

SUMMARY
Single-particle cryo-EM continues to grow into a mainstream structural biology technique. Recent developments in data collection strategies alongside new sample preparation devices heralds a future where users will collect multiple datasets per microscope session. To make cryo-EM data processing more automatic and user-friendly, we have developed an automatic pipeline for cryo-EM data preprocessing and assessment using a combination of deep learning and image analysis tools. We have verified the performance of this pipeline on a number of datasets and extended its scope to include sample screening by the user-free assessment of the qualities of a series of datasets under different conditions. We propose that our workflow provides a decision-free solution for cryo-EM, making data preprocessing more generalized and robust in the high-throughput era as well as more convenient for users from a range of backgrounds.
Keywords: cryo-EM, pipeline, deep-learning
GRAPHICAL ABSTRACT
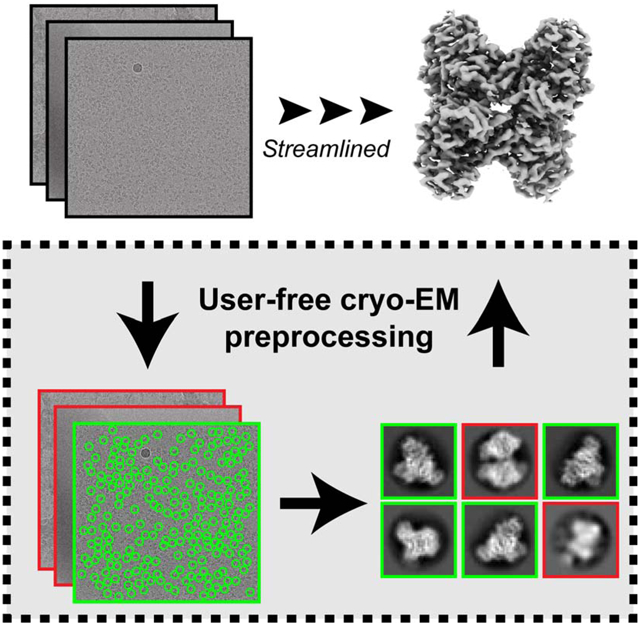
eTOC blurb
Li et al. develop an automatic pre-preprocessing workflow for single-particle cryo-EM. This workflow produces high-resolution particle stacks from micrograph inputs without subjective decision making. They used this workflow to automatically preprocess and analyze 6 challenging datasets, demonstrating that the workflow correctly identified the high-resolution dataset amidst 5 bad datasets.
INTRODUCTION
Single-particle cryo-electron microscopy (cryo-EM) is becoming a mainstream technique for structural biology (Kühlbrandt, 2014). In the past few years, cryo-EM has seen a 20–40% year-to-year growth in structures deposited in the Protein Data Bank. This growth is due to continued developments in sample preparation (Arnold et al., 2017; Cheng et al., 2018; Darrow et al., 2019; Jain et al., 2012; Ravelli et al., 2019; Zivanov et al., 2018), data collection (Fernandez-Leiro and Scheres, 2016; Lyumkis, 2019), and algorithms for data processing (Punjani et al., 2017; Scheres, 2012; Tegunov and Cramer, 2019; Zivanov et al., 2018). These developments have greatly accelerated the speed of data collection for cryo-EM, and have also led to widespread adoption of users across a range of expertise, where experts represent a continually shrinking fraction of cryo-EM users.
With the fast pace of cryo-EM development, several challenges have emerged. First, with new imaging and sample preparation technologies, including the increased frame rate detectors, beam-image shift data collection (Cheng et al., 2018; Zivanov et al., 2018), and robotic sample preparation (Arnold et al., 2017; Darrow et al., 2019; Jain et al., 2012; Ravelli et al., 2019), a single cryo-EM instrument can easily generate 5,000–8,000 movies data per day. These technologies have enabled cryo-EM to become a more high-throughput technique, with more than one dataset collected per day per instrument. Second, although a number of improvements have been made in software development, cryo-EM data processing remains computationally expensive. High-performance computing (HPC) resources and GPUs are typically used (Baldwin et al., 2018; Cianfrocco and Leschziner, 2015). However, since each project requires multiple rounds of human trial and error in the preprocessing steps, these human-driven choices can slow down a project due to a lack of computing resources.
Third, cryo-EM frustrates many users because of its complexity in data processing. The manual and subjective decisions involved in solving a structure, such as the programs, parameters, and determination of good micrographs and good 2D class averages, can affect the final result significantly (Lawson and Chiu, 2018). While an expert can make the correct decisions after a few trials, new users typically find it problematic to perform such monitoring and evaluations. Moreover, due to the variety of samples in the cryo-EM field, it is nearly impossible to create a general guideline for the new users to follow.
Despite the increasing throughput of cryo-EM data collection, the cumbersome nature of cryo-EM preprocessing slows scientists’ ability to ask biological questions from their dataset. For example, during cryo-EM sample screening, scientists may want to assess sample integrity or complex formation. However, in order to compare and contrast multiple grids, the scientist will have to manually interact with the data to perform movie alignment, particle picking, CTF estimation, and 2D classification. Modern cryo-EM needs a tool to streamline data quality assessment and data preprocessing automatically and robustly.
Many approaches have been proposed and developed to address these challenges. For example, Appion (Lander et al., 2009), cryoSPARC (Punjani et al., 2017), SPHIRE (Moriya et al., 2017), Warp (Tegunov and Cramer, 2019), and RELION-3.0 (Fernandez-Leiro and Scheres, 2017; Zivanov et al., 2018) provide preprocessing tools that can be stitched together into pipelines. Despite this ability, easy computation access to these remains an issue. To address the computation resource problem, COSMIC2 (Cianfrocco et al., 2017), a science gateway for cryo-EM, has been developed with the philosophy of bringing popular cryo-EM tools and resources to all scientists in the field, removing the practical limitations that accessing those resources would otherwise entail.
Many algorithms have also been developed to accelerate cryo-EM data preprocessing and minimize subjective decisions and tedious human annotations. Notably, deep learning, especially convolutional neural network (CNN), has greatly changed and improved the step of particle picking (Al-Azzawi et al., 2019; Bepler et al., 2019; Nguyen et al., 2019; Tegunov and Cramer, 2019; Wagner et al., 2019; Wang et al., 2016; Xiao and Yang, 2017; Zhang et al., 2019; Zhu et al., 2017). Nevertheless, the field still lacks a robust tool that will make decisions by evaluating the output from data preprocessing steps, so that human intervention can be removed, making an automatically streamlining workflow possible.
Here, we introduce several deep learning and image analysis tools for automated preprocessing and assessment of cryo-EM datasets. By connecting these tools with state-of-the-art data preprocessing algorithms, we make a general workflow that can achieve expert-level performance on a number of different cryo-EM datasets without any user intervention. Our workflow takes movies or motion-corrected micrographs as the input and outputs a particle stack that contains high-resolution particles that will be used in the following 3D reconstruction steps without any user decisions. Specifically, our workflow can automatically detect bad micrographs using MicAssess, determine the best parameters for particle picking and 2D classification, and identify the good class averages that can be used in 3D reconstruction using 2DAssess. In the workflow, the subjective user decisions are replaced with statistical models based on the features extracted with image processing methods and convolutional neural networks, along with the expert knowledge. We believe that our automatic pipeline helps to establish a framework to accelerate data preprocessing and to perform data assessment at multiple levels in the high-throughput era of cryo-EM.
RESULTS
Overview of the Method
The current routine of cryo-EM data preprocessing consists of a number of subjective user decisions (Fig. 1). First, many users will manually go through all the motion-corrected micrographs to pick out the bad micrographs, and then select an estimated resolution threshold to remove the remaining bad micrographs based on the results of CTF estimation. Next, most particle pickers will require the users to manually pick a few particles, set the estimated particle diameter and determine the picking threshold before automatic particle picking. Then the particles will be extracted with the user-defined box size and pixel size used for 2D classification, where the users need to determine the class number and the diameter of the mask. Finally, the users need to select the good 2D class averages based on their own judgment, and the particles in the selected 2D class averages will be re-extracted and used in the downstream 3D reconstruction steps.
Figure 1. Conventional cryo-EM preprocessing vs. automatic preprocessing pipeline.

Left panel: current workflow describing the preprocessing of cryo-EM datasets, with all the user decisions needed in red. Right panel: the automatic pipeline introduced in this paper. All user decisions are replaced by the new tools developed in blue.
Our general workflow streamlines the preprocessing steps to take either movies or motion-corrected micrographs as the input and output a stack of clean particles that can be used as the input in the subsequent 3D analysis (Fig. 1). During this process, we built statistical models in order to capture human decision-making during the preprocessing steps. Instead of developing new preprocessing tools and algorithms, our workflow takes advantage of these developments and provides evaluations so that expert-level decisions can be made automatically. We provide an overview of the method below.
MicAssess: Automatic micrograph assessment
First, we developed a tool that can assess the quality of motion-corrected micrographs even before CTF estimation: MicAssess. Unlike EMPIAR datasets, which consist of mostly usable micrographs, many realworld data generated from the microscopes are dirty and noisy. Researchers often undertake significant effort to manually eliminate bad micrographs to obtain a clean dataset to work within the downstream preprocessing steps. Although the difference between good and bad micrographs is unambiguous, it is still difficult to find a universal and robust criterion. Many scientists have been using the resolution outputs from CTF estimation for micrograph cleaning, however, there lacks a publicly accepted resolution cutoff, and there are still a number of bad micrographs that will make through using this metric for decision making.
Convolutional neural networks (CNN) are changing the field of computer vision as well as biology in recent years and have been widely applied to image classification, object detection, image segmentation, etc (Moen et al., 2019). In cryo-EM, a number of CNN-based particle picking models have been developed and widely used, including Warp (Tegunov and Cramer, 2019), crYOLO (Wagner et al., 2019), and Topaz (Bepler et al., 2019). With the similar idea, we developed a CNN-based micrograph assessor, MicAssess. The architecture of MicAssess is described in Fig. 2A. Similar to many CNN models, our model consists of a feature extraction convolutional network and a classification network. For the feature extraction network, we used a standard ResNet34 (He et al., 2016), which is a deep and light-weighted fully convolutional residual network with 34 layers. Following the feature extraction, the convolutional network is the classification network, which consists of one fully connected (FC) layers with 512 nodes. Dropout layers with a 0.5 dropout rate and batch normalization are also applied, and leaky rectified linear unit (LReLU) is used as the activation function. Finally, the last layer uses a sigmoid function as the activation function and performs prediction, which is the probability that the input micrograph is considered as “good”.
Figure 2. Deep learning-based tools for cryo-EM micrograph and 2D class average assessment.

(A) The architecture of MicAssess. The motion corrected micrograph will be inputted to a feature extraction convolutional network (a standard ResNet34 in the paper), and after one dropout layer, one fully connected layer and another dropout layer, output the prediction of the micrograph. (B) Examples of the labeled good and bad micrographs in the training set. The good class contains partially good images, images with small or very large proteins, etc.. The bad class contains all different kinds of unusable micrographs, including micrographs that are empty or too dense, contaminated, or with protein aggregates. (C) The architecture of CNN-based model in 2DAssess. The input class average image will be inputted to a feature extraction convolutional network (a standard ResNet50 in the paper), and after one dropout layer, output the prediction of the 2D class average to be one of the four classes. (D) Examples of the labeled 2D class averages in the good, clip, edge, and noise classes in the training set.
Most image classification problems are considered as supervised learning, which means that they need to be trained on labeled datasets. We have collected and manually labeled a total of 4,644 micrographs (2,372 good micrographs and 2,272 bad micrographs) from several EMPIAR datasets in addition to in-house datasets (Table 1). Our good micrograph dataset consists of proteins and complexes ranging from 50 kDa to 4 MDa (Fig. 2B, upper row), while our bad micrograph dataset consists of a variety of unusable micrographs including micrographs that are either empty or too dense, contaminated, or with protein aggregates (Fig. 2B, lower row). The dataset was randomly split into a training set (80 %) and a validation set (20 %). Data augmentation was applied before training to increase the amount of training data and reduce overfitting. The trained model was evaluated on the validation set, and an accuracy of about 97% was achieved. A detailed description can be found in the Methods section.
Table 1.
Sources of the micrographs and the 2D class averages used for developing MicAssess and 2DAssess.
| MicAssess | 2DAssess | ||
|---|---|---|---|
| Particle Name | EMPIAR ID | Particle Name | EMPIAR ID |
| 26S Proteasome | EMPIAR-10072 | 26S Proteasome | EMPIAR-10072 |
| AAV | EMPIAR-10202 | AAV | EMPIAR-10202 |
| E. Coli70S-SelB ribosome | EMPIAR-10077 | E. Coli 70S-SelB ribosome | EMPIAR-10077 |
| Rag complex | EMPIAR-10049 | Rag complex | EMPIAR-10049 |
| NOMPC | EMPIAR-10093 | NOMPC | EMPIAR-10093 |
| GluDH | EMPIAR-10217 | GluDH | EMPIAR-10217 |
| RNA Pol III | EMPIAR-10190 | RNA Pol III | EMPIAR-10190 |
| Spliceosome | EMPIAR-10160 | Spliceosome | EMPIAR-10160 |
| In-house dataset - 160 kDa | N/A | Betagal | EMPIAR-10061 |
| In-house dataset - 480 kDa | N/A | TMEM16 | EMPIAR-10241 |
| In-house dataset - 180 kDa | N/A | In-house dataset - 180 kDa | N/A |
| In-house dataset - 168 kDa | N/A | In-house dataset - Apoferritin | N/A |
| In-house dataset - 80 kDa | N/A | ||
To test the effectiveness of MicAssess, we analyzed a published dataset collected by our lab on the Phosphatidylinositol 3,4,5-trisphosphate (PIP3)-dependent Rac exchanger 1 (P-Rex1) (Cash et al. 2019). This dataset contains 6,736 micrographs and is a combination of untilted and tilted series. Importantly, the training data in MicAssess did not include any P-Rex1 micrographs. As a comparison, we also classified the micrographs using the CTF maximum resolution outputs from CTFFIND4, with determination thresholds being 4 Å for untilted micrographs and 10 Å for tilted micrographs. To quantify the performance of both CTF-based micrograph cleaning and MicAssess, we manually labeled the total 6,736 micrographs and used the labels as the “ground truth” with which to compare.
A comparison of CTF maximum resolution cutoff to the ground truth highlighted a number of discrepancies. As is typical, the distribution of CTF maximum resolution values for tilted or untilted micrographs does not show a bimodal distribution. (Fig. 3A). Therefore, even though 4 Å and 10 Å resolution cutoff thresholds are considered reasonable, such numbers are not obvious from the distribution of the data, but rather arbitrary. Compared to human-labeled “ground truth”, CTF-based micrograph cleaning reached an overall accuracy of 77.5% (Fig. 3B). This indicates that while CTF maximum resolution is a convenient method to remove bad micrographs, there is room for improvement in order to obtain more accurate micrograph assessment.
Figure 3. MicAssess performs equivalently to CTF resolution cutoff on micrograph assessment.
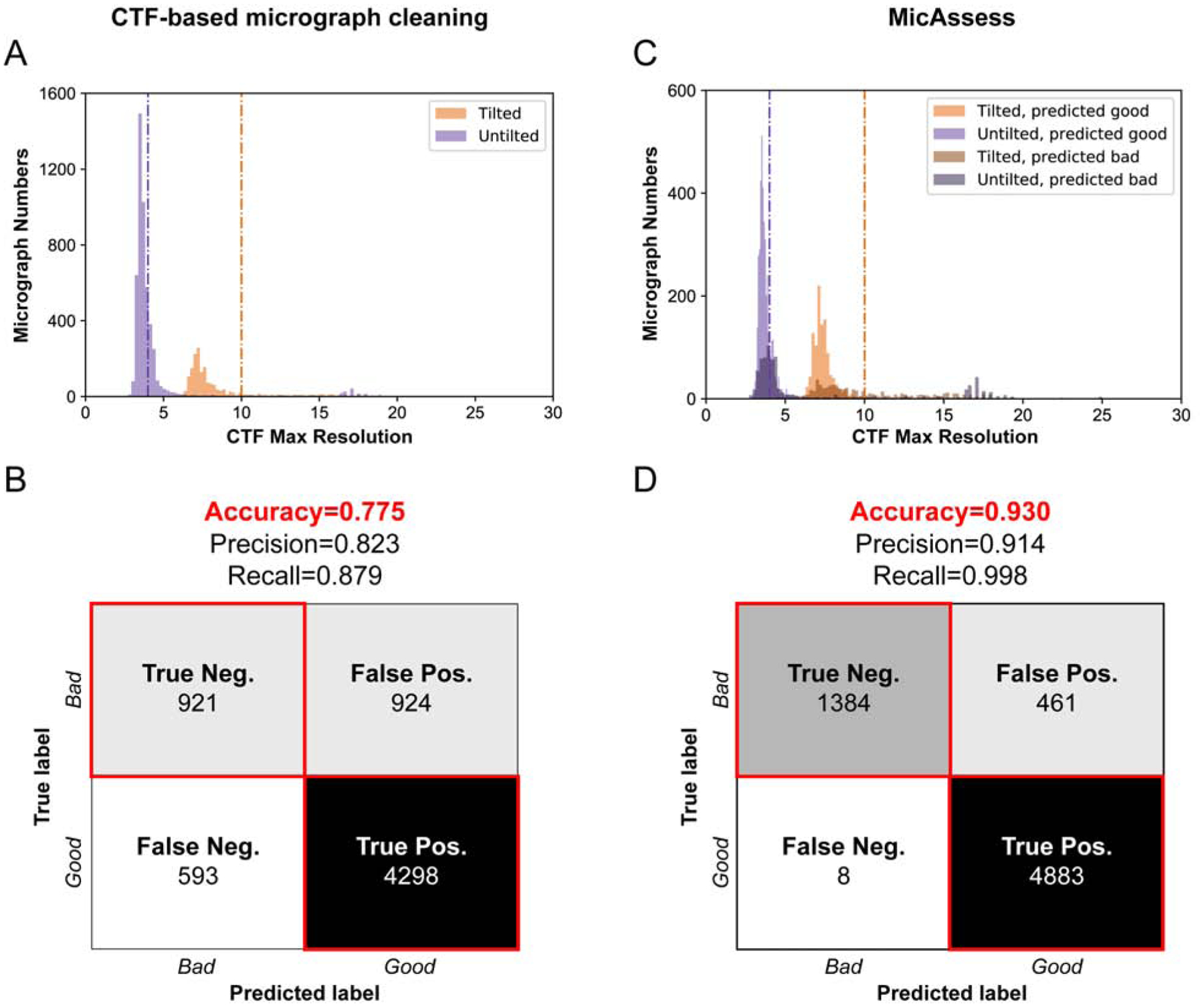
(A) Histograms of the CTF maximum resolutions outputted by CTFFIND4 of the test set. Vertical lines indicate the selected hard thresholds for tilted and untilted micrographs (4 Å and 10 Å respectively). Micrographs higher than the thresholds are considered as bad. (B) Confusion matrix and evaluation metrics for CTF resolution threshold vs. human assessment on P-Rex1:Gβγ dataset. (C) Histograms of the CTF maximum resolutions outputted by CTFFIND4 of the test set, color labeled according to the predictions by MicAssess. Vertical lines indicate 4 Å and 10 Å respectively. (D) Confusion matrix and evaluation metrics of MicAssess on the P-Rex1 test set.
Compared to CTF maximum resolution, MicAssess showed higher accuracy for identifying both good and bad micrographs. To highlight the power of MicAssess, MicAssess was also able to correctly classify many bad micrographs with < 4 Å CTF maximum resolutions (Fig. 3C). Such micrographs will not be captured by the CTF-based micrograph cleaning approach. Overall, MicAssess found 1,388 bad micrographs (Fig. S2) and had an accuracy of 93.0%, with a notably very low false-negative rate (0.12%) (Fig. 3D). In other words, only 8 good micrographs were misclassified to the bad category.
This analysis indicates the MicAssess performs nearly as well as human assessment for the P-Rex1 test dataset. More importantly, MicAssess does not need any arbitrary threshold, and both tilted and untilted micrographs were predicted with the exact same procedure, providing a completely “hands-off” tool for micrograph assessment, which enables the automatic cryo-EM data preprocessing and assessment at the very beginning.
Automatic particle diameter estimation
Since our workflow aims for decision-free preprocessing, the suitable particle picker should not need any human picking beforehand. Therefore, any template-based particle picker or CNN-based particle picker that needs to be trained on manually prelabeled particles cannot be used in the workflow. Fortunately, we are able to use the general model of crYOLO (Wagner et al., 2019), which is a CNN-based particle picker pretrained on a number of EMPIAR and in-house datasets. The two parameters needed for particle picking in crYOLO are box size and threshold.
Optimally, the box size should be the size of the particle. Since this information is usually unclear for a new protein, our workflow will first perform particle picking on a subset of the micrographs with different box sizes. The picked particles will be extracted, low pass filtered and averaged without any alignment. We then find the edge of this averaged image using a Canny edge detector, and the size of the particle is determined based on the edge detected and is dilated by an empirical factor of 1.5 (Fig. S3). After that, the workflow uses crYOLO to pick the particles from all micrographs. The threshold parameter controls the strictness of the decision of a particle. The workflow uses a very low threshold of 0.1 since many false positives can be removed in the following 2D classification step.
2DAssess: Automatic selection of good 2D class averages
After particles are picked and extracted from micrographs with CTF information, particles are subjected to 2D classification, whereby good 2D averages are identified using 2DAssess. Similar to the micrograph classifier, our CNN-based classifier model (Fig. 2C) also requires a labeled dataset for training. We have obtained the 2D class averages from ten different datasets from a range of diameters use in 2D classification, providing 2D averages for optimal masks, masks that are too tight, and masks that are too large (Table 1).
The 2D class averages are preprocessed and labeled in four different classes (Fig. 2D): good, clip, edge, and noise. The good class includes all the good class averages that will be selected and used in the downstream processing steps. The clip class includes the class averages that are clipping the neighboring particles, usually a sign that the diameter is too large. The edge class includes the class averages with “barcode” like patterns, which means that some particles are on the edge of the micrograph or the carbon. The noise class includes all the other bad class averages that are not covered by the clip and edge classes, which contains pure noise, over-aligned, and low-resolution class averages. The dataset was downsampled to account for the class imbalance and then randomly split into a training set (80 %) and a validation set (20 %). We noticed that when the diameter of the mask becomes large, one class average might contain two particles. The CNN-based classifier failed to detect this and would misclassify such 2D class averages to the “good” class. To prevent this, we checked the saliency map (Hou and Zhang, 2007) of the 2D class averages in the predicted “good” class, and re-classify the class averages with two or more objects to the correct “clip” class. The combination of the CNN-based classifier and the saliency map check made up the complete 2D class average assessor, which we named it as 2DAssess.
To further enrich the number of good class averages, we used deep convolutional generative adversarial networks (DCGAN) (Radford et al., 2015) to generate artificial good class averages using the true good class averages in the training set. We then carefully selected 66 artificial good class averages generated by DCGAN (Fig. S4) and added them to the training set. Although the selected images are not from 2D class averages of real proteins, they will most likely be labeled as good class averages without any prior knowledge of the protein. Adding these DCGAN generated images as a data augmentation approach improves the generalizability of the classifier when the good 2D class average samples are limited. Some simple data augmentation (elaborated in the methods section of the paper) was applied in training and validation. Notably, the good class reached a precision of 94% and a recall of 97%.
Testing on EMPIAR Datasets
T20S proteasome (EMPIAR-10025)
First, we tested our workflow on a subset of the published T20S proteasome cryo-EM dataset (EMPIAR-10025) (Campbell et al., 2015) (Fig. 4). This subset contains 87 micrographs, all of which were all being classified as good by MicAssess. Subsequently, the diameter was estimated to be 195 Å. Using this diameter, crYOLO picked 52,153 particles that were used to search a range of diameters during 2D classification (Fig. 4A & B). For each diameter used in RELION 2D classification, 2DAssess was used to estimate the number of good particles. Finally, comparison across all diameters used in 2D classification indicated that the best diameter for T20S was 195 Å (Fig. 4B). For the 195 Å diameter, the good 2D class averages selected by 2DAssess had a 100% prediction accuracy (Fig. 4C), correctly identifying all good and bad 2D averages.
Figure 4. High-resolution cryo-EM structure of T20S proteasome from automatic preprocessing pipeline.
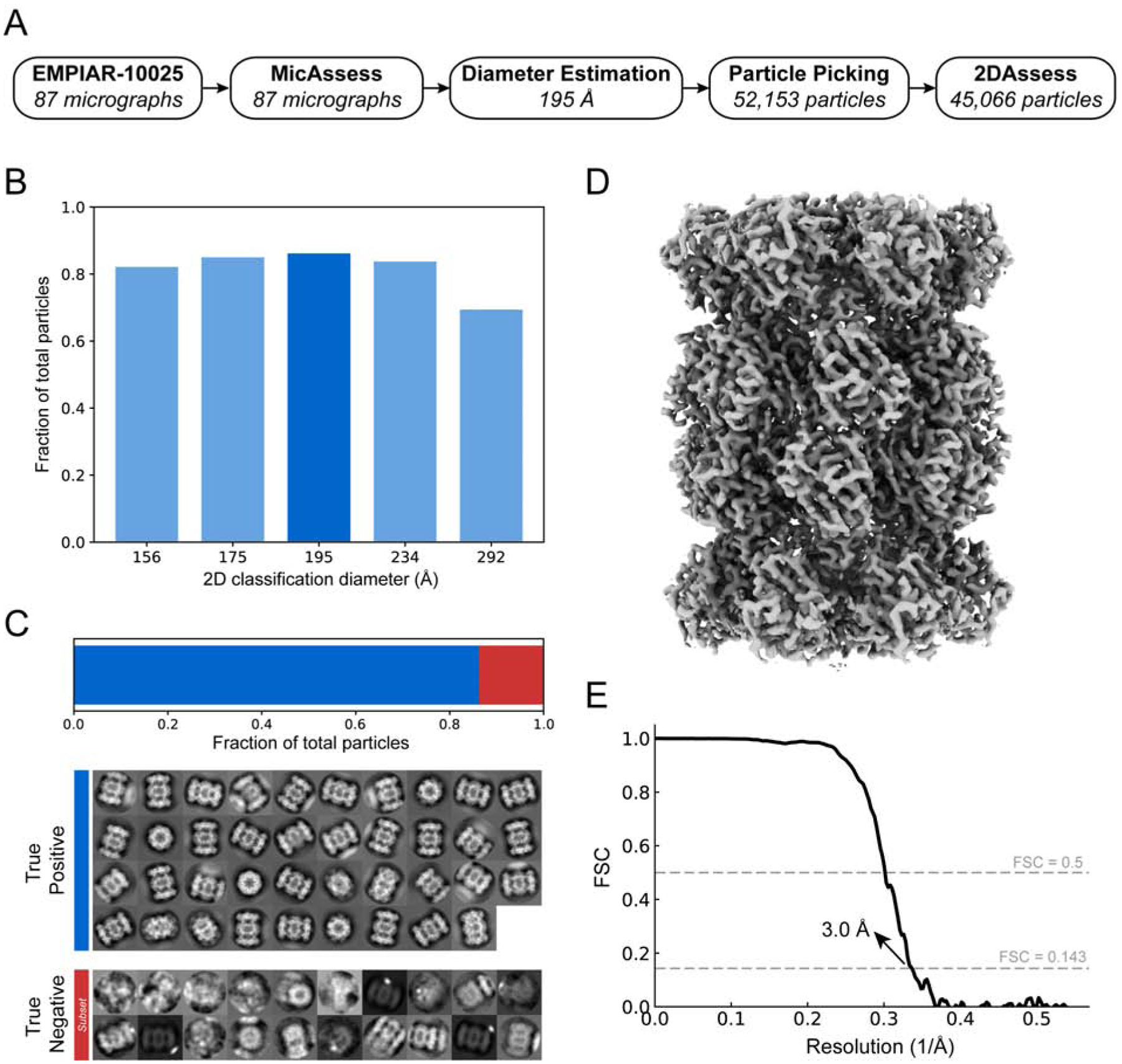
(A) Overview of the intermediate results of automatic pipeline on EMPIAR-10025 dataset. (B) Histogram showing the fractions of the good particles identified by the pipeline with different diameter used in 2D classification. The diameter with the most good particles (195 Å) is selected (darker blue) to be the best diameter, and the corresponding 2D classification result is used to output the final particle stack. (C) 2DAssess achieves 100% prediction accuracy on the EMPIAR-10025 dataset. All the good 2D averages (86.4% of the picked particles) and a subset of the bad 2D averages predicted by 2DAssess are shown. (D) 3D electron density volume using the particle stack outputted by the pipeline as the input for 3D reconstruction steps. (E) FSC curve of the electron density map in panel C, showing a resolution of 3.1 Å.
Using the stack of particles associated with good averages, we then performed 3D refinement to obtain a 3.0 Å structure of the T20S proteasome (Fig. 4D & E). The resolution is slightly lower than the original paper (Campbell et al., 2015) because we used only a small subset of the EMPIAR dataset, and the results were obtained without extensive classification or CTF refinement. This structure demonstrates that the automatic preprocessing pipeline provided a high-resolution stack of particles of T20S without user intervention.
Hemagglutinin (HA) trimer (EMPIAR-10175)
After successfully analyzing T20S, we next wanted to try a more challenging sample. To this end, we selected the influenza hemagglutinin trimer (HA trimer) dataset (EMPIAR-10175) (Noble et al., 2018) due to its extreme orientation differences: end-on views have a diameter of 55Å whereas the side-on views have a diameter of 140Å. After running MicAssess on 1,099 micrographs, MicAssess identified 205 micrographs as bad (examples are shown in Fig. S5), and the rest of 894 micrographs were preprocessed by the downstream pipeline. After 2D classification, the best diameter to be used in 2D classification was selected to be 150 Å (Fig. 5A & B). The good and bad class averages were all correctly classified by 2DAssess (Fig. 5C).
Figure 5. High-resolution cryo-EM structure of HA trimer from automatic preprocessing pipeline.
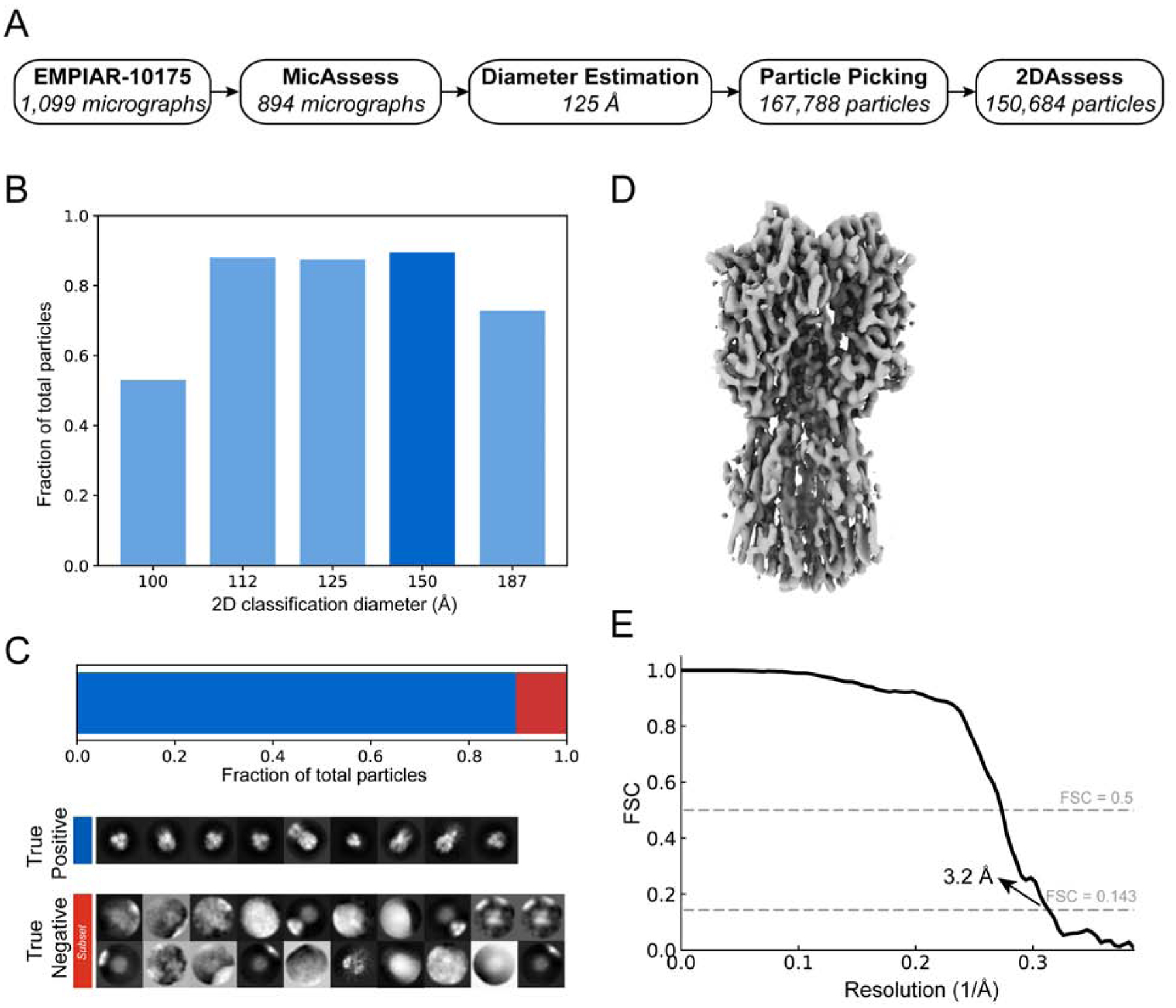
(A) Overview of the intermediate results of automatic pipeline on EMPIAR-10175 dataset. (B) Histogram showing the fractions of the good particles identified by the pipeline with different diameter used in 2D classification. The diameter with the most good particles (150 Å) is selected (darker blue) to be the best diameter, and the corresponding 2D classification result is used to output the final particle stack. (C) 2DAssess achieves 100% prediction accuracy on the EMPIAR-10175 dataset. All the good 2D averages (89.8% of the picked particles) and a subset of the bad 2D averages predicted by 2DAssess are shown. (D) 3D electron density volume using the particle stack outputted by the pipeline as the input for 3D reconstruction steps. (E) FSC curve of the electron density map in panel C, showing a resolution of 3.2 Å.
Using the output stack of good particles, we performed a 3D refinement with the selected 150,684 particles. This allowed us to determine a structure at 3.2 Å resolution (Fig. 5D & E), comparable to what was published previously for HA trimer (Noble et al., 2018). This structure confirmed that the automatic pipeline is capable of handling datasets of varying size and shape, setting the stage for real-world data analysis.
Analysis of real-world data
Aldolase
To extend our preprocessing pipeline, we analyzed an in-house collected aldolase dataset. This dataset contains 1,118 micrographs, in which 1,075 micrographs were predicted as good by MicAssess. The examples of bad micrographs being selected by MicAssess are shown in Fig. S6. After estimating the particle diameter, the 2D classification showed an optimal mask diameter of 108 Å (Fig. 6A & B). 2DAssess correctly predicted all the good class averages. In this dataset, there were two falsely identified good averages that were actually bad, which only accounted for 1.53% of the total particles (Fig. 6C).
Figure 6. High-resolution cryo-EM structure of aldolase from automatic preprocessing pipeline.
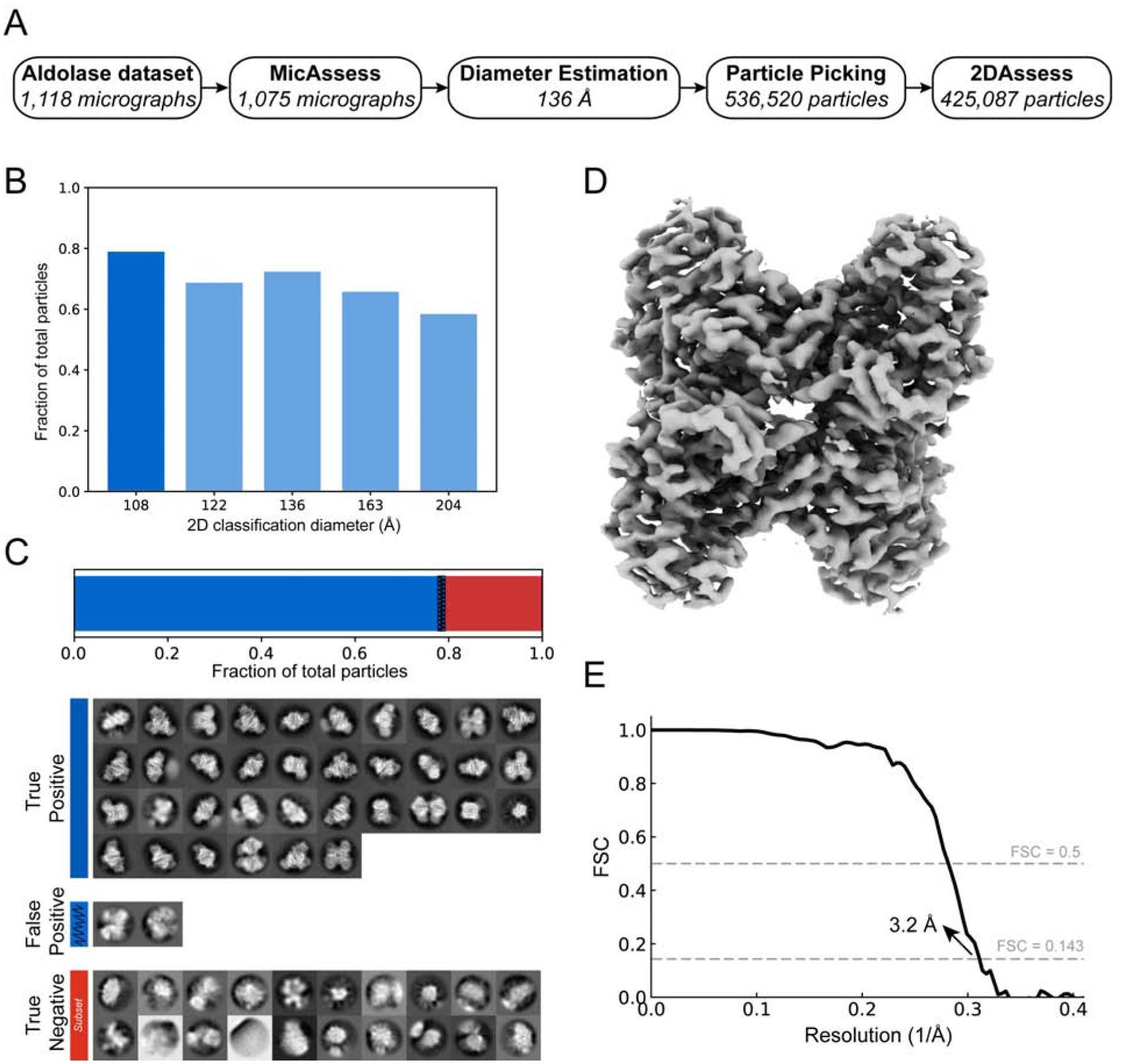
(A) Overview of the intermediate results of automatic pipeline on the aldolase dataset. (B) Histogram showing the fractions of the good particles identified by the pipeline with different diameter used in 2D classification. The diameter with the most good particles (108 Å) is selected (darker blue) to be the best diameter, and the corresponding 2D classification result is used to output the final particle stack. (C) 2DAssess achieves very high prediction accuracy on the aldolase dataset. All the good 2D averages (79.2% of the picked particles) and a subset of the bad 2D averages predicted by 2DAssess are shown. The two false positives (blue shaded) only account for 1.53% of the total picked particles. (D) 3D electron density volume using the particle stack outputted by the pipeline (including the false positives) as the input for 3D reconstruction steps. (E) FSC curve of the electron density map in panel C, showing a resolution of 3.2 Å.
Using the particle stack generated by the pipeline (including all of the false positives), we performed a 3D refinement to obtain a final structure of aldolase at 3.2 Å (Fig. 6D & E). This demonstrates that the preprocessing pipeline successfully handles more realistic data, as expert users also determine a structure to the same resolution.
P-Rex1 - a sample screening case study for high-throughput cryo-EM
Finally, in order to demonstrate the effectiveness of the pipeline, we automatically analyzed multiple datasets to simulate a sample screening experiment. The datasets we used were collected from six cryo-EM sessions of P-Rex1 under different conditions (Fig. 7A), including apo P-Rex1 on different types of grids (18sep06b and 18sep28b), with different additives (18jan09b and 18jan09d), and with a binding partner Gβγ at different concentrations (18jul14a and 18jan18c). The goal of this sample screening case study is to verify that our pipeline provides a robust and user-free approach for automatic data quality assessment at different levels considering that only one dataset (18jan18c) is amenable for highresolution cryo-EM (Cash et al., 2019).
Figure 7. Automatic analysis of multiple P-Rex1 cryo-EM data sets to assess sample quality.

(A) The six datasets analyzed by the automatic pipeline in this case study, including different sample preparations, different additives and whether a binding partner was added. 2D class averages were predicted by 2DAssess and the five good and representative 2D class averages for each dataset are shown for assessment. (B) Fractions of the good micrographs in all the micrographs (orange) and fractions of the good particles outputted by the automatic pipeline in all the picked particles (purple) for each dataset. (C) The numbers of picked particles (brown) and the numbers of good particles outputted by the automatic pipeline (blue) for each dataset.
All six datasets were analyzed with the pre-defined automatic pipeline, where no user input was required other than microscope settings. The outputs of the automatic pipeline were the 2D class averages selected by 2DAssess for each dataset (Fig. 7A & Fig. S7). The datasets were assessed at different levels, from the micrographs to the 2D class averages, throughout the pipeline. At the first step, MicAssess quickly captured that one of the datasets, 18sep28b, contained mostly bad micrographs (70%) (Fig. 7B). All the other five datasets contained mostly good (above 50%) micrographs (Fig. 7B). The particle picker picked 170–350 particles per micrograph for all five datasets, except 18sep28b, which only had an average of 85 picked particles per micrograph, confirming the bad quality of this dataset (Fig. 7C). After 2D classification, the class averages were classified by 2DAssess, where we found that four datasets have over 50% of the picked particles to be good particles outputted by the automatic pipeline (Fig. 7B), and there were 100~200 good particles per micrograph (Fig. 7C).
Although many of the datasets showed promising statistics of good micrograph and good particle fractions, the good 2D class averages selected by 2DAssess revealed that apo P-Rex1 alone and with additives had a very strong preferred orientation on the cryo-EM grids (Fig. 7A). On the other hand, one of the datasets of P-Rex1 with Gβγ (18jul14a) exhibited a sample heterogeneity, where we found Gβγ oligomers in the good 2D class averages (Fig. 7A), indicating that the concentration of Gβγ added was too high. Finally, the 2D class averages output by the automatic pipeline from the last dataset (18jan18c) showed the P-Rex1 and Gβγ interactions, and new orientations were also seen as a result (Fig. 7A). This case study demonstrated that our automatic preprocessing pipeline is an objective, fully automatic approach for sample screening for high-throughput cryo-EM.
DISCUSSION
Cryo-EM is on the verge of becoming a high-throughput technique due to the ability of collecting multiple datasets per microscope session. This new era requires consistent and reproducible methods to assess and preprocess the micrographs directly from the microscopes in a timely manner. Our workflow provides a robust way to assess and preprocess cryo-EM data automatically without any user intervention, and it takes advantage of pre-existing software and preprocessing algorithms. We maintained the flexibility to incorporate any preprocessing algorithms, as long as no subjective user decisions are required. Notably, our workflow also worked on a dataset which used deliberate crowding as a strategy to achieve thinner ice, as shown in our test on EMPIAR-10181 (Herzik et al., 2017) (Fig. S8). While the good results might be expected for the highly curated EMPIAR datasets, our workflow performed equally well on our in-house datasets of aldolase and P-Rex1 screening, indicating that the workflow is likely robust for a variety of sample types. To our knowledge, this is the first fully automatic and generic workflow for cryo-EM data preprocessing.
Instead of competing with state-of-the-art software packages, our workflow uses the deep-learning-based assessment tools we developed and provides a platform to streamline all the preprocessing steps. For example, Warp (Tegunov and Cramer 2019) is a user-friendly preprocessing software which enables the users to interact directly with their data. However, manual inspections and user decisions are still needed in the whole preprocessing with Warp. The assessment tools introduced in this paper can be used in the Warp workflow to help with automatic data preprocessing.
As the initial step in our workflow, it is important that MicAssess can efficiently identify most bad micrographs, but keeping ideally all the good micrographs. Therefore, MicAssess was tuned to tolerate more false positives, reducing the risk of a good micrograph being misclassified. The P-Rex1 benchmark result showed that it can effectively identify most of the bad micrographs from a big real-world dataset. Furthermore, MicAssess also has the potential to be incorporated into the data acquisition step. With the new K3 camera, which can collect as fast as 8,000 movies per day, it is impossible to manually assess the quality of the newly collected micrographs. MicAssess provides a way to assess these micrographs on the fly even before CTF estimation so that the user can get real-time feedback on the qualities of the micrographs.
In our workflow, we only used 2DAssess to predict whether a class average is good or bad, but it can predict four different classes (clip, edge, good, and noise), which contains a lot more information. For example, a large percentage of particles being classified as “clip” usually indicates the mask diameter is too large because neighboring particles are being included in some 2D class averages. This gives the 2DAssess the potential to improve 2D classification by performing automatic diameter searching. Specifically, since most 2D classification algorithms are iterative, intermediate 2D class averages are generated after each iteration. It is possible to apply 2DAssess on the 2D class averages in the early iterations and use the outputted predictions to guide the automatic diameter searching.
Given that MicAssess and 2DAssess are deep-learning based models, both models will continue to improve with more representative training data. Moreover, as deep learning models, these tools can be tuned for specific samples, users, or facilities to aid in sample assessment. Sample tuning could be extended into other parts of the pipeline, including particle picking and, likely, 3D analysis. Further work in this area stands to help streamline initial phases of cryo-EM data processing.
An important aspect of our pipeline centers on creating a workflow that does not depend on user-defined thresholds. These thresholds are typically CTF maximum resolution and particle picking thresholds, but could also apply to how 2D class averages are selected. By developing statistical tools to assess the data, we developed tools that more closely mirror user-based decisions, instead of fixed-value thresholds.
While this pipeline provides an important first step for automated pre-processing, there remains room for improvement. Namely, we continued to use 2D classification as a tool in order to measure particle quality, where belonging to ‘good’ class averages was a criteria for subsequent 3D analysis. Moreover, 2D classification is the bottleneck of the speed of this pipeline, where about 99% of the CPU core hours were spent in the 2D classification step. Future work into particle sorting stands to provide a quick readout of particle quality to enable faster pre-processing routines.
Overall, this work demonstrates that user-free preprocessing is capable to perform in a manner comparable to that of an expert. Future work may extend into automated 3D analysis to enable cryo-EM users to quickly analyze multiple datasets in parallel.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Michael A Cianfrocco (mcianfro@umich.edu).
This study did not generate new unique reagents.
METHOD DETAILS
MicAssess
Motion corrected micrographs in MRC format were low-pass filtered and cropped to downscale to the network input image size of 494×494. Micrographs were then normalized to a mean of zero. A circular mask with diameter 494 pixels was applied to each micrograph, and then rotations and flipping were applied randomly in the training and validation dataset. The model was a 34-layer ResNet connected to two fully connected layers with leaky ReLU as the activation function and 0.5 dropout rate. The final predicting layer used a sigmoid function as the activation function. The loss function used was the binary cross-entropy loss. We used the ADAM optimizer with 0.0001 learning rate in training for optimization. In the real prediction, in order to tolerate more false positives than false negatives, we set the threshold as 0.1 (i.e. only micrographs with probabilities of being good lower than 0.1 will be classified as bad).
MicAssess was written in Python and employed Keras with Tensorflow as the backend. It has been optimized for GPU, but it can be run on CPU-only machines as well, and is compatible with all platforms (Linux, Windows, and macOS). It currently supports data from both K2 and K3 cameras.
CTF estimation
CTF estimation is performed using CTFFIND4 (Rohou and Grigorieff, 2015), with all the parameters, including pixel size, spherical aberration, magnification, and voltage, are related to the experiment given earlier.
2D classification
Picked particles were scaled to about 3 Ångstrom/pixel and extracted using RELION3 (Zivanov et al., 2018). After that, all the particles will be processing with 2D classification in RELION3. The workflow uses the maximum class number, 200, for the best performance in the sacrifice of speed. Multiple 2D classification jobs for one dataset will be submitted, with different diameters of the mask, ranging from 0.5 to 2 times the particle size estimated earlier.
2DAssess
Training and validation data consist of the RELION (Zivanov et al., 2018) outputs of 2D classification from 12 different datasets (Table 1). The EMPIAR datasets were preprocessed by the pipeline, and the outputted 2D class averages were manually labeled to the correct classes. Classes with significantly more samples were downsampled to eliminate the possible problems caused by class imbalance. The final dataset has 527, 550, 898, 1002 images for good, clip, edge, and noise classes respectively, and was randomly split into a training set (80 %) and a validation set (20 %).
Given that the output averaged images from RELION (Zivanov et al., 2018) already contained a mask with diameter d, we cropped all average images to remove mask edges. To do this, we first cropped the images to size dxd which only keep the centers of the images. Images were then normalized to a mean of zero, and resized to 256×256 using Lanczos resampling. Random rotations and flipping were applied in the training and validation dataset.
We used a simple DCGAN (Radford et al., 2015) model to artificially generate images that belong to the good class as a data augmentation approach. The training data used for DCGAN is the 527 images in the good class. The generator of DCGAN was a convolutional neural network implementing upsampling convolutions, organized as input (100-d) -> transpose conv3×3 1024-d, stride 2, batch normalization, ReLU activation -> transpose conv1×1 1024-d, stride 1, batch normalization, ReLU activation -> transpose conv3×3 512-d, stride 2, batch normalization, ReLU activation -> transpose conv1×1 512-d, stride 1, batch normalization, ReLU activation -> transpose conv3×3 256-d, stride 2, batch normalization, ReLU activation -> transpose conv3×3 256-d, stride 2, batch normalization, ReLU activation -> transpose conv3×3 1-d, stride 1, tanh activation -> generated image. The discriminator of DCGAN was a simple convolutional neural network, organized as input -> conv3×3 32-d, stride 2, batch normalization, leaky ReLU activation, dropout rate 0.25 -> conv3×3 64-d, stride 2, batch normalization, leaky ReLU activation, dropout rate 0.25 -> conv3×3 128-d, stride 2, batch normalization, leaky ReLU activation, dropout rate 0.5 -> conv3×3 128-d, stride 2, batch normalization, leaky ReLU activation, dropout rate 0.5 -> fully connected layer with a single output with sigmoid activation. 10,000 epochs were used in training and only the images generated from the last 2,000 were saved. We then carefully selected 66 images and added them to the training set. All the selected images generated by DCGAN are shown in Fig. S4.
The CNN-based classifier failed to correctly classify class averages containing two particles, which is a situation that occurs when the 2D classification mask is too large. Therefore, we confirmed that all images predicted to be in the good class did not have two particles by calculating a saliency map of the 2D class averages. A saliency map is a representation of an image that can highlight the unique features of the image. In our application, we calculated the saliency map with the spectral residual approach and based on the object detected by the saliency map, we checked 1) the number of the object, and 2) whether the center of mass of the detected object is around the center of the image. Only the 2D class averages with one centered object detected will pass this saliency map check. The other class averages, with either more than one object or the object, are typically not well centered (usually due to the case that there are more than one particle but the particles are too close to be differentiated by the saliency map), will be moved to the correct clip class.
2DAssess was written in Python and employed Keras with Tensorflow as the backend. It has been optimized for GPU, but it can be run on CPU-only machines as well, and is compatible with all platforms (Linux, Windows, and macOS).
The number of the good particles that belong to the good 2D class average groups are calculated across all the diameters used in the 2D classification jobs, and the diameter with the best particles is being selected as the best diameter.
T20S single-particle analysis
3D refinement.
After the preprocessing pipeline, 45,066 particles were re-extracted to a pixel size of 0.88 Å/pixel with a box size of 390 Å. Using EMD-6287 as an initial model, we performed a 3D refinement in RELION-v3.0 (Zivanov et al., 2018) using D7 symmetry to obtain a structure at 3.0 Å resolution and B-factor of −94 Å2.
HA Trimer single-particle analysis
3D refinement.
After the preprocessing pipeline, 150,684 particles were re-extracted to a pixel size of 1.275Å/pixel with a box size of 250Å. Using EMD-7792 as an initial model, we performed homogenous 3D refinement in cryoSPARC v2.11.2-live_privatebeta using C3 symmetry to obtain a structure at 3.2 Å resolution and a B-factor of −151 Å2.
Aldolase single-particle analysis
Sample preparation.
Pure aldolase isolated from rabbit muscle was purchased as a lyophilized powder (Sigma Aldrich) and solubilized in 20 mM HEPES (pH 7.5), 50 mM NaCl at 1.6 mg/ml. Sample as dispensed on freshly plasma cleaned UltrAuFoil R1.2/1.3 300-mesh grids (Electron Microscopy Services) and applied to grid in the chamber of a Vitrobot (Thermo Fisher) at ~95% relative humidity, 4°C. The sample was blotted for 4 seconds with Whatman No. #1 filter paper immediately prior to plunge freezing in liquid ethane cooled by liquid nitrogen.
Cryo-EM data acquisition.
Data were acquired using the Leginon automated data-acquisition program (Suloway et al., 2005). Image preprocessing (frame alignment with MotionCor2 (Zheng et al., 2017) and CTF estimation) were done using the Appion processing environment (Lander et al., 2009) for real-time feedback during data collection. Images were collected on a Talos Arctica transmission electron microscope (Thermo Fisher) operating at 200 keV with a gun lens of 6, a spot size of 6, 70 μm C2 aperture and 100 μm objective aperture. Movies were collected using a K2 direct electron detector (Gatan Inc.) operating in counting mode at 45,000x corresponding to a physical pixel size of 0.91 Å/pixel. The dose rate was 4.413 e/pix/sec for a 10 second exposure, which makes for a total dose of 44.13 e/Å2 for the 1118 images collected at a defocus range of 0.8–2 μm.
3D refinement.
After the preprocessing pipeline, 425,087 particles were re-extracted to a pixel size of 1.22 Å/pixel with a box size of 271 Å. Using EMD-8743 as an initial model, we performed a 3D refinement in RELION-v3.0 (Zivanov et al., 2018) using D2 symmetry to obtain a structure at 3.2 Å resolution and B-factor of −110 Å2.
P-Rex1 screening single-particle analysis
P-Rex1 samples were prepared as described (Cash et al., 2019) with the exception of details described in Table 2.
Table 2.
Details of the automatic assessment of multiple P-Rex1 cryo-EM data sets.
| 18sep06b | 18sep28b | 18jan09b | 18jan10d | 18jul14a | 18jan18c | |
|---|---|---|---|---|---|---|
| P-Rex1 concentration (μM) | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 |
| Additive (μM) | - | - | DDM (80) | Lubrol (40) | DDM (80) | DDM (80) |
| Gβγ concentration (μM) | - | - | - | - | 60 | 6.0 |
| Grid type | Quantifoil 1.2/1.3 | Lacey carbon | Quantifoil 1.2/1.3 | Quantifoil 1.2/1.3 | Quantifoil 1.2/1.3 | Quantifoil 1.2/1.3 |
| Microscope | Titan Krios | Talos Arctica | Talos Arctica | Talos Arctica | Titan Krios | Titan Krios |
| Original Pixel Size (Å) | 1 | 0.91 | 0.91 | 0.91 | 1 | 1 |
| Number of total micrographs | 1,716 | 1,491 | 1,206 | 1,110 | 1,352 | 5,011 |
| Number of good micrographs # | 986 | 445 | 841 | 790 | 1,217 | 4,157 |
| Estimated diameter (Å) | 144 | 144 | 135 | 132 | 138 | 151 |
| Number of total picked particles | 178,483 | 37,946 | 177,086 | 205,682 | 424,213 | 921,403 |
| Number of good particles | 94,514 | 9,535 | 114,630 | 141,982 | 145,941 | 492,883 |
| Pixel size for 2D classification (Å) | 4 | 3.59 | 3.64 | 3.67 | 3.94 | 3.97 |
| Best diameter for 2D classification (Å) | 129 | 216 | 108 | 105 | 110 | 120 |
| Workflow CPU core hours (Intel Xeon E5–2660 v3) | 3945 | 4166 | 13712 | 7516 | 4347 | 1251 |
| Workflow GPU hours (NVIDIA GTX 1080 Ti) | ~2 | ~2 | ~2 | ~2 | ~2 | ~2 |
QUANTIFICATION AND STATISTICAL ANALYSIS
The cryo-EM single-particle analysis was performed with published software and tools developed in this study as described in Method Details section.
DATA AND CODE AVAILABILITY
Cryo-EM structures have been deposited to the EMDB under accession codes EMD-21491 (T20S), EMD-21490 (HA Trimer), and EMD-21492 (Aldolase). Aldolase dataset has been deposited to EMPIAR under EMPIAR-10379.
Software tools capable of running MicAssess and 2DAssess arebe available at https://github.com/cianfrocco-lab/Automatic-cryoEM-preprocessing under the MIT license. The preprocessing pipeline will also be incorporated into the freely available COSMIC2 science gateway: https://cosmic2.sdsc.edu:8443/gateway/.
Supplementary Material
Table 3.
Overview of cryo-EM structures.
| T20S Proteosome (EMPIAR-10025) | HA Trimer (EMPIAR-10175) | Aldolase | |
|---|---|---|---|
| Microscope | Titan Krios | Titan Krios | Talos Arctica |
| Detector | Gatan K2 | Gatan K2 | Gatan K2 |
| Voltage (kV) | 300 | 300 | 200 |
| Electron exposure (e−/Å2) | 53 | 73.24 | 44.13 |
| Defocus range (μm) | 0.9 – 2.4 | 1.0–2.1 | 0.8 – 2.0 |
| Original pixel size (Å) | 0.66 | 0.85 | 0.91 |
| Symmetry imposed | D7 | C3 | D2 |
| Initial particle images (no.) | 52,153 | 167,788 | 536,520 |
| Final pixel size (Å) | 0.88 | 1.275 | 1.22 |
| Final particle images (no.) | 45,066 | 150,684 | 425,087 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution (A) | 3.0 | 3.2 | 3.2 |
| B-factor (Å 2) | −94 | −151 | −110 |
| Workflow CPU core hours (Intel Xeon E5–2660 v3) | 1600 | 2353 | 8293 |
| Workflow GPU hours (NVIDIA GTX 1080 Ti) | ~2 | ~2 | ~2 |
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rabbit muscle aldolase | Sigma Aldrich | 89933139 |
| Deposited Data | ||
| Raw, electron micrographs, T20S proteasome | Campbell et al. 2015 | EMPIAR-10025 |
| Raw, electron micrographs, HA Trimer | Noble et al. 2018 | EMPIAR-10175 |
| Raw, electron micrographs aldolase | This paper | EMPIAR-10379 |
| Cryo-EM map of T20S proteasome (Figure 4D) | This paper | EMD-21491 |
| Cryo-EM map of HA Trimer (Figure 5D) | This paper | EMD-21490 |
| Cryo-EM map of aldolase (Figure 6D) | This paper | EMD-21492 |
| Software and Algorithms | ||
| Python | Anaconda | https://www.anaconda.com/distribution |
| Leginon | Suloway et al., 2005 | https://emg.nysbc.org/redmine/projects/leginon |
| Appion | Lander et al., 2009 | https://emg.nysbc.org/redmine/projects/appion |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| MotionCor2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-motioncor2 |
| RELION3 | Zivanov et al., 2018 | https://www3.mrc-lmb.cam.ac.uk/relion |
| UCSF Chimera | Goddard et al., 2007 | https://www.cgl.ucsf.edu/chimera |
| cryoSPARC | Punjani et al., 2017 | https://cryosparc.com |
| Tensorflow | Abadi et al. 2016 | https://www.tensorflow.org |
| Other | ||
| Vitrobot Mark IV | Thermo Fisher Scientific | |
Highlights.
Development of an automatic preprocessing workflow for cryo-EM
Removes subjective decision-making for micrograph and 2D average assessment
Allows users to obtain an automatically curated stack of high-resolution particles
Enables analysis of multiple datasets to allow high-throughput cryo-EM
Acknowledgments
We would like to thank members of the Cianfrocco Laboratory for discussions related to this work. We would also like to thank Dr. Min Su for his help in discussing tilted CTF analysis. This work is supported by NSF-DBI-ABI 1759826 (Y.L. & M.A.C.) and R01-CA-221289 (J.N.C., J.J.G.T, & M.A.C.). The research reported in this publication was supported by the NIH under award number S10OD020011.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests
The authors do not have any competing financial interests for work in this manuscript.
References
- Al-Azzawi A, Ouadou A, R HM, Tanner JJ, Cheng J. 2019. DeepCryoPicker: Fully Automated Deep Neural Network for Single Protein Particle Picking in cryo-EM. bioRxiv. doi: 10.1101/763839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SA, Albiez S, Bieri A, Syntychaki A, Adaixo R, McLeod RA, Goldie KN, Stahlberg H, Braun T. 2017. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. J Struct Biol 197:220–226. [DOI] [PubMed] [Google Scholar]
- Baldwin PR, Tan YZ, Eng ET, Rice WJ, Noble AJ, Negro CJ, Cianfrocco MA, Potter CS, Carragher B. 2018. Big data in cryoEM: automated collection, processing and accessibility of EM data. Curr Opin Microbiol 43:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T, Morin A, Rapp M, Brasch J, Shapiro L, Noble AJ, Berger B. 2019. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Methods. doi: 10.1038/s41592-019-0575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Veesler D, Cheng A, Potter CS, Carragher B. 2015. 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. Elife 4. doi: 10.7554/eLife.06380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash JN, Urata S, Li S, Ravala SK, Avramova LV, Shost MD, Silvio Gutkind J, Tesmer JJG, Cianfrocco MA. 2019. Cryo–electron microscopy structure and analysis of the P-Rex1–Gβγ signaling scaffold. Science Advances 5:eaax8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Eng ET, Alink L, Rice WJ, Jordan KD, Kim LY, Potter CS, Carragher B. 2018. High resolution single particle cryo-electron microscopy using beam-image shift. J Struct Biol 204:270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocco MA, Leschziner AE. 2015. Low cost, high performance processing of single particle cryo-electron microscopy data in the cloud. Elife 4. doi: 10.7554/eLife.06664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocco MA, Wong-Barnum M, Youn C, Wagner R, Leschziner A. 2017. COSMIC2: A Science Gateway for Cryo-Electron Microscopy Structure DeterminationProceedings of the Practice and Experience in Advanced Research Computing 2017 on Sustainability, Success and Impact, PEARC17. New York, NY, USA: ACM; pp. 22:1–22:5. [Google Scholar]
- Darrow MC, Moore JP, John Walker R, Doering K, King RS. 2019. Chameleon: Next Generation Sample Preparation for CryoEM based on Spotiton. Microsc Microanal 25:994–995. [Google Scholar]
- Fernandez-Leiro R, Scheres SHW. 2017. A pipeline approach to single-particle processing in RELION. Acta Crystallographica Section D: Structural Biology 73:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R, Scheres SHW. 2016. Unravelling biological macromolecules with cryo-electron microscopy. Nature 537:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Zhang X, Ren S, Sun J. 2016. Deep residual learning for image recognitionProceedings of the IEEE Conference on Computer Vision and Pattern Recognition. pp. 770–778.
- Herzik MA Jr, Wu M, Lander GC. 2017. Achieving better-than-3-Å resolution by single particle cryo-EM at 200 keV. Nat Methods 14:1075–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Zhang L. 2007. Saliency detection: A spectral residual approach2007 IEEE Conference on Computer Vision and Pattern Recognition. Ieee. pp. 1–8. [Google Scholar]
- Jain T, Sheehan P, Crum J, Carragher B, Potter CS. 2012. Spotiton: a prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J Struct Biol 179:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W 2014. Biochemistry. The resolution revolution. Science. [DOI] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau P-W, Lyumkis D, Potter CS, Carragher B. 2009. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CL, Chiu W. 2018. Comparing cryo-EM structures. J Struct Biol 204:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D 2019. Challenges and opportunities in cryo-EM single-particle analysis. J Biol Chem 294:5181–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen E, Bannon D, Kudo T, Graf W, Covert M, Van Valen D. 2019. Deep learning for cellular image analysis. Nat Methods 16:1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya T, Saur M, Stabrin M, Merino F, Voicu H, Huang Z, Penczek PA, Raunser S, Gatsogiannis C. 2017. High-resolution Single Particle Analysis from Electron Cryo-microscopy Images Using SPHIRE. J Vis Exp. doi: 10.3791/55448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NP, Ersoy I, Gotberg J, Bunyak F, White TA. 2019. DRPnet-Automated Particle Picking In Cryo-Electron Micrographs Using Deep Regression. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble AJ, Wei H, Dandey VP, Zhang Z, Tan YZ, Potter CS, Carragher B. 2018. Reducing effects of particle adsorption to the air-water interface in cryo-EM. Nat Methods 15:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14:290–296. [DOI] [PubMed] [Google Scholar]
- Radford A, Metz L, Chintala S. 2015. Unsupervised Representation Learning with Deep Convolutional Generative Adversarial Networks. arXiv [csLG]. [Google Scholar]
- Ravelli RBG, Nijpels FJT, Henderikx RJM, Weissenberger G, Thewessem S, Gijsbers A, Bart WAM, López-Iglesias C, Peters PJ. 2019. Automated cryo-EM sample preparation by pin-printing and jet vitrification. bioRxiv. doi: 10.1101/651208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N. 2015. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. 2005. Automated molecular microscopy: the new Leginon system. J Struct Biol 151:41–60. [DOI] [PubMed] [Google Scholar]
- Tegunov D, Cramer P. 2019. Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods. doi: 10.1038/s41592-019-0580-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D, Quentin D, Roderer D, Tacke S, Siebolds B, Schubert E, Shaikh TR, Lill P, Gatsogiannis C, Raunser S. 2019. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun Biol 2:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Gong H, Liu G, Li M, Yan C, Xia T, Li X, Zeng J. 2016. DeepPicker: A deep learning approach for fully automated particle picking in cryo-EM. J Struct Biol 195:325–336. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Yang G. 2017. A fast method for particle picking in cryo-electron micrographs based on fast R-CNN. AIP Conf Proc 1836:020080. [Google Scholar]
- Zhang J, Wang Z, Chen Y, Han R, Liu Z, Sun F, Zhang F. 2019. PIXER: an automated particle-selection method based on segmentation using a deep neural network. BMC Bioinformatics 20:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, Agard DA. 2017. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ouyang Q, Mao Y. 2017. A deep convolutional neural network approach to single-particle recognition in cryo-electron microscopy. BMC Bioinformatics 18:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH. 2018. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. doi: 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM structures have been deposited to the EMDB under accession codes EMD-21491 (T20S), EMD-21490 (HA Trimer), and EMD-21492 (Aldolase). Aldolase dataset has been deposited to EMPIAR under EMPIAR-10379.
Software tools capable of running MicAssess and 2DAssess arebe available at https://github.com/cianfrocco-lab/Automatic-cryoEM-preprocessing under the MIT license. The preprocessing pipeline will also be incorporated into the freely available COSMIC2 science gateway: https://cosmic2.sdsc.edu:8443/gateway/.