests that some fungal products might act as adjuvants to promote allergic sensitization to bystander allergens. It is likely that some of this adjuvant activity stems from fungal-encoded proteases, as many allergens have protease activity. Moreover, as discussed below, proteases can promote allergic sensitization to otherwise innocuous bystander proteins, such as OVA55, 70. Although often regarded as being maladaptive, type 2 responses can enhance clearance of fungi through the airway, and eosinophils can directly suppress fungal growth71. Thus, detection of protease activity might have evolved as a strategy to alert the host to fungal invasion and trigger protective type 2 responses against the fungi.
Dust mites are another major source of proteases, which are found in the feces of these insects, suggesting that they likely contribute to food digestion. Dried material from these feces is present in bioaerosols and is therefore easily inhaled. The likely importance of proteases to allergic sensitization and asthma is underscored by the finding that proteases, cysteine proteases in particular, are much more abundant in the allergenic Dermatophagoides genus than in other, non-allergenic mites72. Moreover, genes expressing these proteases in Dermatophagoides are highly expressed, and the encoded proteins very stable73. These findings might account for the high allergenicity of this dust mite genus. Some examples of protease allergens include the house dust mite allergens, Der p 1 and Der p 9, allergens from the mold Aspergillus, and papain (PAP), a cysteine protease found in papaya. A recent study showed that proteases from both fungi and dust mites are present in indoor house dust71. Although that study noted that fungi were the major contributors to the protease activity, it seems plausible that this might depend on the season, region of the country, and lifestyle of the inhabitants.
Mechanisms of protease-mediated allergic sensitization
The mechanism by which proteases promote allergic sensitization remains poorly understood. However, it is clear that this class of molecules triggers distinct pathways from those activated by TLR ligands because proteases do not induce TNF, IL-1α, IL-1β, or GM-CSF. Furthermore, the TNF signaling pathway is dispensable for asthma-like features when proteases are used as the adjuvant for allergic sensitization55. These observations confirm, at least in animal models, that different forms of asthma can arise from distinct signaling pathways, which is precisely the tenet of the endotype hypothesis.
One possible explanation for the ability of proteases to act as adjuvants is their ability to cleave molecules that maintain the integrity of the lung epithelium74. This breach in epithelial integrity might allow allergens to gain access to the underlying interstitium, where they can be captured by immune response-stimulating dendritic cells. Indeed, proteases have been shown to cleave tight junction proteins, such as Occludin and Zona occludins (ZO)-1, leading to increased epithelial permeability74–77. However, while attractive, this hypothesis does not account for the ability of proteases to preferentially drive type 2 responses. Further, attempts to measure impaired barrier function in vivo have not been successful in all models of allergic sensitization, including house dust mite inhalation78 and a mixture of proteases from Aspergillus oryzae55.
A second possible mechanism for protease-mediated allergic sensitization involves the G protein-coupled receptors known as protease activated receptors (PARs). These interesting receptors contain both a ligand and a receptor; in the presence of protease activity, the ligand moiety is cleaved from the remainder of the receptor and then signals through that same receptor. In particular, PAR2 can be activated by a variety of proteases, and antibody-mediated blockade of PAR2 function during allergic sensitization to cockroach extracts abrogates inflammation and reduces AHR following subsequent allergen challenge79.
A third possible mechanism of action for proteases involves the innate immune cytokine, IL-33. This cytokine is constitutively expressed in epithelial cells80, and is initially translated as a precursor protein that normally resides in the nucleus. However, IL-33 can be released from cells by stressors, including aeroallergens, and it can function as an ‘alarmin’ to alert the host to epithelial cell damage81, 82. The precursor protein has relatively weak immune activity, but is activated by proteolytic cleavage, and by binding to its receptor IL1RL1 (also known as ST2), drives the production of type 2 cytokines by group 2 innate lymphoid cells (ILC2s). Intriguingly, environmental allergens can also cleave IL-33, thereby activating this cytokine83. Thus, IL-33 might serve as a protease activity sensor that evolved to detect fungal infection, but can also be activated by otherwise harmless protease allergens, leading to maladaptive allergic responses. In addition to this cellular and molecular evidence suggesting a role for IL-33 in protease-mediated asthma, strong genetic evidence supports this association. Thus, single nucleotide polymorphisms in both IL33 and IL1RL1 are strongly and reproducibly associated with asthma in both single gene and genome-wide association studies84, 85.
Several cell types can express ST2, and it remains unclear whether these different cell types are differentially important in distinct forms of asthma. IL-33 is a particularly potent activator of ILC2s, and might therefore be important in non-allergic, eosinophilic asthma. However, IL-33 might also promote Th2 development during allergic sensitization. Thus, in a model of protease (papain)-induced allergic sensitization, IL-33 activates ILC2s, which in turn produce IL-13 that promotes the trafficking of dendritic cells from the lung to regional LNs86. In addition, IL-33 also increases cell surface display of OX40L on ILC2s, which drive the expansion of both Th2 and Treg cells87. Although Tregs are normally thought to suppress inflammation, recent evidence suggests that IL-33 can upregulate GATA3 in these cells, thereby converting them to a pro-inflammatory cell type88. Thus, IL-33 can activate multiple pathways that are associated with heightened type 2 responses to allergens.
Conclusion and Perspectives
Understanding how different environmental components act as adjuvants to promote allergic sensitization is important, not only to more fully understand the origins of asthma, but to also inform public health policies aimed at reducing the incidence of this disease. Administration of relatively large amounts of purified products, such as TLR ligands and proteases, have provided great insight into their potential mechanisms of action, but it is unlikely that these treatments are representative of the quantities of compounds that are normally inhaled by humans. A major advantage of using house dust extracts is that they represent, at least to a first approximation, what humans are actually inhaling in their homes. Further, the ability of adjuvant activity in these extract to promote allergic sensitization to naturally-occurring indoor allergens in the dust, highlights the utility of dust samples as tools to understand allergic sensitization. It is likely that these different extracts will be even more informative when associations can be made between the components that comprise them and the type of immune responses they promote (Figure 2). Ultimately, integrating genetic and epigenetic data on asthmatics with distinct endotypes together with a comprehensive analysis of the dust they inhale daily should provide an even deeper understanding of how gene-by-environment interactions drive allergic sensitization and susceptibility to asthma.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Compliance with Ethical Standards The author declares that he has no competing interests.
Human and Animal Rights and Informed Consent This article includes studies performed in the author’s laboratory at the National Institute of Environmental Health Sciences (NIEHS), Research Triangle Park, North Carolina. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at the NIEHS.
Literature Cited
- 1.••.Murrison LB, Brandt EB, Myers JB, Hershey GKK. Environmental exposures and mechanisms in allergy and asthma development. J Clin Invest 2019; 129:1504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]; Excellent review on environmental exposures and asthma.
- 2.••.Burbank AJ, Sood AK, Kesic MJ, Peden DB, Hernandez ML. Environmental determinants of allergy and asthma in early life. J Allergy Clin Immunol 2017; 140:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Excellent review on long-lasting effects of environmental exposures in early life.
- 3.Martinez FD, Vercelli D. Asthma. Lancet 2013; 382:1360–72. [DOI] [PubMed] [Google Scholar]
- 4.Cabieses B, Uphoff E, Pinart M, Anto JM, Wright J. A systematic review on the development of asthma and allergic diseases in relation to international immigration: the leading role of the environment confirmed. PLoS One 2014; 9:e105347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.•.Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The Role of Lung and Gut Microbiota in the Pathology of Asthma. Immunity 2020; 52:241–55. [DOI] [PMC free article] [PubMed] [Google Scholar]; Excellent review on the impact of microbiota on asthma.
- 6.Barnthouse M, Jones BL. The Impact of Environmental Chronic and Toxic Stress on Asthma. Clin Rev Allergy Immunol 2019; 57:427–38. [DOI] [PubMed] [Google Scholar]
- 7.Borbet TC, Zhang X, Muller A, Blaser MJ. The role of the changing human microbiome in the asthma pandemic. J Allergy Clin Immunol 2019; 144:1457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brehm JM, Ramratnam SK, Tse SM, Croteau-Chonka DC, Pino-Yanes M, Rosas-Salazar C, et al. Stress and Bronchodilator Response in Children with Asthma. Am J Respir Crit Care Med 2015; 192:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol 2018; 141:1169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood LG. Diet, Obesity, and Asthma. Ann Am Thorac Soc 2017; 14:S332–S8. [DOI] [PubMed] [Google Scholar]
- 11.Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 2002; 57:875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGrath KW, Icitovic N, Boushey HA, Lazarus SC, Sutherland ER, Chinchilli VM, et al. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med 2012; 185:612–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomson NC. Novel approaches to the management of noneosinophilic asthma. Ther Adv Respir Dis 2016; 10:211–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology 2006; 11:54–61. [DOI] [PubMed] [Google Scholar]
- 15.Wang M, Gao P, Wu X, Chen Y, Feng Y, Yang Q, et al. Impaired anti-inflammatory action of glucocorticoid in neutrophil from patients with steroid-resistant asthma. Respir Res 2016; 17:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol 2010; 125:1028–36 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008; 372:1107–19. [DOI] [PubMed] [Google Scholar]
- 18.Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol 2003; 3:405–12. [DOI] [PubMed] [Google Scholar]
- 19.Larche M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol 2003; 111:450–63; quiz 64. [DOI] [PubMed] [Google Scholar]
- 20.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 2008; 181:4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med 2003; 97:726–33. [DOI] [PubMed] [Google Scholar]
- 22.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol 2003; 28:42–50. [DOI] [PubMed] [Google Scholar]
- 23.Kaminska M, Foley S, Maghni K, Storness-Bliss C, Coxson H, Ghezzo H, et al. Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J Allergy Clin Immunol 2009; 124:45–51 e1–4. [DOI] [PubMed] [Google Scholar]
- 24.Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 2001; 108:430–8. [DOI] [PubMed] [Google Scholar]
- 25.Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic Sensitization Through the Airway Primes Th17-dependent Neutrophilia and Airway Hyperresponsiveness. Am J Respir Crit Care Med 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 2009; 123:1185–7. [DOI] [PubMed] [Google Scholar]
- 27.Chien JW, Lin CY, Yang KD, Lin CH, Kao JK, Tsai YG. Increased IL-17A secreting CD4+ T cells, serum IL-17 levels and exhaled nitric oxide are correlated with childhood asthma severity. Clin Exp Allergy 2013; 43:1018–26. [DOI] [PubMed] [Google Scholar]
- 28.Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, et al. Role for NLRP3 Inflammasome-mediated, IL-1beta-Dependent Responses in Severe, Steroid-Resistant Asthma. Am J Respir Crit Care Med 2017; 196:283–97. [DOI] [PubMed] [Google Scholar]
- 29.Illi S, von Mutius E, Lau S, Niggemann B, Gruber C, Wahn U, et al. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet 2006; 368:763–70. [DOI] [PubMed] [Google Scholar]
- 30.Rubner FJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, et al. Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol 2017; 139:501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strachan DP. Hay fever, hygiene, and household size. BMJ 1989; 299:1259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Illi S, von Mutius E, Lau S, Bergmann R, Niggemann B, Sommerfeld C, et al. Early childhood infectious diseases and the development of asthma up to school age: a birth cohort study. BMJ 2001; 322:390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman AS, He Y, Moore ML, Hershenson MB, Hartert TV. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am J Respir Crit Care Med 2015; 191:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med 2002; 347:869–77. [DOI] [PubMed] [Google Scholar]
- 35.Gehring U, Bischof W, Schlenvoigt G, Richter K, Fahlbusch B, Wichmann HE, et al. Exposure to house dust endotoxin and allergic sensitization in adults. Allergy 2004; 59:946–52. [DOI] [PubMed] [Google Scholar]
- 36.Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, et al. Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N Engl J Med 2016; 375:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carnes MU, Hoppin JA, Metwali N, Wyss AB, Hankinson JL, O’Connell EL, et al. House Dust Endotoxin Levels Are Associated with Adult Asthma in a U.S. Farming Population. Ann Am Thorac Soc 2017; 14:324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol 2017; 18:1076–83. [DOI] [PubMed] [Google Scholar]
- 39.Thorne PS, Kulhankova K, Yin M, Cohn R, Arbes SJ, Jr., Zeldin DC. Endotoxin exposure is a risk factor for asthma: the national survey of endotoxin in United States housing. Am J Respir Crit Care Med 2005; 172:1371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science 2010; 327:291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med 2002; 196:1645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeon SG, Oh SY, Park HK, Kim YS, Shim EJ, Lee HS, et al. TH2 and TH1 lung inflammation induced by airway allergen sensitization with low and high doses of double-stranded RNA. The Journal of allergy and clinical immunology 2007; 120:803–12. [DOI] [PubMed] [Google Scholar]
- 43.Redecke V, Hacker H, Datta SK, Fermin A, Pitha PM, Broide DH, et al. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. Journal of immunology 2004; 172:2739–43. [DOI] [PubMed] [Google Scholar]
- 44.Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll-like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nature medicine 2012; 18:1705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nature medicine 2009; 15:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitehead GS, Thomas SY, Cook DN. Modulation of distinct asthmatic phenotypes in mice by dose-dependent inhalation of microbial products. Environ Health Perspect 2014; 122:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitehead GS, Wilson RH, Nakano K, Burch LH, Nakano H, Cook DN. IL-35 production by inducible costimulator (ICOS)-positive regulatory T cells reverses established IL-17-dependent allergic airways disease. The Journal of allergy and clinical immunology 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shalaby KH, Jo T, Nakada E, Allard-Coutu A, Tsuchiya K, Hirota N, et al. ICOS-expressing CD4 T cells induced via TLR4 in the nasal mucosa are capable of inhibiting experimental allergic asthma. Journal of immunology 2012; 189:2793–804. [DOI] [PubMed] [Google Scholar]
- 49.Wilson RH, Maruoka S, Whitehead GS, Foley JF, Flake GP, Sever ML, et al. The Toll-like receptor 5 ligand flagellin promotes asthma by priming allergic responses to indoor allergens. Nat Med 2012; 18:1705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKinley L, Kim J, Bolgos GL, Siddiqui J, Remick DG. Reproducibility of a novel model of murine asthma-like pulmonary inflammation. Clin Exp Immunol 2004; 136:224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fulde M, Sommer F, Chassaing B, van Vorst K, Dupont A, Hensel M, et al. Neonatal selection by Toll-like receptor 5 influences long-term gut microbiota composition. Nature 2018; 560:489–93. [DOI] [PubMed] [Google Scholar]
- 52.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010; 328:228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 2014; 41:478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitehead GS, Hussain S, Fannin R, Trempus CS, Innes CL, Schurman SH, et al. TLR5 Activation Exacerbates Airway Inflammation in Asthma. Lung 2020; 198:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.••.Whitehead GS, Thomas SY, Shalaby KH, Nakano K, Moran TP, Ward JM, et al. TNF is required for TLR ligand-mediated but not protease-mediated allergic airway inflammation. J Clin Invest 2017; 127:3313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; TLR ligands and proteases promote allergic sensitizaion through distinct immune pathways.
- 56.Ying S, Robinson DS, Varney V, Meng Q, Tsicopoulos A, Moqbel R, et al. TNF alpha mRNA expression in allergic inflammation. Clin Exp Allergy 1991; 21:745–50. [DOI] [PubMed] [Google Scholar]
- 57.Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med 2006; 354:697–708. [DOI] [PubMed] [Google Scholar]
- 58.Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 2005; 60:1012–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, et al. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med 2009; 179:549–58. [DOI] [PubMed] [Google Scholar]
- 60.McAlees JW, Whitehead GS, Harley IT, Cappelletti M, Rewerts CL, Holdcroft AM, et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol 2015; 8:863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan AM, Chen HC, Pochard P, Eisenbarth SC, Herrick CA, Bottomly HK. TLR4 signaling in stromal cells is critical for the initiation of allergic Th2 responses to inhaled antigen. J Immunol 2010; 184:3535–44. [DOI] [PubMed] [Google Scholar]
- 62.Hahn I, Klaus A, Maus R, Christman JW, Welte T, Maus UA. Dendritic cell depletion and repopulation in the lung after irradiation and bone marrow transplantation in mice. Am J Respir Cell Mol Biol 2011; 45:534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Citrin DE, Shankavaram U, Horton JA, Shield W 3rd, Zhao S, Asano H, et al. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J Natl Cancer Inst 2013; 105:1474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomas SY, Whitehead GS, Takaku M, Ward JM, Xu X, Nakano K, et al. MyD88-dependent dendritic and epithelial cell crosstalk orchestrates immune responses to allergens. Mucosal Immunol 2018; 11:796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharpe RA, Bearman N, Thornton CR, Husk K, Osborne NJ. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol 2015; 135:110–22. [DOI] [PubMed] [Google Scholar]
- 66.Tischer CG, Hohmann C, Thiering E, Herbarth O, Muller A, Henderson J, et al. Meta-analysis of mould and dampness exposure on asthma and allergy in eight European birth cohorts: an ENRIECO initiative. Allergy 2011; 66:1570–9. [DOI] [PubMed] [Google Scholar]
- 67.Iossifova YY, Reponen T, Ryan PH, Levin L, Bernstein DI, Lockey JE, et al. Mold exposure during infancy as a predictor of potential asthma development. Ann Allergy Asthma Immunol 2009; 102:131–7. [DOI] [PubMed] [Google Scholar]
- 68.Thacher JD, Gruzieva O, Pershagen G, Melen E, Lorentzen JC, Kull I, et al. Mold and dampness exposure and allergic outcomes from birth to adolescence: data from the BAMSE cohort. Allergy 2017; 72:967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z, Biagini Myers JM, Brandt EB, Ryan PH, Lindsey M, Mintz-Cole RA, et al. beta-Glucan exacerbates allergic asthma independent of fungal sensitization and promotes steroid-resistant TH2/TH17 responses. J Allergy Clin Immunol 2017; 139:54–65 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol 2002; 169:5904–11. [DOI] [PubMed] [Google Scholar]
- 71.Porter P, Susarla SC, Polikepahad S, Qian Y, Hampton J, Kiss A, et al. Link between allergic asthma and airway mucosal infection suggested by proteinase-secreting household fungi. Mucosal Immunol 2009; 2:504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acevedo N, Zakzuk J, Caraballo L. House Dust Mite Allergy Under Changing Environments . Allergy Asthma Immunol Res 2019; 11:450–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Randall TA, London RE, Fitzgerald MC, Mueller GA. Proteases of Dermatophagoides pteronyssinus. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, et al. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J Allergy Clin Immunol 2011; 128:1216–24 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol 2014; 134:509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vinhas R, Cortes L, Cardoso I, Mendes VM, Manadas B, Todo-Bom A, et al. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy 2011; 66:1088–98. [DOI] [PubMed] [Google Scholar]
- 77.Wan H, Winton HL, Soeller C, Taylor GW, Gruenert DC, Thompson PJ, et al. The transmembrane protein occludin of epithelial tight junctions is a functional target for serine peptidases from faecal pellets of Dermatophagoides pteronyssinus. Clin Exp Allergy 2001; 31:279–94. [DOI] [PubMed] [Google Scholar]
- 78.Turi GJ, Ellis R, Wattie JN, Labiris NR, Inman MD. The effects of inhaled house dust mite on airway barrier function and sensitivity to inhaled methacholine in mice. Am J Physiol Lung Cell Mol Physiol 2011; 300:L185–90. [DOI] [PubMed] [Google Scholar]
- 79.Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, et al. Mucosal allergic sensitization to cockroach allergens is dependent on proteinase activity and proteinase-activated receptor-2 activation. J Immunol 2011; 186:3164–72. [DOI] [PubMed] [Google Scholar]
- 80.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 2008; 3:e3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem 2012; 287:6941–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol 2011; 186:4375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.••.Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol 2018; 19:375–85. [DOI] [PubMed] [Google Scholar]; Environmental protease allergens act on IL-33 to promote type 2 responses.
- 84.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med 2010; 363:1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol 2014; 134:170–7. [DOI] [PubMed] [Google Scholar]
- 86.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014; 40:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity 2018; 48:1195–207 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.•.Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol 2017; 140:1351–63 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]; IL-33 can convert regulatory T cells to effector T cells