

Abstract
In organic device applications, a high contact resistance between metal electrodes and organic semiconductors prevents an efficient charge injection and extraction, which fundamentally limits the device performance. Recently, various contact doping methods have been reported as an effective way to resolve the contact resistance problem. However, the contact doping has not been explored extensively in organic field effect transistors (OFETs) due to dopant diffusion problem, which significantly degrades the device stability by damaging the ON/OFF switching performance. Here, the stability of a contact doping method is improved by incorporating “dopant‐blockade molecules” in the poly(2,5‐bis(3‐hexadecylthiophen‐2‐yl)thieno[3,2‐b]thiophene) (PBTTT) film in order to suppress the diffusion of the dopant molecules. By carefully selecting the dopant‐blockade molecules for effectively blocking the dopant diffusion paths, the ON/OFF ratio of PBTTT OFETs can be maintained over 2 months. This work will maximize the potential of OFETs by employing the contact doping method as a promising route toward resolving the contact resistance problem.
Keywords: 2,3,5,6‐tetrafluoro‐7,7,8,8‐tetracyanoquinodimethane; contact doping; dopant‐blockade; poly(2,5‐bis(3‐hexadecylthiophen‐2‐yl)thieno[3,2‐b]thiophene); solid‐state diffusion; tetracyanoquinodimethane
A new strategy to enhance the device stability of doped‐contact organic field transistors (DC‐OFETs) by adopting dopant‐blockade molecules is proposed. These electrically neutral molecules are carefully selected to be readily bulk‐incorporated in the active channel region to effectively block the diffusion paths of the dopants from the contact regions, while maintaining the excellent electrical characteristics of the DC‐OFETs.

1. Introduction
Developing high‐performance organic field effect transistors (OFETs) has been a bottleneck in exploiting the merits of organic semiconductors (OSCs) such as solution‐processability, mechanical flexibility, and bio‐compatibility, for realizing practical organic optoelectronic device applications.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] One of the main challenges remaining for utilizing OFETs is a high contact resistance originating from an injection barrier at the OSC–metal interface.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] An inefficient charge injection across such non‐ohmic contacts not only lowers the effective mobility of OFETs but limits the range of operation voltage due to a highly non‐linear (S‐shaped) output characteristic curve at low bias voltage,[ 15 , 16 ] which is not desirable for analog circuit applications.[ 17 , 18 ]
To overcome the contact resistance problem in OFETs, there have been a diverse range of approaches for lowering of injection barrier height between metal electrodes and transport level of OSCs such as work function modification of metal electrodes by self‐assembled monolayer (SAM) treatment[ 19 , 20 , 21 , 22 , 23 , 24 , 25 ] and insertion of charge injection layer that bridges the energy gap between metal electrodes and OSCs.[ 26 , 27 , 28 , 29 ] In addition, contact doping (i.e., introduction of external dopant molecules near contact regions) has been studied as an effective method for improving contact properties.[ 15 , 30 , 31 , 32 , 33 , 34 , 35 ] An ideal contact doping technique in OFETs would meet two criteria; first, a high maximum doping level for enhancing charge injection and second, a spatial confinement of dopants for device stability. However, these two requirements cannot often be simultaneously achieved since achieving a high doping level requires a large dopant density, which leads to a faster diffusion of dopant molecules within the host semiconductor. This dopant diffusion problem results in a rise of the OFF current due to an unintentional doping of the active channel. Confining the dopant molecules within the selective regions near the contacts is especially challenging in the case of bulk‐doping since a large concentration gradient of dopant density at the edges of the doped regions accelerates the dopant diffusion. The dopants from the contact regions diffuse toward the active channel region, which should remain undoped in order to maintain a sharp ON/OFF switching characteristic in OFET. Therefore, the dopant diffusion has limited the contact doping techniques to be employed extensively in OFETs.
Despite a significant level of attention given in the dopant diffusion problem in organic solar cells and organic light emitting diodes,[ 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 ] there have been relatively few studies which have focused on OFETs. Recently, we have developed a surface etching treatment for suppressing the dopant diffusion in doped‐contact poly(2,5‐bis(3‐tetradecylthiophen‐2‐yl)thieno[3,2‐b]thiophene) (PBTTT) OFETs.[ 44 ] This system exploited a facile and efficient bulk‐doping of PBTTT via solid‐state diffusion of 2,3,5,6‐tetrafluoro‐7,7,8,8‐tetracyanoquinodimethane (F4‐TCNQ) which resulted in high‐conductivity and high carrier‐concentration regions in spatially selected regions in OFETs.[ 45 ] This doping method effectively reduced the contact resistance (by a factor of 5) and showed its potential by demonstrating the low‐voltage operation organic transistor. However, despite an improved device stability by the surface etching treatment, suppressing the diffusion of dopant molecules from the contact regions to the channel region of PBTTT OFETs could not be completely avoided, and thereby resulting in degradation of the switching characteristics of OFETs.
In this study, the stability of the contact doping method in PBTTT OFETs was significantly improved by introducing a novel technique for suppressing the dopant diffusion. The diffusion pathways of the dopants within the active channel region were spatially blocked by incorporating 7,7,8,8‐tetracyanoquinodimethane (TCNQ) as “dopant‐blockade molecules” in PBTTT OFETs. The dopant‐blockade molecules were carefully chosen such that, they are electrically inactive and they readily locate themselves in the diffusion paths of the dopants. This technique effectively constructed barriers against the motion of dopant molecules by incorporating the dopant‐blockade molecules. This concept can be considered analogous to a recent work, which achieved a high operational and environmental stability by filling voids that can act as water adsorption sites in the polymer film of OFETs by using a specific range of organic solvents and molecules.[ 46 ] In our work, we demonstrated that introducing TCNQ as dopant‐blockade molecules in PBTTT film remarkably increase the device stability against dopant diffusion by comparing the OFETs with and without the TCNQ incorporation.
2. Results and Discussion
Figure 1a shows the molecular structures of the organic materials used in this work. In order to suppress the dopant diffusion in the channel region of the PBTTT OFETs, we adopted dopant‐blockade molecules in the F4‐TCNQ‐doped‐contact PBTTT OFETs, which are fabricated in the same way as our previous work.[ 44 ] The dopant‐blockade molecules should be selected to avoid the charge transfer reaction with PBTTT molecules for maintaining the electrical properties of the doped‐contact PBTTT OFETs (denoted as “DC‐FET”) after introducing dopant‐blockade molecules in the PBTTT channel. Figure 1b,c shows the values of the highest occupied molecular orbital (HOMO) level and lowest unoccupied molecular orbital (LUMO) level of F4‐TCNQ and TCNQ. TCNQ was adopted as the dopant‐blockade molecule because it has a similar molecular structure with F4‐TCNQ but the LUMO level is higher than the HOMO level of PBTTT, which energetically prohibits the charge transfer reaction with PBTTT.[ 47 ] On the contrary, the charge transfer reaction between PBTTT and F4‐TCNQ is energetically favorable because the HOMO level of PBTTT is higher than the LUMO level of F4‐TCNQ. Therefore, electrons transfer easily from PBTTT to F4‐TCNQ, whereas the charge transfer between PBTTT and TCNQ is energetically unfavorable. Based on this energetic mismatch between the HOMO level of PBTTT and LUMO level of TCNQ, it was expected that introducing TCNQ molecules in the channel region hardly affect the electrical properties of PBTTT films. To verify this idea, we compared the pristine PBTTT OFET and the PBTTT OFET with TCNQ molecules deposited on the entire PBTTT channel. As a result, the transistor characteristics of the PBTTT OFETs was well preserved (see Figure S1, Supporting Information), compared to the entire‐doped PBTTT OFET with F4‐TCNQ which became a conductor rather than a transistor due to doping.[ 44 ]
Figure 1.
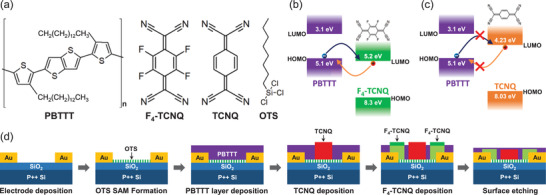
a) Molecular structures of PBTTT, F4‐TCNQ, TCNQ, and OTS. b) HOMO and LUMO level of PBTTT and F4‐TCNQ. c) HOMO and LUMO level of PBTTT and TCNQ. d) Schematic images of the fabrication process of the dopant‐blockade PBTTT OFET. The red regions in the middle of transistors are the TCNQ‐incorporated regions. The dark green regions represent the neutral F4‐TCNQ molecules, which are not involved in the charge transfer reaction with PBTTT molecules. The bright green regions are the doped‐PBTTT regions by using F4‐TCNQ dopant molecules.
Figure 1d shows how the TCNQ molecules were incorporated in the doped‐contact PBTTT FETs by entailing the step‐by‐step fabrication process of the TCNQ‐incorporated doped‐contact PBTTT OFET (dopant‐blockaded doped‐contact‐FET, denoted as “DB/DC‐FET”). First, Ti/Au was deposited as source and drain electrodes on a cleaned SiO2 (270 nm)/Si substrate using an electron‐beam evaporator. Then, the substrates with the source and drain electrodes were transferred to N2 filled glove box and immersed in octyltrichlorosilane (OTS) solution to form SAM on SiO2 surface for enhancing the morphology of PBTTT films to be deposited on the SiO2. PBTTT solution was made by PBTTT dissolved in 1,2‐dichlorobenzene and spin coated to deposit the PBTTT films on the OTS treated substrates. After that, the PBTTT deposited substrates were annealed at 180 °C to form a clear terrace morphology (see Figure S2, Supporting Information).[ 48 , 49 , 50 , 51 ] After the PBTTT film deposition, TCNQ molecules were incorporated selectively in the middle of the channel region of the devices by using a thermal evaporator with a shadow mask, and then F4‐TCNQ molecules were deposited near the metal contact regions of the PBTTT film. The thermal evaporation method used for both the TCNQ and F4‐TCNQ deposition ensures the spatial selectivity, that is, the position of the dopant‐blockade molecules (TCNQ) are limited in the conduction channel region of OFET, separated from the contact regions near the source and drain electrodes (see Section S3, Supporting Information for detailed top‐view schematic illustrations of DB/DC‐FET). This is important since the presence of the TCNQ molecules near the contact regions could interrupt the charge transfer between the dopant molecules (F4‐TCNQ) and the host PBTTT molecules, and therefore resulting in a lower doping efficiency. Finally, the devices were etch‐treated by argon plasma for further enhancing the device stability by removing the neutral F4‐TCNQ molecules, which are highly diffusive compared to their charged counterparts.[ 44 , 52 , 53 ]
The absence of the charge transfer reaction between TCNQ and PBTTT was confirmed by UV–vis absorption spectroscopy measurements. Figure 2a displays the UV–vis absorption data of a pristine PBTTT film (black line), a TCNQ‐incorporated PBTTT film (PBTTT/TCNQ, shown as a red line), and a F4‐TCNQ‐doped PBTTT film (PBTTT/F4‐TCNQ, shown as a green line). The pristine PBTTT film (black line) showed a clear π–π* transition peak near 550 nm.[ 44 , 45 ] On the other hand, the PBTTT/F4‐TCNQ film (green line) displayed a clear bleaching of a PBTTT π–π* transition peak. In addition, the F4‐TCNQ anion peaks near 800 and 900 nm appeared and a broad polaron absorption of charged PBTTT was observed from 600 to 1000 nm. These changes of the absorption spectrum were caused by the charge transfer between PBTTT and F4‐TCNQ.[ 44 ] However, for the PBTTT/TCNQ film (red line), the absorption data almost unchanged, compared with the pristine film. This good agreement between the pristine film and PBTTT/TCNQ film supported the absence of the charge transfer reaction between PBTTT and TCNQ.
Figure 2.
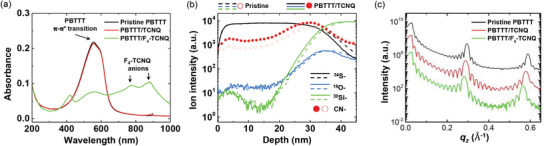
a) UV–vis spectroscopy data for the pristine PBTTT film (black line), PBTTT/TCNQ film (red line), and PBTTT/F4‐TCNQ film. b) The depth profiles of ion intensity for the pristine PBTTT film (dashed lines and empty symbols) and PBTTT/TCNQ film (solid lines and filled symbols); the sulfur ion signals are black solid and dashed lines, the oxygen ion blue solid and dashed lines, silicon ion green solid and dashed lines and the cyanide ion signal red empty and filled circle symbols. c) The XRR data of the pristine PBTTT film (black line), the PBTTT/TCNQ film (red line), and the PBTTT/F4‐TCNQ film (green line). The intervals between large q z peaks of each film represent the out‐of‐plane lamellar stacking distance.
In order to effectively block the diffusion paths of the dopant molecules, the dopant blockade molecules would ideally be bulk‐incorporated in the PBTTT film. Considering that F4‐TCNQ undergoes solid‐state diffusion by penetrating all the way down to the substrate from the top surface of the PBTTT films, it was expected that TCNQ would also undergo solid‐state diffusion in PBTTT due to a similar molecular structure. Figure 2b shows elemental depth profiles of the pristine PBTTT film (dashed lines and empty symbols) and the PBTTT/TCNQ film (solid lines and filled symbols) by time of flight secondary ion mass spectroscope (ToF‐SIMS). The sulfur ion signal (34S−, black solid and dashed lines) is a representative signal of PBTTT, both the oxygen ion (18O−, blue solid and dashed lines) and silicon ion (30Si−, green solid and dashed lines) signals for the silicon substrates and the cyanide ion signal (CN−, red empty and filled circle symbols) for TCNQ. Comparing the pristine PBTTT film and PBTTT/TCNQ film, the sulfur, oxygen, and silicon ions signals showed very similar depth profiles due to the same constituents of the both samples from PBTTT and silicon substrates. On the contrary, the PBTTT/TCNQ film showed a higher cyanide ion signal than the pristine PBTTT film down to the depth of nearly 40 nm, which is the thickness of the PBTTT film (see Figure S4, Supporting Information for a clearer comparison).[ 45 ] Therefore, the depth profile of the elemental analysis indicates that TCNQ molecules become structurally incorporated in PBTTT films all the way down to the interface between the PBTTT film and the silicon substrate by solid‐state diffusion.
Figure 2c shows the X‐ray reflectivity (XRR) data of the pristine PBTTT film (black line), the PBTTT/TCNQ film (red line), and the PBTTT/F4‐TCNQ film (green line). Previous studies showed that F4‐TCNQ molecules were located at the side chain regions in the PBTTT film when F4‐TCNQ molecules were deposited on a PBTTT polymer film.[ 45 , 54 , 55 ] Upon the molecular intercalation of F4‐TCNQ, the out‐of‐plane lamellar stacking distance of the PBTTT/F4‐TCNQ film becomes lager than that of the pristine PBTTT film. The expansion of the out‐of‐plane lamellar stacking distance after F4‐TCNQ doping was indicated by a shorter q z spacing between the (h00) diffraction peaks of the PBTTT/F4‐TCNQ film compared to that of the pristine film. If the TCNQ molecules are located at the side chain region in the PBTTT by solid‐state diffusion, the deposition of TCNQ molecules would also cause the expansion of the out‐of‐plane lamellar stacking distance in the PBTTT/TCNQ film. This is indeed observed in Figure 2c and confirms the presence of the TCNQ molecules in the side chain region of the PBTTT. The extracted out‐of‐plane lamellar spacing was 21.64 Å for the PBTTT/TCNQ film and 21.37 Å for the pristine PBTTT film. The expansion of the lamellar spacing was even larger for the PBTTT/F4‐TCNQ film for which the extracted value was 22.21 Å. The larger expansion in the lamellar spacing of the PBTTT/F4‐TCNQ film could be correlated with the bigger size of F4‐TCNQ molecules relative to TCNQ molecules. Based on the results of the UV–vis absorption, ToF‐SIMS, and XRR measurements, we deduced that TCNQ molecules diffused into the bulk of PBTTT film by solid‐state diffusion all the way down to the bottom substrate, without a significant charge transfer reaction with PBTTT molecules.
Figure 3a shows the schematic images of the DC‐FET (w/o TCNQ) and the DB/DC‐FET (with TCNQ). Figure 3b,c shows the transfer curves of the doped‐contact PBTTT OFET (DC‐FET) and TCNQ‐incorporated doped‐contact PBTTT OFET (DB/DC‐FET) in linear (red line) and saturation (blue line) regimes. Transfer curves of the both devices show similar electrical characteristics. The DB/DC‐FET showed typical p‐type OFET device characteristics with the maximum mobility value of 0.058 cm2 V−1 s−1 (see Figure 3c) which was extracted by the following equation, in a saturation regime, where μ, W ch, L ch, V th, and C ox = ε0εr/d = 1.28 × 10−4F/m−2 denote the mobility, the channel width, the channel length, the threshold voltage and the unit‐area capacitance of the DB/DC‐FET. The average mobility of DB/DC‐FET was found to be 0.037 ± 0.013 cm2 V−1 s−1 (extracted from eight different devices) which was similar to the average mobility of DC‐FET of 0.033 ± 0.008 cm2 V−1 s−1 (extracted from five different devices, with the maximum value of 0.045 cm2 V−1 s−1), which supports the non‐invasive nature of the dopant‐blockade molecules. A slight increase in the OFF current could be due to unintentional air exposure during extra device fabrication steps required for DB/DC‐FET. Figure 3d shows the output curves of the 50 µm channel length DB/DC‐FET. The device shows a clear ohmic behavior in a low V DS region, providing evidence of a low contact resistance. To extract the contact resistance of the DB/DC‐FET, we used Y‐function method. The extracted contact resistance of the DB/DC‐FET was 4.55 kΩ cm, which is comparable to our previous result for the DC‐FET (the details of the Y‐function method are provided in Section S5, Supporting Information). Despite of the additional incorporation of the TCNQ molecules in the PBTTT channel before the contact doping with F4‐TCNQ molecules, the contact doping remained effective for achieving a low contact resistance; the pristine PBTTT OFET had more than five times larger contact resistance value of 24.5 kΩ cm.[ 44 ] Therefore, we deduced that the incorporation of the dopant‐blockade molecules did not affect the operation properties of the DC‐FET.
Figure 3.
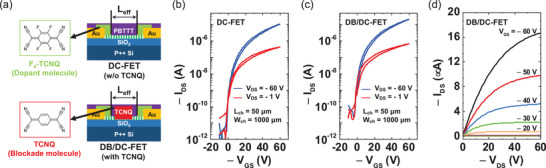
a) Schematic images of the DC‐FET (w/o TCNQ) and DB/DC‐FET (with TCNQ). The transfer curves of b) the DC‐FET and c) the DB/DC‐FET. d) The output curves of the DB/DC‐FET.
Figure 4a,b shows the change of the transfer curves of the DC‐FET and the DB/DC‐FET, respectively, with the channel length of 50 µm over 2 months (100 µm channel data are provided in Figure S6, Supporting Information). From Figure 4a, it is clear that the ON/OFF ratio of the DC‐FET decreases due to the rise of the OFF current. On the other hand, the rise of the OFF current of the DB/DC‐FET is significantly less, which is presented in Figure 4b. Figure 4c shows the traces of the ON/OFF ratio of the both kinds of the devices for 2 months; the DC‐FET is denoted as empty symbols, the DB/DC‐FET as filled symbols, 100 µm channel length devices as black lines, and 50 µm channel length devices as red lines. For 100 µm channel devices, the traces of the both kinds of devices had stable ON/OFF ratio greater than 104 over the 2 months. The stability of the ON/OFF ratio of the 100 µm channel length devices reproduces our previous results for the DC‐FET.[ 44 ] On the contrary, for the 50 µm channel length devices, the ON/OFF ratio stability was affected significantly by the TCNQ incorporation. The trace of the DC‐FET decreases steeply from 20 to 40 days. However, the trace of the DB/DC‐FET shows the ON/OFF ratio decaying much slower and the ON/OFF ratio remained higher than 103 for 2 months. A similar trend could be seen from the change of subthreshold swing, threshold voltage, and mobility values both of which increase steeply from after 20 days for the 50 µm DC‐FET (see Section S7, Supporting Information), which is supportive of the dopant‐diffusion‐induced instability. Therefore, based on these traces of the ON/OFF ratio of devices, we have found that the dopant‐blockade method is effective for achieving stable DC‐FETs.
Figure 4.
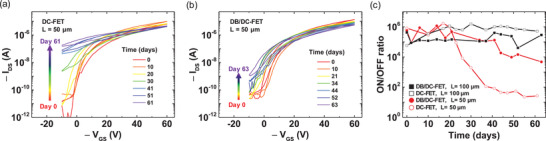
Time evolution of the transfer curves of a) the DC‐FET and b) the DB/DC‐FET with the channel length of 50 µm over 2 months. c) The ON/OFF ratio of the DC‐FET and the DB/DC‐FET over time with the channel length of 50 and 100 µm.
In order to describe the dopant‐blockade effect on the dopant diffusion in details, we first considered the diffusion of the dopant and dopant‐blockade molecules independently within PBTTT OFETs. Figure 5a,b illustrates the propagation of the dopant molecules within the channel regions of the DC‐FET and that of the dopant‐blockade molecules in the TCNQ‐incorporated PBTTT OFET (denoted as “TCNQ only”), respectively. Figure 5a describes the expansion of the doped PBTTT regions; purple regions represent neutral PBTTT regions, green for doped PBTTT regions, darker green for higher dopant concentration, and black dashed lines define the initially doped regions by F4‐TCNQ molecules (Figure 5a, top). As time passes, the doped regions expand by the diffusion of dopant molecules into the channel and create conduction paths between the two electrodes (Figure 5a, bottom), and therefore the OFF current increases. Figure 5b depicts the diffusion of TCNQ molecules in the channel region of “TCNQ only” device. The red regions represent the TCNQ molecules in the PBTTT channel, more vivid red for higher TCNQ concentration. The TCNQ molecules are initially confined in the middle of the channel (Figure 5b, top) as described in the fabrication process (Figure 1d), which is denoted as black dashed lines. The TCNQ molecules diffuse toward the contact regions from the middle of the channel along the concentration gradient as time passes (Figure 5b, bottom).
Figure 5.
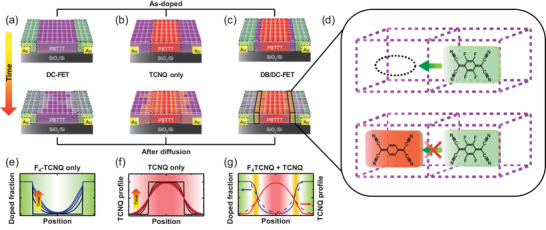
Schematic diagrams of a) the propagation of doped regions in the DC‐FET, b) the diffusion of TCNQ molecules in the channel region of the TCNQ‐incorporated PBTTT OFET, and c) the diffusion of F4‐TCNQ and TCNQ molecules in the opposite directions for the DB/DC‐FET. d) The illustration for hindrance by the dopant‐blockade molecules against the diffusion of the dopant molecules in the void spaces of the PBTTT film. e) The predicted profiles of the doped fraction of PBTTT molecules at different positions of the channel in the DC‐FET. f) The spatial diffusion profile of TCNQ molecules in the PBTTT channel of the TCNQ‐incorporated PBTTT OFET. g) The predicted changes in the profiles of the doped fraction of PBTTT molecules and the spatial diffusion of TCNQ molecules for the DB/DC‐FET as blue and red solid lines. Dashed lines are taken from (e) and (f) for comparison.
In DB/DC‐FETs, both the F4‐TCNQ molecules (dopant) and TCNQ molecules (dopant‐blockade) diffuse together in the opposite directions, affecting the motion of each other, as shown in Figure 5c. The orange regions (Figure 5c, bottom) represent the channel regions where the dopant and the dopant‐blockade molecules co‐exist and both molecules hinder the diffusion of one another by occupying the potential diffusion sites in the PBTTT film. This process is depicted in Figure 5d. If there is no TCNQ dopant‐blockade molecule, the dopant molecules move easily into the neighboring empty site. However, if a dopant‐blockade molecule already occupies the neighboring empty site, the dopant molecule is sterically hindered from moving to the next site.
The effect of the dopant‐blockade molecules on the dopant diffusion can be summarized by the predicted molecular diffusion profiles by the same numerical simulation method used in our previous study.[ 44 ] The method is based on 1D modified Fick's diffusion equation with a correction term that accounts for capturing of neutral F4‐TCNQ molecules via charge‐transfer, which predicts a gradual propagation of the doped PBTTT fraction toward the center of the channel in DC‐FET over time (Figure 5e,“F4‐TCNQ only”). On the other hand, the diffusion of TCNQ molecules can be assumed to follow a free Fick's diffusion equation (i.e., no correction term), considering that TCNQ molecules diffuse without electrostatic attraction with PBTTT molecules due to the lack of charge transfer. In TCNQ‐incorporated PBTTT device (Figure 5f, “TCNQ only”), the TCNQ molecules initially concentrated at the center of the channel (black line) gradually diffuse away over time as shown in spatial diffusion profiles in Figure 5f. When the two species diffuse together in DB/DC‐FET (Figure 5g, “F4‐TCNQ + TCNQ”), the dopant‐blockade effect suppresses the diffusion of one another as shown from the doped PBTTT fraction (solid blue line) and TCNQ (solid red line) profiles, compared to the diffusion profiles of the individual species (dashed lines taken from Figure 5e,f for comparison). In our simulation, the dopant‐blockade effect was emulated by reduction in the diffusion constants of the two species when the two species co‐exist at a given position (orange regions). A clear reduction of the doped PBTTT fraction at the center of the channel shows that dopant‐blockade effect illustrated here remarkably suppresses the dopant diffusion and improves the stability of DC‐FETs.
3. Conclusion
In conclusion, we improved the device stability of the F4‐TCNQ doped‐contact PBTTT OFETs by incorporating dopant‐blockade molecules (TCNQ). The dopant‐blockade molecules were carefully selected to avoid the charge transfer reaction with the host materials, which was supported by the UV–vis absorption spectroscopy. Furthermore, the selected dopant‐blockade molecules were readily incorporated in PBTTT film by solid‐state diffusion, investigated by elemental and structural analysis. By incorporating the dopant‐blockade molecules, we demonstrated a more stable switching property of the doped‐contact PBTTT OFET devices without affecting their electrical characteristics. The dopant‐blockade molecules filled the dopant diffusion paths in PBTTT and effectively suppressed the diffusion of the dopant molecules. This study proposes a new strategy to enhance the stability of molecular doping methods in OSCs, and reinforce the potential of OFETs by employing the contact doping techniques for resolving the contact resistance problem.
4. Experimental Section
Device Fabrication
SiO2 (270 nm)/Si substrates were cleaned by sonication with de‐ionized water, isopropanol, and acetone for 10 min in each cleaning solvent. Ti/Au (2/30 nm) electrodes were deposited on the cleaned substrates as source and drain electrodes by using an electron‐beam evaporator under 10−6 torr, patterned by shadow masks. 30 mm OTS solution was prepared by dissolving OTS in toluene solvent. The substrates were then transferred to N2 filled glove box and immersed in the OTS solution over 12 h to form the OTS SAM on SiO2 surfaces. The OTS‐treated substrates were cleaned again by isopropanol, acetone, and toluene for 10 min in each solvent with sonication to remove residual OTS molecules on the surface. PBTTT solution was made by dissolving PBTTT in 1,2‐dichlorobenzene with the concentration of 9 mg mL−1 and heated at 110 °C before the spin‐coating. Then, the PBTTT solution was spin‐coated on the OTS‐treated substrates with 1500 rpm for 45 s. The PBTTT deposited substrates were annealed at 180 °C for 20 min and cooled down slowly. After that, TCNQ molecules were selectively incorporated in the middle of the channel region of the PBTTT transistor with the nominal thickness of 10 nm by using a thermal evaporator under 5 × 10−6 torr with a shadow mask. Then, F4‐TCNQ molecules were deposited near the metal contact regions of the device with the nominal thickness of 10 nm by the same method for TCNQ deposition. The shadow mask for patterning the F4‐TCNQ molecules was carefully aligned under an optical microscope in order to avoid the spatial overlap between the F4‐TCNQ‐doped region and TCNQ‐incorporated region. Approximately 10 µm error margin was present in the manual alignment process due to a limited control in manually operating the sample stage of the microscope. Finally, the devices were etch‐treated by argon plasma for 1 s. The samples for UV–vis absorption, TOF‐SIMS, and XRR measurements were prepared in the same way as above without the patterning on the different substrates; fused silica windows for UV–vis absorption measurement and bare Si substrates for TOF‐SIMS and XRR measurements.
Device and Film Characterization
Electrical measurement was performed by a semiconductor parameter analyzer (Keithley 4200 SCS) under vacuum condition (≈10−3 torr). UV–vis absorptions were measured by UV–vis spectroscopy (JASCO V‐770). Element depth profiles were acquired by a time‐of‐flight secondary ion mass spectrometry (TOF.SIMS5, Iontof). XRR scans were measured on a home diffractometer (3303TT, GE) using Cu Kα‐radiation (λ = 1.5406Å) and a 1D detector (Meteor 1D, XRD Eigenmann).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors appreciate the financial support of the National Creative Research Laboratory program (Grant No. 2012026372) through the Korean National Research Foundations (NRF) funded by the Korean Ministry of Science and ICT. S.C. appreciates the financial support from the Korean Ministry of Trade, Industry & Energy and Korea Display Research Consortium support program (10051541). H.S. acknowledges funding from the European Research Council (ERC) through a Synergy Grant (Grant No. 610116). K.K. appreciates the financial support by Postdoctoral Science Fellowship from POSCO TJ Park Foundation. A spelling error in an author's name (H.A.) was corrected on July 9, 2020 after initial online publication.
Kim Y., Broch K., Lee W., Ahn H., Lee J., Yoo D., Kim J., Chung S., Sirringhaus H., Kang K., Lee T., Highly Stable Contact Doping in Organic Field Effect Transistors by Dopant‐Blockade Method. Adv. Funct. Mater. 2020, 30, 2000058 10.1002/adfm.202000058
Contributor Information
Keehoon Kang, Email: keehoon.kang@snu.ac.kr.
Takhee Lee, Email: tlee@snu.ac.kr.
References
- 1. Lingstedt L. V., Ghittorelli M., Brückner M., Reinholz J., Crăciun N. I., Torricelli F., Mailänder V., Gkoupidenis P., Blom P. W. M., Adv. Healthcare Mater. 2019, 8, 1900128. [DOI] [PubMed] [Google Scholar]
- 2. Lipomi D. J., Bao Z., MRS Bull. 2017, 42, 93. [Google Scholar]
- 3. Lee W.‐Y., Wu H.‐C., Lu C., Naab B. D., Chen W.‐C., Bao Z., Adv. Mater. 2017, 29, 1605166. [DOI] [PubMed] [Google Scholar]
- 4. Kasparek C., Blom P. W. M., Appl. Phys. Lett. 2017, 110, 023302. [Google Scholar]
- 5. Someya T., Bao Z., Malliaras G. G., Nature 2016, 540, 379. [DOI] [PubMed] [Google Scholar]
- 6. Sirringhaus H., Adv. Mater. 2014, 26, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaltenbrunner M., Sekitani T., Reeder J., Yokota T., Kuribara K., Tokuhara T., Drack M., Schwödiauer R., Graz I., Bauer‐Gogonea S., Bauer S., Someya T., Nature 2013, 499, 458. [DOI] [PubMed] [Google Scholar]
- 8. Kang H., Kitsomboonloha R., Jang J., Subramanian V., Adv. Mater. 2012, 24, 3065. [DOI] [PubMed] [Google Scholar]
- 9. Sekitani T., Someya T., Mater. Today 2011, 14, 398. [Google Scholar]
- 10. Yan H., Chen Z., Zheng Y., Newman C., Quinn J. R., Dötz F., Kastler M., Facchetti A., Nature 2009, 457, 679. [DOI] [PubMed] [Google Scholar]
- 11. Subramanian V., Chang P. C., Lee J. B., Molesa S. E., Volkman S. K., IEEE Trans. Compon. Packag. Technol. 2005, 28, 742. [Google Scholar]
- 12. Murphy A. R., Fréchet J. M. J., Chang P., Lee J., Subramanian V., J. Am. Chem. Soc. 2004, 126, 1596. [DOI] [PubMed] [Google Scholar]
- 13. Sekitani T., Someya T., MRS Bull. 2012, 37, 236. [Google Scholar]
- 14. Zhou L., Wanga A., Wu S.‐C., Sun J., Park S., Jackson T. N., Appl. Phys. Lett. 2006, 88, 083502. [Google Scholar]
- 15. Ante F., Kälblein D., Zaki T., Zschieschang U., Takimiya K., Ikeda M., Sekitani T., Someya T., Burghartz J. N., Kern K., Klauk H., Small 2011, 7, 1186. [DOI] [PubMed] [Google Scholar]
- 16. Yamamura A., Watanabe S., Uno M., Mitani M., Mitsui C., Tsurumi J., Isahaya N., Kanaoka Y., Okamoto T., Takeya J., Sci. Adv. 2018, 4, eaao5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Natali D., Caironi M., Adv. Mater. 2012, 24, 1357. [DOI] [PubMed] [Google Scholar]
- 18. Zaki T., Ante F., Zschieschang U., Butschke J., Letzkus F., Richter H., Klauk H., Burghartz J., IEEE J. Solid‐State Circuits 2012, 47, 292. [Google Scholar]
- 19. Youn J., Dholakia G. R., Huang H., Hennek J. W., Facchetti A., Marks T. J., Adv. Funct. Mater. 2012, 22, 1856. [Google Scholar]
- 20. Singh K. A., Nelson T. L., Belot J. A., Young T. M., Dhumal N. R., Kowalewski T., McCullough R. D., Nachimuthu P., Thevuthasan S., Porter L. M., ACS Appl. Mater. Interfaces 2011, 3, 2973. [DOI] [PubMed] [Google Scholar]
- 21. Noh Y.‐Y., Cheng X., Tello M., Lee M.‐J., Sirringhaus H., Semicond. Sci. Technol. 2011, 26, 034003. [Google Scholar]
- 22. Gholamrezaie F., Asadi K., Kicken R. A. H. J., Langeveld‐Voss B. M. W., de Leeuw D. M., Blom P. W. M., Synth. Met. 2011, 161, 2226. [Google Scholar]
- 23. Boudinet D., Benwadih M., Qi Y., Altazin S., Verilhac J.‐M., Kroger M., Serbutoviez C., Gwoziecki R., Coppard R., Le Blevennec G., Kahn A., Horowitz G., Org. Electron. 2010, 11, 227. [Google Scholar]
- 24. Cheng X., Noh Y.‐Y., Wang J., Tello M., Frisch J., Blum R.‐P., Vollmer A., Rabe J. P., Koch N., Sirringhaus H., Adv. Funct. Mater. 2009, 19, 2407. [Google Scholar]
- 25. Marmont P., Battaglini N., Lang P., Horowitz G., Hwang J., Kahn A., Amato C., Calas P., Org. Electron. 2008, 9, 419. [Google Scholar]
- 26. He D., Qiao J., Zhang L., Wang J., Lan T., Qian J., Li Y., Shi Y., Chai Y., Lan W., Ono L. K., Qi Y., Xu J.‐B., Ji W., Wang X., Sci. Adv. 2017, 3, e1701186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang C. G., Ang M. C. Y., Choo K.‐K., Keerthi V., Tan J.‐K., Syafiqah M. N., Kugler T., Burroughes J. H., Png R.‐Q., Chua L.‐L., Ho P. K. H., Nature 2016, 539, 536. [DOI] [PubMed] [Google Scholar]
- 28. Kano M., Minari T., Tsukagoshi K., Appl. Phys. Lett. 2009, 94, 143304. [Google Scholar]
- 29. Cho S., Seo J. H., Lee K., Heeger A. J., Adv. Funct. Mater. 2009, 19, 1459. [Google Scholar]
- 30. Xu Y., Sun H., Li W., Lin Y.‐F., Balestra F., Ghibaudo G., Noh Y.‐Y., Adv. Mater. 2017, 29, 1702729. [DOI] [PubMed] [Google Scholar]
- 31. Xu Y., Sun H., Shin E.‐Y., Lin Y.‐F., Li W., Noh Y.‐Y., Adv. Mater. 2016, 28, 8531. [DOI] [PubMed] [Google Scholar]
- 32. Günther A. A., Sawatzki M., Formánek P., Kasemann D., Leo K., Adv. Funct. Mater. 2016, 26, 768. [Google Scholar]
- 33. Khim D., Baeg K.‐J., Caironi M., Liu C., Xu Y., Kim D.‐Y., Noh Y.‐Y., Adv. Funct. Mater. 2014, 24, 6252. [Google Scholar]
- 34. Minari T., Darmawan P., Liu C., Li Y., Xu Y., Tsukagoshi K., Appl. Phys. Lett. 2012, 100, 093303. [Google Scholar]
- 35. Nicht S., Kleemann H., Fischer A., Leo K., Lüssem B., Org. Electron. 2014, 15, 654. [Google Scholar]
- 36. Reiser P., Benneckendorf F. S., Barf M.‐M., Müller L., Bäuerle R., Hillebrandt S., Beck S., Lovrincic R., Mankel E., Freudenberg J., Jänsch D., Kowalsky W., Pucci A., Jaegermann W., Bunz U. H. F., Müllen K., Chem. Mater. 2019, 31, 4213. [Google Scholar]
- 37. Liu Y., Cole M. D., Jiang Y., Kim P. Y., Nordlund D., Emrick T., Russell T. P., Adv. Mater. 2018, 30, 1705976. [DOI] [PubMed] [Google Scholar]
- 38. Larrain F. A., Fuentes‐Hernandez C., Chou W.‐F., Rodriguez‐Toro V. A., Huang T.‐Y., Toney M. F., Kippelen B., Energy Environ. Sci. 2018, 11, 2216. [Google Scholar]
- 39. Kolesov V. A., Fuentes‐Hernandez C., Chou W.‐F., Aizawa N., Larrain F. A., Wang M., Perrotta A., Choi S., Graham S., Bazan G. C., Nguyen T.‐Q., Marder S. R., Kippelen B., Nat. Mater. 2017, 16, 474. [DOI] [PubMed] [Google Scholar]
- 40. Li J., Rochester C. W., Jacobs I. E., Friedrich S., Stroeve P., Riede M., Moulé A. J., ACS Appl. Mater. Interfaces 2015, 7, 28420. [DOI] [PubMed] [Google Scholar]
- 41. Dai A., Wan A., Magee C., Zhang Y., Barlow S., Marder S. R., Kahn A., Org. Electron. 2015, 23, 151. [Google Scholar]
- 42. Lüssem B., Riede M., Leo K., Phys. Status Solidi A 2013, 210, 9. [Google Scholar]
- 43. Bruder I., Watanabe S., Qu J., Müller I. B., Kopecek R., Hwang J., Weis J., Langer N., Org. Electron. 2010, 11, 589. [Google Scholar]
- 44. Kim Y., Chung S., Cho K., Harkin D., Hwang W.‐T., Yoo D., Kim J.‐K., Lee W., Song Y., Ahn H., Hong Y., Sirringhaus H., Kang K., Lee T., Adv. Mater. 2019, 31, 1806697. [DOI] [PubMed] [Google Scholar]
- 45. Kang K., Watanabe S., Broch K., Sepe A., Brown A., Nasrallah I., Nikolka M., Fei Z., Heeney M., Matsumoto D., Marumoto K., Tanaka H., Kuroda S., Sirringhaus H., Nat. Mater. 2016, 15, 896. [DOI] [PubMed] [Google Scholar]
- 46. Nikolka M., Nasrallah I., Rose B., Ravva M. K., Broch K., Sadhanala A., Harkin D., Charmet J., Hurhangee M., Brown A., Illig S., Too P., Jongman J., McCulloch I., Bredas J.‐L., Sirringhaus H., Nat. Mater. 2017, 16, 356. [DOI] [PubMed] [Google Scholar]
- 47. Méndez H., Heimel G., Winkler S., Frisch J., Opitz A., Sauer K., Wegner B., Oehzelt M., Röthel C., Duhm S., Többens D., Koch N., Salzmann I., Nat. Commun. 2015, 6, 8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Himmelberger S., Dacuña J., Rivnay J., Jimison L. H., McCarthy‐Ward T., Heeney M., McCulloch I., Toney M. F., Salleo A., Adv. Funct. Mater. 2013, 23, 2091. [Google Scholar]
- 49. Wang C., Jimison L. H., Goris L., McCulloch I., Heeney M., Ziegler A., Salleo A., Adv. Mater. 2010, 22, 697. [DOI] [PubMed] [Google Scholar]
- 50. Umeda T., Tokito S., Kumaki D., J. Appl. Phys. 2007, 101, 054517. [Google Scholar]
- 51. McCulloch I., Heeney M., Bailey C., Genevicius K., MacDonald I., Shkunov M., Sparrowe D., Tierney S., Wagner R., Zhang W., Chabinyc M. L., Kline R. J., McGehee M. D., Toney M. F., Nat. Mater. 2006, 5, 328. [DOI] [PubMed] [Google Scholar]
- 52. Reiser P., Müller L., Sivanesan V., Lovrincic R., Barlow S., Marder S. R., Pucci A., Jaegermann W., Mankel E., Beck S., J. Phys. Chem. C 2018, 122, 14518. [Google Scholar]
- 53. Li J., Koshnick C., Diallo S. O., Ackling S., Huang D. M., Jacobs I. E., Harrelson T. F., Hong K., Zhang G., Beckett J., Mascal M., Moulé A. J., Macromolecules 2017, 50, 5476. [Google Scholar]
- 54. Kang K., Schott S., Venkateshvaran D., Broch K., Schweicher G., Harkin D., Jellett C., Nielsen C. B., McCulloch I., Sirringhaus H., Mater. Today Phys. 2019, 8, 112. [Google Scholar]
- 55. Patel S. N., Glaudell A. M., Peterson K. A., Thomas E. M., O'Hara K. A., Lim E., Chabinyc M. L., Sci. Adv. 2017, 3, e1700434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information