

Abstract
Identifying effective drugs to delay the progression of aortic aneurysms is a formidable challenge in vascular medicine. Methyltransferase-like 3 (METTL3) plays a key role in catalyzing the formation of N6-methyladenosine (m6A), but despite the functional importance of METTL3 and m6A in various fundamental biological processes, their roles in abdominal aortic aneurysm (AAA) are unknown. Here, we found that METTL3 knockdown in apolipoprotein E-deficient (ApoE−/−) mice treated with angiotensin II suppressed the formation of AAAs, while METTL3 overexpression exerted the opposite effects. Similar results were obtained in a calcium chloride (CaCl2)-induced mouse AAA model. Mechanistically, METTL3-dependent m6A methylation promoted primary microRNA-34a (miR-34a, pri-miR34a) maturation through DGCR8. Moreover, miR-34a overexpression significantly decreased SIRT1 expression and aggravated AAA formation, while miR-34a deficiency produced the opposite effects. In a rescue experiment, miR-34a knockdown or forced expression of SIRT1 partially attenuated the protective effects of METTL3 deficiency against AAA formation. Our studies reveal an important role for METTL3/m6A-mediated miR-34a maturation in AAA formation and provide a novel therapeutic target and diagnostic biomarker for AAA treatment.
Graphical Abstract
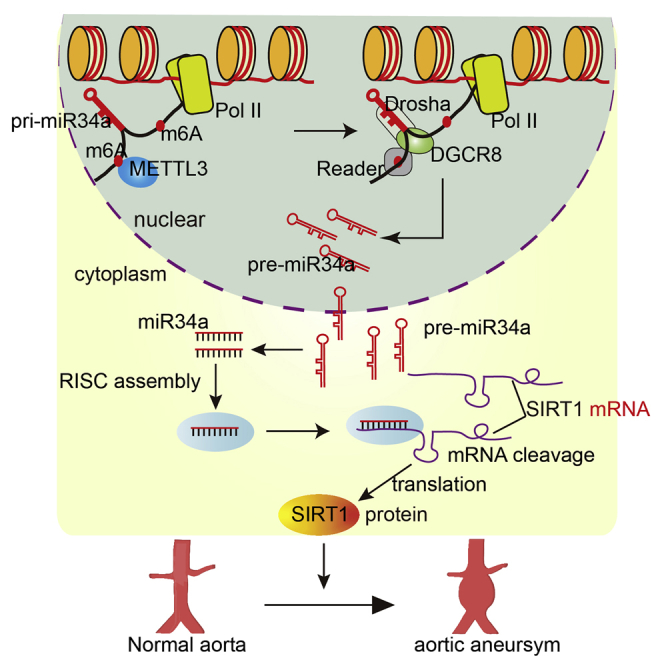
Despite the functional importance of METTL3 and m6A in various fundamental biological processes, their roles in abdominal aortic aneurysm (AAA) are unknown. Zhong et al. reveal an important role for METTL3/m6A-mediated miR-34a maturation in AAA formation and provide a novel therapeutic target and diagnostic biomarker for AAA treatment.
Introduction
Abdominal aortic aneurysm (AAA) is a devastating disease with an overall prevalence of 6% in men and 1.6% in women and a mortality rate exceeding 80% after rupture despite surgical advancements.1,2 Deferring the development of AAA is key to curbing the catastrophic consequences of this condition. In the clinic, lifestyle and metabolic improvements such as smoking cessation and control of high blood pressure and lipid and glucose levels are conventional measures to slow AAA development.2 Controlling these risk factors does yield some benefits; however, there has been limited progress in reducing AAA morbidity and rupture rates because this approach is not drastic enough to effectively slow AAA development and is difficult to implement successfully in the real world. Blocking the pathogenesis of AAA formation has been deemed a potential strategy for preventing or deferring AAA development and has gained attention in the endeavor to address AAA.
Current studies on the pathogenetic mechanism AAA indicate that AAA is induced by comprehensive aberrant expression of coding genes and noncoding genes.3, 4, 5 Previous studies seeking proper targets for AAA therapy have focused on controlling AAA progression by manipulating the expression of coding genes and noncoding genes via regulation of the content of mRNAs or ncRNAs in tissues or organs.4, 5, 6 Theoretically, the expression of genes is determined not only by the content of mRNAs or non-coding RNAs (ncRNAs) but also by posttranscriptional regulations; such regulations can lead to aberrant gene expression by regulating pre-mRNA maturation, mRNA stability, and mRNA translation in multiple pathophysiological processes. Posttranscriptional regulation-mediated maturation of pre-mRNA participates in vascular pathological processes including neointima formation after vascular injury,7 atherosclerosis, and hypertension. In addition, in some cases, posttranscriptional modification even determines whether genes can be expressed in different biological processes. Splicing factors mediate the posttranscriptional regulating of CaV1.2 pre-mRNA to produce multiple functionally distinct mature mRNAs, namely, alternative splicing transcripts, which participate in various biological processes8 such as atherosclerosis,9 hypertension,10 and cerebral arterial contraction.11 However, the role of posttranscriptional regulation in AAA development remains unknown.
N6-methyladenosine (m6A) methylation, an important RNA posttranscriptional regulatory mechanism, is regarded as the most common posttranscriptional modification for mRNA12 and as a general modification for ncRNA in eukaryotic organisms. m6A modification of RNA exists within disease pathophysiological processes, promoting pri-microRNA (miRNA) maturation13 and mRNA translation14 and enhancing RNA stability.15 Importantly, recent studies have also supported critical roles for m6A RNA modification in cardiovascular disorders and mechanisms such as cardiac hypertrophy16 and cardiomyocyte regeneration.17 Interestingly, our preliminary experiments revealed that RNA m6A methylation levels were upregulated in AAA samples compared to control samples and that this upregulation was induced by elevations in the expression of the m6A methyltransferase methyltransferase-like 3 (METTL3), indicating that m6A of RNA may participate in the progression of AAA. Recent studies have reported that METTL3 increased the expression of inflammatory cytokines and activated the nuclear factor κB (NF-κB) signaling pathway,18,19 which are implicated in AAA development. Moreover, METTL3 potentiates multiple pri-miRNA splicing to form mature miRNAs,13 which are regarded as crucial and upregulated in aortic aneurysm formation. Given these findings, we hypothesize that METTL3 facilitates pri-miRNA maturation by promoting m6A RNA modification, and thus aggravates vascular inflammation and AAA formation.
Results
m6A Modification and METTL3 Are Upregulated in AAA Tissues
To determine the potential role of m6A modification in AAA, we constructed an AAA model (Figure 1A) and examined the levels of m6A in the total RNA of angiotensin II (Ang II)-induced AAA tissues, CaCl2-induced AAA tissues and control aortic tissues. Using a colorimetric m6A quantification strategy, we found that there were more m6A modifications in AAA tissues than in control aortic tissues (Figure 1B). Then, we isolated poly(A)+ RNA in AAA and adjacent control tissues and examined the m6A modifications by RNA immunoblotting. In addition, upon further rRNA depletion, we found that there were more m6A modifications of rRNA-free poly(A)+ RNA in both AAA tissues than in control tissues (Figures 1C and 1D). Together, these results indicated that m6A modifications were upregulated in AAA. m6A modifications primarily depend on the balance between m6A methyltransferase and demethylase genes. Therefore, we investigated the expression levels of m6A methyltransferase and demethylase genes in AAA tissues and corresponding control aortic tissues. We found that the m6A methyltransferases METTL3 and METTL14 were significantly upregulated in AAAs (Figures 1E and 1F). To determine the localization of METTL3 in aortic tissues, we performed immunofluorescent staining for METTL3 and α-SMA. The results showed that METTL3 was mainly located in the nuclei of vascular smooth muscle cells (VSMCs) (Figures 1G and 1H).
Figure 1.

m6A Modification and METTL3 Expression Are Upregulated in AAA
(A) Representative photographs showing the macroscopic features of 4-week Ang II-induced AAAs in male ApoE−/− mice or 6-week CaCl2-induced AAAs in male C57BL/6J mice. (B) m6A modification of total RNA from AAA tissues and control aortic tissues (n = 5). (C) m6A modification of rRNA-free poly(A)+ RNA from AAA tissues and control aortic tissues as detected by m6A immunoblot analysis. (D) Equal amounts of RNA from AAA tissues and control aortic tissues were stained with ethidium bromide. (E) mRNA levels of METTL3, METTL14, and Wilms tumour 1-associating protein (WTAP) in saline- or Ang II-induced mouse AAA samples (n = 4). (F) Western blot analysis of monocyte chemoattractant protein 1 (MCP1), matrix metalloproteinase 2 (MMP2), P21, and METTL3 in saline- or Ang II-induced mouse AAA homogenates (n = 4). (G) Representative immunofluorescent staining of METTL3, smooth muscle 22α (SM22α, green), and 4’,6-diamidino-2-phenylindole (DAPI) in suprarenal aortas from saline- or Ang II-induced mouse AAAs (scale bars, 50 μm). (H) Number of accumulated METTL3-positive cells in the suprarenal aortic walls of saline- or Ang II-infused mice (n = 4). Three different visual fields were captured for each section. (I) Representative western blot analyses of MCP1, MMP2, P21, and METTL3 in human AAA and adjacent nonaneurysmal aortic samples. (J–M) Representative immunostaining (J) and densitometric analysis of METTL3 (K), SM22α (L), and α-SMA (M) in human AAA samples and adjacent control aortas (n = 4; scale bars, 50 μm). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
We further investigated the expression of METTL3 in human AAA tissues. Human AAA tissues and corresponding adjacent normal aortic sections were obtained from patients undergoing AAA resection surgery. In agreement with the above mouse results, we found that the protein expression of METTL3 was markedly higher in AAA tissues than in control tissues (Figure 1I). In addition, immunohistochemical staining consistently showed that the expression levels of METTL3 were substantially higher in human aortic smooth muscle cells (SMCs) than in adjacent control aortic SMCs (Figures 1J to 1M). Taken together, our data showed upregulated METTL3 expression in both human and animal AAA.
Reductions in METTL3 Suppress Ang II-Induced AAA Formation
To further determine whether there was a causative link between the reduction in METTL3 and AAA development, we used adeno-associated viruses (AAVs) carrying METTL3 small interfering RNA (siRNA; the sh-METTL3 group), overexpression plasmids (the AAV-METTL3 group) and control sequences (the scrambled RNA [Scr-RNA]) group, and the AAV-green fluorescent protein (GFP) group to perform gain- and loss-of-function experiments on Ang II-treated C57BL/6J or apolipoprotein E-deficient (ApoE−/−) mice. First, four METTL3 siRNA sequences and pcDNA3.1-METTL3 were separately and successfully transfected into cultured VSMCs. Analysis of the METTL3 mRNA levels in VSMCs showed that siRNA 1, siRNA 2, and pcDNA3.1-METTL3 were effective in inhibiting and promoting METTL3 expression (Figures S1A and S1B). Next, AAV9 vectors carrying METTL3 siRNA 1, overexpression plasmids, and control sequences were injected into the tail veins of ApoE−/− mice and C57BL/6J mice. Compared with that in the saline group, the GFP signal was significantly enhanced in the virus intervention groups, indicating that the virus successfully infected the aorta (Figures S2A and S2B). Moreover, from the 30th day to the 60th day, the sh-METTL3 group and the AAV-METTL3 group demonstrated significantly reduced and enhanced aortic METTL3 expression, respectively, compared with the corresponding control groups (Figures S2C and S2D; Figures 2A and 3A). Accordingly, on the 30th day, METTL3-deficient mice and METTL3-overexpressing mice were randomly selected to start receiving Ang II infusion for 4 weeks. After 2 weeks of Ang II treatment, the change in abdominal aortic diameter was monitored by ultrasound (Figure 2B). After 28 days of Ang II infusion, AAA formation was significantly reduced in the sh-METTL3 group (16 of 40 mice, 40%; 20 AAAs) compared with the Scr-RNA group (30 of 40 mice, 75%; 30 AAAs; Figures 2C and 2D). Compared with those in the Scr-RNA mice, the maximal abdominal aortic diameter and the elastin degradation score were remarkably reduced in Ang II-treated sh-METTL3 ApoE−/− mice (Figures 2E and 2F), although there were no noticeable effects on the systolic blood pressure of the mice (Figure S1E). In addition, knockdown of METTL3 markedly attenuated the levels of vascular macrophage infiltration (Figure 2E); the expression of MMP2 (Figures 2H and 2I; Figure 3A), MCP-1/CCL2 (Figure 2H; Figure S3B), and P21 (Figure 2H); and the activity of MMPs (Figure 2J) in Ang II-treated ApoE−/− mice.
Figure 2.

METTL3 Reduction Prevents Ang II-Induced AAA Formation
(A) Western blots showing the expression of METTL3 over time after sh-METTL3 transfection in the suprarenal aortas of ApoE−/− mice (n = 3). (B) Two-dimensional color-coded ultrasound imaging of AAAs after 14 days of Ang II treatment. (C) Representative photographs of the macroscopic features of AAAs in Ang II-infused ApoE−/− mice. (D) Statistical analysis of AAA incidence in Ang II-infused ApoE−/− mice. (E) Maximal aortic diameters in Ang II-infused ApoE−/− mice. (F) Representative elastin staining and elastin degradation scores in suprarenal aortas from Ang II-infused ApoE−/− mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). The data are presented as the medians and quartiles. ∗p < 0.05. (G) Representative immunostaining for galectin 3 (MAC2, scale bars, 200 and 50 μm) and corresponding densitometric analysis (n = 4). (H and I) Western blots (H) and densitometric analysis (I) of the protein expression of METTL3, P21, MMP2, and MCP1 in Ang II-induced male ApoE−/− mice (n = 4). (J) Representative in situ zymography photographs of MMP activity in Ang II-induced male ApoE−/− mice (scale bars, 50 μm; n = 4). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Figure 3.

METTL3 Overexpression Promotes Ang II-Infused AAA Formation
(A) Western blots showing the expression of METTL3 over time after AAV-METTL3 transfection in the suprarenal aortas of Ang II-treated C57BL/6J mice (n = 3). (B) Two-dimensional color-coded ultrasound imaging of AAAs after 14 days of Ang II treatment. (C) Representative photographs of the macroscopic features of AAAs in Ang II-infused C57BL/6J mice. (D) Statistical analysis of AAA incidence in Ang II-infused C57BL/6J mice. (E) Maximal aortic diameters in Ang II-infused C57BL/6J mice. (F) Representative elastin staining and elastin degradation scores in suprarenal aortas from Ang II-infused C57BL/6J mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). The data are presented as the medians and quartiles. ∗∗p < 0.01. (G) Representative immunostaining for MAC2 (scale bars, 200 and 50 μm) and corresponding densitometric analysis (n = 4). (H and I) Western blots (H) and densitometric analysis (I) of the protein expression of METTL3, P21, MMP2, and MCP1 in Ang II-induced male C57BL/6J mice (n = 4). (J) Representative in situ zymography photographs of MMP activity in Ang II-induced male C57BL/6J mice (scale bars, 50 μm; n = 4). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Moreover, a gain-of-function experiment was carried out in C57BL/6J mice. The dilation of the abdominal aorta was monitored by ultrasound on the 14th day of Ang II infusion (Figure 3B). On the 28th day of Ang II treatment, METTL3-overexpressing mice had significantly higher AAA incidence (18 of 40 mice, 45%; 15 AAAs) than control mice (5 of 40 mice, 12.5%; 5 AAAs; Figures 3C and 3D). Correspondingly, the maximal aortic diameter and the elastin degradation score were evidently increased in the AAV-METTL3 group compared with the AAV-GFP group (Figures 3E and 3F). Additionally, compared with those in Ang II-treated C57BL/6J mice, the levels of vascular macrophage infiltration (Figure 3G), MMP2 expression (Figures 3H and 3I; Figure S4A), MCP-1/CCL2 expression (Figures 3H and 3I; Figure S4B), P21 expression (Figures 3H and I), and MMP activity (Figure 3J) in Ang II-treated METTL3-overexpressing mice were increased, but there were no marked effects on systolic blood pressure (Figure S1F). Based on these findings, METTL3 deficiency ameliorates AAA formation and related pathological changes, while METTL3 overexpression elicits the opposite effects.
Disruption of METTL3 Inhibits Calcium-Chloride-Induced AAA Formation
Furthermore, the role of vascular METTL3 in AAA formation independent of Ang II was also determined in a CaCl2-induced AAA model. 6 weeks after CaCl2 treatment in the infrarenal aorta, METTL3-deficient mice demonstrated decreased maximal abdominal aortic diameters (Figures S5A and S5B). As expected, mouse aortic METTL3 protein expression was clearly downregulated in the sh-METTL3 group compared to the control group (Figure S5C). The elastin degradation scores were also lower in the sh-METTL3 group than in the control group (Figures S5D and S5E). Moreover, METTL3, MCP1, MMP2, and P21 expression (Figures S5H, S6A, and S6B) and macrophage infiltration (Figures S5F and S5G), as detected by immunohistochemical staining, were also significantly reduced in METTL3-deficient mice compared with control mice. In contrast, 3 weeks after CaCl2 treatment, METTL3-overexpressing mice displayed markedly higher maximal abdominal aortic diameters and elastin degradation scores than AAV-GFP mice (Figures S7A, S7B, S7D, and S7E); substantial increases in the expression of METTL3, MCP1, MMP2, and P21 (Figures 7C, 7H, S8A, and S8B) and in the levels of macrophage infiltration (Figures S7F and S7G) were also observed. These results indicate that knockdown of METTL3 exerts a protective effect against CaCl2-induced AAA formation.
Figure 7.

miR34a Promotes AAA by Inhibiting SIRT1
(A) Representative photographs of the macroscopic features of AAAs in Ang II-infused DMSO- or EX527-treated C57BL/6J mice after anti-miR34a virus infection. (B) Statistical analysis of AAA incidence in Ang II-infused DMSO- or EX527-treated C57BL/6J mice. (C) Maximal aortic diameters in the Ang II-infused C57BL/6J mice in the two groups. (D and F) Representative elastin staining (D) and elastin degradation scores (F) in suprarenal aortas from Ang II-infused C57BL/6J mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). (E and G) Representative immunostaining for MAC2 (E) (scale bars, 200 and 50 μm) and the corresponding densitometric analysis (G) (n = 4). (H–K) Representative immunostaining and densitometric analysis for MMP2 (H and I) and MCP1 (J and K) (scale bars, 200 and 50 μm; n = 4). (L) Relative mRNA expression of MMP2, MCP1, and P21 in Ang II-infused DMSO- or EX527-treated C57BL/6J mouse aortas. (n = 4). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
METTL3-Dependent m6A Methylation Regulates the Processing of miR34a
Previous studies have demonstrated that m6A modification can mark pri-miRNAs for processing in a manner dependent on METTL3/m6A/DGCR8 or METTL14/m6A/DGCR8, suggesting that alteration of METTL3/m6A or METTL14/m6A might lead to abnormal expression of miRNAs in many pathophysiological processes, including vascular disease. Given the key roles of miRNAs in AAA,20,21 we hypothesized that METTL3 might influence AAA formation by affecting AAA-related miRNAs through m6A-dependent pri-miRNA processing. We confirmed that METTL3 was required for the binding of pri-miRNAs to the microprocessor protein DGCR8 in VSMCs. As expected, the results of the immunoprecipitation assay showed that METTL3 coprecipitated with DGCR8 from VSMCs. In addition, ribonuclease treatment weakened the interaction, suggesting that this interaction might be partly mediated by RNA (Figure 4A). Then, we determined whether METTL3 alters the binding of methylated RNAs to DGCR8 in VSMCs. Consistently, we observed significant decreases in the amount of methylated RNA bound to DGCR8 in METTL3-knockdown cells compared to control cells (Figure 4B). As expected, RNA m6A dot blot analyses showed that METTL3 has a key role in catalyzing the formation of m6A (Figure 4C). These findings suggest that METTL3 might also manipulate pri-miRNA processing by regulating the recognition and binding of DGCR8 to pri-miRNAs in VSMCs. Furthermore, we found that many miRNAs were aberrantly expressed in AAA, and we sought to identify the pri-miRNAs modified by the m6A mark through m6A-seq of nuclear RNA. We collected and mined m6A-seq data for nuclear RNA and miRNA microarray data for AAA tissues from the Gene Expression Omnibus database of the National Center for Biotechnology Information (GEO: GSE60213, GSE51228, and GSE54943). We found that 10 miRNAs were significantly upregulated and 2 were significantly downregulated in AAA samples compared to control samples, and the pri-miRNA of all the differentially regulated miRNAs contained m6A tags. We then measured the miRNAs in METTL3-knockdown VSMCs and found that miR34a was significantly downregulated in METTL3-knockdown VSMCs (Figure 4D). Moreover, based on previous reports of the important function of miR-34a in promoting vascular senescence and age-associated proinflammatory secretory factor expression and the finding that miR-34a could inhibit SIRT1 in VSMCs, we inferred that METTL3 might participate in AAA formation via miR-34a.
Figure 4.

METTL3-Dependent m6A Methylation Regulates the Processing of miR34a
(A) Coimmunoprecipitation of the METTL3-interacting protein DGCR8 in VSMCs. An IgG antibody was used as a control for immunoprecipitation. (B) Immunoprecipitation of DGCR8, METTL3, and associated RNA from control VSMCs or METTL3-deficient VSMCs. (C) Poly(A)+ RNA isolated from METTL3-deficient or METTL3-overexpression VSMCs was used in RNA m6A dot blot analyses with an m6A antibody. (D and E) quantitative real-time PCR analysis to detect the expression of miR34a (D) and pri-miR34a (E) in sh-METTL3 cells, METTL3-overexpressing cells and corresponding control cells. (F) Immunoprecipitation of DGCR8-associated RNA from METTL3-knockdown VSMCs or control cells followed by quantitative real-time PCR analysis of pri-miR34a binding to DGCR8. Pri-let-7e was used as a positive control (n = 3). (G) Immunoprecipitation of m6A-modified RNA in METTL3-knockdown VSMCs or control cells followed by quantitative real-time PCR analysis of pri-let-7e (left) and m6A modification site1 of pri-miR34a (right) m6A modification levels. (H) Immunoprecipitation of m6A-modified RNA in METTL3-knockdown VSMCs or control cells followed by quantitative real-time PCR analysis of m6A modification site 2 (left) and m6A modification site 3 (right) of pri-miR34a m6A modification levels. (I and J) quantitative real-time PCR analysis of the expression of miR34a (I) and pri-miR34a (J) in human AAA tissues and control aortic tissues. The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Moreover, we examined the expression of miR34a and pri-miR34a in METTL3-knockdown or METTL3-overexpressing VSMCs. As expected, pri-miR34a levels were increased in METTL3-knockdown cells and decreased in METTL3-overexpressing cells. Compared with pri-miR34a, mature miR34a exhibited the opposite expression patterns in METTL3-knockdown and METTL3-overexpressing cells (Figures 4D and 4E). We also observed increased binding of pri-miR34a to DGCR8 in METTL3-overexpressing cells when we immunoprecipitated DGCR8 from these cells, and we measured pri-miR34a expression by qRT-PCR (Figure 4F). Moreover, when we immunoprecipitated m6A from RNA of control and METTL3-overexpressing cells, we found that METTL3 overexpression significantly increased the amount of pri-miR34a modified by m6A at three m6A sites (Figures 4G and 4H). Consistent with those of previous studies, our results showed that miR34a was upregulated in AAA. Together, these results indicated that m6A marks facilitate pri-miR34a recognition by DGCR8 and subsequent processing into mature miRNA and that METTL3 promotes AAA formation via miR34a.
Reductions in miR34a Suppress Ang II-Induced AAA Formation
We further explored the role of miR-34a in AAA development as a downstream target of METTL3. We treated miR-34a-deficient mice and miR-34a-overexpressing mice with AAV9 vectors carrying miR34a mimic (AAV-miR-34a), inhibitor (anti-miR-34a), or control sequences (AAV-GFP and scr-miR) by tail vein injection. As expected, the GFP signal was markedly increased in virus-treated mice compared with saline-treated mice, indicating that the virus successfully infected the aorta (Figure S10). In addition, compared with the corresponding control groups, significant reductions and enhancements in aortic miR-34a expression were observed in the anti-miR-34a group and the AAV-miR-34a group, respectively, from the 30th day to the 60th day (Figures 5A and 6A). Therefore, on the 30th day, miR-34a-deficient mice were randomly selected to start receiving 4 weeks of Ang II infusion. After Ang II infusion, the miR-34a-deficient mice exhibited a lower incidence of AAA development (29 of 40 mice, 72.5%; 29 AAAs) than the control mice (19 of 40 mice, 47.5%; 19 AAAs; Figures 5B and 5C). Consistently, the maximal abdominal aortic diameters, the elastin degradation scores and vascular macrophage infiltration were markedly reduced in Ang II-treated miR-34a-deficient ApoE−/− mice (Figures 5D to 5F). As expected, Ang II treatment increased systolic blood pressure in both groups, and there was no notable difference in systolic blood pressure between the two groups (Figure 5G). Moreover, miR-34a deficiency markedly inhibited SIRT1 expression, MMP2 expression, MCP-1/CCL2 expression, and P21 expression, and MMP activity (Figures 5G to 5J; Figures S11A and S11B). These results indicate that knockdown of miR-34a suppresses Ang II-induced AAA formation and related pathological changes.
Figure 5.

Reductions in miR34a Suppress Ang II-Induced AAA Formation
(A) miR34a levels after anti-miR34a virus infection of the suprarenal aortas of Ang II-infused ApoE−/− mice (n = 4). (B) Representative photographs of the macroscopic features of AAAs in Ang II-infused ApoE−/− mice. (C) Statistical analysis of AAA incidence in Ang II-infused ApoE−/− mice. (D) Maximal aortic diameters in Ang II-infused ApoE−/− mice. (E) Representative elastin staining and elastin degradation scores in suprarenal aortas from Ang II-infused ApoE−/− mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). The data are presented as the medians and quartiles. ∗p < 0.05. (F) Representative immunostaining for MAC2 (scale bars, 200 and 50 μm) and corresponding densitometric analysis (n = 4). (G) Systolic blood pressure at baseline, and at 7, 10, 14, 21, and 28 days after Ang II infusion (n = 10 in each group). (H and I) Western blots (H) and densitometric analysis (I) of the protein expression of MMP2, MCP1, P21, and SIRT1 in Ang II-infused male ApoE−/− mice (n = 4). (J) Representative in situ zymography photographs of MMP activity in Ang II-infused male ApoE−/− mice (scale bars, 50 μm; n = 4). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Figure 6.

Overexpression of miR34a Aggravates Ang II-Induced AAA Formation
(A) Quantitative real-time PCR analysis of the expression of miR34a after AAV-miR34a virus infection of the suprarenal aortas of Ang II-infused C57BL/6J mice (n = 3). (B) Representative photographs of the macroscopic features of AAAs in Ang II-infused C57BL/6J mice. (C) Statistical analysis of AAA incidence in Ang II-infused C57BL/6J mice. (D) Maximal aortic diameters in Ang II-infused C57BL/6J mice. (E) Representative elastin staining and elastin degradation scores in suprarenal aortas from Ang II-infused C57BL/6J mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). The data are presented as the medians and quartiles. ∗∗p < 0.01. (F) Representative immunostaining for MAC2 (scale bars, 200 and 50 μm) and the corresponding densitometric analysis (n = 4). (G) Systolic blood pressure at baseline and at 7, 10, 14, 21, and 28 days after Ang II infusion (n = 10 in each group). (H and I) Western blots (H) and densitometric analysis (I) of the protein expression of MMP2, MCP1, P21, and SIRT1 in Ang II-infused male C57BL/6J mice (n = 4). (J) Representative in situ zymography photographs of MMP activity in Ang II-infused male C57BL/6J mice (scale bars, 50 μm; n = 4). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Furthermore, a gain-of-function experiment was performed in C57BL/6J mice. We treated miR-34a-overexpressing mice and corresponding control mice with Ang II for 4 weeks. Ang II infusion for 4 weeks caused AAAs in 11.4% (4/35) of the C57BL/6J mice (Figures 6B and 6C); AAA development was promoted by forced expression of miR-34a (14 of 35 mice, 40%; 14 AAAs). Moreover, miR-34a-overexpressing mice had larger maximal abdominal aortic diameters, higher elastin degradation scores, and more vascular macrophage infiltration (Figures 6D to 6F) than control mice. Accordingly, we found that forced expression of miR-34a enhanced MMP2 expression, MCP-1/CCL2 expression, P21 expression, and MMP activity (Figures 6H to 6J; Figures S12A and S12B). These results indicate that miR34a exacerbates AAA formation and related pathological changes.
miR34a Promotes AAA by Inhibiting SIRT1
Next, we sought to determine the potential mechanism by which miR34a promotes AAA formation. Studies have shown that miR34a induces VSMC senescence through downregulation of SIRT1 and promotion of the expression of age-associated proinflammatory secretory factors22 and that miR-34a can inhibit SIRT1 in VSMC or vascular disease.23,24 Thus, we sought to determine whether miR34a promotes AAA formation by inhibiting SIRT1 expression. We carried out rescue experiments by using the SIRT1 antagonist EX527 1 day before the beginning of 28 days of Ang II treatment in C57BL/6J mice. We observed that compared with DMSO administration, EX527 administration attenuated the promoting effect of miR34a overexpression on AAA formation, aortic expansion, elastin degradation, and vascular macrophage infiltration (Figures 7A–7F). Correspondingly, lower expression of MMP2, MCP1, and P21 was observed in EX527-treated mice than in DMSO-treated mice (Figures 7H–7L). The above results indicate that miR34a exacerbates AAA formation by inhibiting SIRT1.
METTL3 Overexpression Promotes AAA via miR34a/SIRT1
To determine the essential role of miR34a/SIRT1 in METTL3 overexpression-mediated AAA formation, we carried out rescue experiments in METTL3-overexpressing mice. METTL3-overexpressing mice were separately treated with anti-miR or anti-miR34a followed by 28 days of Ang II infusion. After 28 days, we observed that compared with anti-miRNA administration, anti-miR34a administration attenuated the promoting effect of METTL3 overexpression on AAA formation, aortic expansion, elastin degradation, and vascular macrophage infiltration (Figures 8A–8F). Correspondingly, lower expression of MMP2, MCP1, and P21 was observed in anti-miR34a-treated mice than in anti-miRNA-treated mice (Figures 8H–8L). In addition, METTL3-overexpressing mice were separately treated with AAV-GFP or AAV-SIRT1 followed by 28 days of Ang II infusion. After 28 days, compared with AAV-GFP administration, AAV-SIRT1 administration also inhibited the promoting effect of METTL3 overexpression on AAA formation, aortic expansion, elastin degradation, and vascular macrophage infiltration (Figure S13A–S13G). SIRT1 overexpression inhibited the increased expression of MMP2, MCP1, and P21 caused by METTL3 overexpression (Figure S13H). As expected, our results showed that SIRT1 was substantially upregulated in METTL3 knockdown samples but downregulated in METTL3 overexpression samples. The higher expression of SIRT1 was found in human AAA tissues than in normal human aortic tissues. Taken together, our results indicate that miR34a/SIRT1 is involved in METTL3 overexpression-promoted AAA development.
Figure 8.

METTL3 Overexpression Promotes AAA via miR34a/SIRT1
(A) Representative photographs of the macroscopic features of AAAs in AAV-METTL3 transfected Ang II-infused C57BL/6J mice in the AAV-anti-NC group or the AAV-anti-miR34a group. (B) Statistical analysis of AAA incidence in AAV-METTL3 transfected Ang II-infused C57BL/6J mice. (C) Maximal aortic diameters in the AAV-METTL3-transfected Ang II-infused C57BL/6J mice in the two groups. (D and F) Representative elastin staining (D) and elastin degradation scores (F) in suprarenal aortas from AAV-METTL3 transfected Ang II-infused C57BL/6J mice. The magnified photographs were taken at the position where the most severe elastin degradation occurred (scale bars, 200 and 50 μm; magnified photographs). The data are presented as the medians and quartiles. ∗∗p < 0.01. (E and G) Representative immunostaining for MAC2 (E) (scale bars, 200 and 50 μm) and the corresponding densitometric analysis (G) (n = 3). (H–K) Representative immunostaining and densitometric analysis for MMP2 (H and I) and MCP1 (J and K) (scale bars, 200 and 50 μm; n = 3). (L) Relative mRNA expression of MMP2, MCP1, and P21 in AAV-METTL3 transfected Ang II-infused C57BL/6J mouse aortas (n = 3). The data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Discussion
In the present study, we revealed that METTL3-mediated pri-miRNA maturation is a potent instigator of AAA formation. We showed that the m6A levels of RNA were upregulated in AAA samples and that METTL3 was the main factor in the aberrant m6A modification in AAAs. Downregulation of METTL3 reduced vascular inflammation and AAA formation in two AAA models. Mechanistically, METTL3 promoted AAA formation by accelerating the pri-miR34a maturation process in a DGCR8-dependent manner and thus inhibiting the expression of SIRT1, the target gene of miR34a.
In our study, we fully demonstrated that the m6A methyltransferase METTL3, an important mediator of RNA posttranslational modification, promoted AAA formation in two accepted AAA models. We found that METTL3 deficiency significantly reduced Ang II-induced AAA formation in ApoE−/− mice with hyperlipidemia. Moreover, METTL3 overexpression substantially alleviated the progression of both Ang II- and CaCl2-induced AAA even in mice with nonhyperlipidemic background. Taken together, these results show that the role of METTL3 in AAA formation is independent of a specific pathological environment. Furthermore, we found that METTL3 knockdown inhibited aortic enlargement, elastin degradation, and inflammatory activation, which are major drivers of the progression of AAA.25,26 Lending support to our findings, METTL3 has previously been shown to contribute to the activation of inflammation in bladder cancer18 and the lipopolysaccharide inflammatory response.19 Notably, METTL3 deficiency or overexpression had no effect on blood pressure, indicating that the effect of METTL3 on Ang II-induced AAA incidence is independent of alterations in blood pressure. In addition, with the innovation of detection technology, m6A modification can easily be detected, which enables the RNA m6A modification to be used as a diagnostic biomarker of AAA. Compared to the strategy used in previous studies to estimate AAA progression, which involved assessment of alterations in the expression levels of certain genes, assessment of the levels of global RNA m6A modification has unique advantages. RNA m6A modification, which is controlled by multiple genes, tends to exhibit less heterogeneity among individuals than single gene expression levels. Taken together, these findings suggest that METTL3 might be a novel therapeutic target and diagnostic biomarker for AAA.
We further demonstrated that METTL3 aggravated AAA formation by participating in the posttranscriptional modification of pri-miRNAs, namely, by promoting the maturity of AAA-related miRNAs. In recent years, an increasing number of studies have demonstrated that miRNAs reduce target gene expression to play relevant roles in various pathophysiological processes, including AAA.27,28 Importantly, mature miRNAs, but not unprocessed miRNAs, are loaded onto the RNA-inducing silencing complex (RISC) to silence target genes in biological processes, strongly supporting our hypothesis that targeting RNA maturation might be an attractive therapeutic strategy AAA. In the current study, we demonstrated that the role of METTL3 in AAA formation relies on facilitation of miR34a maturation through m6A/DGCR8-dependent pri-miRNA processing. After mining m6A-seq data from nuclear RNA and miRNA microarray data from AAA, we found that ten upregulated miRNAs and two downregulated miRNAs contained m6A tags in their pri-miRNAs; thus, these miRNAs have the potential to be regulated by METTL3. Among these miRNAs, we found that mature miR-34a was the most downregulated miRNA in METTL3-knockdown mice and that overexpression of METTL3 promoted its expression. Correspondingly, pri-miR-34a accumulated upon METTL3 deficiency and was reduced in METTL3-overexpressing mice. These results suggest that METTL3 may promote the processing of mature miR34a in an m6A-dependent manner, as supported by several other experiments. First, we predicted m6A sites based on the sequence of pri-miR34a with Database:SRAMP (http://www.cuilab.cn/sramp).29 The methylated RNA immunoprecipitation and qRT-PCR results demonstrated that the m6A levels of pri-miR34a were increased in AAAs and METTL3-overexpressing VSMCs compared to control samples, while those in METTL3-deficient VSMCs were decreased. Moreover, the immunoprecipitation experiments confirmed that METTL3 knockdown significantly increased the expression of m6A-modified pri-miR34a, suggesting that formation of mature miRNAs was associated with DGCR8-dependent pri-miRNA processing. Immunoprecipitation of DGCR8 demonstrated decreased levels of pri-miR34a bound to DGCR8 in METTL3-deficient VSMCs compared to control VSMCs. These results demonstrated that METTL3 promoted AAA formation by targeting miR34a through m6A/DGCR8-dependent pri-miRNA processing, indicating that the maturity and content of RNA determines its biological function in AAA formation. Our results might indicate that compared with control of RNA content alone, dual manipulation of both RNA content and maturity may be a better intervention strategy. Although radical changes in RNA content through gene knockout can maximally inhibit the biological functions of an RNA during disease progression, complete knockout of certain genes, which is unlikely to be suitable for future clinical translation, often causes side effects due to compensatory overexpression of other genes.
We further demonstrated that miR34a promoted AAA formation by reducing the expression of SIRT1. Our results show that miR34a promotes AAA formation by exacerbating aortic inflammation and VSMC senescence, as supported by our findings that miR34a overexpression upregulated aortic expression of MMP2 and MCP-1 and increased macrophage infiltration, which are well-known factors affecting AAA development, and that miR34a repression exerted the opposite effects. Consistently, other studies have reported that miR34a induces the senescence of VSMCs and the expression of age-associated proinflammatory secretory factors, which are the main predisposing factors for AAA. Mechanistically, a luciferase activity assay verified that the target gene of miR34a was SIRT1, which has been reported to reduce AAA formation by inhibiting p21-dependent vascular cell senescence, proinflammatory molecule secretion, and vascular inflammation.5 Accordingly, the expression of SIRT1 was increased in miR34a-deficient mice and METTL3-deficient mice compared to control mice. More importantly, we verified that miR34a or SIRT1 is indispensable in the association of METTL3 overexpression with AAA formation through rescue experiments. We found that miR34a knockdown or SIRT1 overexpression attenuated METTL3 overexpression-exacerbated AAA formation, as well as related vascular pathophysiological changes, namely, elevations in the levels of aortic MMP-2 and proinflammatory molecules. Taken together, our findings reveal that the METTL3/miR-34a/SIRT1 pathway plays a critical role in AAA formation.
Although conditional gene knockout mice are perfect animal models when investigating disease molecular mechanisms, we chose to use AAV9-mediated METTL3 intervention in adult arteries. Our previous study and other studies have demonstrated that adult arteries,30 especially arterial SMCs, are amenable to transfection with AAV9. Second, our study concentrated on the METTL3/miR34a pathway in AAA. Nonetheless, as METTL3 has theoretical importance in regulating mRNAs and other ncRNAs containing m6A tags, whether there are other potential downstream targets of METTL3 should be explored. Although there may be other downstream targets of METTL3, we demonstrated that miR34a is indispensable in the association of METTL3 overexpression with AAA formation through rescue experiments.
In summary, our work revealed that METTL3/m6A participates in AAA formation by promoting the expression of mature miR34a and thus reducing SIRT1 expression. Our study highlights the functional importance of pri-miRNA maturation mediated by METTL3 in AAA progression. Our findings suggest the potential of METTL3 as a novel therapeutic target and diagnostic biomarker for AAA.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Human Aortic Samples
All protocols using human aortic samples were approved by the Research Ethics Committees of Zhongshan People’s Hospital, Sun Yat-sen Hospital, and Guangzhou General Hospital of Guangzhou Military Region. AAA samples were obtained from patients undergoing open surgical repair according to the protocols. Control samples were trimmed from adjacent nonaneurysmal aortic segments from the same patients. All human aortic samples were analyzed by western blotting and immunohistochemistry (IHC). All of the human procedures conformed to the principles outlined in the Declaration of Helsinki (clinical information on the patients is available in Table S1).
Experimental Animals
The Institutional Animal Care and Use Committee at the Southern Medical University approved all animal procedures. The protocols followed the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals. Only male mice were used in this study considering the low incidence of experimental AAA in female mice. Male C57BL/6J mice and male apolipoprotein E-deficient (ApoE−/−) mice on a C57BL/6J background were purchased from the Laboratory Animal Center of Southern Medical University. All mice were housed under pathogen-free conditions with water and a normal chow diet at a temperature of 22°C and a humidity of 60%–65% under a 12-h dark/light cycle (lights on at 08:00).
Ang II-Induced AAA Model
Ang II-induced AAA model was performed as previously described.31 Male wild-type mice aged 10 to 12 weeks and male ApoE−/− mice aged 12 to 16 weeks were used. Ang II (A9525, Sigma, St. Louis, MO, USA) or normal saline was administered subcutaneously via an implanted osmotic minipump (ALZET, Model 2004, DURECT Corporation, Cupertino, CA, USA) at a rate of 1 μg/kg/min for 28 days. Each minipump was inserted subcutaneously into an anesthetized mouse via a small incision in the dorsum of the neck.
CaCl2-Induced AAA Model
CaCl2-induced AAA model was performed as previously described.32 Male C57BL/6J mice were anesthetized with an intraperitoneal injection of pentobarbital (40 mg/kg). The abdominal aorta between the renal arteries and the bifurcation of the iliac arteries was disassociated from the surrounding structures. Video microscopy was used to assay the diameter of the aorta in triplicate. After the measurements were taken, a small piece of gauze dipped in 0.5 mol/L CaCl2 was spread perivascularly onto the aortic passage for 15 min. Control mice received substitute treatment with NaCl (0.9%)-soaked gauze for 15 min. Then, the aorta was rinsed with 0.9% sterile saline, and the incision was sutured. After 3 or 6 weeks, all the animals were sacrificed, and the aortas were harvested for further analysis.
Aneurysm Quantification
To confirm the existence of AAA, we euthanized mice and cut them open ventrally. A volume of 10 mL of phosphate-buffered saline (PBS) was injected into the left cardiac ventricle and exited through the severed right atrium. Under a dissecting microscope, the periadventitial tissue was scraped, and the aorta was photographed. The suprarenal aorta was identified as the passage below the last pair of intercostal arteries and above the right renal branch. To quantify AAA size, we assessed the maximum outer width of the abdominal aorta with Image-Pro Plus (IPP) software (Media Cybernetics, USA). To quantify aneurysm severity, we adopted the accepted human aneurysm definition, which required a more than 50% increase in the outer aortic diameter of treated mice compared with control mice. If mice died before being euthanized, necropsies were performed to confirm whether there were aortic ruptures. Aortic rupture was defined as the existence of blood clots in either the thoracic cavity or the retroperitoneal cavity. Prematurely deceased animals were used only for analysis of mortality and rupture rates and were excluded from the other analyses. The diameter of the abdominal aorta was measured as previously described. Judgment of AAAs was performed by an independent investigator blinded to the experimental processes. A second independent investigator matched the diameters to the different mouse treatments.
Ultrasonography for AAA
Upon completion of Ang II infusion, the surviving mice were anesthetized intraperitoneally with pentobarbital (40 mg/kg), and the aortas were evaluated by two-dimensional color-coded ultrasound imaging with a Sequoia ultrasound system with a linear array ultrasound transducer (15 L8-S; mechanical index, 0.17; frequency, 14 MHz; Siemens Medical Systems).
Histological Analyses
Mice were sacrificed, and whole aortas were perfused with normal saline, fixed with 4% paraformaldehyde at physiological pressure for 5 min and isolated. Then, the aortas were segmented to obtain suprarenal abdominal aortas (for Ang II-induced AAA models; from the ascending aorta to the suprarenal abdominal aortas) or infrarenal abdominal aortas (for CaCl2-induced AAA models; to the entrance of both iliac arteries). Aortic samples were harvested, fixed for 24 h, and embedded in paraffin. Cross-sections (5 μm each) at intervals of approximately 500 μm were prepared. At least 10 sections were analyzed per mouse. Immunostaining or elastin van Gieson staining was performed.
Cell Culture and Treatment
Mouse aortic VSMCs were purchased from Guangzhou Geneseed Biotech. Human aortic VSMCs were purchased from Cellbio Company (Shanghai, China). The VSMCs were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin and were kept at 37°C in a humidified atmosphere containing 5% CO2. Cells at passages 3 to 6 grown to 70%–80% confluence were used.
RNA Interference, Cell Transfection, and Injection of AAVs
Specific siRNAs against METTL3 and scrambled negative control siRNAs (NCs) were designed by GenePharma (Shanghai, China). The overexpression plasmid pEnter-METTL3 (GenBank: NM_019721.2) was designed by Vigene Biosciences (Jinan, Shandong, China; the sequences are available in the Supplemental Information). For ex vivo experiments, VSMCs were cultured for 24 h. Meanwhile, 50 nM siRNA, 50 nM NCs, or 4 μg of pEnter-METTL3 was randomly added to 250 μL of Opti-MEM (GIBCO BRL, Paisley, UK); 5 μL of Lipofectamine 3000 (L3000015, Invitrogen, USA) was then added, and the mixture was incubated at room temperature for 0.5 h. Then, the mixture was added to the cells, and the cells were incubated for 6 h before replacement of the medium with the same volume of DMEM and further incubation for 48 h. The cells were then subjected to analyses. For in vivo experiments, a METTL3 AAV was constructed and packaged by Vigene Biosciences. Specific siRNAs against METTL3 and the NCs were separately constructed into a vector, and an AAV9 harboring these sequences was generated by Vigene Biosciences (Jinan, China). AAV-SIRT1 virus comes from the research published in our group.33 Mice were infected with one of the above viruses (1 × 1011 vg) through tail vein injection. 30 days later, the mice were subjected to Ang II, CaCl2, or appropriate control treatments as previously indicated. (Information about sequence of specific siRNAs against METTL3 and miR34a is available in Table S2.)
Immunohistochemical Analysis
Immunohistochemical analysis was carried out as previously described.34 Mouse slides were deparaffinized, endogenous peroxidase activity was blocked with 3% (vol/vol) hydrogen peroxide, and nonspecific binding sites were blocked by preincubation with 10% bovine serum in PBS. The slides were then incubated at 4°C overnight with primary antibodies (in 1% bovine serum albumin [BSA]) and a biotinylated secondary antibody (in 1% BSA) before being incubated with a horseradish peroxidase (HRP)-labeled streptavidin solution. Next, the slides were dyed with diaminobenzidine and counterstained with hematoxylin. The primary antibodies were METTL3, monocyte chemoattractant protein 1 (MCP1)/CCL2, smooth muscle 22 α (SM22α), α-smooth muscle actin (SMA), matrix metalloproteinase 2 (MMP2), galectin 3 (MAC2), and rabbit immunoglobulin G (IgG). For negative controls from Ang II-treated male ApoE−/− mice, the primary antibody was omitted, and the slides were incubated with secondary antibody only. All negative control experiments showed nonsignificant staining. (Information about the antibodies is available in Table S3.)
Immunofluorescent Staining
Immunofluorescent staining was carried out as previously described.4 Mouse suprarenal abdominal aortas were subjected to immunofluorescent staining. Frozen slides of 5 μm sections were fixed with acetone, blocked with 1% bovine serum in PBS for 1 h, and incubated at 4°C overnight with diluted primary antibodies against METTL3 (diluted in AAPR67-100, PanEra). Then, the slides were incubated with Alexa Fluor-labeled secondary antibodies at 37°C for 45 min in the dark (at maximum excitation wavelengths of 488 nm [green] and 568 nm [red]). The slides were then rinsed with PBS, and images were obtained with a fluorescence inverted microscope (IX83, Olympus, USA). (Information about the antibodies is available in Table S4.)
Elastin Staining and Degradation
Suprarenal aortic samples were embedded in paraffin, cut, and then measured via Victoria blue van Gieson (VVG) staining with a commercial kit (GenMed, Shanghai). Fields viewed under 40× magnification were used to evaluate elastin degradation. Elastin preservation was graded according to a previously established standard: grade 1 indicated no elastin degradation and a well-organized elastin lamina; grade 2 indicated elastin degradation with interruptions or breaks in the lamina; grade 3 indicated elastin degradation with multiple interruptions or breaks in the lamina; and grade 4 indicated severe elastin fragmentation or loss or aortic rupture.
Western Blot Analysis
Western blots were performed as described previously.35 Aortic protein concentrations were determined using western blot analysis with the Bradford method. Samples containing identical amounts of protein were denatured, subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a 10% running gel, dissociated with 10% SDS-PAGE, and probed with primary antibodies for METTL3, monocyte chemotactic protein 1 (MCP1), MMP9, SIRT1, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), or β-actin (all diluted in AAPR67-100 [PanEra]) at 4°C overnight. Next, the membranes were washed and incubated with an HRP-conjugated anti-rabbit secondary antibody (1:5,000 dilution, DAKO) for 2 h. GAPDH or β-Actin was used as a negative control. Bands were detected using enhanced chemiluminescence (ECL Advance; #RPN2235, GE Healthcare Life Sciences). The signals were recorded using a ChemiDoc imaging system (Bio-Rad Laboratories) and analyzed with ImageJ analysis software (NIH, Bethesda, MD, USA). All experiments were performed in triplicate. (The antibodies are listed in the Table S5.)
MAC2, MCP1, and MMP2 Quantification
MAC2, MCP1, and MMP2 quantification was performed with IPP software based on the integrated optical density (IOD) value, the chroma sum and the positive staining area (brown). The mean IOD was defined as the IOD value divided by the area of the samples. Three to four low-power sections and 9–12 high-power sections were obtained for each sample; the latter were used for calculation of the mean IOD value.
Quantitative Real-Time PCR
Total RNA was isolated from ex vivo VSMCs or mouse aortic wall tissues. Homogenates were resuspended in TRIzol reagent (Invitrogen), and the total RNA was purified. Then, the total RNA was converted into cDNA by reverse transcriptase (Takara Biotechnology, Dalian, China) using primer sequences for the molecules of interest. Quantitative real-time PCR was performed with a SYBR Green RT-PCR Kit (Takara Biotechnology, Dalian, China) and a LightCycler 480 II system (Roche Diagnostics, Basel, Switzerland). Quantification of the relative mRNA levels of the target genes was performed using the 2-ΔΔCt method, with β-actin as a normalization reference. The mRNA levels are expressed as the fold increases over control values after normalization. The primers were synthesized by Saicheng Biotech (Guangzhou, China). (The primers for quantitative real-time PCR are listed in Table S6.)
In Situ Zymography
The detection of MMP activity was carried out as previously reported. Mouse suprarenal abdominal aortas were subjected to in situ detection of gelatinolytic activity. First, reagent A and reagent B were mixed according to the kit instructions (GMS80062, Genmed Scientifics, USA). Then, in a dark environment, serial 10-mm fresh-frozen sections were incubated at 4°C with the mixture of reagent A and reagent B. When the mixture solidified, the frozen sections were transferred to a 37°C dark chamber and incubated for 1 h. Finally, the sections were photographed under a fluorescence inverted microscope (IX83, Olympus, Japan). Proteolytic activity was detected as bright green fluorescence (530 nm).
RNA m6A Immunoblotting
RNA m6A immunoblotting was carried out as previously reported.36 Total RNA was isolated as described above and then treated with deoxyribonuclease I (AMPD1-1KT, Sigma). A GenElute mRNA Miniprep Kit (MRN70, Sigma-Aldrich) and a RiboMinus Transcriptome Isolation Kit (K155002, human/mouse, Thermo Fisher Scientific) were used for m6A immunoblotting analysis. Poly(A)+ RNA (500 ng) and ribosomal RNA (rRNA)-free poly(A)+ RNA (200 ng) were mixed 1:1 with glyoxal loading dye (Ambion). After denaturation at 65°C for 15 min, the samples were run on a 1% agarose gel for 1 h at 70 V and transferred to nylon membranes (GE Healthcare Life Biosciences) for approximately 2 to 3 h using a NorthernMax Kit (Ambion) according to the manufacturer’s protocols. The membranes with RNA were ultraviolet-crosslinked and blocked in blocking buffer (5% nonfat dry milk in 0.1% PBST; 0.1% Tween-20 in 1× PBS, pH 7.4) for 1 h at room temperature. Next, the membranes were incubated with rabbit anti-m6A antibody (202 003; Synaptic Systems) diluted 1:1,000 in 0.1% PBST at 4°C overnight. Following washing with 0.1% PBST, HRP-conjugated donkey anti-rabbit IgG (Cell Signaling Technology) diluted 1:2,500 in blocking buffer was added to the membranes, which were incubated at room temperature for 1 h. Then, the membranes were again washed adequately with 0.1% PBST and detected using a 3,3′-diaminobenzidine peroxidase substrate kit (Yeasen Biotechnology, Shanghai, China). For visualization of the total RNA, equal amounts of RNA were run on agarose gels and then stained using ethidium bromide.
RNA m6A Quantification
RNA m6A quantification was carried out as previously reported.36 Total RNA was isolated using TRIzol (Omerga, USA) as described above and treated with deoxyribonuclease I (AMPD1-1KT, Sigma). The RNA was quantified using a NanoDrop spectrophotometer. The m6A content of the total RNA was detected using an m6A RNA methylation quantification kit (ab185912; Abcam). Briefly, 200 ng of RNA was coated onto the wells of an assay plate. Then, a capture antibody solution and a detection antibody solution were added to the assay wells at suitable dilutions following the manufacturer’s instructions. The m6A levels were quantified colorimetrically by reading the absorbance of each well at a wavelength of 450 nm and calculating the concentrations based on a standard curve.
RNA m6A Dot Blot Assays
RNA m6A dot blot assays were carried out as previously reported.37 Dot blots were performed in a manner generally consistent with the m6A immunoblotting analysis described above. Briefly, 400 ng of poly(A)+ RNA was double-diluted and spotted on the surface of a nylon membrane (11209299001, Sigma-Aldrich). Then, the membranes with RNA were ultraviolet-crosslinked and blocked with blocking buffer. After incubation with m6A antibody and HRP-conjugated anti-rabbit IgG (ab6802; Abcam), the membranes were detected using a 3,3′-diaminobenzidine peroxidase substrate kit as described above. For visualization of the RNA, 400 ng of the same poly(A)+ RNA was spotted onto a membrane, which was then stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) for 2 h and washed with ribonuclease-free water for 5 h.
RNA Immunoprecipitation
RNA m6A RNA immunoprecipitation was carried out as previously reported.36 Human VSMCs overexpressing METTL3 and control cells were UV-irradiated at 254 nm/cm2 and 400 mJ/cm2 (Stratagene Stratalinker) and lysed with radioimmunoprecipitation (RIP) lysis buffer (17-700; Magna RIP Kit; Millipore, MA) at 4°C through disruptive sonication. Immunoprecipitation of endogenous DGCR8 was performed using an anti-DGCR8 antibody (ab191875, Abcam) overnight at 4°C. After washing, the immunoprecipitated protein-RNA complex was analyzed by western blotting and treated with proteinase K. The RNA was extracted with phenol:chloroform:isoamyl alcohol and subjected to quantitative real-time PCR using primers for primary microRNAs (pri-miRNAs) with normalization to the input. For m6A RNA binding experiments, similar to the methods for DGCR8 RNA immunoprecipitation, RNA from human VSMCs overexpressing METTL3 and control cells was isolated and treated with deoxyribonuclease I (AMPD1-1KT, Sigma). The RNA was fragmented by sonication for 10 s on an ice-water mixture. Immunoprecipitation was performed by incubating the DNA-free fragmented RNA using a rabbit anti-m6A antibody (202 003; Synaptic Systems) that had previously been bound to magnetic Dynabeads (Life Technologies) in RIP Immunoprecipitation Buffer (17-700; Magna RIP Kit; Millipore, MA, USA). The beads were then treated with proteinase K (20 mg/mL) for 1.5 h at 42°C. The RNA was extracted with phenol:chloroform:isoamyl alcohol and subjected to cDNA synthesis and quantitative real-time PCR using primers for pri-miRNAs with normalization to the input. (Information about the RIP-specific primer pairs for the miR34AHG gene is available in the online Tables S7 and S8.)
Statistical Analysis
The data are expressed as the mean ± standard deviation (SD) or medians with interquartile range. Statistical analysis was performed with SPSS version 20.0 (SPSS, Chicago, IL, USA). A normal distribution test was first performed. If variance equality among the different groups was confirmed, Student’s t test was used to analyze differences between 2 independent groups, while one or two-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test was used to analyze differences among 3 or more groups. When data failed to pass the tests for both normality and equal variance, nonparametric Mann-Whitney U tests were performed for 2 independent groups. Fisher’s exact test was performed for the aneurysm incidence analysis. Differences were considered statistically significant at p <0.05.
Author Contributions
The contribution of each author is as follows: L.Z. wrote the paper, performed the experiments, and analyzed the data, X.H., H.S., Y.S., G.C., and X.S. performed the experiments and analyzed the data, J.S. and X.C. analyzed the data and wrote the paper, W.L. and Y.L. designed the research, and J.B. designed the research and wrote the paper. All authors read and approved the final version of the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by grants to J.B. from the National Natural Science Foundation of China (nos. 81771857 and 81571698) and Guangzhou Regenerative Medicine and Health Laboratory of Guangdong (2018GZR110105009).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.005.
Supplemental Information
References
- 1.Li X., Zhao G., Zhang J., Duan Z., Xin S. Prevalence and trends of the abdominal aortic aneurysms epidemic in general population--a meta-analysis. PLoS ONE. 2013;8:e81260. doi: 10.1371/journal.pone.0081260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golledge J., Norman P.E. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. 2011;217:57–63. doi: 10.1016/j.atherosclerosis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Maegdefessel L. Non-coding rna contribution to thoracic and abdominal aortic aneurysm disease development and progression. Front. Physiol. 2017;8:429. doi: 10.3389/fphys.2017.00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong L., He X., Si X., Wang H., Li B., Hu Y., Li M., Chen X., Liao W., Liao Y., Bin J. Sm22alpha (smooth muscle 22alpha) prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching through suppressing reactive oxygen species/nf-kappab (nuclear factor-kappab) Arterioscler. Thromb. Vasc. Biol. 2019;39:e10–e25. doi: 10.1161/ATVBAHA.118.311917. [DOI] [PubMed] [Google Scholar]
- 5.Chen H.Z., Wang F., Gao P., Pei J.F., Liu Y., Xu T.T., Tang X., Fu W.Y., Lu J., Yan Y.F. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ. Res. 2016;119:1076–1088. doi: 10.1161/CIRCRESAHA.116.308895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li D.Y., Busch A., Jin H., Chernogubova E., Pelisek J., Karlsson J., Sennblad B., Liu S., Lao S., Hofmann P. H19 induces abdominal aortic aneurysm development and progression. Circulation. 2018;138:1551–1568. doi: 10.1161/CIRCULATIONAHA.117.032184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Veer E.P., de Bruin R.G., Kraaijeveld A.O., de Vries M.R., Bot I., Pera T., Segers F.M., Trompet S., van Gils J.M., Roeten M.K. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013;113:1065–1075. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- 8.Tang Z.Z., Zheng S., Nikolic J., Black D.L. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol. Cell. Biol. 2009;29:4757–4765. doi: 10.1128/MCB.00608-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiwari S., Zhang Y., Heller J., Abernethy D.R., Soldatov N.M. Atherosclerosis-related molecular alteration of the human CaV1.2 calcium channel alpha1C subunit. Proc. Natl. Acad. Sci. USA. 2006;103:17024–17029. doi: 10.1073/pnas.0606539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tajada S., Cidad P., Colinas O., Santana L.F., López-López J.R., Pérez-García M.T. Down-regulation of CaV1.2 channels during hypertension: how fewer CaV1.2 channels allow more Ca(2+) into hypertensive arterial smooth muscle. J. Physiol. 2013;591:6175–6191. doi: 10.1113/jphysiol.2013.265751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannister J.P., Bulley S., Narayanan D., Thomas-Gatewood C., Luzny P., Pachuau J., Jaggar J.H. Transcriptional upregulation of alpha2delta-1 elevates arterial smooth muscle cell voltage-dependent ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension. 2012;60:1006–1015. doi: 10.1161/HYPERTENSIONAHA.112.199661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C.X., Cui G.S., Liu X., Xu K., Wang M., Zhang X.X., Jiang L.Y., Li A., Yang Y., Lai W.Y. METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol. 2018;16:e2004880. doi: 10.1371/journal.pbio.2004880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alarcón C.R., Lee H., Goodarzi H., Halberg N., Tavazoie S.F. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. (6)-methyladenosine modulates messenger rna translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorn L.E., Lasman L., Chen J., Xu X., Hund T.J., Medvedovic M., Hanna J.H., van Berlo J.H., Accornero F. The n(6)-methyladenosine mrna methylase mettl3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139:533–545. doi: 10.1161/CIRCULATIONAHA.118.036146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathiyalagan P., Adamiak M., Mayourian J., Sassi Y., Liang Y., Agarwal N., Jha D., Zhang S., Kohlbrenner E., Chepurko E. Fto-dependent n(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139:518–532. doi: 10.1161/CIRCULATIONAHA.118.033794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng M., Sheng L., Gao Q., Xiong Q., Zhang H., Wu M., Liang Y., Zhu F., Zhang Y., Zhang X., Yuan Q., Li Y. The m(6)a methyltransferase mettl3 promotes bladder cancer progression via aff4/nf-kappab/myc signaling network. Ocogene. 2019;38:3667–3680. doi: 10.1038/s41388-019-0683-z. [DOI] [PubMed] [Google Scholar]
- 19.Feng Z., Li Q., Meng R., Yi B., Xu Q. Mettl3 regulates alternative splicing of myd88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J. Cell Mol. Med. 2018;22:2558–2568. doi: 10.1111/jcmm.13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joviliano E.E., Ribeiro M.S., Tenorio E.J.R. Micrornas and current concepts on the pathogenesis of abdominal aortic aneurysm. Rev. Bras. Cir. Cardiovasc. 2017;32:215–224. doi: 10.21470/1678-9741-2016-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar S., Boon R.A., Maegdefessel L., Dimmeler S., Jo H. Role of noncoding rnas in the pathogenesis of abdominal aortic aneurysm. Circ. Res. 2019;124:619–630. doi: 10.1161/CIRCRESAHA.118.312438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Badi I., Burba I., Ruggeri C., Zeni F., Bertolotti M., Scopece A., Pompilio G., Raucci A. Microrna-34a induces vascular smooth muscle cells senescence by sirt1 downregulation and promotes the expression of age-associated pro-inflammatory secretory factors. J. Gerontol. A Biol. Sci. Med. Sci. 2015;70:1304–1311. doi: 10.1093/gerona/glu180. [DOI] [PubMed] [Google Scholar]
- 23.Badi I., Mancinelli L., Polizzotto A., Ferri D., Zeni F., Burba I., Milano G., Brambilla F., Saccu C., Bianchi M.E. Mir-34a promotes vascular smooth muscle cell calcification by downregulating sirt1 (sirtuin 1) and axl (axl receptor tyrosine kinase) Arterioscler. Thromb. Vasc. Biol. 2018;38:2079–2090. doi: 10.1161/ATVBAHA.118.311298. [DOI] [PubMed] [Google Scholar]
- 24.Guo Y., Li P., Gao L., Zhang J., Yang Z., Bledsoe G., Chang E., Chao L., Chao J. Kallistatin reduces vascular senescence and aging by regulating microRNA-34a-SIRT1 pathway. Aging Cell. 2017;16:837–846. doi: 10.1111/acel.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Krishna S., Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis. 2013;226:29–39. doi: 10.1016/j.atherosclerosis.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Saraff K., Babamusta F., Cassis L.A., Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 27.Maegdefessel L., Azuma J., Toh R., Merk D.R., Deng A., Chin J.T., Raaz U., Schoelmerich A.M., Raiesdana A., Leeper N.J. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J. Clin. Invest. 2012;122:497–506. doi: 10.1172/JCI61598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Gregoli K., Mohamad Anuar N.N., Bianco R., White S.J., Newby A.C., George S.J., Johnson J.L. Microrna-181b controls atherosclerosis and aneurysms through regulation of timp-3 and elastin. Circ. Res. 2017;120:49–65. doi: 10.1161/CIRCRESAHA.116.309321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bostick B., Ghosh A., Yue Y., Long C., Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14:1605–1609. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- 31.Daugherty A., Manning M.W., Cassis L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Longo G.M., Xiong W., Greiner T.C., Zhao Y., Fiotti N., Baxter B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Sun Y., Huang S., Chen Y., Chen X., Li M., Si X., He X., Zheng H., Zhong L. Inhibition of AZIN2-sv induces neovascularization and improves prognosis after myocardial infarction by blocking ubiquitin-dependent talin1 degradation and activating the Akt pathway. EBioMedicine. 2019;39:69–82. doi: 10.1016/j.ebiom.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jing Y., Hu Y., Li H., Wang J., Si X., Zheng H., Liu J., Liao W., Liao Y., Bin J. Assessment of thrombotic risk in atrial fibrillation with ultrasound molecular imaging of p-selectin. Thromb. Haemost. 2018;118:388–400. doi: 10.1160/TH17-02-0103. [DOI] [PubMed] [Google Scholar]
- 35.Xie J., Cui K., Hao H., Zhang Y., Lin H., Chen Z., Huang X., Cao S., Liao W., Bin J. Acute hyperglycemia suppresses left ventricular diastolic function and inhibits autophagic flux in mice under prohypertrophic stimulation. Cardiovasc. Diabetol. 2016;15:136. doi: 10.1186/s12933-016-0452-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma J.Z., Yang F., Zhou C.C., Liu F., Yuan J.H., Wang F., Wang T.T., Xu Q.G., Zhou W.P., Sun S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6 -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–543. doi: 10.1002/hep.28885. [DOI] [PubMed] [Google Scholar]
- 37.Li Z., Weng H., Su R., Weng X., Zuo Z., Li C., Huang H., Nachtergaele S., Dong L., Hu C. Fto plays an oncogenic role in acute myeloid leukemia as a n(6)-methyladenosine rna demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.