

Abstract
Oxidative stress plays an essential role in the pathogenesis of chronic diseases such as cardiovascular diseases, diabetes, neurodegenerative diseases, and cancer. Long term exposure to increased levels of pro-oxidant factors can cause structural defects at a mitochondrial DNA level, as well as functional alteration of several enzymes and cellular structures leading to aberrations in gene expression. The modern lifestyle associated with processed food, exposure to a wide range of chemicals and lack of exercise plays an important role in oxidative stress induction. However, the use of medicinal plants with antioxidant properties has been exploited for their ability to treat or prevent several human pathologies in which oxidative stress seems to be one of the causes. In this review we discuss the diseases in which oxidative stress is one of the triggers and the plant-derived antioxidant compounds with their mechanisms of antioxidant defenses that can help in the prevention of these diseases. Finally, both the beneficial and detrimental effects of antioxidant molecules that are used to reduce oxidative stress in several human conditions are discussed.
Keywords: reactive oxygen species, oxidative stress, natural antioxidants, neurological disorders, cardiovascular diseases, cancer, aging, antioxidant defense
Introduction
Many natural biological processes in our bodies, such as breathing, digesting food, metabolize alcohol and drugs, and turning fats into energy produce harmful compounds called free radicals. Free radicals are usually destroyed by our body’s natural antioxidant system. If this system an not cope properly, free radicals can trigger a negative chain reaction in the body, a reaction that can destroy the cell membrane, block the action of major enzymes, prevent cellular processes necessary for proper functioning of the body, prevent normal cell division, destroy deoxyribonucleic acid (DNA), and block energy generation (Kurutas, 2015).
Oxidative stress is reported to associate with the development of several metabolic, chronic disorders or cancers (Finkel and Holbrook, 2000; Reuter et al., 2010; Aminjan et al., 2019).
The theory of free radicals of oxygen has been known for over 50 years, however, it is only in the last two decades that their role in the development of diseases were discovered and, thus, the beneficial effects of antioxidants have been widely studied (Liu, 2019).
Free radicals play an essential role in several biological processes. Many of these are necessary for life, such as the intracellular destruction of bacteria by phagocytes, especially by granulocytes and macrophages. Researchers believe that free radicals are also involved in some cellular signaling processes, known as redox signaling (Finkel and Holbrook, 2000). At low-to-moderate amounts, ROS are beneficial both in regulating processes involving the maintenance of homeostasis as well as a wide variety of cellular functions (Finkel and Holbrook, 2000; Bhattacharyya et al., 2014).
Excessive ROS production determines structural modification of cellular proteins and the alteration of their functions, leading to cellular dysfunction and disruption of vital cellular processes (Finkel and Holbrook, 2000; Kaminski et al., 2002). High ROS levels cause lipid, protein, and DNA damage. In particular, ROS can break the lipid membrane and increase membrane fluidity and permeability. Protein damage involves site-specific amino acid modification, peptide chain fragmentation, cross-linked reaction products aggregation, electric charge alteration, enzymatic inactivation, and proteolysis susceptibility (Ayala et al., 2014). Finally, ROS can damage DNA through oxidizing deoxyribose, breaking strand, removing nucleotides, modifying bases and crosslinking DNA-protein (Sharma et al., 2012; Cadet and Wagner, 2013; Cadet et al., 2017; Liang et al., 2020).
Primary oxygen free radicals are superoxide and hydroxyl radical. They are derived from molecular oxygen under chemical reduction conditions. Excessive amounts of these free radicals can lead to cell damage and apoptosis, contributing to many diseases such as cancer, stroke (Tsatsakis A. et al., 2019), myocardial infarction, diabetes, and other significant conditions (Padureanu et al., 2019). Many cancers are thought to be the result of interactions between free radicals and DNA that lead to mutations that affect the cell cycle and which then leads to neoplasia (Reuter et al., 2010).
Because free radicals are necessary for life, the body has several enzymatic mechanisms to minimize radically induced damage and to protect against excessive production of free radicals. Antioxidants play a vital role in these defense mechanisms. In healthy organisms, protection against the harmful effects of reactive oxygen species is achieved by maintaining a delicate balance between oxidants and antioxidants. The continuous production of free radicals in aerobic organisms must therefore be equalized by a similar rate of antioxidant consumption. Enzymatic or non-enzymatic, antioxidants are substances that prevent the formation of free radicals, and seek and neutralize or repair the damage caused by them (Clark et al., 1985). The protection against oxidative damage and chronic diseases is achieved through a variety of endogenous and exogenous antioxidants (Cadet et al., 2012).
ROS homeostasis is ensured by various antioxidant systems present both in plants (Sharma et al., 2012) and the human body (Birben et al., 2012). Natural ROS production through the mitochondrial respiratory chain is involved since ROS can be metabolically beneficial, but, at the same time, harmful to cells in some conditions (Hsu et al., 2000; Poli et al., 2004; Valko et al., 2006).
Conversely, in pathological or stress conditions, ROS overwhelms antioxidant systems leading to an imbalance, which, in turn, causes oxidative stress and irreversible changes in cell compounds, including proteins, carbohydrates and lipids, in addition to being able to disrupt normal cellular-signaling mechanisms (Birben et al., 2012; Zal et al., 2014; Salehi et al., 2018; Sharifi-Rad et al., 2018).
In autoimmune diseases, free radicals can change the expression of self-antigen-type proteins, increasing their immune response or changing their antigenic profile. The immune response can also be influenced by external antioxidants such as allergens in susceptible individuals. Pollen from some plant species has been shown to contain nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), which induces an inflammatory response in the airways with specific symptoms due to infiltration with proinflammatory cytokines, TNF-alpha and interleukins from epithelial cells. The appearance and accumulation of intracellular prooxidant factors has the long-term effect of altering the immune response by altering the structure and, implicitly, the function of proteins or enzymes such as: interferon−gamma (IFN−γ), cluster of differentiation antigen 14 (CD14), and tumor necrosis factor-a (TNF-α) (Tsoukalas et al., 2019c).
In cancers, alteration of purine or pyrimidine in the structure of cellular DNA, which is associated with a number of other reactions that produce oxides and free radicals, may be the cause of neoplasms. If the intracellular mechanisms of repair of oxidative defects are insufficient or disturbed in turn by the oxidative factors present, there are definitive consequences in some genes or products resulting from the expression of these genes, which causes mutagenesis and modification of the apoptotic mechanism of the cell, thus resulting in the tumor cell (Buj and Aird, 2018).
In the long term, the changes spread and self-sustain with the permanent activation of the autoimmune response and the accumulation of local proinflammatory factors, for example: TNF-alpha, proteases, kinases. These factors favor tissue necrosis and accelerate tissue growth with the appearance of new modified cells that maintain the immune response and propagate the initial genetic defects with chaotic and extensive multiplication; also, oxidative stress produces structural changes of cell membranes with decreased adhesion, and the migration of altered tumor cells in neighboring tissues or in distant blood and lymph (Forni et al., 2019).
In cellular aging, two theories on the mechanisms of cellular aging are currently accepted: the mitochondrial theory and the free radical theory. They support the hypothesis that mitochondria are affected by an increased level of intracellular free radicals, which leads to the alteration of their function and a decreased cellular regenerative capacity. At the same time, the progressive accumulation of intracellular oxidizing factors that exceed the antioxidant capacity is also accepted. Under these conditions, the biological decline of the respective tissue and the reduction of the adaptive c pacity to stress appear. Subsequently, regardless of the mechanism involved, in mitochondrial DNA damage or in the direct involvement of prooxidant factors in cellular mechanisms, the cellular response to stress will produce an overexpression of proinflammatory genes with increasing levels of prooxidant factors (Liguori et al., 2018).
Oxidative stress stimulates the immune response and causes allergic diseases, such as asthma, allergic rhinitis, atopic dermatitis, or food allergies. This means that the antioxidant protection system of patients with allergic diseases is outdated compared to that of healthy individuals (Sackesen et al., 2008). Supplementation with antioxidants could therefore compensate for the increased inflammatory and oxidative stress processes in asthma patients. However, Murr et al. (2005) have shown that too much antioxidant supplementation can increase the susceptibility to allergic diseases and thus asthma, by decreasing the Th1-type immune response and increasing the Th2-type response with immunoglobulin synthesis.
The modern lifestyle associated with an unhealthy diet, lack of physical exercise, exposure to a combination of chemicals from different sources pesticides (Tsatsakis A.M. et al., 2019), heavy metals, food additives, and environmental pollution can influence the appearance of oxidative stress. It can contribute to the increasing burden of chronic diseases, as is suggested by several experimental and human studies (Fenga et al., 2017; Docea et al., 2018; Fountoucidou et al., 2019; Kostoff et al., 2020). This comprehensive review aims to provide strong evidence that antioxidants may contribute to the amelioration of some chronic-degenerative conditions, in addition to being able to promote healthy aging.
Chronic Diseases Influenced by ROS-Modalities of Action
The ROS Sources
Free radicals are generally produced as a result of the influence of external factors, such as pollution, cigarette smoke, or internally, as a result of intracellular metabolism if the antioxidant mechanisms are overwhelmed (Figure 1).
FIGURE 1.
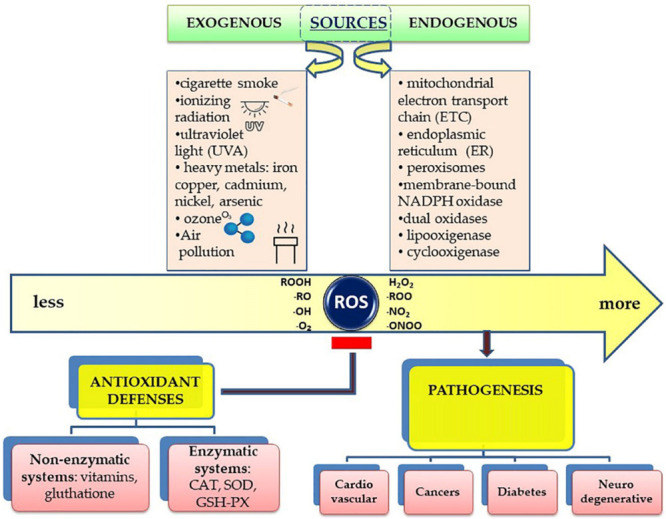
Schematic presentation of the sources of free radicals and their effects on the human body.
Exogenous ROS
Environmental triggers, such as exposure to cigarette smoke, UV radiation, heavy metal ions, ozone, allergens, drugs or toxins, pollutants, pesticides, or insecticides, may all contribute to the increase of ROS production in cells (Antunes dos Santos et al., 2018; Mahajan et al., 2018; Oke et al., 2019).
Ionizing radiation acts by converting hydroxyl radicals, superoxides and organic radicals into organic hydroperoxides and hydrogen peroxide. Subsequently, the peroxides react with the metal ions of Fe and Cu at the cellular level through redox reactions with secondary oxidative activity. Several studies have shown that the exposure of fibroblasts to alpha particles has led to an intracellular increase of oxygen and an accelerated production of peroxide at this level (Spitz et al., 2004; Spitz and Hauer-Jensen, 2014).
Ultraviolet radiation (UVA) triggers oxidative reactions by stimulating riboflavin, porphyrins and NADPH-oxidase, with the production of 8-oxo-guanine as the main result and the decrease of intracellular glutathione (GSH) level with a return to normal after cessation of exposure (Marchitti et al., 2011).
Heavy metals play an essential role in the production of free radicals (Ściskalska et al., 2014). Iron, copper, cadmium, nickel, arsenic, and lead can induce free radicals by Fenton or Haber-Weiss type reactions, but also by direct reactions between metal ions and cellular compounds with similar effects – for example, the production of thiol type radicals. Lead triggers lipid peroxidation and increases glutathione peroxidase concentration in brain tissue. Arsenic induces the production of peroxides, superoxides, nitric oxide and inhibits antioxidant enzymes such as glutathione-transferase, glutathione-peroxidase, and glutathione-reductase by binding to the sulfhydryl group. The free radicals generated from these reactions can affect DNA, with substitutions of some DNA bases such as guanine with cytosine, guanine with thymine and cytosine with thymine (Jan et al., 2015). Exposure to ozone can affect lung function even in healthy individuals by increasing inflammatory infiltrate in the respiratory epithelium (Wu X. et al., 2019).
Endogenous ROS Production
The main endogenous sites of cellular redox-reactive species generation-including ROS and reactive nitrogen species (RNS) comprise mitochondrial electron transport chain (ETC), endoplasmic reticulum (ER), peroxisomes, membrane-bound NADPH oxidase (NOX) isoforms 1–5, dual oxidases (Duox) 1 and 2 complexes, and nitric oxide synthases isoforms 1–5 (NOS1–3). The complexes I and III of mitochondrial ETC produces superoxide anion (Rodriguez and Redman, 2005).
The mitochondrial ETC is considered to be the primary endogenous source of ROS but other internal sources are also present. Other sources of ROS, primarily H2O2, are microsomes and peroxisomes. Immune cells, such as macrophages and neutrophils, can also generate ROS due to their oxygen-dependent mechanisms to fight against invading microorganisms based on NOX2 isoform (Curi et al., 2016). Furthermore, dysregulated ROS signaling may contribute to a multitude of diseases associated with oxidative stress (Finkel, 2011).
ROS are produced in mitochondria during aerobic metabolism (Rodriguez and Redman, 2005). ROS generation within mitochondria (oxidative metabolism) is closely associated with ATP synthesis (oxidative phosphorylation). In aerobic organisms, the coupling of these reactions is the primary source of energy (Papa et al., 2012).
Mitochondria serve as a major ROS generator and, at the same time, as a ROS receptor. Covalent and enzymatic changes in proteins during or after protein biosynthesis as well as during protein cleavage or degradation promote disease through oxidative damage and mitochondrial dysfunction. These post-translational changes participate in the regulation of mitochondrial function through free radical species and other messengers (Hu and Ren, 2016).
Since oxidative phosphorylation is a leaky process, 0.2–5% of the electrons circulate through ETC in each round of ATP production. This produces an incompletely O2 reduction (Hamanaka et al., 2013).
Superoxide radicals are produced by NADPH oxidases (NOX) and, to a minor extent, as by-products of a wide number of metabolic enzymes such as cyclooxygenase (COX) 1/2, lipoxygenase, xanthine oxidoreductase (XOR) and cytochrome p450 (Finkel, 2003).
Because of the anionic properties of superoxide radicals, they diffuse through biological lipid membranes at the meager extent. They are sequentially reduced inside cells to form hydrogen peroxide and hydroxyl radical (Bartosz, 2009). Furthermore, peroxyl and alkoxyl radicals, as well as hypochlorite ions, are also formed (Valko et al., 2007).
All these types of ROS can be very harmful to cells; in fact, they can oxidize and subsequently inactivate several functions of cell components and even DNA (Valko et al., 2007). All these processes may trigger irreversible apoptotic and necrotic cell death.
Several studies indicate that human cells can also actively trigger ROS production at small doses, as part of signaling pathways, regulating cell survival and proliferation, as a defense mechanism against invaders (Bartosz, 2009; Sena and Chandel, 2012). In particular, specific enzymatic systems, such as the NOX family, dedicated explicitly to superoxide radical production with physiological signaling purposes, are developed by cells (Bedard and Krause, 2007).
In normal situations, electrons are transferred through mitochondrial ETC for oxygen reduction to water, but approximately 1–3% of electrons escape from this system and produce superoxide (Ramsay, 2019). Beyond this, other internally generated sources of ROS are present in humans, including:
(i) oxidative burst from phagocytes (white blood cells) during bacteria and virus killing and foreign proteins denaturation;
(ii) xanthine oxidoreductase (XOR) metabolism;
(ii) arachidonate pathways;
(iii) peroxisomes metabolism;
(iv) detoxification of toxic substances (i.e., vigorous exercise, chronic inflammation, and infections) (Birben et al., 2012).
ROS decrease phosphatase activity, by inhibiting catalytic regions susceptible to oxidation, and, thus, enhance protein tyrosine phosphatase (PTP) phosphorylation and influences signal transduction (Bedard and Krause, 2007). ROS can also improve signal transduction pathways that disturb the nuclear factor-κB (NF-κB) activation and translocation of this into the nucleus. The DNA binding potential of oxidized NF-κB is significantly reduced. However, NF-κB may be decreased by TR or redox factor 1 (Kabe et al., 2005). The above provokes ROS and RNS so it can strongly affect NF-κB-dependent inflammatory signals. Cyclopentenones are electrophilic anti-inflammatory prostaglandins which are conjugated with the reactive thiols of ROS-modified peptides and proteins and thus dampens ROS-mediated NF-κB signaling (Homem de Bittencourt and Curi, 2001). On the other hand, endogenous stress has an intracellular origin. Several studies have highlighted the role of cultural cell conditions, altering gene expression patterns of different genes and their DNA stability. Metabolic processes trigger different types of ROS, that are able to, if present at inadequate levels, oxidize DNA and induce various damage, such as double-stranded DNA breaks and deficiencies, often found in human tumors (De Bont and van Larebeke, 2004). Moreover, there are non-enzymatic reactions, like the mitochondrial respiratory chain which involves NADPH oxidase, XOR, uncoupled endothelial NOS, cytochrome P450 enzymes, lipoxygenase and COX (Sena and Chandel, 2012; Battelli et al., 2014a).
Cellular oxidative metabolism produces free radicals and organic peroxides as by-products during cellular mitochondrial electron transport or through metal-catalyzed oxidation of metabolites and oxidoreductases (Forman and Torres, 2002; Hussain et al., 2016).
Moreover, nitric oxide is produced in hypoxic conditions in a respiratory chain reaction, and RNS may trigger reactive species production, such as reactive aldehydes, malondialdehyde (MDA) and 4-hydroxy-2-non-enal (Hussain et al., 2016). ROS can alter the cell’s redox status and, thus, send a signal. However, an imbalance in this protective mechanism can lead to damage in cell molecules, such as DNA, proteins and lipids, resulting in cell death by necrotic and apoptotic processes (Bhattacharyya et al., 2014; Hussain et al., 2016). Stimulated ROS production was first described in phagocytic cells, including neutrophils and macrophages, during phagocytosis or stimulation with a wide variety of agents through NADPH oxidase activation. This was named “the respiratory burst” due to transient oxygen consumption (Lamy et al., 1996; Peake and Suzuki, 2004). The respiratory burst of neutrophils, as well as their degranulation, constitute a defensive response to host tissue damage, whether induced by mechanical (muscle damage during exercise, thermal stress), chemical or infectious stimuli (Lamy et al., 1996; Peake and Suzuki, 2004).
Nowadays, ROS production has also been observed in a variety of cells other than phagocytes, and their implication in physiologic signaling is well documented (Di Meo et al., 2016).
The Role of Lifestyle in Oxidative Stress Response
Lifestyle: smoking, alcohol consumption, adequate or inappropriate diet, exercise, training or untrained condition, contribute to oxidative stress. Some research has shown the presence of reactive oxygen species and muscle level and their role in regulating muscle activity. Skeletal muscle fibers continuously generate reactive oxygen species at a low level, which increases during muscle contraction. They exert multiple direct and indirect effects on muscle activity (contractility, excitability, metabolism, and calcium homeostasis) and are involved in skeletal muscle fatigue during strenuous exercise (Pingitore et al., 2015).
Exhausting exercises, long exercises, overtraining syndrome, and overcoming limits as a phase of the initial onset of overtraining syndrome, induce a significant response to oxidative stress. Instead, moderate exercise, low intensity training, and prolonged training, improve endogenous antioxidant status. Reactive oxygen species play an important role in cell signaling and in regulating the expression of antioxidant genes. Physical exertion produces a hyperregulation of the nuclear factor kappa B and mitogen-activated protein kinase that activates gene expression of a number of enzymes and proteins with an important role in maintaining oxidative/antioxidant intracellular homeostasis (Antonioni et al., 2019).
Physical exercise is considered the main treatment of non-pharmacological therapies along with lifestyle changes for various chronic diseases, especially cardiovascular diseases (Ren and Taegtmeyer, 2015). The results of some experimental studies have highlighted the role of autophagy, a conservative process of catabolism for the degradation and recycling of cellular organs and nutrients, in the cardiovascular benefits offered by training (Wu N. N. et al., 2019). Regular exercise as a unique form of physiological stress is able to trigger adaptation, while autophagy, especially selective mitochondrial autophagy, also called mitophagy, allows for such cardiovascular adaptation (Wu N. N. et al., 2019).
Cigarette smoke comprises a series of oxidants, free radicals, as well as organic components (e.g., nitric oxide and nitric superoxide) where the lung level activates the accumulation of neutrophils and macrophages, which increases the production of oxidants locally (Valavanidis et al., 2009).
Biochemical/Molecular Targets and Chronic Diseases Mechanistically Linked to ROS
Endogenous ROS comprises the by-products of cellular metabolism in aerobic organisms.
At low concentrations, they are usually involved in different cell processes, such as proliferation, differentiation, and apoptosis, like a second messenger in cell signaling (Salehi et al., 2018). ROS production within cells under physiological condition is dependent on mitochondria respiration, NOX, uncoupled NOS and XOR.
The increase in ROS levels, its production in inappropriate cellular compartments or its production with defective forms during oxidative processes can trigger the development of numerous chronic-degenerative disorders, leading to severe damage to bio macromolecules (Chen et al., 2012; Gandhi and Abramov, 2012; Salehi et al., 2018). Oxidative stress, as a result of the imbalance between oxidative and antioxidative processes in cells, therefore plays an essential role in the pathogenesis of numerous chronic-degenerative disorders.
ROS and Cardiovascular Diseases
The main cardiovascular risk factors, such as hypertension and hypercholesterolemia contribute to enhancing ROS generation, leading to oxidative stress (Li et al., 2014). From all these cardiovascular risk factors, hypertension is an essential factor in the development of cardiovascular diseases (CVD) (Elahi et al., 2009).
ROS has a dual role in cardiovascular physiopathology.
Small amounts of ROS in the cardiovascular system could provide remarkable benefits: anti-atherosclerotic, pro-angiogenesis and endogenous cardioprotective effects (Taverne et al., 2013). Large numbers of ROS induce the loss of cell viability, since oxidative stress is involved in the development and/or progression of CVD, such as endothelial dysfunction, atherosclerosis, myocardial ischemia/reperfusion damage, heart failure, arrhythmias (Taverne et al., 2013; Sharifi-Rad J. et al., 2020).
In CVD, gene expression is altered due to oxidative stress. Increased ROS levels modulate transcription factor activity, especially NF-κB, activator protein-1 (AP-1) and the peroxisome proliferators-activated receptor (PPAR) family of transcriptional activators (Elahi et al., 2009).
As a result of increasing ROS generation, one of the first events in atherogenesis, as well as in other CVDs correlated with endothelial dysfunction, is the oxidative modification of low-density lipoprotein (LDL) (Singh et al., 2002; Perrotta and Aquila, 2015). Indeed, both cell membranes and LDL, enriched with phospholipids, are highly sensitive to oxidative modification. Oxidized phospholipids, through receptor-mediated or receptor-independent pathways, can therefore then activate endothelial cells, induce endothelium adhesion molecules expression, attract monocytes, have endothelium cytotoxic effects, and increase proinflammatory gene activity and cellular growth factors (Esper et al., 2006). All of these processes provoke endothelial dysfunction, platelet aggregation, and metalloproteinase expression and favor thrombogenesis (Esper et al., 2006).
In atherosclerotic plaque, increased matrix metalloproteinase expression and activity triggered by oxidative stress lead to its rupture and consequent thrombosis (He and Zuo, 2015). The NF-κB activity in atherosclerosis is mainly due to oxidized LDL (Singh et al., 2002). At the same time, upregulated NF-κB is detected in smooth muscle cells, endothelial cells, macrophages and T cells of atherosclerotic plaques (Mach et al., 1998).
In the blood vessel wall, all layers can produce ROS under pathological conditions, and most of them are primarily derived from NOX (Reid, 2001). Due to increased ROS levels, NO bioavailability is decreased, and consequently, endothelium-dependent relaxation is reduced (Chen J.Y. et al., 2018).
Cardiac myocytes have a more significant number of mitochondria than other cells and use higher oxygen levels for energy production in the form of ATP. In myocytes, ROS trigger cardiac injury, both oxidizing essential proteins for excitation-contraction and decreasing NO bioactivity (Hare and Stamler, 2005).
In myocardial ischemia, mitochondrial electron transport is imbalanced, leading to ATP depletion, acidosis, mitochondrial depolarization, intracellular Ca2+ overload and, then, apoptosis (Wattanapitayakul and Bauer, 2001). Furthermore, oxidative stress produced in mitochondria induces mitochondrial DNA (mtDNA) damage and leads to CVD. In myocardial ischemia, hypoxia and reoxygenation trigger an increase in free radical production in cardiac tissue (Elahi et al., 2009). ROS produced during reoxygenation cause direct oxidative damage to cellular components and lead to indirect damage through the activation of localized inflammation (Gutteridge and Halliwell, 2000). In heart failure, excessive ROS production is based on increased activity of XOR and NOX (Battelli et al., 2014b). Increased ROS production is a consequence of prolonged endoplasmic reticulum stress and mitochondrial-derived oxidative stress in cardio-metabolic disorders.
The function of these organelles is correlated with Ca2+ release and uptake and, as a result of oxidative stress, abnormal Ca2+ handling may cause arrhythmias. Furthermore, some disturbance in these organelles activates signaling pathways that alter cardiac ion channels function or expression, involved in the generation of an action potential that promotes arrhythmogenesis (Tse et al., 2016).
The administration of cytostatics to humans is followed by cardiotoxicity due to increased plasma levels of ROS and lipid peroxidation products and decreased plasma and tissue levels of antioxidants. Myocardial changes that occur after treatment include: myocyte loss through apoptosis or necrosis, loss of myofibrils, distension of the sarcoplasmic reticulum, and mitochondrial ballooning. Recent studies on transgenic mice have shown that in cardiotoxicity induced by Doxorubicin, free radicals can be counteracted by metallothionein and liensinine (Kang, 1996; Liang et al., 2020).
ROS and Cancers
Cancer development in humans is a complex process that includes cellular and molecular changes mediated by various endogenous and exogenous stimuli (Docea et al., 2012; Sani et al., 2017; Mishra et al., 2018). It has been established that oxidative DNA damage is one of the key characteristics of carcinogenesis (Smith et al., 2016). Cancer initiation and promotion are associated with chromosomal defects and activation of oncogenes by free radicals (Glasauer and Chandel, 2014). A common form of injury is the formation of hydroxylated DNA bases, considered an important event in chemical carcinogenesis. They interfere with healthy cell growth by causing genetic mutations and altering normal gene transcription. Oxidative lesions also produce many changes in the structure of DNA (Li et al., 2015).
ROS involvement in a different stage of carcinogenesis has been shown in various model systems. Excessive amounts of these free radicals can lead to cell damage and apoptosis. Many forms of cancer are considered to be the result of free radicals and DNA reactions, leading to mutations that can affect the cell cycle and lead to neoplasia (Pizzino et al., 2014, 2017).
ROS overproduction has an impact on cancer cell proliferation, metastatic potential, and it is associated with invasiveness and poor prognosis (Liou et al., 2016). ROS contributes to cancer cell migration through various mechanisms: (i) matrix degradation, (ii) cell-cell contact, (iii) cytoskeleton remodeling, regulation of gene expression, (iv) invadopodia formation (Pizzino et al., 2014).
For example, mitochondria-derived ROS has an impact on initial extracellular matrix contact, NOX-derived ROS are involved in invadopodia formation. At the same time, ROS increase in cytosol plays a significant role in cytoskeleton remodeling (Herrera et al., 2004). The effect of ROS on cancers depends on the type of organ, as well as on the grade of disease progression.
Skin carcinogenesis and exposure to UVA: the ultraviolet component A sunlight (UV-A with the wavelength 320–400 nm) has the potential to generate oxidative stress in cells and tissues, so that endogenous and exogenous antioxidants strongly influence the biological effects of UVA (Sage et al., 2012). The physiological doses of UVA determine the expression of some genes (collagenase, hem oxygenase-1, and nuclear oncogenes), whose effects can be significantly increased by removing intracellular GSH or by increasing the lifetime of molecular oxygen. Repeated exposure of human skin to UV radiation leads not only to skin carcinogenesis but also to photo-aging through DNA damage (Cortat et al., 2013; Saha et al., 2017).
Hydroxyl radicals can bind to DNA and produce 8-OH deoxyguanosine (8-OHdG), which consequently increases the risk of mutation. 8-OHdG can also initiate cancer-inducing mutagenesis by transforming GC pairs to TA pairs during DNA replication (Sova et al., 2010). Therefore, 8-OHdG molecules may be used as indicators for free radicals’ detection during DNA mutagenesis (Forcados et al., 2017). Additionally, increased cancer cell proliferation requires high ATP levels that lead to ROS accumulation, particularly at initial stages of cancer genesis.
In cancer cells, there is the condition of constant oxidative stress induced by mitochondrial dysfunction and metabolic changes. In fact, under normal circumstances, increased ROS levels stimulate cell death, but cancer cells overcome that by activating numerous oncogenes, which then induce nuclear factor erythroid 2-related factor 2 (NRF2) expression. NRF2 is the primary regulator of cell survival that raises cancer progression by protecting cancer cells from ROS and DNA damage (Jaramillo and Zhang, 2013).
ROS are implicated in cancer progression, promoting cyclin D1 expression, extracellular signal-regulated kinase (ERK) and JUN N-terminal kinase (JNK) phosphorylation, and MAPK activation (Saha et al., 2017). However, cancer cells enable proliferation, avoiding ROS-induced apoptosis, despite high mutagenesis.
In neoplastic disorders, ROS promote protein oxidation and lipid peroxidation. Moreover, ROS trigger toxic protein carbonyls formation which has a significant impact on other proteins or lipids (Benfeitas et al., 2017). In addition, as a result of lipid peroxidation, cancer cells accumulate products, such as 4-hydroxy-2-non-enal, one of the most studied products of phospholipid peroxidation, owing to its reactivity and cytotoxicity.
ROS and Neurodegenerative Disorders
In the brain, not all neuronal groups are equally sensitive to oxidative stress. For instance, neurons with longer axons and multiple synapses require more energy for axonal transport or long-term plasticity (Salehi et al., 2019c; Tsatsakis A. et al., 2019). High ATP demand, in combination with dysfunctional mitochondria, make these neuron groups more sensitive to degeneration (Wang and Michaelis, 2010). Correctly, dopaminergic neurons are exposed to additional oxidative stress produced by the dopamine metabolism, generating H2O2 and dopamine autoxidation, which generates superoxide (Delcambre et al., 2016; Buga et al., 2019).
Alzheimer’s, Parkinson’s, Huntington’s, amyotrophic lateral sclerosis and Friedreich’s ataxia comprise the most common neurodegenerative disorders (Reddy, 2009; Nussbaum et al., 2017; Salehi et al., 2020a). During aging, mutations in mtDNA accumulate, cytosolic calcium dysregulates, and ETC function decreases, making aging one of the major risk factors contributing to neurodegeneration (Payne and Chinnery, 2015). The oxidized molecules of DNA, proteins and lipids found in the brain tissue of post-mortem patients with neurodegenerative disorders highlight the role of oxidative stress in these diseases (Sharifi-Rad M. et al., 2020). Another cause of neurodegenerative diseases is a defective use of metals by the brain, by the intervention of mutant proteins, formed as a result of oxidative stress (Niedzielska et al., 2016). In the case of Alzheimer disease, a protein called amyloid beta (Aβ), consisting of 40 amino acid residues, is present in all the cells of the body, under normal, harmless and even beneficial conditions, as it is a natural antioxidant (Danielson and Andersen, 2008; Li et al., 2013). It has been found that in modified protein formations Aβ plaques, which are formed in the case of Alzheimer’s disease outside the affected neurons, in areas that control cognitive functions, quantities of three, up to four times higher than normal copper, zinc and iron are fixed (Riederer et al., 1989).
One explanation is the accumulation in the brain of a modified form of the Ab protein (consisting of 42 amino acid residues), which fails to properly bind metals, promotes oxidative processes; by reacting in self-defense, neurons produce antioxidants in increased quantities, including the modified form of the Aβ protein, which thus becomes an antioxidant pro-oxidant, amplifying oxidative disasters by initiating chain reactions (Danielson and Andersen, 2008).
Mutations of the superoxide dismutase 1 (SOD1) protein have been linked to another neurodegenerative disease that affects motility (familial amyotrophic lateral sclerosis) (Huai and Zhang, 2019).
In its unmodified form, SOD1 is a natural antioxidant that prevents the formation of peroxide anion as a dangerous reactive form of oxygen (Saccon et al., 2013).
The mutant forms of this protein fixate a much smaller amount of metals than the usual form, which results in the formation of an excess of peroxynitrite (ONOO–) affecting the motor neurons required for normal functioning, causing severe motor disorders (Pasinelli et al., 2004).
The excessive use of glucose for energy production makes the brain especially susceptible to oxidative stress, and mitochondrial ETC is the primary ROS source (Cobley et al., 2018). Most of the ROS present in the brain derive from mitochondrial ETC complex I and III (ETC I and III), as O2– by-products (Andreyev et al., 2005).
Monoamine oxidase (MAO) is also a great source of ROS, especially in Parkinson’s. Indeed, the main targets for mitochondria-generated ROS are mitochondrial permeability transition pore (MPTP), poly (ADP-ribose) polymerase (PARP), and mtDNA (Gandhi and Abramov, 2012).
Other oxidant sources arise from NADPH oxidase, present in astrocytes, microglia and neurons, while NOS inhibition has shown neuroprotective effects (Abramov et al., 2005).
In the pathogenesis of neurodegeneration, many processes are included, such as protein misfolding and aggregation, abnormal kinase-signaling pathways, neuronal calcium dysregulation, and even impaired synaptic transmission (Gandhi and Abramov, 2012). Detached abnormally aggregated proteins that are morphologically characteristic of many neurodegenerative diseases in neurons and/or glial cells, as well as the rupture of metal ions homeostasis, are highly associated with oxidative stress (Chen et al., 2012). Mechanisms of action of ROS: these affect proteins by modifying them in oxidative forms, which tend to form aggregates (Blokhuis et al., 2013). Protein aggregates then inhibit proteasomes, the main organelles in the cell for degradation of abnormal proteins (Chen et al., 2012; Takalo et al., 2013). Accumulation of modified proteins with an inability to be destroyed in the proteasome stimulate more ROS formation and form a vicious cycle, a phenomenon included in neurodegenerative diseases related to oxidative stress (Chen et al., 2012; Takalo et al., 2013).
ROS, Diabetes, and Metabolic Syndrome
Many metabolic contexts can lead to conditions of oxidative stress. A condition in which oxidation is an important pathogenetic link is type 2 diabetes. In this disease, insulin resistance is the basic component, to which a compensatory hypersecretion of insulin is linked. Reactive oxygen species can induce inactivation of signaling mechanisms between insulin receptors and the glucose transport system, leading to insulin resistance (Chen X.F. et al., 2018).
On the other hand, diabetes itself is a generator of oxidative stress, with atherogenetic consequences. Hyperglycemia induces the generation of superoxide ions in endothelial cells at the mitochondrial level. In diabetes, electron transfer and oxidative phosphorylation are decoupled, resulting in the production of superoxide anions and inefficient ATP synthesis. Therefore, preventing the damage caused by oxidation is a therapeutic strategy in diabetes. Increased levels of free fatty acids with consecutive accumulation of intramyocellular lipids were thought to be the cause of insulin resistance and beta-pancreatic cell death.
Studies have shown that both glucose and free fatty acids can initiate the formation of free radicals through mitochondrial mechanisms and NADPH oxidase in muscles, adipocytes, beta cells and other cell types. Free fatty acids penetrate cellular organs, including mitochondria, where high levels of reactive oxygen species can cause peroxidation and damage. Recent studies show that type II diabetes and insulin resistance are associated with a decrease in mitochondrial oxidative function in skeletal muscle. Moreover, in this type of diabetes, the mitochondria are smaller, rounder and more likely to produce superoxide. Disorders of the mitochondrial transport chain, excessive generation of reactive species and lipoperoxides, as well as decreases in antioxidant mechanisms have also been observed in diabetes and obesity.
Diabetes has a number of complications over time, of which macrovasculopathy is very important. The increase in cardiovascular risk in patients with diabetes can be explained by the association between diabetes hypertension, dyslipidemia and coronary atherosclerotic disease. However, other mechanisms are also involved, such as the effects of hyperglycemia on endothelial function, the effects of glucose and fatty acids on myocardial cells, at the structural level but also of gene expression (Aroor et al., 2012; Karam et al., 2017).
Diabetic cardiovascular complications are caused by impaired cardiac microvascular function. In addition to the structural and functional changes that occur in diabetic cardiomyopathy, other mechanisms can be targeted pharmacologically. Sodium-glucose co-transporter-2 (SGLT2) inhibitors are the first class of antidiabetic drugs that have reduced the risk of heart failure in type 2 diabetes (Karam et al., 2017). Empagliflozin has an indication to reduce cardiovascular mortality in patients with diabetes and atherosclerotic disease. A recent study demonstrated the beneficial effect of empagliflozin on cardiac microvascular injury in diabetes and the protective mechanism against oxidative stress in mitochondria (Zhou et al., 2018).
Another recent study showed that aminoguanidine has a beneficial effect on diabetes-induced heart abnormalities. Aminoguanidine saves contractile abnormalities and diabetes-induced cardiac remodeling. This was explained by inhibition of endoplasmic reticulum stress and induction of autophagy (Pei et al., 2018).
Insulin resistance, abdominal obesity, atherogenic dyslipidemia, endothelial dysfunction, high blood pressure, hypercoagulability, genetic predisposition and chronic stress are the main factors underlying the metabolic syndrome. Metabolic syndrome is often characterized by oxidative stress, a condition in which there is an imbalance between the production and inactivation of reactive oxygen species. Increased generation of reactive oxygen species, decreased activity of antioxidant systems or both mechanisms may be involved in the occurrence of oxidative stress (Karam et al., 2017).
A study showed that lenalidomide attenuates oxidative cardiovascular tissue damage and apoptosis in obese mice by inhibiting tumor necrosis factor (Zhu et al., 2014).
ROS and Aging
Postulated over 60 years ago, the “free radical theory of aging” pointed to ROS as a cause of damage accumulation in cell constituents and connective tissues. This accumulation of losses in cells would be the reason for aging and aging-associated degenerative diseases (Tsoukalas et al., 2019a). Aging can be caused by both genetic and external factors, such as incorrect diet, improper physical exercise, chronic drug use, untreated inflammatory conditions, smoking, and alcohol abuse.
Today, while there are several theories of aging, the basic principle of most of them is still oxidative stress (Finkel and Holbrook, 2000; Payne and Chinnery, 2015). The major systems involved in overproduction of oxidative stress in cells are mitochondria and NOX (Bedard and Krause, 2007). NOX comprises a family of several membrane-associated enzymes, located in mitochondria of many cell types, and it has been shown in different studies that its enhanced activity and/or expression is included in age-associated diseases (Zhang et al., 2004; Park et al., 2008; Egea et al., 2017).
In the aging process, it has been noticed that high-molecular protein aggregates accumulate in cells (Davalli et al., 2016). Predominantly, these aggregates are made from proteins, with the remainder consisting of various lipids (Barrera, 2012; Takalo et al., 2013; Tsoukalas et al., 2019b). Most of the proteins aggregated are oxidized/modified by different reactive metabolites, and they could bind to cellular proteins. Thus, the crucial point for protein homeostasis maintenance is the degradation of these aggregates.
The central place for cell damaged protein degradation is the proteasome, which recognizes only unfolded proteins as degradation targets (Saez and Vilchez, 2014). Proteasome inhibition prevents further degradation of newly formed oxidized proteins and increases protein aggregation formation in cells (Takalo et al., 2013; Saez and Vilchez, 2014). Besides that, proteasome becomes dysfunctional during aging.
While proteasomal dysfunction is correlated with age progression and protein aggregation, proteasome activation slows the aging progress down and increases longevity (Chondrogianni et al., 2014). In many invertebrate models and cell lines, it has been shown that the overexpression of different proteasomal regulatory or catalytic subunits or treatment with specific compounds has positive effects on proteasome activity (Saez and Vilchez, 2014).
Along with the “free radical theory of aging” popularization, this has started the trend of antioxidant supplement consumption (Conti et al., 2016). Recently, most of the data have indicated that antioxidant supplementation does not decrease the incidence of age-related diseases (Schottker et al., 2015).
Antioxidant Defenses
Antioxidants break radical chain reactions, preventing oxidative stress-related damage (Da Pozzo et al., 2018; Figure 2).
FIGURE 2.
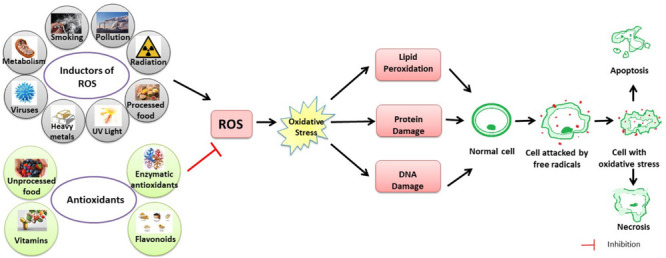
Schematic figure of the link between ROS, oxidative stress and their effects on the human body. Oxidative stress is the imbalance that occurs when there is an increased production of free radicals that exceeds the body’s ability to neutralize it. Alteration of chemical reactions at the cellular level leads to the appearance of free radicals and peroxides that affect the intracellular structures – proteins, lipids, DNA, with the disruption of intrinsic mechanisms at this level. Free radicals are normally produced in the body due to the influence of external factors, such as pollution, cigarette smoke, or internal, due to intracellular metabolism when antioxidant mechanisms are exceeded.
Their role requires acting both in hydrophilic and hydrophobic cellular environments, so their chemical structure is quite heterogeneous.
There are enzymatic and non-enzymatic antioxidants (Banafsheh and Sirous, 2016), as shown in Figure 1. but, from a nutritional perspective, a more informative classification can be made between endogenous and exogenous classes.
The first class comprises all antioxidants that cells can synthesize from smaller building blocks. Accordingly, all enzymatic antioxidants are endogenous, as well as some non-enzymatic ones (i.e., thiols antioxidants and coenzyme Q10, Figure 3).
FIGURE 3.
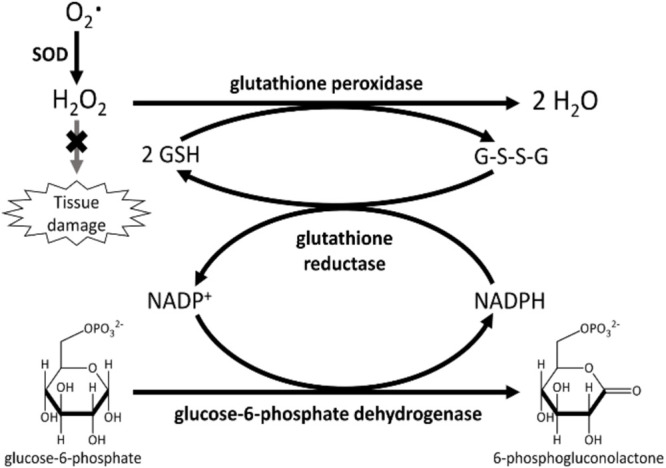
Primary enzymes (SOD or peroxidases) act directly in scavenging ROS. Secondary enzymes, such as glutathione reductase and glucose-6-phosphate dehydrogenase, support the action of primary enzymes regenerating NAPDH and reduced glutathione.
On the contrary, exogenous antioxidants have to be ingested through the diet, since their synthesis is impossible in eukaryotic cells. So, particular attention should be paid on this latter class, since this is the most unpredictable component in cellular redox balance.
Antioxidants can be divided into two categories depending on their solubility: water soluble and liposoluble (Lazzarino et al., 2019).
Water soluble antioxidants are best absorbed in the body because the vegetables and fruits that contain such antioxidants, also contain water. On the other hand, they are rapidly eliminated from the body through the urine. Water-soluble antioxidants include polyphenols, but also vitamin C (Lazzarino et al., 2019).
Liposoluble antioxidants, fat-soluble antioxidants are those that are absorbed in the presence of fats. Therefore, in the absence of fats, the body cannot absorb and use these antioxidants. It is important to note, however, that they are not easily removed from the body and can accumulate over time, exceeding the healthy level. Vitamin E is an example of a fat-soluble antioxidant (Lazzarino et al., 2019).
Enzymatic Antioxidants
Several enzymes obstruct free radicals’ formation, some of them act directly in scavenging ROS (primary enzymes), whereas “secondary enzymes” play an indirect role by supporting other endogenous antioxidants (Banafsheh and Sirous, 2016). This is the case, for instance, for glucose-6-phosphate dehydrogenase that regenerates NADPH, essential for primary enzyme action (Figure 2).
Primary Enzymes
Primary enzymes act directly on the main ROS arising from incomplete O2 reduction, O2– and H2O2. SOD scavenges the former, whereas CAT and GPX remove the latter. SOD (E.C. 1.15.1.1) is a metalloenzyme, catalyzing superoxide anion dismutation to H2O2 and molecular oxygen, as shown in reaction 1 (Singh et al., 2017). In turn, H2O2 can be removed by the other enzymatic antioxidant systems.
| (1) |
SODs can be divided into four groups, with different metal cofactors. Copper-zinc SOD is most abundant in chloroplasts, cytosol and extracellular space. Iron SOD is found in plant cytosol and in microbial cells, whereas manganese SODs are mitochondrial (Perera et al., 2017). SOD also competes for superoxide anion with NO. Therefore, SOD also indirectly reduces the formation of another deleterious ROS, peroxynitrite (ONOO–, reaction 2), and increases the NO biological availability, an essential modulator for endothelial function.
| (2) |
CAT (E.C. 1.11.1.6) is a tetrameric ferriheme oxidoreductase, which catalyzes H2O2 dismutation to water and gaseous oxygen, as shown in reaction 3 (Grigoras, 2017).
| (3) |
CAT is mainly located in peroxisomes, and despite being ubiquitous, the highest activity is present in liver and red blood cells. CAT works with a two-step mechanism, somewhat resembling the formation in the first step of a peroxidase-like compound I intermediate, CpdI (reaction 4) (Alfonso-Prieto et al., 2009; Zucca et al., 2014), which in turn decomposes to resting-state upon reaction with a second hydrogen peroxide molecule (reaction 5):
| (4) |
| (5) |
A NADPH molecule is bound to each subunit, minimizing H2O2–mediated inactivation [105]. CAT is one of the enzymes with the highest known kcat(more than 106 s–1) in all known proteins, close to a diffusion-controlled reaction (Tovmasyan et al., 2015). The role of CAT versus peroxynitrite has been much debated, but recent advances suggested CAT’s ability to scavenge ONOO– (Gebicka and Didik, 2009).
GPX (E.C. 1.11.1.19) is a selenium-dependent oxidoreductase, which uses H2O2 or organic hydroperoxide as the oxidant, and the tripeptide GSH as the electron donor (Cardoso et al., 2017), in a typical class I peroxidase catalytic cycle (reactions 6 and 7).
| (6) |
| (7) |
The GPX family is composed of eight isoenzymes (GPX1-8). Each enzyme presents peculiar features. GPX1, 2, 3, and 4 incorporate selenocysteine (a non-standard amino acid, where the sulfur atom of cysteine is replaced by selenium). GPX6 contains selenium only in humans, but not in rodents; whereas GPX5, 7, and 8 do not have selenium, but a “normal” cysteine instead (Cardoso et al., 2017).
During the catalytic cycle, selenocysteine is converted from selenol (Enz-SeH) to selenenic acid (Enz-SeOH), with concomitant reduction of H2O2 or ROOH. Then, the first GSH molecules yield selenenyl sulfide intermediate (Enz-Se-SG). An incoming second GSH molecule attacks Enz-Se-SG, regenerating the enzymatic resting form Enz-SeH, releasing the oxidized and dimerized GSSG (Cardoso et al., 2017).
Another important class of enzymatic peroxide scavenger is PRDX. Six different classes of PRDX have been identified (Poole and Nelson, 2016), showing either one (1-Cys PRDX) or two (2-Cys PRDX) redox-active cysteine residues (Park et al., 2016). The PRDX catalytic cycle involves H2O2 decomposition and the subsequent regeneration of the resting enzyme, using a small cysteine protein thioredoxin (Trx) as the reductant (reactions 8 and 9). Trx shows two vicinal cysteines (in the typical CXXC motif), forming, in turn, a disulfide internal bridge upon oxidation. In the case of PRDX6 isoform, Trx can be replaced by GSH.
| (8) |
| (9) |
Secondary Enzymes
All the enzymatic activities described above rely on the continuous regeneration of the reduced form of reductants (mainly GSH and Trx). This is usually performed by some reductases, NADPH-dependent (such as glutathione reductase E.C. 1.8.1.7 and thioredoxin reductase E.C. 1.8.1.9). However, as shown in Figure 2, reduced NADPH is, in turn, needed by these reductases for their continuous action. So, enzymes responsible for the constant NADPH production can be considered secondary antioxidants, as their misfunction could affect the whole ROS balance. The main NADPH metabolic source is the pentose phosphate pathway, through the first two enzymatic activities: glucose-6-phosphate dehydrogenase (E.C. 1.1.1.49) and 6-phosphogluconate dehydrogenase (E.C. 1.1.1.44). However, other contributions come from the malic enzyme (E.C. 1.1.1.40) and a recently identified folate-dependent mechanism (Fan et al., 2014).
Non-enzymatic Antioxidants
Some chemical molecules of low-molecular-weight can also directly act as antioxidants. In this case, their action is not catalytic, always needing antioxidant regeneration or its supply from the diet. Non-enzymatic antioxidants can therefore be divided into endogenous (if the eukaryotic cell is able to synthesize it) and exogenous (if the antioxidant needs to be ingested mandatorily through the diet).
Endogenous Non-enzymatic Antioxidants
Both GSH and Trx can act also act potent ROS scavengers (including ⋅OH, H2O2, organic peroxides, and ONOO–) directly, without enzymatic help.
GSH (γ-glutamyl-cysteinyl-glycine, Figure 4) is a tripeptide, mainly distributed in cytosol, but also in nuclei, peroxisomes and mitochondria. Despite being ubiquitous, the liver is the leading site for its synthesis (Banafsheh and Sirous, 2016). GSH biosynthesis is an endergonic process (ATP hydrolysis is coupled), in which firstly glutamate and cysteine condense to yield γ-glutamylcysteine (reaction catalyzed by glutamate-cysteine ligase, E.C. 6.3.2.2). This unusual γ-peptidic bond protects it from the common peptidases action. In the final step, GSH synthetase (E.C. 6.3.2.3) adds a glycine residue to the α-amino group of cysteine (Hasanuzzaman et al., 2017).
FIGURE 4.
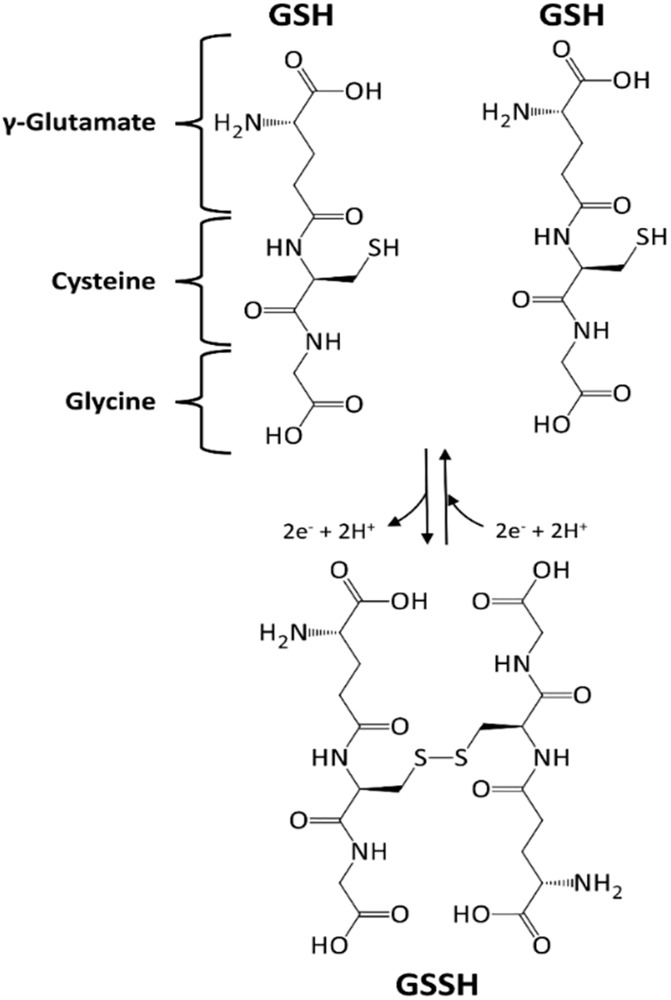
Glutathione (GSH), a tripeptide with an active –SH function. GSH undergoes a redox cycle, dimerizing with a disulfide bridge formation.
α-Lipoic acid (1,2-dithiolane-3-pentanoic acid, Figure 4) is a disulfide compound that undergoes a redox cycle similar to GSH. Accordingly, it scavenges reactive ROS, and regenerate vitamins C and E, and GSH in their active forms (Kucukgoncu et al., 2017). Lipoic acid also has a role in metal chelation, preventing Fenton-like radical reactions (Zhang and McCullough, 2016). Nevertheless, even small proteins, such as Trx and glutaredoxin can similarly function as thiol antioxidants, showing redox-active mono- or di-cysteine motif (CXXC). Both proteins can be in turn reduced back to their active form, directly by GSH or indirectly by NADPH (Banafsheh and Sirous, 2016).
Melatonin (N-acetyl-5-methoxytryptamine, Figure 5) is a neurohormone derived from amino acid tryptophan. It is involved in circadian rhythms but also acts as a potent antioxidant, protecting cell membranes against lipid peroxidation (Beyer et al., 1998).
FIGURE 5.

Chemical structures of Lipoic acid, Melatonin, Coenzyme Q10.
It has been described to be more effective in ROS scavenging than vitamin E, GSH, vitamin C and β-carotene (Watson, 1998).
Coenzyme Q10 or ubiquinone (2,3-dimethoxy-5-methyl-6-polyisoprene parabenzoquinone, Figure 5) is an isoprenoid antioxidant present in cell membranes, essential for ETC (Tafazoli, 2017). Its synthesis starts from oligomerization of isoprenoid building blocks, isopentenyl pyrophosphate and dimethylallyl pyrophosphate (both arising from the mevalonate pathway and the key enzyme 3-hydroxy-3-methyl-glutaryl-CoA reductase (E.C. 1.1.1.88). The resulting decaprenyl diphosphate is then conjugated with a tyrosine derivative to yield the active form of the coenzyme. It is one of the few liposoluble antioxidants, ensuring lipoproteins and lipids protection from radical chain reactions, peroxidation and oxidative damage (Lee et al., 2017). In its active form (quinol), coenzyme Q10 can scavenge several ROS or regenerate other oxidized antioxidants (including vitamins C and E). In turn, the quinone form can be reduced back by several NAD(P)H-dependent enzymatic systems.
Exogenous Non-enzymatic Antioxidants
Exogenous antioxidants need to be supplemented continuously through the diet since their synthetic pathways are usually present only in microbial or plant cells. Vitamins, two of which show prominent antioxidant effects, such as vitamins C and E, belong to essential class of molecules.
Vitamin C (ascorbic acid) exists in two redox forms: ascorbic acid (AA) is the reduced form, which is deprotonated at physiological pH (thus, occurring in its anion form, ascorbate). Due to its high electron-donating power, AA can undergo two-electron oxidation, yielding dehydroascorbic acid (DHA). One-electron oxidation of AA is also possible, generating a semi-dehydro-ascorbyl radical (Kocot et al., 2017). DHA can be regenerated to the active AA form by GSH- or Trx-dependent mechanisms. Humans do not express the enzyme L-gulonolactone oxidase (E.C. 1.1.3.8), avoiding the possibility of endogenous synthesis. Thus, AA must be ingested by food (or supplements), particularly tomatoes, pineapples, watermelons and all citrus fruits (Banafsheh and Sirous, 2016). AA effectively quenches ROS, both directly and cooperatively regenerating oxidized vitamin E, GSH, and carotenoids. Scavenged ROS can be superoxide anion, 1O2, H2O2, organic peroxides, ●OH or hypochlorous acid (HClO).
Vitamin E is a fat-soluble vitamin, mostly found in several vegetable oils, nuts, broccoli and fish. Eight different forms have been reported (α-, β-, γ-, and δ-tocopherol, and α-, β-, γ-, and δ-tocotrienol), but α-tocopherol has the highest antioxidant activity, especially in cell membranes (Salehi et al., 2020b). A variously methyl-substituted chromanol ring characterizes tocopherols. A long phytyl chain gives the hydrophobicity (Figure 6).
FIGURE 6.
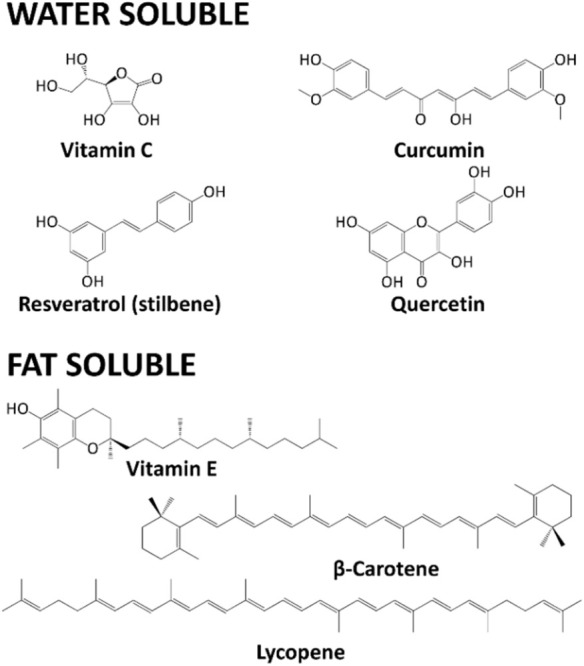
Chemical structures of Vitamin C, Curcumin, Resveratrol, Quercetin, Vitamin E, β-carotene, Lycopene.
On the contrary, tocotrienols bear an unsaturated isoprenoid chain. α-Tocopherol is able to undergo hydrogen transfer to several ROS, including 1O2, superoxide anion and peroxyl radicals. The oxidized and radical derivative of vitamin E is then reduced by the AA.
Carotenoids are a broad class of tetraterpenes, widely distributed among plants. Carotenoids are divided into unoxygenated “carotenes.” On the contrary, if oxygen atoms are present, “xanthophylls” is the correct term (Meybodi et al., 2017). Carotenes are also vitamin A precursors. Carotenoids protect plant chlorophyll, acting as accessory pigments during photosynthesis. Thus, they are intensely colored (red, orange, or yellow) molecules.
Carotenoids have been suggested to be chemopreventive agents in cancer (Marti et al., 2016; Salehi et al., 2019a), even if the real effect is still debated (Panic et al., 2017). Their biological activities also include ROS scavenging (Hernández-Almanza et al., 2016; Salehi et al., 2019b). β-Carotene comprises one of the most diffused carotenes, being the primary pro-vitamin A precursor, and it is found mainly in carrots, pumpkins, mangoes and apricots. Lycopene is another well-known acyclic carotene, not being a precursor of vitamin A, and is found primarily in tomatoes and other red fruits, but not in strawberries and cherries. Indeed, carotenoids are strong ROS scavengers, operating a very particular physical and chemical 1O2 quenching (Banafsheh and Sirous, 2016). In the physical mechanism, the carotenoid electron-rich structure absorbs 1O2 excess energy, reaching an excited state. The conjugated double bond structure in carotenoids is responsible for this ability. The excited state then decays to the ground state, losing the surplus energy as heat. During this cycle, the structure of this molecule stays unchanged.
Polyphenols are a large class of plant secondary metabolites, whose synthesis is usually possible only in these organisms (Sanjust et al., 2008; Salehi et al., 2019a). The key enzyme [phenylalanine ammonia-lyase (PAL), EC 4.3.1.5] is only present in plants (Rasouli et al., 2016). PAL catalyzes the non-oxidative deamination of phenylalanine to trans-cinnamic acid, which is the fundamental building block for polyphenol synthesis in the phenylpropanoid pathway (Ertani et al., 2016). Several biological functions have been ascribed to polyphenols, including anti-inflammatory, antioxidant, antimicrobial and antimelanogenesis effects (Zucca et al., 2016; Barbieri et al., 2017; Da Pozzo et al., 2018), suggesting their use in the human diet and as food supplements. The most widely recognized polyphenols’ classes are flavonoids, phenolic acids, stilbenes (including resveratrol), tannins, coumarins, curcuminoids and lignans. For instance, one of the most studied polyphenols has been curcumin, gaining a lot of attention also for nutraceutical applications. This phytochemical has, in fact, potent activity as a scavenger for superoxide anions, H2O2, lipid peroxides, ●OH, and several RNS (Imam et al., 2017). Curcumin can also increase GSH cellular levels (Banafsheh and Sirous, 2016).
Epigallocatechin-3-gallate (EGCG) is a well-known antioxidant. The green tea catechins include catechin, epicatechin, epigallocatechin, epicatechin gallate, and epigallocatechin gallate (Barbieri et al., 2017). Catechins can react with superoxide anions and ●OH, and is also able to chelate metal ions (Imam et al., 2017; Roychoudhury et al., 2017).
Flavonoids, in addition to its strong antioxidant properties, quench ROS formation inhibiting several enzymes and chelating metals involved in radical chain reactions (Banafsheh and Sirous, 2016). In fact, flavonoids present shallow redox potentials, allowing them to directly reduce highly reactive ROS with high redox potentials, such as superoxide anion, peroxyl radicals and ●OH by hydrogen atom transfer mechanism (HAT) (Salehi et al., 2019d). Furthermore, flavonoids can also affect free metal ion concentrations. In the cell environment, these can promote ROS formation reacting with H2O2 and generating highly reactive ●OH, in a series of Fenton and Fenton-like reactions. Indeed, flavonoids have the well-known capacity to chelate several metal ions (such as iron and copper), blocking free radical generation (Kumar and Pandey, 2013). For instance, quercetin is one of the most diffused flavonols present in broccoli, apples, grapes, onions and soybeans, with both iron-chelating and iron-stabilizing abilities (Kumar and Pandey, 2013). On the other hand, catechol and galloyl-derivatives are generally well-known metal chelators (Jomova and Valko, 2011). So, they can all exert their antioxidant activity by blocking Fenton-like reactions.
Organosulfur compounds have also been suggested as potent antioxidants. The most studied are probably some sulfur-containing metabolites present in garlic (mainly S-allyl-mercapto cysteine, S-allyl cysteine, and diallyl sulfide, diallyl trisulfide) (Kimura et al., 2017). These organosulfur are also responsible for typical garlic flavor. Their antioxidant actions include scavenging ROS and inhibiting lipids peroxidation (Borek, 2001; Miltonprabu et al., 2017).
Several minerals, in small amounts, are also essential for some enzymatic antioxidant activities. They are therefore sometimes regarded as antioxidants themselves. For instance, selenium is a necessary component of GPX (Battin and Brumaghim, 2009), while copper, zinc, and manganese are fundamental for SOD activity.
Pro-Oxidative Role of Antioxidants: Double-Sided Sword
The balance between ROS production and purification maintains homeostasis of the body, but is most often directed to the formation of free radicals and involvement in the pathophysiology of chronic diseases. The use of antioxidant supplements containing multivitamins and minerals has always grown in popularity among consumers. But some recent studies have not shown any beneficial effect of antioxidant therapy. Oxidative stress has a dual character: it is both harmful and beneficial to the body, because some ROS are signaling molecules on cellular signaling pathways. Lowering the level of oxidative stress through antioxidant supplements is therefore not beneficial in such cases (Ye et al., 2004; Ray et al., 2012).
Antioxidants are also prone to oxidation since oxidation and reduction reactions do not happen in isolation. AA, a potent antioxidant, mediates several physiological responses. Still, it can also act as a pro-oxidant when it combines with iron and copper, reducing Fe3+ to Fe2+ (or Cu2+ to Cu+), which in turn reduces H2O2 to hydroxyl radicals (Duarte and Lunec, 2005). This reaction is responsible for oxidative stress-produced DNA damage. However, the role of AA as anti- or pro-oxidant depends on the dose used, as observed in the case of ischemia-induced oxidative stress (Seo and Lee, 2002).
On the other hand, the pro-oxidant/antioxidant activity of β-carotene and lycopene has been reported to be dependent on its interaction with biological membranes and other co-antioxidant molecules, like vitamins C or E (Young and Lowe, 2001). With increased oxygen tension, carotenoids tend to lose their antioxidant potential. Otherwise, α-tocopherol, a powerful antioxidant, becomes pro-oxidant at high concentrations (Cillard and Cillard, 1980). Interestingly, when it reacts with a free radical, it becomes a radical in itself. If there is not enough AA for its regeneration, it will remain in that highly reactive state (Lü et al., 2010).
Flavonoids can also act as pro-oxidants depending on the concentrations used (Prochazkova et al., 2011). Indeed, dietary phenolics may act as pro-oxidants in the presence of O2 and transition metals, like iron and copper, catalyzing phenolics’ redox cycling and leading to ROS and phenoxyl radicals’ formation, with consequent damage in DNA, lipids and other biological components (Galati and O’Brien, 2004). Nevertheless, the extent to which these phytochemicals are capable of acting as anti- or pro-oxidants in vivo is still poorly understood, and this topic undoubtedly requires further research.
Clinical Studies Related to Efficacy of Antioxidants in Prevention of Chronic Diseases
The hypothesis that antioxidants could protect against cancer because they can neutralize reactive oxygen species (ROS) that can damage DNA has long been issued. In laboratory and animal studies, the presence of elevated levels of exogenous antioxidants has been shown to prevent the types of free radicals that have been associated with the development of cancer.
A few randomized studies evaluating the role of antioxidant supplements for cancer prevention were conducted in collaboration with the National Cancer Institute (Goodman et al., 2011). No data were obtained to justify that they are effective in primary cancer prevention. An analysis in the United States concluded that there is no clear scientific evidence for the benefits of vitamin and mineral supplements in cancer prevention.
It is important to point out that there have been cases where people who have resorted to these types of supplements have encountered an unfavorable evolution of the disease. Preclinical studies also report that antioxidants have contributed to the expansion of tumor processes in animal models. A well-known case is that of vitamin A, for which the administration of high doses in supplements has been associated with an increased risk of cancer. Vitamin A can be obtained preformed from animal sources or plant products, derived from β-carotene. Supplements are recommended only in special cases, at the doctor’s advice and especially in areas where there are frequent deficits (Jerome-Morais et al., 2011).
β-Carotene is an orange pigment found in fruits and vegetables (carrots, sweet potatoes, mangoes, apricots), and in the body it is converted to vitamin A. A normal intake has a beneficial effect against the risk of cancer. However, studies have shown a correlation between the administration of β-carotene supplements and the risk of bladder cancer, as well as the risk of lung cancer in smokers (Lin et al., 2009).
In another study, the administration of α-tocopherol and β-carotene for lung cancer did not change the incidence of lung cancer. However, α-tocopherol supplements have been shown to be effective in prostate cancer whose incidence is reduced (Goodman et al., 2011).
A trial evaluated the effectiveness of long-term supplementation with vitamin E and vitamin C in the risk of developing cancer. One of the findings of the study was that these types of supplements do not reduce the risk of prostate cancer or the overall risk of cancer in men of middle age or older. No significant results were obtained regarding the risk of colorectal or lung cancer (Gaziano et al., 2009).
Vitamin E and C supplements showed poor results in many studies. According to a clinical study entitled Physicians’ Health Study II conducted between 1997 and 2007, which included over 14,000 men over 50, the risk of major cardiovascular events was not reduced by supplementation with vitamins E or C (Lin et al., 2009).
Women’s Health Study (WHS) is another comprehensive study in the United States involving nearly 40,000 healthy women, with a minimum of 45 years of age who received 600 IU of vitamin E administered on alternate days, and who were followed for a period of 10 years (Lee et al., 2005). There was a reduction in cardiovascular mortality, but no significant effect was observed on overall mortality. The authors concluded that vitamin E supplementation for the prevention of cardiovascular disease among healthy women is not justified. Moreover, cancer mortality is not significantly influenced by vitamin E supplementation (Lee et al., 2005).
The Selenium and Vitamin E Cancer Prevention Trial (SELECT) which included over 35,000 men over the age of 50, showed that selenium and vitamin E supplements do not prevent prostate cancer. In addition, a study even suggsted that vitamin E supplements increase the risk of prostate cancer by 17% compared to a placebo (Klein et al., 2011).
Discussion
This article summarizes the evidence from a large number of meta-analyzes covering the pathophysiological impact of antioxidants on the most common chronic diseases. The main criticism of the review is that the data were extracted from meta-analyzes and not from individual studies, but this can be considered an advantage because meta-analyzes provide the highest degree of evidence.
In the case of antioxidants, studies show that more does not necessarily mean better. Consuming superfoods does not compensate for other unhealthy eating habits or an unbalanced lifestyle. Free radicals, as well as antioxidants, can have beneficial effects on the body. Therefore, we are talking about a balance and not a negative role attributed to free radicals and a positive one to antioxidants.
Degradation of nucleic acids, proteins, lipids or other cellular components are among the effects that an excessive concentration of free radicals can generate. Many diseases that are intensely discussed today are associated with the influence of oxidative stress – cancer, cardiovascular disease, Alzheimer’s disease, autoimmune diseases. Risk factors leading to free radicals include air pollution, ionizing radiation, prolonged exercise, infections, excessive consumption of polyunsaturated fatty acids (Poprac et al., 2017).
On the other hand, antioxidants are considered to be the solution to these problems – substances that neutralize free radicals. In reality, the term “antioxidant” refers to a chemical property of a substance to donate electrons. In some situations, some substances act as antioxidants, in other situations they become prooxidants, depending on the chemical composition of the environment in which they are. There are many types of antioxidants, and the role in the body and the mechanisms by which they act are different. One misconception is that one antioxidant can be replaced with another, having the same effect. In fact, each has its own unique biological properties (Chen X.F. et al., 2018).
There is also a significant difference between taking antioxidants from food and administering an isolated substance as a supplement. Many substances that demonstrate beneficial effects in the laboratory do not work when introduced into the human body. Many antioxidants do not have good bioavailability. The concentration of antioxidants such as polyphenols is sometimes so low in the blood that no significant effect is observed (Fernández-García et al., 2012). Fruits and vegetables contain bioactive substances that in many cases do not work as antioxidants if we consider them outside of the body. But they work as antioxidants when they are in the body, because they activate their own antioxidant mechanisms. These bioactive substances are the secret behind vegetable consumption (Kurutas, 2015).
Antioxidant supplements may have different health benefits. On the one hand, it is possible that other substances present in food are responsible for the positive effects on health, not necessarily a certain type of antioxidant, but the synergistic effect of several substances. On the other hand, the chemical structure of antioxidants in food is often different from that identified in supplements. An example is vitamin E. There are eight variants of vitamin E in the foods we eat, while the supplements used in most studies contain only one form (Firuzi et al., 2011).
Studies also frequently include healthy people, for whom oxidative stress on the body is not significant to determine a risk of disease. Antioxidants can benefit certain categories of patients in whom there is a real, documented imbalance, but it may not bring anything extra for a person who gets a sufficient amount of nutrients from their diet.
Observational studies analyze the trends, or habits of certain large population groups. In many, all the risk factors that could influence the course of the study can be controlled, and demonstrating a cause-effect relationship is difficult. We also cannot rely on small studies, carried out over a short period of time and using very concentrated substances extracted from different plant or animal products, to say that we have a superfood. Nutrition is a complex science, and at the moment we can only rely on the evidence accumulated so far. A food rich in antioxidants will not compensate for an unhealthy lifestyle.
Concluding Remarks and Perspectives
Oxidative stress can be reduced by approaching a balanced lifestyle. Nutrition plays a critical role, and the best treatment against oxidative stress is antioxidants. Oxidative stress plays an important role in the pathogenesis of potentially severe conditions. In the long term, increasing the level of prooxidant factors can cause structural defects in mitochondrial DNA and alterations in enzymatic functionality or cellular structures, with the appearance of functional, structural abnormalities or aberrations in gene expression. It has also been shown that in addition to metabolic products, other external agents can have a prooxidant effect, which has led to the conclusion that lifestyle and diet can play an important role in controlling oxidative stress.
Plant-derived bioactive molecules have gained pivotal attention in recent years, given their therapeutic relevance in both disease prevention and treatment, whether using the whole plants, plant extracts or even the isolated constituents with full phytochemical profiles. The daily intake of a wide variety of phytochemicals has shown to be chemopreventive. It might hold promise for add-on treatment for several diseases, including cancer, diabetes, cardiovascular disease and neurodegenerative disorders. Larger randomized trials are needed to obtain clear scientific evidence on the benefits or risks of antioxidant supplementation during cancer treatment.
Antioxidants are also prone to oxidation, and therefore their use as foods (or supplements) should be carefully considered because oxidation and reduction reactions do not happen in isolation. The intake of high doses of antioxidants has been increasingly highlighted since there is increasing evidence of some detrimental effects. The study of their chemical components as future prophylactic and therapeutic agents would be of particular interest, as they are more effective and safer than those widely available.
In conclusion, oxidative stress is an important pathogenetic link for humans and studies in this field may be important elements in the future, to better understand and manage various diseases.
Author Contributions
JS-R and MS-R contributed to the conceptualization. NA, PZ, EV, and LD contributed to the validation investigation. EP, JR, PT, EA, IP, YE, and MB contributed to the resources. AP, MN, and AD: data curation. MS-R, AD, LP, MI, NM, MM, WS, DC, WC, and JS-R contributed to the review and editing. All authors contributed to the writing of the manuscript. All authors read and approved the final manuscript and contributed equally to the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
NM would like to thank the Portuguese Foundation for Science and Technology (FCT–Portugal) for the Strategic project ref. UID/BIM/04293/2013 and “NORTE2020 – Programa Operacional Regional do Norte” (NORTE-01-0145- FEDER-000012).
Footnotes
Funding. This work was supported by the CONICYT PIA/APOYO CCTE AFB170007.
References
- Abramov A. Y., Jacobson J., Wientjes F., Hothersall J., Canevari L., Duchen M. R. (2005). Expression and modulation of an NADPH oxidase in mammalian astrocytes. J. Neurosci. 25 9176–9184. 10.1523/jneurosci.1632-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Prieto M., Biarnes X., Vidossich P., Rovira C. (2009). The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 131 11751–11761. 10.1021/ja9018572 [DOI] [PubMed] [Google Scholar]
- Aminjan H. H., Abtahi S. R., Hazrati E., Chamanara M., Jalili M., Paknejad B. (2019). Targeting of oxidative stress and inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity mitigation. Life Sci. 232:116607. 10.1016/j.lfs.2019.116607 [DOI] [PubMed] [Google Scholar]
- Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry 70 200–214. [DOI] [PubMed] [Google Scholar]
- Antonioni A., Fantini C., Dimauro I., Caporossi D. (2019). Redox homeostasis in sport: do athletes really need antioxidant support? Res. Sports Med. 27 147–165. 10.1080/15438627.2018.1563899 [DOI] [PubMed] [Google Scholar]
- Antunes dos Santos A., Ferrer B., Marques Gonçalves F., Tsatsakis A. M., Renieri E. A., Skalny A. V., et al. (2018). Oxidative stress in methylmercury-induced cell toxicity. Toxics 6:47. 10.3390/toxics6030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroor A. R., Mandavia C., Ren J., Sowers J. R., Pulakat L. (2012). Mitochondria and oxidative stress in the cardiorenal metabolic syndrome. Cardiorenal Med. 2 87–109. 10.1159/000335675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A., Munoz M. F., Arguelles S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014:360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banafsheh A. A., Sirous G. (2016). Studies on oxidants and antioxidants with a brief glance at their relevance to the immune system. Life Sci. 146 163–173. 10.1016/j.lfs.2016.01.014 [DOI] [PubMed] [Google Scholar]
- Barbieri R., Coppo E., Marchese A., Daglia M., Sobarzo-Sanchez E., Nabavi S. F., et al. (2017). Phytochemicals for human disease: An update on plant-derived compounds antibacterial activity. Microbiol. Res. 196 44–68. 10.1016/j.micres.2016.12.003 [DOI] [PubMed] [Google Scholar]
- Barrera G. (2012). Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012 137289–137289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosz G. (2009). Reactive oxygen species: destroyers or messengers? Biochem. Pharmacol. 77 1303–1315. 10.1016/j.bcp.2008.11.009 [DOI] [PubMed] [Google Scholar]
- Battelli M. G., Bolognesi A., Polito L. (2014a). Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim. Biophys. Acta 1842 1502–1517. 10.1016/j.bbadis.2014.05.022 [DOI] [PubMed] [Google Scholar]
- Battelli M. G., Polito L., Bolognesi A. (2014b). Xanthine oxidoreductase in atherosclerosis pathogenesis: not only oxidative stress. Atherosclerosis 237 562–567. 10.1016/j.atherosclerosis.2014.10.006 [DOI] [PubMed] [Google Scholar]
- Battin E. E., Brumaghim J. L. (2009). Antioxidant activity of sulfur and selenium: a review of reactive oxygen species scavenging, glutathione peroxidase, and metal-binding antioxidant mechanisms. Cell Biochem. Biophys. 55 1–23. 10.1007/s12013-009-9054-7 [DOI] [PubMed] [Google Scholar]
- Bedard K., Krause K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev 87 245–313. 10.1152/physrev.00044.2005 [DOI] [PubMed] [Google Scholar]
- Benfeitas R., Uhlen M., Nielsen J., Mardinoglu A. (2017). New challenges to study heterogeneity in cancer redox metabolism. Front. Cell Dev. Biol. 5:65. 10.3389/fcell.2017.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer C. E., Steketee J. D., Saphier D. (1998). Antioxidant properties of melatonin–an emerging mystery. Biochem. Pharmacol. 56 1265–1272. 10.1016/s0006-2952(98)00180-4 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A., Chattopadhyay R., Mitra S., Crowe S. E. (2014). Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 94 329–354. 10.1152/physrev.00040.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birben E., Sahiner U. M., Sackesen C., Erzurum S., Kalayci O. (2012). Oxidative stress and antioxidant defense. World Allergy Organ. J. 5 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokhuis A. M., Groen E. J., Koppers M., Van Den Berg L. H., Pasterkamp R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125 777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borek C. (2001). Antioxidant health effects of aged garlic extract. J. Nutr. 131 1010s–1015s. 10.1093/jn/131.3.1010s [DOI] [PubMed] [Google Scholar]
- Buga A.-M., Docea A. O., Albu C., Malin R. D., Branisteanu D. E., Ianosi G., et al. (2019). Molecular and cellular stratagem of brain metastases associated with melanoma. Oncol. Lett. 17 4170–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buj R., Aird K. M. (2018). Deoxyribonucleotide triphosphate metabolism in cancer and metabolic disease. Front. Endocrinol. 9:177. 10.3389/fendo.2018.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J., Davies K. J. A., Medeiros M. H. G., Di Mascio P., Wagner J. R. (2017). Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med. 107 13–34. 10.1016/j.freeradbiomed.2016.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J., Ravanat J. L., Tavernaporro M., Menoni H., Angelov D. (2012). Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett. 327 5–15. 10.1016/j.canlet.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Cadet J., Wagner J. R. (2013). DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 5:a012559. 10.1101/cshperspect.a012559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso B. R., Hare D. J., Bush A. I., Roberts B. R. (2017). Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 22 328–335. 10.1038/mp.2016.196 [DOI] [PubMed] [Google Scholar]
- Chen J.-Y., Ye Z.-X., Wang X.-F., Chang J., Yang M.-W., Zhong H.-H., et al. (2018). Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 97 423–428. 10.1016/j.biopha.2017.10.122 [DOI] [PubMed] [Google Scholar]
- Chen X.-F., Wang L., Wu Y.-Z., Song S.-Y., Min H.-Y., Yang Y., et al. (2018). Effect of puerarin in promoting fatty acid oxidation by increasing mitochondrial oxidative capacity and biogenesis in skeletal muscle in diabetic rats. Nutr. Diabetes 8 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Guo C., Kong J. (2012). Oxidative stress in neurodegenerative diseases. Neural. Regen. Res. 7 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondrogianni N., Sakellari M., Lefaki M., Papaevgeniou N., Gonos E. S. (2014). Proteasome activation delays aging in vitro and in vivo. Free Radic. Biol. Med. 71 303–320. 10.1016/j.freeradbiomed.2014.03.031 [DOI] [PubMed] [Google Scholar]
- Cillard J., Cillard P. (1980). [Prooxidant effect of alpha-tocopherol on essential fatty acids in aqueous media]. Ann. Nutr. Aliment. 34 579–591. [PubMed] [Google Scholar]
- Clark I. A., Cowden W. B., Hunt N. H. (1985). Free radical-induced pathology. Med. Res. Revi. 5 297–332. [DOI] [PubMed] [Google Scholar]
- Cobley J. N., Fiorello M. L., Bailey D. M. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15 490–503. 10.1016/j.redox.2018.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti V., Izzo V., Corbi G., Russomanno G., Manzo V., De Lise F., et al. (2016). Antioxidant supplementation in the treatment of aging-associated diseases. Front. Pharmacol. 7:24. 10.3389/fphar.2016.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortat B., Garcia C. C. M., Quinet A., Schuch A. P., De Lima-Bessa K. M., Menck C. F. M. (2013). The relative roles of DNA damage induced by UVA irradiation in human cells. Photochem. Photobiol. Sci. 12 1483–1495. [DOI] [PubMed] [Google Scholar]
- Curi R., Newsholme P., Marzuca-Nassr G. N., Takahashi H. K., Hirabara S. M., Cruzat V., et al. (2016). Regulatory principles in metabolism-then and now. Biochem. J. 473 1845–1857. 10.1042/bcj20160103 [DOI] [PubMed] [Google Scholar]
- Da Pozzo E., De Leo M., Faraone I., Milella L., Cavallini C., Piragine E., et al. (2018). Antioxidant and antisenescence effects of bergamot juice. Oxid. Med. Cell. Longev. 2018:9395804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson S. R., Andersen J. K. (2008). Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic. Biol. Med. 44 1787–1794. 10.1016/j.freeradbiomed.2008.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalli P., Mitic T., Caporali A., Lauriola A., D’arca D. (2016). ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell. Longev. 2016:3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bont R., van Larebeke N. (2004). Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19 169–185. 10.1093/mutage/geh025 [DOI] [PubMed] [Google Scholar]
- Delcambre S., Nonnenmacher Y., Hiller K. (2016). “Dopamine metabolism and reactive oxygen species production,” in Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease, ed. Buhlman L. (Cham: Springer; ). [Google Scholar]
- Di Meo S., Reed T. T., Venditti P., Victor V. M. (2016). Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016:1245049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docea A. O., Gofita E., Goumenou M., Calina D., Rogoveanu O., Varut M., et al. (2018). Six months exposure to a real life mixture of 13 chemicals’ below individual NOAELs induced non monotonic sex-dependent biochemical and redox status changes in rats. Food Chem. Toxicol. 115 470–481. 10.1016/j.fct.2018.03.052 [DOI] [PubMed] [Google Scholar]
- Docea A. O., Mitruţ P., Grigore D., Pirici D., Cãlina D. C., Gofiţã E. (2012). Immunohistochemical expression of TGF beta (TGF-β), TGF beta receptor 1 (TGFBR1), and Ki67 in intestinal variant of gastric adenocarcinomas. Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 53 683–692. [PubMed] [Google Scholar]
- Duarte T. L., Lunec J. (2005). Review: when is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic. Res. 39 671–686. 10.1080/10715760500104025 [DOI] [PubMed] [Google Scholar]
- Egea J., Fabregat I., Frapart Y. M., Ghezzi P., Gorlach A., Kietzmann T., et al. (2017). European contribution to the study of ROS: a summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 13 94–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi M. M., Kong Y. X., Matata B. M. (2009). Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell. Longev. 2 259–269. 10.4161/oxim.2.5.9441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertani A., Pizzeghello D., Francioso O., Tinti A., Nardi S. (2016). Biological activity of vegetal extracts containing phenols on plant metabolism. Molecules 21:205. 10.3390/molecules21020205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper R. J., Nordaby R. A., Vilarino J. O., Paragano A., Cacharron J. L., Machado R. A. (2006). Endothelial dysfunction: a comprehensive appraisal. Cardiovasc. Diabetol. 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Ye J., Kamphorst J. J., Shlomi T., Thompson C. B., Rabinowitz J. D. (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510 298–302. 10.1038/nature13236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenga C., Gangemi S., Teodoro M., Rapisarda V., Golokhvast K., Docea A. O., et al. (2017). 8-Hydroxydeoxyguanosine as a biomarker of oxidative DNA damage in workers exposed to low-dose benzene. Toxicol. Rep. 4 291–295. 10.1016/j.toxrep.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-García E., Carvajal-Lérida I., Jarén-Galán M., Garrido-Fernández J., Pérez-Gálvez A., Hornero-Méndez D. (2012). Carotenoids bioavailability from foods: from plant pigments to efficient biological activities. Food Res. Int. 46 438–450. 10.1016/j.foodres.2011.06.007 [DOI] [Google Scholar]
- Finkel T. (2003). Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 15 247–254. 10.1016/s0955-0674(03)00002-4 [DOI] [PubMed] [Google Scholar]
- Finkel T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T., Holbrook N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408 239–247. 10.1038/35041687 [DOI] [PubMed] [Google Scholar]
- Firuzi O., Miri R., Tavakkoli M., Saso L. (2011). Antioxidant therapy: current status and future prospects. Curr. Med. Chem. 18 3871–3888. 10.2174/092986711803414368 [DOI] [PubMed] [Google Scholar]
- Forcados G. E., James D. B., Sallau A. B., Muhammad A., Mabeta P. (2017). Oxidative stress and carcinogenesis: potential of phytochemicals in breast cancer therapy. Nutr. Cancer 69 365–374. 10.1080/01635581.2017.1267777 [DOI] [PubMed] [Google Scholar]
- Forman H. J., Torres M. (2002). Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 166 S4–S8. [DOI] [PubMed] [Google Scholar]
- Forni C., Facchiano F., Bartoli M., Pieretti S., Facchiano A., D’arcangelo D., et al. (2019). Beneficial role of phytochemicals on oxidative stress and age-related diseases. BioMed Res. Int. 2019 1–16. 10.1155/2019/8748253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountoucidou P., Veskoukis A. S., Kerasioti E., Docea A. O., Taitzoglou I. A., Liesivuori J., et al. (2019). A mixture of routinely encountered xenobiotics induces both redox adaptations and perturbations in blood and tissues of rats after a long-term low-dose exposure regimen: The time and dose issue. Toxicol. Lett. 317 24–44. 10.1016/j.toxlet.2019.09.015 [DOI] [PubMed] [Google Scholar]
- Galati G., O’Brien P. J. (2004). Potential toxicity of flavonoids and other dietary phenolics: significance for their chemopreventive and anticancer properties. Free Radic. Biol. Med. 37 287–303. 10.1016/j.freeradbiomed.2004.04.034 [DOI] [PubMed] [Google Scholar]
- Gandhi S., Abramov A. Y. (2012). Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012:428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziano J. M., Glynn R. J., Christen W. G., Kurth T., Belanger C., Macfadyen J., et al. (2009). Vitamins e and c in the prevention of prostate and total cancer in men: the physicians’ health study ii randomized controlled trial. Jama 301 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebicka L., Didik J. (2009). Catalytic scavenging of peroxynitrite by catalase. J. inorg. Biochem. 103 1375–1379. 10.1016/j.jinorgbio.2009.07.011 [DOI] [PubMed] [Google Scholar]
- Glasauer A., Chandel N. S. (2014). Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 92 90–101. 10.1016/j.bcp.2014.07.017 [DOI] [PubMed] [Google Scholar]
- Goodman M., Bostick R. M., Kucuk O., Jones D. P. (2011). Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic. Biol. Med. 51 1068–1084. 10.1016/j.freeradbiomed.2011.05.018 [DOI] [PubMed] [Google Scholar]
- Grigoras A. G. (2017). Catalase immobilization—A review. Biochem. Eng. J. 117 1–20. 10.1016/j.bej.2016.10.021 [DOI] [Google Scholar]
- Gutteridge J. M., Halliwell B. (2000). Free radicals and antioxidants in the year 2000. A historical look to the future. Ann. N.Y.Acad. Sci. 899 136–147. 10.1111/j.1749-6632.2000.tb06182.x [DOI] [PubMed] [Google Scholar]
- Hamanaka R. B., Glasauer A., Hoover P., Yang S., Blatt H., Mullen A. R., et al. (2013). Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal. 6:ra8. 10.1126/scisignal.2003638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare J. M., Stamler J. S. (2005). NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Invest. 115 509–517. 10.1172/jci200524459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanuzzaman M., Nahar K., Anee T. I., Fujita M. (2017). Glutathione in plants: biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 23 249–268. 10.1007/s12298-017-0422-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Zuo L. (2015). Redox roles of reactive oxygen species in cardiovascular diseases. Int. J. Mol. Sci. 16 27770–27780. 10.3390/ijms161126059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Almanza A., Montañez J., Martínez G., Aguilar-Jiménez A., Contreras-Esquivel J. C., Aguilar C. N. (2016). Lycopene: progress in microbial production. Trends Food Sci. Technol. 56 142–148. 10.1016/j.tifs.2016.08.013 [DOI] [Google Scholar]
- Herrera B., Murillo M. M., Alvarez-Barrientos A., Beltran J., Fernandez M., Fabregat I. (2004). Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic. Biol. Med. 36 16–26. 10.1016/j.freeradbiomed.2003.09.020 [DOI] [PubMed] [Google Scholar]
- Homem de Bittencourt P. I., Jr., Curi R. (2001). Antiproliferative prostaglandins and the MRP/GS-X pump role in cancer immunosuppression and insight into new strategies in cancer gene therapy. Biochem. Pharmacol. 62 811–819. 10.1016/s0006-2952(01)00738-9 [DOI] [PubMed] [Google Scholar]
- Hsu T. C., Young M. R., Cmarik J., Colburn N. H. (2000). Activator protein 1 (AP-1)- and nuclear factor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. Free Radic. Biol. Med. 28 1338–1348. 10.1016/s0891-5849(00)00220-3 [DOI] [PubMed] [Google Scholar]
- Hu N., Ren J. (2016). Reactive oxygen species regulate myocardial mitochondria through post-translational modification. React. Oxyg. Species 2 264–271. [Google Scholar]
- Huai J., Zhang Z. (2019). Structural properties and interaction partners of familial ALS-associated SOD1 mutants. Front. Neurol. 10:527. 10.3389/fneur.2019.00527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain T., Tan B., Yin Y., Blachier F., Tossou M. C. B., Rahu N. (2016). Oxidative stress and inflammation: what polyphenols can do for us? Oxid. Med. Cell. Longev. 2016:7432797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam M. U., Zhang S., Ma J., Wang H., Wang F. (2017). Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients 9:671. 10.3390/nu9070671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan A. T., Azam M., Siddiqui K., Ali A., Choi I., Haq Q. M. (2015). Heavy metals and human health: mechanistic insight into toxicity and counter defense system of antioxidants. Int. J. Mol. Sci. 16 29592–29630. 10.3390/ijms161226183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo M. C., Zhang D. D. (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27 2179–2191. 10.1101/gad.225680.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome-Morais A., Diamond A. M., Wright M. E. (2011). Dietary supplements and human health: for better or for worse? Mol. Nutr. Food Res. 55 122–135. 10.1002/mnfr.201000415 [DOI] [PubMed] [Google Scholar]
- Jomova K., Valko M. (2011). Advances in metal-induced oxidative stress and human disease. Toxicology 283 65–87. 10.1016/j.tox.2011.03.001 [DOI] [PubMed] [Google Scholar]
- Kabe Y., Ando K., Hirao S., Yoshida M., Handa H. (2005). Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox. Signal. 7 395–403. 10.1089/ars.2005.7.395 [DOI] [PubMed] [Google Scholar]
- Kaminski K. A., Bonda T. A., Korecki J., Musial W. J. (2002). Oxidative stress and neutrophil activation—the two keystones of ischemia/reperfusion injury. Int. J. Cardiol. 86 41–59. 10.1016/s0167-5273(02)00189-4 [DOI] [PubMed] [Google Scholar]
- Kang Y. (1996). Chen Y, and epstein PN. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 271 12610–12616. 10.1074/jbc.271.21.12610 [DOI] [PubMed] [Google Scholar]
- Karam B. S., Chavez-Moreno A., Koh W., Akar J. G., Akar F. G. (2017). Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 16:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Tung Y. C., Pan M. H., Su N. W., Lai Y. J., Cheng K. C. (2017). Black garlic: a critical review of its production, bioactivity, and application. J. Food Drug Anal. 25 62–70. 10.1016/j.jfda.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E. A., Thompson I. M., Jr., Tangen C. M., Crowley J. J., Lucia M. S., Goodman P. J. (2011). Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Jama 306 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot J., Luchowska-Kocot D., Kielczykowska M., Musik I., Kurzepa J. (2017). Does vitamin C influence neurodegenerative diseases and psychiatric disorders? Nutrients 9:659. 10.3390/nu9070659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostoff R. N., Heroux P., Aschner M., Tsatsakis A. (2020). Adverse health effects of 5G mobile networking technology under real-life conditions. Toxicol. Lett. 232 35–40. 10.1016/j.toxlet.2020.01.020 [DOI] [PubMed] [Google Scholar]
- Kucukgoncu S., Zhou E., Lucas K. B., Tek C. (2017). Alpha-lipoic acid (ALA) as a supplementation for weight loss: results from a meta-analysis of randomized controlled trials. Obes. Rev. 18 594–601. 10.1111/obr.12528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Pandey A. K. (2013). Chemistry and biological activities of flavonoids: an overview. Sci. World J. 2013:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurutas E. B. (2015). The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr. J. 15 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy M., Mathy-Hartert M., Deby-Dupont G. (1996). “Neutrophil-induced Oxidative Stress,” in Yearbook of Intensive Care and Emergency Medicine, ed. Vincent J. L. (Berlin: Springer; ), 83–95. [Google Scholar]
- Lazzarino G., Listorti I., Bilotta G., Capozzolo T., Amorini A. M., Longo S., et al. (2019). Water- and fat-soluble antioxidants in human seminal plasma and serum of fertile males. Antioxidants 8:96. 10.3390/antiox8040096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. M., Cook N. R., Gaziano J. M., Gordon D., Ridker P. M., Manson J. E., et al. (2005). Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. Jama 294 56–65. [DOI] [PubMed] [Google Scholar]
- Lee S. Q., Tan T. S., Kawamukai M., Chen E. S. (2017). Cellular factories for coenzyme Q10 production. Microb. Cell Fact. 16:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Horke S., Forstermann U. (2014). Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237 208–219. 10.1016/j.atherosclerosis.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Li J., Wuliji O., Li W., Jiang Z.-G., Ghanbari H. A. (2013). Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 14 24438–24475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Cao L., Han L., Xu Q., Ma Q. (2015). Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2/ERK/NF-κB axis. Int. J. Oncol. 46 2613–2620. 10.3892/ijo.2015.2938 [DOI] [PubMed] [Google Scholar]
- Liang X., Wang S., Wang L., Ceylan A. F., Ren J., Zhang Y. (2020). Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of drp1-mediated maladaptive mitochondrial fission. Pharmacol. Res. 157:104846. 10.1016/j.phrs.2020.104846 [DOI] [PubMed] [Google Scholar]
- Liguori I., Russo G., Aran L., Bulli G., Curcio F., Della-Morte D., et al. (2018). Sarcopenia: assessment of disease burden and strategies to improve outcomes. Clin. Interv. Aging 13:913. 10.2147/cia.s149232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Cook N. R., Albert C., Zaharris E., Gaziano J. M., Van Denburgh M., et al. (2009). Vitamins C and E and beta carotene supplementation and cancer risk: a randomized controlled trial. J. Natl. Cancer Inst. 101 14–23. 10.1093/jnci/djn438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G. Y., Doppler H., Delgiorno K. E., Zhang L., Leitges M., Crawford H. C., et al. (2016). Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14 2325–2336. 10.1016/j.celrep.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.-Q. (2019). Bridging free radical chemistry with drug discovery: a promising way for finding novel drugs efficiently. Eur. J. Med. Chem. 189:112020. 10.1016/j.ejmech.2019.112020 [DOI] [PubMed] [Google Scholar]
- Lü J.-M., Lin P. H., Yao Q., Chen C. (2010). Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. J. Cell. Mol.Med. 14 840–860. 10.1111/j.1582-4934.2009.00897.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach F., Schonbeck U., Libby P. (1998). CD40 signaling in vascular cells: a key role in atherosclerosis? Atherosclerosis 137(Suppl.), S89–S95. [DOI] [PubMed] [Google Scholar]
- Mahajan L., Verma P. K., Raina R., Pankaj N. K., Sood S., Singh M. (2018). Alteration in thiols homeostasis, protein and lipid peroxidation in renal tissue following subacute oral exposure of imidacloprid and arsenic in Wistar rats. Toxicol. Rep. 5 1114–1119. 10.1016/j.toxrep.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchitti S. A., Chen Y., Thompson D. C., Vasiliou V. (2011). Ultraviolet radiation: cellular antioxidant response and the role of ocular aldehyde dehydrogenase enzymes. Eye Contact Lens 37:206. 10.1097/icl.0b013e3182212642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti R., Rosello S., Cebolla-Cornejo J. (2016). Tomato as a source of carotenoids and polyphenols targeted to cancer prevention. Cancers 8:58. 10.3390/cancers8060058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meybodi N. M., Mortazavian A. M., Monfared A. B., Sohrabvandi S., Meybodi F. A. (2017). Phytochemicals in Cancer prevention: a review of the evidence. Int. J. Cancer Manag. 10:e7219. [Google Scholar]
- Miltonprabu S., Sumedha N. C., Senthilraja P. (2017). Diallyl trisulfide, a garlic polysulfide protects against As-induced renal oxidative nephrotoxicity, apoptosis and inflammation in rats by activating the Nrf2/ARE signaling pathway. Int. Immunopharmacol. 50 107–120. 10.1016/j.intimp.2017.06.011 [DOI] [PubMed] [Google Scholar]
- Mishra A. P., Salehi B., Sharifi-Rad M., Pezzani R., Kobarfard F., Sharifi-Rad J., et al. (2018). Programmed Cell death, from a cancer perspective: an overview. Mol. Diagn. Ther. 22 281–295. [DOI] [PubMed] [Google Scholar]
- Murr C., Schroecksnadel K., Winkler C., Ledochowski M., Fuchs D. (2005). Antioxidants may increase the probability of developing allergic diseases and asthma. Med. Hypotheses 64 973–977. 10.1016/j.mehy.2004.11.011 [DOI] [PubMed] [Google Scholar]
- Niedzielska E., Smaga I., Gawlik M., Moniczewski A., Stankowicz P., Pera J., et al. (2016). Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 53 4094–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum L., Hogea L. M., Cãlina D., Andreescu N., Grãdinaru R., ?tefãnescu R., et al. (2017). Modern treatment approaches in psychoses. Pharmacogenetic, neuroimagistic and clinical implications. Farmacia 65 75–81. [Google Scholar]
- Oke G. O., Abiodun A. A., Imafidon C. E., Monsi B. F. (2019). Zingiber officinale (Roscoe) mitigates CCl4-induced liver histopathology and biochemical derangements through antioxidant, membrane-stabilizing and tissue-regenerating potentials. Toxicol. Rep. 6 416–425. 10.1016/j.toxrep.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padureanu R., Albu C. V., Mititelu R. R., Bacanoiu M. V., Docea A. O., Calina D., et al. (2019). Oxidative stress and inflammation interdependence in multiple sclerosis. J. Clin. Med. 8:1815. 10.3390/jcm8111815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panic N., Nedovic D., Pastorino R., Boccia S., Leoncini E. (2017). Carotenoid intake from natural sources and colorectal cancer: a systematic review and meta-analysis of epidemiological studies. Eur. J. Cancer Prev. 26 27–37. 10.1097/cej.0000000000000251 [DOI] [PubMed] [Google Scholar]
- Papa S., Martino P. L., Capitanio G., Gaballo A., De Rasmo D., Signorile A., et al. (2012). The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 942 3–37. [DOI] [PubMed] [Google Scholar]
- Park L., Zhou P., Pitstick R., Capone C., Anrather J., Norris E. H., et al. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. US.A. 105 1347–1352. 10.1073/pnas.0711568105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M. H., Jo M., Kim Y. R., Lee C. K., Hong J. T. (2016). Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 163 1–23. 10.1016/j.pharmthera.2016.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P., Belford M. E., Lennon N., Bacskai B. J., Hyman B. T., Trotti D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43 19–30. 10.1016/j.neuron.2004.06.021 [DOI] [PubMed] [Google Scholar]
- Payne B. A. I., Chinnery P. F. (2015). Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim. Biophys. Acta 1847 1347–1353. 10.1016/j.bbabio.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peake J., Suzuki K. (2004). Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc. Immunol. Rev. 10 129–141. [PubMed] [Google Scholar]
- Pei Z., Deng Q., Babcock S. A., He E. Y., Ren J., Zhang Y. (2018). Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicol. Lett. 284 10–20. 10.1016/j.toxlet.2017.11.018 [DOI] [PubMed] [Google Scholar]
- Perera N. C. N., Godahewa G. I., Lee S., Kim M. J., Hwang J. Y., Kwon M. G., et al. (2017). Manganese-superoxide dismutase (MnSOD), a role player in seahorse (Hippocampus abdominalis) antioxidant defense system and adaptive immune system. Fish Shellfish Immunol. 68 435–442. 10.1016/j.fsi.2017.07.049 [DOI] [PubMed] [Google Scholar]
- Perrotta I., Aquila S. (2015). The role of oxidative stress and autophagy in atherosclerosis. Oxid. Med. Cell. Longev. 2015:130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingitore A., Lima G. P. P., Mastorci F., Quinones A., Iervasi G., Vassalle C. (2015). Exercise and oxidative stress: potential effects of antioxidant dietary strategies in sports. Nutrition 31 916–922. 10.1016/j.nut.2015.02.005 [DOI] [PubMed] [Google Scholar]
- Pizzino G., Bitto A., Interdonato M., Galfo F., Irrera N., Mecchio A., et al. (2014). Oxidative stress and DNA repair and detoxification gene expression in adolescents exposed to heavy metals living in the Milazzo-Valle del Mela area (Sicily. Italy). Redox. Biol. 2 686–693. 10.1016/j.redox.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzino G., Irrera N., Cucinotta M., Pallio G., Mannino F., Arcoraci V., et al. (2017). Oxidative stress: harms and benefits for human health. Oxid. Med. Cell. Longev. 2017:8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G., Leonarduzzi G., Biasi F., Chiarpotto E. (2004). Oxidative stress and cell signalling. Curr. Med. Chem. 11 1163–1182. 10.2174/0929867043365323 [DOI] [PubMed] [Google Scholar]
- Poole L. B., Nelson K. J. (2016). Distribution and features of the six classes of peroxiredoxins. Mol. Cells 39 53–59. 10.14348/molcells.2016.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poprac P., Jomova K., Simunkova M., Kollar V., Rhodes C. J., Valko M. (2017). Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol. Sci. 38 592–607. 10.1016/j.tips.2017.04.005 [DOI] [PubMed] [Google Scholar]
- Prochazkova D., Bousova I., Wilhelmova N. (2011). Antioxidant and prooxidant properties of flavonoids. Fitoterapia 82 513–523. 10.1016/j.fitote.2011.01.018 [DOI] [PubMed] [Google Scholar]
- Ramsay R. R. (2019). Electron carriers and energy conservation in mitochondrial respiration. Chem. Texts 5:9. [Google Scholar]
- Rasouli H., Farzaei M. H., Mansouri K., Mohammadzadeh S., Khodarahmi R. (2016). Plant cell cancer: may natural phenolic compounds prevent onset and development of plant cell malignancy? A literature review. Molecules 21:1104. 10.3390/molecules21091104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P. D., Huang B.-W., Tsuji Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24 981–990. 10.1016/j.cellsig.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P. H. (2009). Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectr. 14 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid M. B. (2001). Invited Review: redox modulation of skeletal muscle contraction: what we know and what we don’t. J. Appl. Physiol. 90 724–731. 10.1152/jappl.2001.90.2.724 [DOI] [PubMed] [Google Scholar]
- Ren J., Taegtmeyer H. (2015). Too much or not enough of a good thing—The Janus faces of autophagy in cardiac fuel and protein homeostasis. J. Mol. Cell. Cardiol. 84 223–226. 10.1016/j.yjmcc.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Reuter S., Gupta S. C., Chaturvedi M. M., Aggarwal B. B. (2010). Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 49 1603–1616. 10.1016/j.freeradbiomed.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer P., Sofic E., Rausch W. D., Schmidt B., Reynolds G. P., Jellinger K., et al. (1989). Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 52 515–520. 10.1111/j.1471-4159.1989.tb09150.x [DOI] [PubMed] [Google Scholar]
- Rodriguez R., Redman R. (2005). Balancing the generation and elimination of reactive oxygen species. Proc. Natl. Acad. Sci. 102 3175–3176. 10.1073/pnas.0500367102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychoudhury S., Agarwal A., Virk G., Cho C. L. (2017). Potential role of green tea catechins in the management of oxidative stress-associated infertility. Reprod. Biomed. Online 34 487–498. 10.1016/j.rbmo.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Saccon R. A., Bunton-Stasyshyn R. K., Fisher E. M., Fratta P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136 2342–2358. 10.1093/brain/awt097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackesen C., Ercan H., Dizdar E., Soyer O., Gumus P., Tosun B. N., et al. (2008). A comprehensive evaluation of the enzymatic and nonenzymatic antioxidant systems in childhood asthma. J. Allergy Clin. Immunol. 122 78–85. 10.1016/j.jaci.2008.03.035 [DOI] [PubMed] [Google Scholar]
- Saez I., Vilchez D. (2014). The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 15 38–51. 10.2174/138920291501140306113344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage E., Girard P.-M., Francesconi S. (2012). Unravelling UVA-induced mutagenesis. Photochem. Photobiol. Sci. 11 74–80. 10.1039/c1pp05219e [DOI] [PubMed] [Google Scholar]
- Saha S. K., Lee S. B., Won J., Choi H. Y., Kim K., Yang G. M., et al. (2017). Correlation between oxidative stress, nutrition, and cancer initiation. Int. J. Mol. Sci. 18:1544. 10.3390/ijms18071544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Calina D., Docea A. O., Koirala N., Aryal S., Lombardo D., et al. (2020a). Curcumin’s nanomedicine formulations for therapeutic application in neurological diseases. J. Clin. Med. 9:430. 10.3390/jcm9020430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Rescigno A., Dettori T., Calina D., Docea A. O., Singh L., et al. (2020b). Avocado–soybean unsaponifiables: a panoply of potentialities to be exploited. Biomolecules 10:130. 10.3390/biom10010130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Capanoglu E., Adrar N., Catalkaya G., Shaheen S., Jaffer M., et al. (2019a). Cucurbits plants: a key emphasis to its pharmacological potential. Molecules 24:1854. 10.3390/molecules24101854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Lopez-Jornet P., Pons-Fuster López E., Calina D., Sharifi-Rad M., Ramírez-Alarcón K., et al. (2019b). Plant-derived bioactives in oral mucosal lesions: a key emphasis to curcumin, lycopene, chamomile, aloe vera, green tea and coffee properties. Biomolecules 9:106. 10.3390/biom9030106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Sestito S., Rapposelli S., Peron G., Calina D., Sharifi-Rad M., et al. (2019c). Epibatidine: a promising natural alkaloid in health. Biomolecules 9:6. 10.3390/biom9010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Shivaprasad Shetty M. V., Anil Kumar N., Živkoviæ J., Calina D., Oana Docea A., et al. (2019d). Veronica Plants—Drifting from farm to traditional healing, food application, and phytopharmacology. Molecules 24:2454. 10.3390/molecules24132454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B., Martorell M., Arbiser J. L., Sureda A., Martins N., Maurya P. K., et al. (2018). Antioxidants: positive or negative actors? Biomolecules 8:124. 10.3390/biom8040124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sani T. A., Mohammadpour E., Mohammadi A., Memariani T., Yazdi M. V., Rezaee R., et al. (2017). Cytotoxic and apoptogenic properties of Dracocephalum kotschyi aerial part different fractions on calu-6 and mehr-80 lung cancer cell lines. Farmacia 65 189–199. [Google Scholar]
- Sanjust E., Mocci G., Zucca P., Rescigno A. (2008). Mediterranean shrubs as potential antioxidant sources. Nat. Prod. Res. 22 689–708. 10.1080/14786410801997125 [DOI] [PubMed] [Google Scholar]
- Schottker B., Brenner H., Jansen E. H., Gardiner J., Peasey A., Kubinova R., et al. (2015). Evidence for the free radical/oxidative stress theory of ageing from the CHANCES consortium: a meta-analysis of individual participant data. BMC Med. 13:300. 10.1186/s12916-015-0537-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ściskalska M., Zalewska M., Grzelak A., Milnerowicz H. (2014). The influence of the occupational exposure to heavy metals and tobacco smoke on the selected oxidative stress markers in smelters. Biol. Trace Element Res. 159 59–68. 10.1007/s12011-014-9984-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena L. A., Chandel N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48 158–167. 10.1016/j.molcel.2012.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M. Y., Lee S. M. (2002). Protective effect of low dose of ascorbic acid on hepatobiliary function in hepatic ischemia/reperfusion in rats. J. Hepatol. 36 72–77. 10.1016/s0168-8278(01)00236-7 [DOI] [PubMed] [Google Scholar]
- Sharifi-Rad J., Rodrigues C. F., Sharopov F., Docea A. O., Can Karaca A., Sharifi-Rad M., et al. (2020). Diet, lifestyle and cardiovascular diseases: linking pathophysiology to cardioprotective effects of natural bioactive compounds. Int. J. Environ. Res. Public Health 17:2326. 10.3390/ijerph17072326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi-Rad M., Lankatillake C., Dias D. A., Docea A. O., Mahomoodally M. F., Lobine D., et al. (2020). Impact of natural compounds on neurodegenerative disorders: from preclinical to pharmacotherapeutics. J. Clin. Med. 9:1061. 10.3390/jcm9041061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi-Rad J., Sharifi-Rad M., Salehi B., Iriti M., Roointan A., Mnayer D., et al. (2018). In vitro and in vivo assessment of free radical scavenging and antioxidant activities of Veronica persica Poir. Cell. Mol. Biol. 64 57–64. [PubMed] [Google Scholar]
- Sharma P., Jha A. B., Dubey R. S., Pessarakli M. (2012). reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012:26. [Google Scholar]
- Singh R. B., Mengi S. A., Xu Y. J., Arneja A. S., Dhalla N. S. (2002). Pathogenesis of atherosclerosis: a multifactorial process. Exp. Clin. Cardiol. 7 40–53. [PMC free article] [PubMed] [Google Scholar]
- Singh Y. P., Patel R. N., Singh Y., Butcher R. J., Vishakarma P. K., Singh R. K. B. (2017). Structure and antioxidant superoxide dismutase activity of copper(II) hydrazone complexes. Polyhedron 122 1–15. 10.1016/j.poly.2016.11.013 [DOI] [Google Scholar]
- Smith M. T., Guyton K. Z., Gibbons C. F., Fritz J. M., Portier C. J., Rusyn I., et al. (2016). Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 124 713–721. 10.1289/ehp.1509912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sova H., Jukkola-Vuorinen A., Puistola U., Kauppila S., Karihtala P. (2010). 8-Hydroxydeoxyguanosine: a new potential independent prognostic factor in breast cancer. Br. J. Cancer 102 1018–1023. 10.1038/sj.bjc.6605565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz D. R., Azzam E. I., Li J. J., Gius D. (2004). Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metast. Rev. 23 311–322. 10.1023/b:canc.0000031769.14728.bc [DOI] [PubMed] [Google Scholar]
- Spitz D. R., Hauer-Jensen M. (2014). Ionizing Radiation-Induced Responses: Where Free Radical Chemistry Meets Redox Biology and Medicine. Rochelle, NY: Mary Ann Liebert, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafazoli A. (2017). Coenzyme Q10 in breast cancer care. Future Oncol. 13 1035–1041. 10.2217/fon-2016-0547 [DOI] [PubMed] [Google Scholar]
- Takalo M., Salminen A., Soininen H., Hiltunen M., Haapasalo A. (2013). Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegenerat. Dis. 2 1–14. [PMC free article] [PubMed] [Google Scholar]
- Taverne Y. J., Bogers A. J., Duncker D. J., Merkus D. (2013). Reactive oxygen species and the cardiovascular system. Oxid. Med. Cell. Longev. 2013:862423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovmasyan A., Maia C. G., Weitner T., Carballal S., Sampaio R. S., Lieb D., et al. (2015). A comprehensive evaluation of catalase-like activity of different classes of redox-active therapeutics. Free Radic. Biol. Med. 86 308–321. 10.1016/j.freeradbiomed.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsakis A., Docea A. O., Calina D., Tsarouhas K., Zamfira L.-M., Mitrut R., et al. (2019). A mechanistic and pathophysiological approach for stroke associated with drugs of abuse. J. Clin. Med. 8:1295. 10.3390/jcm8091295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsakis A. M., Docea A. O., Calina D., Buga A. M., Zlatian O., Gutnikov S., et al. (2019). Hormetic Neurobehavioral effects of low dose toxic chemical mixtures in real-life risk simulation (RLRS) in rats. Food Chem. Toxicol. 125 141–149. 10.1016/j.fct.2018.12.043 [DOI] [PubMed] [Google Scholar]
- Tse G., Yan B. P., Chan Y. W., Tian X. Y., Huang Y. (2016). Reactive oxygen species, endoplasmic reticulum stress and mitochondrial dysfunction: the link with cardiac arrhythmogenesis. Front. Physiol. 7:313. 10.3389/fphys.2016.00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukalas D., Fragkiadaki P., Docea A. O., Alegakis A. K., Sarandi E., Thanasoula M., et al. (2019a). Discovery of potent telomerase activators: unfolding new therapeutic and anti-aging perspectives. Mol. Med. Rep. 20 3701–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukalas D., Fragkiadaki P., Docea A. O., Alegakis A. K., Sarandi E., Vakonaki E., et al. (2019b). Association of nutraceutical supplements with longer telomere length. Int. J. Mol. Med. 44 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoukalas D., Fragoulakis V., Sarandi E., Docea A. O., Papakonstantinou E., Tsilimidos G., et al. (2019c). Targeted metabolomic analysis of serum fatty acids for the prediction of autoimmune diseases. Front. Mol. Biosci. 6:120. 10.3389/fmolb.2019.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A., Vlachogianni T., Fiotakis K. (2009). Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 6 445–462. 10.3390/ijerph6020445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., Telser J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39 44–84. 10.1016/j.biocel.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. (2006). Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 160 1–40. 10.1016/j.cbi.2005.12.009 [DOI] [PubMed] [Google Scholar]
- Wang X., Michaelis E. K. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2:12. 10.3389/fnagi.2010.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R. R. (1998). Melatonin in the Promotion of Health, 2nd Edn Boca Raton, FL: Taylor & Francis Group. [Google Scholar]
- Wattanapitayakul S. K., Bauer J. A. (2001). Oxidative pathways in cardiovascular disease: roles, mechanisms, and therapeutic implications. Pharmacol. Ther. 89 187–206. 10.1016/s0163-7258(00)00114-5 [DOI] [PubMed] [Google Scholar]
- Wu N. N., Tian H., Chen P., Wang D., Ren J., Zhang Y. (2019). Physical exercise and selective autophagy: benefit and risk on cardiovascular health. Cell 8:1436. 10.3390/cells8111436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Liu X., Huang H., Li Z., Xiong T., Xiang W., et al. (2019). Effects of major ozonated autoheamotherapy on functional recovery, ischemic brain tissue apoptosis and oxygen free radical damage in the rat model of cerebral ischemia. J. Cell. Biochem. 120 6772–6780. 10.1002/jcb.27978 [DOI] [PubMed] [Google Scholar]
- Ye G., Metreveli N. S., Donthi R. V., Xia S., Xu M., Carlson E. C., et al. (2004). Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 53 1336–1343. 10.2337/diabetes.53.5.1336 [DOI] [PubMed] [Google Scholar]
- Young A. J., Lowe G. M. (2001). Antioxidant and prooxidant properties of carotenoids. Arch. Biochem. Biophys. 385 20–27. 10.1006/abbi.2000.2149 [DOI] [PubMed] [Google Scholar]
- Zal F., Taheri R., Khademi F., Keshavarz E., Rajabi S., Mostafavi-Pour Z. (2014). The combined effect of furosemide and propranolol on GSH homeostasis in ACHN renal cells. Toxicol. Mech. Methods 24 412–416. 10.3109/15376516.2014.926437 [DOI] [PubMed] [Google Scholar]
- Zhang J., McCullough P. A. (2016). Lipoic acid in the prevention of acute kidney injury. Nephron 134 133–140. 10.1159/000448666 [DOI] [PubMed] [Google Scholar]
- Zhang W., Wang T., Qin L., Gao H. M., Wilson B., Ali S. F., et al. (2004). Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. Faseb. J. 18 589–591. 10.1096/fj.03-0983fje [DOI] [PubMed] [Google Scholar]
- Zhou H., Wang S., Zhu P., Hu S., Chen Y., Ren J. (2018). Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 15 335–346. 10.1016/j.redox.2017.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Jiang S., Hu N., Luo F., Dong H., Kang Y. M., et al. (2014). Tumour necrosis factor-α inhibition with lenalidomide alleviates tissue oxidative injury and apoptosis in ob/ob obese mice. Clin. Exp. Pharmacol. Physiol. 41 489–501. 10.1111/1440-1681.12240 [DOI] [PubMed] [Google Scholar]
- Zucca P., Argiolas A., Nieddu M., Pintus M., Rosa A., Sanna F., et al. (2016). Biological activities and nutraceutical potentials of water extracts from different parts of cynomorium coccineum L. (Maltese Mushroom). Polish J. Food Nutr.l Sci. 66 179–188. 10.1515/pjfns-2016-0006 [DOI] [Google Scholar]
- Zucca P., Rescigno A., Rinaldi A. C., Sanjust E. (2014). Biomimetic metalloporphines and metalloporphyrins as potential tools for delignification: molecular mechanisms and application perspectives. J. Mol. Catal. A Chem. 388-389 2–34. 10.1016/j.molcata.2013.09.010 [DOI] [Google Scholar]