

Abstract
Many emerging applications in microscale engineering rely on the fabrication of 3D architectures in inorganic materials. Small‐scale additive manufacturing (AM) aspires to provide flexible and facile access to these geometries. Yet, the synthesis of device‐grade inorganic materials is still a key challenge toward the implementation of AM in microfabrication. Here, a comprehensive overview of the microstructural and mechanical properties of metals fabricated by most state‐of‐the‐art AM methods that offer a spatial resolution ≤10 μm is presented. Standardized sets of samples are studied by cross‐sectional electron microscopy, nanoindentation, and microcompression. It is shown that current microscale AM techniques synthesize metals with a wide range of microstructures and elastic and plastic properties, including materials of dense and crystalline microstructure with excellent mechanical properties that compare well to those of thin‐film nanocrystalline materials. The large variation in materials' performance can be related to the individual microstructure, which in turn is coupled to the various physico‐chemical principles exploited by the different printing methods. The study provides practical guidelines for users of small‐scale additive methods and establishes a baseline for the future optimization of the properties of printed metallic objects—a significant step toward the potential establishment of AM techniques in microfabrication.
Keywords: additive manufacturing, 3D printing, mechanical properties, micro, microstructure, metals, nano
The synthesis of device‐grade inorganic materials is a key challenge for small‐scale additive manufacturing. The presented comparison of microstructure and mechanical properties of metals fabricated by most current techniques shows a variation in material quality closely related to the individual method deposition principles. Importantly, it is shown that some approaches offer metals with properties equal to those of thin films used in microfabrication.
![]()
1. Introduction
Complex 3D structures with feature sizes in the micro‐ and nanometer range currently enable new areas of exciting growth in materials science and engineering. 3D geometries are often superior to their established, planar counterparts and enable a wide range of emerging applications in fields ranging from microelectromechanical systems (MEMS) and electronics to biomedicine and metamaterials.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ] As progress in 3D engineering follows from the ability to realize relevant structures from suitable materials,[ 4 ] the development of innovative small‐scale 3D fabrication methods is essential. At the required scales, additive manufacturing (AM) promises the greatest flexibility and closest control of synthesized geometries in comparison to alternative fabrication approaches, for example, self assembly and stress‐controlled deformation.[ 4 ] Hence, a large number of microscale AM techniques with a minimum feature size <10 µm have emerged in the past decade,[ 1 , 2 , 5 , 9 ] paving the way to additively manufactured 3D metamaterials,[ 10 , 11 , 12 , 13 , 14 ] printed electrical circuit elements,[ 15 , 16 , 17 , 18 , 19 ] or small‐scale sensors.[ 20 , 21 , 22 , 23 ]
Yet, to enable the above‐mentioned progress, AM techniques must provide the materials that today's microfabrication processes require: combinations of device‐grade, inorganic materials that provide structural, electronic, and optical functionality. This need is illustrated by the widespread use of two‐photon lithography (TPL) as a mere templating tool: after structuring an organic photoresist, the photosensitive material is exchanged with inorganic materials necessary for proper functioning of the final structure, for example, materials with a high dielectric constant for optical metamaterials,[ 10 , 11 ] or materials with a high stiffness or strength for mechanical metamaterials.[ 12 , 13 , 14 ] These materials are invariably deposited by established microfabrication processes such as atomic layer deposition[ 13 ] or electrodeposition.[ 10 , 12 ] While this symbiosis is an ideal approach for many applications, direct patterning of high‐performance materials is preferable for a number of reasons, including a reduction in the number of processing steps, the realization of geometries that are not dictated by requirements for subsequent template inversion (proper filling of complex templates can be demanding[ 24 ]) and—maybe most interesting for advanced designs of devices and materials—the possibility of spatially varying properties (not accessible with homogeneous coating or filling).
Unfortunately, direct AM of inorganic materials often results in materials that cannot yet satisfy the standards of modern microfabrication.[ 5 ] This shortcoming, in combination with challenging scalability and limited throughput, is a major handicap for incorporating AM in advanced micro‐ and nanofabrication routines. Materials engineering is thus necessary to improve the quality of printed inorganic materials. A first and crucial step toward this goal is the knowledge of the deposited materials' microstructure and its effect on the materials' properties. Here, we identify a deficit in existing literature: while some recent studies have covered materials optimization of particular AM techniques,[ 22 , 23 , 25 , 26 ] characterization of general microstructure‐property relationships of printed inorganic materials is in its infancy—a shortfall the present paper tackles, focusing on the mechanical performance of metals.
The indispensability of metals for the microfabrication of high‐performance 3D devices has motivated the development of multiple direct metal AM processes in the past decade.[ 5 ] As these techniques explore different physico‐chemical principles, the microstructure of the deposited metals varies greatly with respect to purity, defect density and porosity. As a result, a large variance in materials properties is observed. Studies on electrical properties report typical conductivities of 10−5 – 0.55 × bulk conductivity[ 15 , 16 , 25 , 27 , 28 , 29 , 30 , 31 ]. Similarly, elastic and plastic properties of additively fabricated metals vary over a range of ≈0.05 – 1 × bulk values (Table 1 ). While these studies enable isolated insights into the materials synthesized by different laboratories and techniques, their entirety does not allow for a properly founded overview of today's materials quality and related issues—the exemplary nature of previous work, the large variation in sample geometry and the different characterization techniques used prevent a fair comparison between techniques and their materials.
Table 1.
Studied small‐scale metal AM techniques. Techniques and printed metals tested in this study, including literature values and measured data for the mechanical performance of the printed materials: E: Young's modulus, H: hardness, σy: yield stress, σ0.07: flow stress at 7% strain. For inks, as‐deposited (ad.) and thermally annealed (ann.) metals are discriminated. References in the first column refer to historically important publications and notable review articles. Consult Table S1, Supporting Information, for average values of the mechanical properties of all tested samples including standard deviations. *Note: Pt nanoparticles embedded in a carbonaceous matrix
| Technique | Printed metal | Mechanical properties | ||||||
|---|---|---|---|---|---|---|---|---|
| (This study) | Literature | This study | ||||||
| E [GPa] | H [GPa] | σy [GPa] | E [GPa] | H [GPa] | σ0.07 [GPa] | |||
| Transfer techniques | ||||||||
| Colloidal inks | DIW (s.t.) – Direct ink writing of shear‐thinning inks[ 15 , 19 ] | Ag | 12.6 – 33.3 (ad.), 31.1 – 42.1 (ann.) | 0.141 (ad.), 0.435 – 0.461 (ann.) | 0.265 (ad.), 0.168 – 0.289 (ann.) | |||
| DIW (N.) – Direct ink writing of Newtonian inks[ 38 ] | Ag | 4.56 – 35.7 (ad.), 49.2 – 65.7 (ann.) | 0.066 (ad.), 0.855 (ann.) | 0.315 (ad.), 0.422 (ann.) | ||||
| EHDP – Electrohydrodynamic printing of nanoparticle inks[ 1 , 39 , 40 ] | Au | 2 × 10−4 (ad.),[ 41 ] 6 (ann.)[ 42 ] | 0.52 – 1.09 (ad.), 31.3 – 39.8 (ann.) | 0.048 (ad.) | 0.017 (ad.), 0.246 – 0.411 (ann.) | |||
| LAEPD – Laser‐assisted electrophoretic deposition of nanoparticles[ 43 ] | Au | 1.5 (ad.)[ 43 ] | 7.29 – 9.09 (ad.), 47.1 – 54.2 (ann.) | 0.369 (ad.), 1.42 (ann.) | 0.146 (ad.), 0.366 (ann.) | |||
| LIFT (ink) – Laser‐induced forward transfer of nanoparticle inks[ 17 , 44 , 45 , 46 ] | Ag | 54.8[ 47 ] | 1.37[ 47 ] | 1.16 – 1.94 (ad.), 12.8 – 36.4 (ann.) | 0.108 (ad.), 0.400 – 0.661 (ann.) | 0.013 (ad.), 0.088 – 0.118 (ann.) | ||
| Melts | LIFT (melt) – Laser‐induced forward transfer of thin films melts[ 48 ] | Au, Cu | 12 (Cu), 9 (Au)[ 49 ] | 49.8 –73.2 (Cu), 24.3 – 28.3 (Au) | 1.66 (Cu), 0.293 (Au) | 0.415 (Cu), 0.186 (Au) | ||
| Synthesis techniques | ||||||||
| Electrochemical | MCED – Meniscus‐confined electrodeposition[ 16 , 26 , 50 , 51 , 52 ] | Cu | 128[ 51 ] | 2[ 51 ] | 0.63 – 0.962[ 26 , 53 ] | 114.2 – 121.8 | 2.71 | 0.774 |
| FluidFM – Confined electrodeposition in liquid[ 54 , 55 , 56 ] | Cu | 0.7 – 0.9[ 54 ] | 134.0 – 138.4 | 2.28 | 0.962 | |||
| EHD‐RP – Electrohydrodynamic redox printing[ 57 ] | Cu | 81[ 57 ] | 1 – 1.5[ 57 ] | 80.4 – 81.7 | 2.22 | 1.10 – 1.38 | ||
| Electron/ion‐induced CVD | FIBID – Focused ion beam‐induced deposition[ 29 ] | Pt* | 95.3 – 140.0 | 9.42 | 2.64 | |||
| FEBID – Focused electron beam‐induced deposition[ 22 , 29 , 58 ] | Pt* | 10 – 100[ 22 , 59 , 60 , 61 ] | 3.7 – 7.6[ 62 ] | 1 – 2 (tensile) [ 59 ] | 59.3 – 75.5 | 6.01 | 2.65 | |
| cryo‐FEBID – FEBID at cryogenic temperatures[ 63 ] | Pt* | 3.85 – 13.8 | 0.843 | 0.100 | ||||
Here, we present a comprehensive overview of the microstructure of metals printed by most contemporary small‐scale AM processes, and relate the microstructure of these metals to their mechanical performance. Mechanical properties of 3D metal structures are a key for many applications at small scales, both in traditional MEMS[ 32 ] and for projected implementations of printed metals, for example, unsuspended interconnects for flexible electronics,[ 15 ] out‐of‐plane, high‐aspect‐ratio switches,[ 33 , 34 ] sensors[ 21 , 22 ] and probes,[ 20 , 23 ] or printed actuators and manipulators.[ 34 , 35 ] Studying a standardized set of samples by cross‐sectional electron microscopy, nanoindentation and microcompression, we show that a range of characteristic microstructures causes a significant variation in elastic and plastic properties within one order of magnitude. On one hand, the standardized approach used in this study allows a fair comparison of the capabilities of the various small‐scale AM methods (within the boundaries discussed). On the other, the presented overview is the groundwork for future optimization of materials performance.
2. Overview of Included AM Methods and Samples
The microscale metal AM techniques included in this study are listed in Table 1, and schematics of the respective working principles are shown in Figure 1 . The processes are classified according to their underlying principle of material deposition to emphasize the fundamental similarities that determine the properties of the resulting materials. We group the techniques in two main categories: transfer and synthesis techniques. Transfer techniques require the previous synthesis of metallic materials before the actual AM process—the subsequent deposition simply transfers the pre‐synthesized material to the location of interest. In contrast, the synthesis methods rely on the growth of the metal at the location of interest during the AM process. Both categories are further divided into the sub‐groups of methods that do or do not use colloidal inks (transfer techniques), and methods that use wet electrochemistry or local electron/ion‐initiated surface reactions with physisorbed molecules supplied from the gas phase (synthesis methods). Relevant techniques not covered in this paper are two‐photon‐induced reduction of metal ions,[ 30 ] pyrolysis of TPL‐structured, metal‐containing resins,[ 36 ] and implosion fabrication.[ 37 ]
Figure 1.
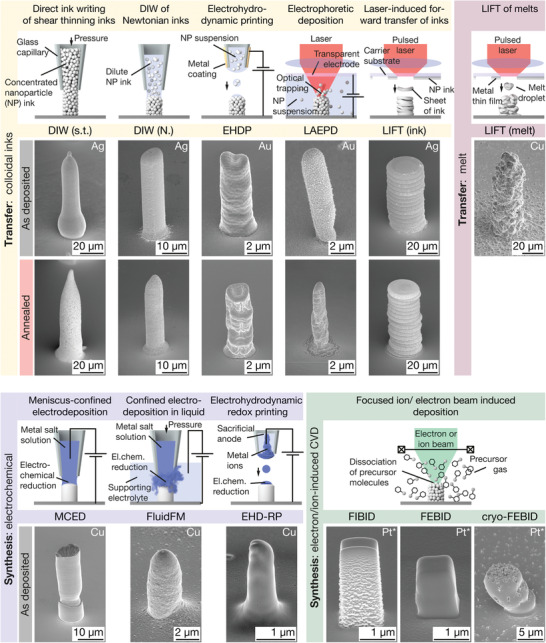
Small‐scale metal AM methods included in this study and pillars printed by these techniques. Small‐sale AM methods are grouped into transfer and synthesis methods, based on their principle of metal deposition. Subgroups include: Transfer of colloids, transfer of melts, electrochemical synthesis and synthesis via electron/ion‐induced CVD. The SE micrographs show representative pillars printed with each of the techniques tested in this study. As‐printed and annealed pillars are shown if thermal annealing was performed (not the same samples). Samples for DIW and LIFT (ink) are printed from Ag inks, and samples from EHDP and LAEPD from suspensions of Au nanoparticles. The LIFT (melt) pillar is Cu (for Au, see Figure S1, Supporting Information), as are the structures for MCED, the FluidFM and EHD‐RP. FIBID and (cryo‐)FEBID pillars were deposited from Methylcyclopentadienyl platinum (IV) trimethyl (MeCpPt(Me)3). *: Pt nanoparticles embedded in a carbonaceous matrix. Tilt angle of all micrographs: 55°.
For a detailed description of the working principle of the different AM techniques and their performance characteristics, please refer to our review.[ 5 ] In brief, printing metals from colloidal inks is typically a two‐step process that requires post‐print thermal annealing to render metallic materials, burning off organic components and sintering the particles. The principles of ink transfer to the substrate vary between the techniques (DIW, EHDP, LAEPD, and LIFT (ink), see Table 1 for an explanation of the acronyms), but resulting materials characteristics are comparable: an agglomeration of surfactant‐coated micro‐ or nanoparticles in the as‐deposited state, and a crystalline, sintered microstructure in the annealed state. LIFT of metallic melts is an ink‐free transfer method that does not demand any post‐print treatment, as it transfers molten droplets of metals that solidify as pure metals on the substrate. Electrochemical methods (MCED, FluidFM, EHD‐RP) use different approaches to localize growth, but all rely on the electrochemical reduction of metal ions and generally offer as‐deposited dense and crystalline metals. Focused particle beam methods (FIBID, FEBID, cryo‐FEBID) make use of electron‐induced dissociation of physisorbed gaseous precursors (typically organometallic compounds) to synthesize metal‐carbon composites with metallic characteristics—as the process can be likened to a localized non‐thermal CVD process, we use the term ”electron/ion‐induced CVD” to summarize the working principle.
Samples for this study consisted of a standardized set of pads and pillars that was printed with each technique to assess both the microstructural as well as the mechanical properties of the deposited metals. Materials presented here were optimized in the authors' laboratories to the best of the authors' knowledge, but likely do not yet present the optimal microstructure that could possibly be achieved by the individual methods. Due to current process limitations, the elemental nature of the studied metals varies between techniques: Au and Ag for ink‐based methods, Cu for electrochemical methods, and Pt for FEBID and FIBID (Table 1). Further, the dimensions of the samples were adjusted to the spatial resolution and volumetric print speed of the individual technique. Pads ranged from 10 to 200 µm in width and 400 nm to >10 µm in thickness. Pillar diameters were typically ≥1 µm, with maximum diameters of ≈45 µm. A set of as‐deposited and annealed samples was prepared for nanoparticle inks, because thermal annealing is generally required for functional performance of these materials. In contrast, only as‐deposited but no post‐processed (annealed, cured, purified) FIBID and FEBID structures were included, because the vast majority of reported applications is limited to as‐deposited materials[ 5 ] and existing purification and annealing procedures[ 28 ] are not yet applied by a broad range of practitioners.
3. Results
3.1. Morphology of Printed Samples
Representative samples printed with each of the techniques are displayed in Figure 1 and 2 . If multiple annealing states were prepared, the sample annealed the longest at the highest temperature is shown. Cu and Au structures were tested for LIFT (melt), but only Cu is shown here (for Au, refer to Figure S1, Supporting Information). Note that the dimensions of the printed structures were dictated by the requirements of mechanical testing: pads were required to be at least a few hundred nanometers in thickness and ≥10 µm in width (without any upper boundaries for thickness and size) and were supposed to have a smallest possible surface roughness. Pillars had to be at least 1 µm in diameter and 2 – 3 µm in height (with a maximum diameter of 50 µm). No flat top was required. Any conclusions regarding resolution and smallest printable geometries should be made with these boundary conditions in mind—none of the methods strived to synthesize the smallest geometries for this study.
Figure 2.

Printed pads. SE micrographs of representative pads printed with each of the techniques. If thermal annealing was performed, samples of as‐printed and annealed pads are shown. Metals: DIW, LIFT (ink): Ag; EHDP, LAEPD: Au. LIFT (melt), MCED, FluidFM, EHD‐RP: Cu. FIBID, (cryo‐)FEBID: MeCpPt(Me)3. For Au deposited by LIFT (melt), see Figure S1, Supporting Information. *: Pt nanoparticles embedded in a carbonaceous matrix. Tilt angle: 45°.
In general, the morphology and volume of the printed pads and pillars reflect the deposition principle of each individual technique. FEBID and FIBID geometries show highest resolution and lowest surface roughness but their build volume is limited to a few cubic micrometers. In contrast, structures printed by LIFT (melt) feature the highest microscale surface roughness but their volume is only rivaled by DIW samples. DIW pillars drawn from a single filament are smooth, but the pads' surface modulation with a modulation length of tens of micrometers clearly reveals the in‐plane hatch pattern. Techniques with submicrometer feature size (EHDP, EHD‐RP, FIBID, FEBID, and cryo‐FEBID) enable printing of micrometer‐sized pillars with in‐plane hatching and hence control the pillar's shape (for example square in the case of EHD‐RP, FIBID and FEBID, Figure S2, Supporting Information). Commonly, pads were fabricated with in‐plane hatching. The few pads deposited without in‐plane hatching show lowest surface roughness (LIFT (ink), MCED), but the stacking of layers results in a pronounced roughness of LIFT (ink) printed pillars. In cryo‐FEBID pillars, substrate drift during the cycling between precursor deposition and patterning caused a shift between individual layers.
Thermal annealing of DIW and LIFT (ink) geometries exhibits good shape retention. In contrast, sintering of EHDP and LAEPD structures caused shrinkage and warping. In the case of EHDP, the shrinkage even resulted in unsuspended pads that prohibited reliable indentation testing (Figure S9, Supporting Information). The warping of EHDP pillars was less pronounced for small diameters (printed without in‐plane hatching and lower flow rates, Figure S10, Supporting Information).
3.2. Microstructure
The microstructure of printed metals was assessed by cross‐sectional electron microscopy of both pads (Figure 3 ) and pillars (Figure 4 ). If multiple annealing states were prepared, the sample annealed the longest at the highest temperature is shown. Only Cu is shown for LIFT (melt). For a complete collection of the microstructure of all samples and micrographs of pads with lower magnification, please refer to Section S3 and Figure S3, Supporting Information. Figure S5, Supporting Information, provides a qualitative overview of the chemical composition of printed pads analyzed by EDX spectroscopy. Quantitative microstructure analysis of grain size, porosity, or texture is omitted as a consequence of the comprehensive character of the study: the focus is on common features or distinctions between the materials synthesized by the different methods rather than on detailed structure‐property relationships. We note that a thorough analysis will be necessary for future materials optimization.
Figure 3.
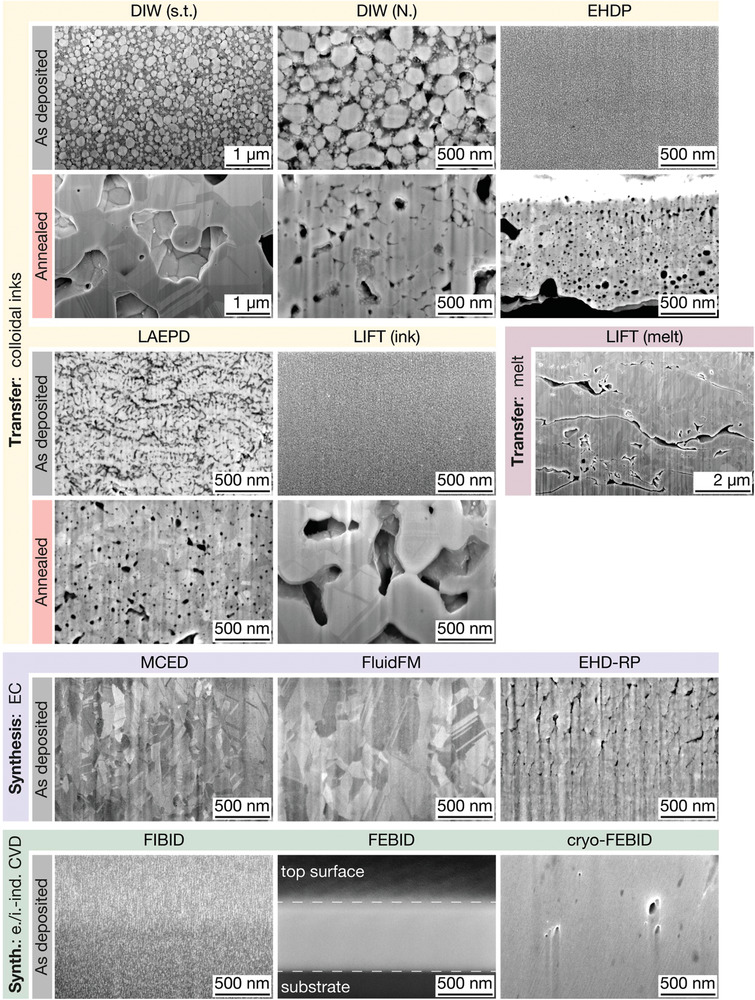
Microstructure of printed pads. Representative cross‐section micrographs of printed pads. Four distinctive microstructures are observed: 1) Agglomerations of metal colloids and organic constituents (as‐deposited inks), 2) polycrystalline and porous metals (transfer techniques, annealed), 3) polycrystalline and dense metals (electrochemical techniques), and 4) dense metal‐carbon composites (FIBID and FEBID). Note: FIB curtaining effects are observed in the following micrographs and are imaging artefacts rather than real features: DIW (N.), LAEPD, LIFT (ink), LIFT (melt), EHD‐RP and cryo‐FEBID. For micrographs of lower magnification, see Figure S3, Supporting Information. All images are tilt‐corrected.
Figure 4.

Microstructure of printed pillars. Representative cross‐section micrographs of printed pillars. The microstructure of the printed pillars is generally comparable to the microstructure of the corresponding pads. However, radial microstructure gradients are obvious in most annealed pillars printed from colloidal inks (DIW, EHDP, LIFT (ink)), whereas pillars printed by synthesis methods and LIFT (melt) are more homogeneous. The line feature in the FEBID pillar was caused by an interruption of the automated exposure pattern. All images are tilt‐corrected.
Four types of microstructure are identified if the micrographs are reduced to their common denominators: 1) agglomerations of metal colloids and organic constituents, 2) polycrystalline and porous metals, 3) polycrystalline and dense metals, and 4) dense carbon‐metal composites. All as‐printed colloidal inks belong to category (1). The non‐metallic contrast (dark) in the micrographs in Figures 3 and 4 is interpreted as organic components, based on the chemical analysis that suggests significant amounts of carbon in as‐printed inks (Figure S5, Supporting Information) and on the fact that all inks in this study are composed of micro‐ and nanoparticles coated with organic surfactants and mixed with organic solvents. All transfer methods eventually synthesize materials of category (2) (crystalline and porous), although the degree and homogeneity of the observed porosity is subject to large variations. The microstructure synthesized upon thermal consolidation of colloidal inks is pronouncedly porous. The pore size ranges from tens of nanometers to approximately one micron and pores do not show a preferential orientation. The absolute pore fraction varies between techniques (Figure 3) and annealing states (Section S3, Supporting Information), and can range from ≈5 – 35% (estimated areal fraction by threshold analysis of cross‐section micrographs, not shown). Metals printed by LIFT of Au and Cu melts are also porous, although the density of deposited Cu is higher than that of Au (Figure S1, Supporting Information). In contrast to annealed inks, the pore distribution is pronouncedly inhomogeneous, the pore size in the microscale range, and the pore shape non‐spherical (pores are often elongated inter‐droplet gaps). Additionally, the grain size can vary by one order of magnitude within a printed structure. Category (3) (polycrystalline and dense) is populated by Cu synthesized by additive electrochemical methods, with a typical microstructure that is nano‐ to micro‐crystalline and dense (that is, no obvious porosity is detected at the studied length scale, except two isolated pores approximately 500 nm in size in the MCED pillar). Yet, EHD‐RP pillars feature nanoscale voids. In contrast, pads may start to grow with higher density but evolve mostly vertical gaps at either a certain height (≈500 nm in Figure 3) or already after a few layers (Figure S17, Supporting Information). Room‐temperature FEBID and FIBID deposits belong to category (4) (carbon‐metal composites): they are dense, but, as established in literature, consist of metal nanoparticles embedded in an amorphous carbon matrix.[ 29 ] A typical metal content achieved with commercial setups at room temperature is approximately 10 – 15 at%[ 28 ] for MeCpPt(Me)3, the precursor used in this study. FEBID deposits grown at cryogenic temperatures have been reported to have a similar composition.[ 64 ] In contrast to room‐temperature FEBID materials, cryo‐FEBID pillars feature pronounced, vertically elongated pores (Figure 4).
The homogeneity of the deposited microstructure, that is the spatial distribution of grain and pore size and pore fraction, varies between techniques and also printed structures. Annealed inks often feature gradients in porosity and variations of grain sizes within a deposit. This phenomenon is observed mostly in pillars (Figure 4), and less frequently in pads (Figure S4, Supporting Information). The micrographs in Figure 4 specifically suggest that the outer region of pillars is often denser and features smaller pore sizes (DIW, EHDP, LIFT (ink)). In extreme cases, annealed pillars developed hollow centers (EHDP, LIFT (ink)). In contrast, synthesis techniques produce a homogeneous microstructure in both pads and pillars (apart from the large pores observed in the pillars by MCED, cryo‐FEBID, and the change in microstructure as a function of height in the pads by EHD‐RP).
3.3. Mechanical Properties
The mechanical properties of the printed materials were analyzed by nanoindentation of pads and microcompression of pillars, deriving the Young's modulus E (nanoindentation, and microcompression), the hardness H (nanoindentation) and the flow stress at 7% strain σ0.07 (microcompression). Figure 5 presents indentation and microcompression data of samples printed by three techniques: DIW (s.t.) (representing transfer techniques), FluidFM (electrochemical synthesis) and FIBID (electron/ion‐induced CVD). The complete dataset including all samples of all techniques is provided in Section S3, Supporting Information. The averaged values of E, H, and σ0.07 are plotted in Figure 6a,b and listed in Table 1 and, in more detail, in Table S1, Supporting Information. Figure 6c,d normalizes the measured values to literature values of thin films fabricated by traditional deposition techniques used in microfabrication (PVD and electrodeposition, denoted with the subscript PVD, Table S3, Supporting Information). These data are presented for a qualitative comparison of the state‐of‐the‐art performance of printed metals to that of metals established in microfabrication. Additionally, the normalization enables the comparison between the different metal elements deposited by the various additive techniques. As a note: the Young's modulus of thin films E PVD is consistent in literature and usually comparable to the bulk Young's modulus E, because the elastic properties are mostly independent of a metal's microstructure (apart from porosity). Hence, the normalized data for E are representative. In contrast, literature values for H PVD easily vary by a factor of two due to variations in microstructure of the deposited films (H Ag: 0.7 – 1.5,[ 65 , 66 ] H Au: 1 – 2 GPa,[ 67 , 68 ] H Cu: 1.6 – 3.5 GPa,[ 69 , 70 , 71 , 72 ] H Pt: 1.5 – 8.6[ 73 , 74 , 75 ]). Consequently, the normalized H data should be treated with care, although we tried to select a value that is most representative of the available literature (Ag: 1.2 GPa, Au: 1.2 GPa, Cu: 2 GPa, Pt: 4 GPa) to normalize our data.
Figure 5.
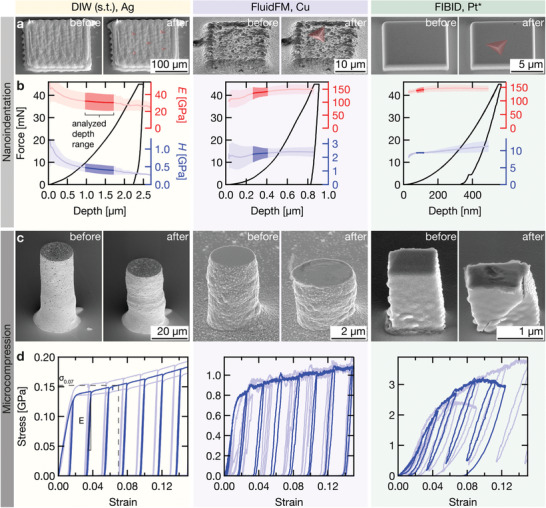
Mechanical testing. Nanoindentation and microcompression data of samples printed by three exemplary techniques: DIW (s.t.) representing transfer techniques, FluidFM for electrochemical techniques, and FIBID for electron/ion‐induced CVD. a) Representative pads before (left) and after (right) nanoindentation. The indents are highlighted in red. Tilt angle: 45°. b) Single indentation curves (black) and average Young's modulus E (red) and hardness H (blue) as a function of indentation depth. The solid line is the mean value derived from all measured indents, the shaded area the standard deviation. Elastic and plastic properties are reported from the highlighted depth range. c) Pillars before (left) and after (right) microcompression. Tilt angle: 55°. d) Three representative stress‐strain curves measured for each technique. One curve is highlighted for clarity. *: Pt nanoparticles embedded in a carbonaceous matrix.
Figure 6.
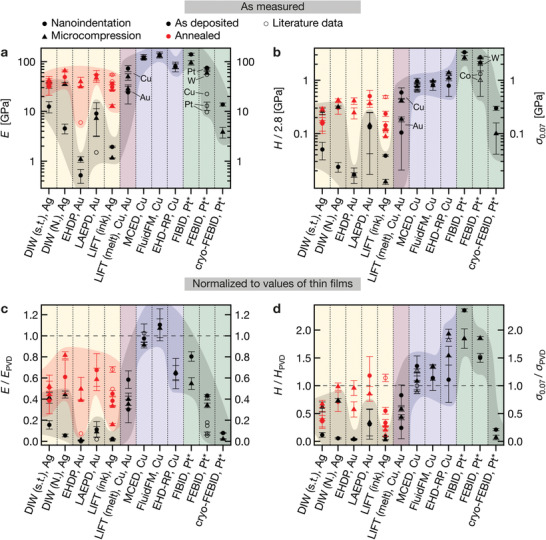
Young's modulus E, hardness H and flow stress σ0.07 of printed metals. Element symbols indicate the printed metals. a) E measured by nanoindentation (circles) and microcompression (triangles) for as‐deposited (black) and annealed (red) samples of all techniques. Error bars are one measured standard deviation. Literature data are open symbols. Element labels of FEBID data points refer to literature data. b) H (circles) and σ0.07 (triangles). Hardness data is divided by a constraint factor of 2.8 for a direct comparison with the flow stress. Literature data: open symbols. Note that these graphs intend to summarize the measured data and should not be perceived as a quantitative ranking of the absolute capabilities of the techniques, as future materials optimization may improve the microstructure of the printed materials. c, d) Measured data normalized by literature data for thin films deposited by traditional microfabrication methods (Table S3, Supporting Information). Note that the Young's modulus of thin films E PVD is consistent in literature. Hence, the normalized data for E is representative. In contrast, literature values for H PVD are subjected to considerable variations. The normalized H data should be treated with corresponding care, although we tried to select a value that is most representative of the available literature. References for literature data: EHDP,[ 42 ] LAEPD,[ 43 ] LIFT (ink),[ 47 , 78 , 79 ] MCED,[ 26 , 51 ] FluidFM,[ 54 ] EHD‐RP,[ 57 ] FEBID.[ 22 , 59 , 60 , 61 , 62 ] *: Pt nanoparticles embedded in a carbonaceous matrix.
Both the elastic and plastic properties vary by two orders of magnitude between the individual techniques if all as‐printed samples are considered, and by one order of magnitude if as‐printed inks are excluded. The normalized modulus E/E PVD of annealed inks and samples printed by LIFT is always <1, while their hardness H/H PVD can be >1. Electrochemical techniques enable E/E PVD = 1 (except EHD‐RP with E/E PVD < 1) and H/H PVD > 1. Room‐temperature FEBID and FIBID structures exhibit a lower elastic modulus than Pt, but a hardness that is higher than that of most metals routinely used in microfabrication (H: 6 – 9 GPa, typical hardness of Pt thin films ≈1.5 – 5 GPa[ 73 , 75 ]). Thus, the normalized hardness H/H PVD of FEBID and FIBID structures is >1, although E/E PVD < 1 (A comparison to hardest metals, for example W with a hardness of 6 – 18 GPa[ 76 , 77 ] would yield H PVD < 1). An exception is cryo‐FEBID deposits, which are pronouncedly more compliant and softer than their room‐temperature counterparts.
4. Discussion
4.1. Limitations of the Study and Validity and General Applicability of the Results
Generally, material properties are a function of a material's chemistry and microstructure. This implies that no absolute value for any property can be assigned to the individual small‐scale AM methods, because each technique can potentially synthesize materials with a range of microstructures and compositions by making adjustments to the feedstock materials and the process parameters. Thus, the generalizations of the microstructural and mechanical data reported here are constrained by the following considerations. Within the limits discussed below, the reported values are representative and reproducible for each technique. Yet, these limits—imposed by the narrow range of metals, the absence of materials' optimization, and the non‐ideal test geometries—underline the necessity to treat the here‐reported data as ranges rather than absolute numbers. The measured values are mere snapshots of the materials synthesized today. These should be perceived as a visualization of the commonalities and differences between the individual techniques rather than as absolute and static maxima for any given method.
Three factors constrain the interpretation of the reported results. First, the materials tested in this study are representative materials synthesized in the authors' laboratories, but were not optimized for high mechanical strength. Hence, within the limitations of each technique, higher modulus and strength might be expected once printing and annealing processes are optimized. Such optimization, however, is beyond the scope of this study. Second, the range of metals most commonly studied is very limited and different for the various deposition methods: Ag or Au for ink‐based techniques, Cu for electrochemical deposition, and Pt, Co, Fe or W in the case of FEBID/ FIBID. As the study had to comply to these limits, the obtained data cannot entirely be decoupled from the fact that different elements were used. Consequently, the results cannot be linked exclusively to the process characteristics of the individual AM principles. However, general microstructural features directly related to the deposition principles typically remain unaffected by the chemical nature (porosity in Au and Ag inks, inter‐droplet gaps in LIFT printed Au and Cu structures, Figure S1, Supporting Information). Third, an accurate measurement of mechanical properties requires well‐defined geometries of the test specimens. In this respect, many of the used small‐scale AM methods struggle to deposit samples with ideal geometries. Due to the limited deposition rates of some techniques and little control of the surface roughness, samples for nanoindentation analysis were sometimes characterized by a low pad volume—manifested as a sample thickness <1 – 2 µm (FIBID, FEBID) or a limited sample width, which itself caused distances from an indent to the edge of a pad <10 × the indentation depth (EHDP, LAEPD, FluidFM, EHD‐RP, FIBID, and FEBID: pad width only 10 – 20 µm)—or a high surface roughness ≫100 nm (LIFT (melt)). No model for correcting the influence of the limited geometry has been applied to the data. The data are shown as measured. Due to problematic combinations of roughness and limited pad thickness, some nanoindentation data were analyzed at a depth >1/10 of the pad thickness without accounting for substrate effects and should be treated with corresponding care (Table S2, Supporting Information). To improve the reliability of the data, the nanoindentation measurements were complemented by microcompression results. Microcompression analysis was complicated by inhomogeneous pillar diameters and pillar tilt (LAEPD, EHDP [annealed only], MCED, cryo‐FEBID). For some AM methods, these factors result in a pronounced scatter between individual samples: measured standard deviations typically range between 10 – 20% but can be as high as 40 – 80% for a few techniques, especially for nanoindentation data (Section S3, Supporting Information). Despite these difficulties, a qualitative comparison of the data derived from the two methods suggest that the influence of the sample geometry is not dominant, compared to any of the other measurement uncertainties. The elastic moduli measured with both methods, as well as the hardness of the pads (divided by a constraint factor of 2.8) compared to the strength of the pillars, are often in similar ranges or identical if standard deviations are respected.
4.2. Morphology
The samples of this study were not fabricated for benchmarking the resolution or surface roughness of the individual small‐scale AM techniques. For a general overview of the geometrical capabilities of the respective techniques, please refer to our recent review article.[ 5 ] Nevertheless, two points are noteworthy and further discussed in Section S1, Supporting Information. First, thermal annealing of inks can cause distortion of printed objects due to shrinkage upon coalescence of individual particles, but such a distortion is not always observed. Second, the different printing strategies utilized by the different techniques—with or without in‐plane hatching depending on whether the technique offers an adaptable feature size—not only affect the print speed but also the sample morphology.
4.3. Microstructure and Resulting Mechanical Properties
Four types of microstructure were identified:
-
1.
Agglomeration of metal colloids and organic constituents (as‐printed inks)
-
2.
Polycrystalline and porous metals (annealed inks and LIFT of melts)
-
3.
Polycrystalline and dense metals (electrochemical synthesis)
-
4.
Dense carbon‐metal composites (electron/ion‐induced CVD)
These microstructures result in four distinctive classes of mechanical performance:
-
1.
E/E PVD ≪ 1, H/H PVD ≪ 1 (as‐printed inks)
-
2.
E/E PVD < 1, H/H PVD ≤ 1 (annealed inks and LIFT of melts)
-
3.
E/E PVD ≤ 1, H/H PVD ≥ 1 (electrochemical synthesis)
-
4.
E/E PVD < 1, H/H PVD > 1 (electron/ion‐induced CVD)
4.3.1. Transfer Techniques: Inks
As‐deposited inks are composed of individual micro‐ or nanoparticles. The volumetric metal content of the inks is a function of particle size distribution and amount of nonvolatile organic ink constituents. Due to organic capping layers and residual solvent, the filling factors of these colloidal systems are necessarily below the theoretical maximal values for hard spheres. Especially in the case of nanoparticles, the surfactant layers can occupy a considerable volume. For illustration, Richner et al.[ 80 ] have calculated theoretic volume filling factors as small as 0.21 – 0.26 for nanoparticle inks (random loose packing, 5 nm particle dia.). Nevertheless, inks can be compared to colloidal crystals in a first approximation, despite their lower filling factors. The mechanical behavior of all as‐deposited inks (E: 0.5 – 10 GPa, H: 30 – 600 MPa) is similar to that of nanoparticle colloidal crystals (E: 0.1 – 6 GPa,[ 81 , 82 ] H: 10 – 450 MPa[ 81 ]). The considerable variation is presumably owed to the large difference in particle packing density, the differences in particle shape and size distribution which will both affect the flow properties, the chemical instability of as‐printed inks (and thus changing properties over time, also when kept in inert atmosphere), and the differences in ink preparation (some inks were actively dried after printing, others not). Two exceptions from this general picture of colloidal inks were observed. First, the strength of DIW inks determined by microcompression was significantly higher than the respective hardness derived from nanoindentation of the same materials (briefly discussed in Section S3.1, Supporting Information). Second, we hypothesize that electrophoretically printed Au nanoparticles are fused upon laser‐assisted deposition (Figure 3), resulting in the high strength observed for as‐deposited LAEPD samples (Section S2.1., Supporting Information).
The microstructure of annealed inks is dominated by inter‐granular porosity (ranging from ≈5 – 35%) resulting from incomplete densification upon sintering, although the absolute pore fraction varies between techniques (Figure 3) and annealing states (Section S3, Supporting Information). Additional to the mere presence of porosity, it is frequently inhomogeneously distributed in printed pillars (Figure 4). These gradients in pore fraction, pore size, and grain size are probably a result of local variations in sintering conditions (heat conductance and heat capacitance) in combination with mechanical constraints exerted by both the substrate and previously sintered material (as discussed below). Apart from the microstructure, the shrinkage upon annealing can affect the printed geometries by warping or partial delamination.
In general, the observed behavior of annealed inks is in qualitative and quantitative agreement with previous mechanical studies of Ag inks reporting moduli and hardnesses of 10 – 100 GPa and 0.03 – 1.4 GPa, respectively[ 47 , 83 , 84 ] and decreasing strength with longer annealing due to coarsening of the porosity (ligament size effect).[ 85 ] The porosity and its characteristic length scale in combination with the nanoscale grain size results in the peculiar observation that the modulus of annealed inks is invariably inferior to that of dense thin films, but their normalized strength can be close to unity or larger. While the elastic properties of a porous material are mainly determined by the fraction and shape of its pores,[ 86 , 87 ] resulting in a decreased stiffness with increased porosity, the yield strength is additionally modulated by the grain size as well as the ligament size (in highly porous solids such as inks after short annealing[ 85 , 88 ]). Consequently, a porous material can be strong if the grain size or ligament size is sufficiently small so that Hall‐Petch strengthening or ligament size effects compensate for the loss in strength due to the lower density. The strength of these materials typically decreases upon prolonged annealing due to coarsening of grains and ligaments (compare microcompression results in Figures S6 and S7, Supporting Information and nanoindentation hardness in Figures S12 and S13, Supporting Information).
The pronounced porosity and the large spread in elastic and plastic properties reported in this study, as well as in the literature, highlight a major challenge for ink‐based techniques: shrinkage upon annealing is inherent to the concept of colloidal inks. Accompanying effects, such as the evolution of porosity, warping of printed geometries or stress‐induced failure, predominantly characterize the properties of the printed materials and structures. Thus, the management of the densification and coarsening processes during annealing is required for most applications. The same conclusions apply to metals synthesized by pyrolysis of metal‐containing resists structured by TPL,[ 36 ] one of the metal AM methods not included in this study. Here, a linear shrinkage of 80% upon pyrolysis highlights the challenge for dealing with volume loss in extremis.
The observed porosity of the annealed inks contrasts with the low residual porosity usually reported for macroscale structures fabricated by DIW of metal and metal oxide inks (with 50% isotropic linear shrinkage).[ 89 ] Nearly 100% density is achieved in a range of sintered metal colloids, including Fe and Ni,[ 90 ] W,[ 91 , 92 ] or CoCrFeNi high‐entropy alloys.[ 93 ] To explain the marked difference, a comparison of the boundary conditions of micro‐ and macroscale sintering is insightful. At large scales, the absence of mechanical constraints is an important factor in preventing residual porosity. Without the mechanical constraint of a substrate, the sintering of macroscale DIW geometries facilitates unconstrained, homogeneous shrinkage and thus densification. In contrast, at small scales, printed microscale objects are not free to shrink and densify as they are (and usually need to be) invariably attached to a substrate. Accordingly, shrinkage is constrained both by their support and also the material that has been sintered before and is already firmly attached to the substrate (such as the outer regions of annealed ink pillars or the surface of EHDP pads). Hence, complete densification cannot be achieved, as the evolving stresses counteract the coalescence of pores or result in failure or delamination. In this light, other concepts that typically enable high densification at larger scales are probably also less effective at small scales, including higher sintering temperatures (homologous temperatures used for sintering of large‐scale structures are typically in the range of ≈0.35 – 0.9,[ 90 , 92 ] and were ≈0.2 – 0.4 for the present study) and the use of sintering aids.[ 91 ]
Consequently, a compromise between maximum densification and minimal shrinkage is probably necessary for ink‐printed geometries at small scales. For the highest strength combined with the best shape retention, a brief annealing that guarantees neck formation between individual nanoparticles, but avoids coarsening of the microstructure, is probably the most promising approach, as it avoids large shrinkage but offers high strength via ligament size effect strengthening. High densification via thermal sintering will, in most cases, be accompanied by pronounced grain growth and hence a weakening of the metal. Thus, alternative strengthening mechanisms such as precipitation hardening should be considered for high strength if nanoporous structures are not an option. Unfortunately, it is likely that sintering protocols need to be adjusted to every geometry and ink‐substrate combination. The observed gradients in microstructure and differences between the microstructure of pillars and pads subjected to the same thermal treatment (Figure S4, Supporting Information) suggest that the local densification is influenced by local boundary conditions, which vary for every geometry and materials combination. Nevertheless, although this optimization is not a trivial task, a large body of established sintering knowledge from powder metallurgy could, in principle, guide strategies for improved annealing of printed inks.
4.3.2. Transfer Techniques: LIFT of Melts
LIFT of metal melt droplets renders as‐printed functional materials that do not require any post‐print processing. Nevertheless, as with inks, residual porosity is a characteristic feature of the synthesized materials. This observation is in agreement with literature reports of porosities of 5 – 15% in Cu[ 25 , 94 , 95 ] and up to 50% in Au.[ 95 ] Grain and pore sizes measured in this study are large compared to that of inks (early annealing stages), but grain sizes <100 nm have been reported for LIFT‐printed Cu.[ 96 ] The porosity in combination with a medium grain size results in normalized elastic and plastic properties that are lower than those of dense thin films, although the measured elastic moduli of 50 – 75 GPa (Cu) and 25 – 30 GPa (Au) are higher than previously reported values (Cu: 12 GPa, Au: 9 GPa[ 49 ]).
The large pore size, the inhomogeneous pore distribution, and the comparably high surface roughness are challenges for the use of these materials in mechanical applications at small scales. To some extent, the porosity and roughness can be minimized by alternative printing algorithms and a higher laser fluence, increasing the impact energy and temperature of the droplets.[ 25 ] As a complication, the observed microstructural inhomogeneities (variations in density and grain size) suggest that the boundary conditions for solidification are subjected to strong local variations. Consequently, microstructure engineering is a task with many variables. Nevertheless, optimization for higher strength by increasing the defect density of the melt‐solidified metal could be achieved, especially in Cu with its comparably low porosity. Because direct control of the local cooling rates and hence the evolving grain size is inaccessible, alternative approaches to microstructure engineering should be considered. As the LIFT process facilitates alloying,[ 94 ] solid solution hardening, or precipitation of an immiscible phase for hardening or grain boundary pinning could be a viable path to increase the defect density and thus the strength, regardless of local cooling rates.
4.3.3. Synthesis Techniques: Electrochemical
Cu synthesized by MCED and the FluidFM, the two techniques that use traditional aqueous electrochemistry, is dense and crystalline with grain sizes of <100 – 500 nm. Isolated pores were observed in the pillars grown by MCED. Such pores do not exist in similar studies in the literature,[ 26 , 51 ] suggesting that they can be eliminated by adjusting deposition parameters. In general, microstructural data comparable to our observations have been published earlier.[ 26 , 51 , 54 , 97 , 98 ] As a result of the high density, the measured elastic and plastic properties (E: 114 – 138 GPa, H: 2.3 – 2.7 GPa) can easily compete with properties of Cu films prepared by sputtering or electroplating (E: 130 GPa, H: 2 GPa for a grain size of 100 nm[ 72 ]). The high materials quality of electrochemically 3D printed Cu is confirmed by previous studies of its mechanical[ 26 , 51 , 53 , 54 ] and electrical[ 16 , 98 ] properties.
In contrast, samples printed by EHD‐RP are not completely dense. Pillars are homogeneous but feature nanoscale pores, and all pads developed vertical gaps (often resembling columnar growth) either after a few layers or only after an initially mostly dense growth (Figure S3, Supporting Information). Grain sizes in both geometries are <50 nm and thus pronouncedly smaller than those by the other electrochemical techniques. The combination of residual porosity and nanoscale grain size results in high strength (H: 2.2 GPa, σ0.07: 1.1 – 1.38 GPa) but a lowered elastic modulus (E: ≈80 GPa). These findings are in line with previously reported mechanical data.[ 57 ] Despite the use of an organic solvent, the measured carbon content is as low as that of MCED and FluidFM (Figure S5, Supporting Information). Thus, it is assumed that the porosity or potentially oxide phases, rather than residual organic solvents, are responsible for the low stiffness. Thermal annealing would possibly promote densification but has not yet been studied. While previous results showed a dependence of the synthesized grain size on the deposition parameters,[ 57 ] the present study adds the observation that the microstructure is further influenced by the printing strategy itself. The evolution of vertical gaps in pads is likely the result of growth instabilities in combination with preferred growth at protrusions due to field‐focusing effects. The reason for the exclusive presence of these instabilities in the pads is unclear. Yet, the initial deposition of Cu of high density observed in some pads indicate that the synthesis of dense material is possible given the printing parameters are optimized.
In general, electrochemical small‐scale AM offers extended options for microstructure engineering. First steps in this direction have already been made, demonstrating a range of grain sizes,[ 26 ] twin densities,[ 51 , 53 ] and local alloying and tuning of local porosity within printed geometries.[ 57 ] In its extent, the range of microstructures that could be accessed with electrochemical AM is unique amongst all small‐scale AM methods. This advantage could and should be exploited for maximizing materials properties and adjusting the synthesized microstructure to the specific needs of different applications (for example electrical conductivity versus mechanical strength).
In principle, the high density and purity and resulting high stiffness and strength of electrodeposited metals makes these materials the obvious choice for mechanical and also electrical applications. Unfortunately, electrochemical methods are currently limited in broad applicability. First, the electrochemical reduction necessitates an electrically conductive substrate. Second, the techniques are typically restricted to small build volumes due to a low deposition speed compared to transfer methods. However, the low volumetric throughput has been addressed recently: an increase in deposition speed by one order of magnitude by EHD‐RP,[ 57 ] parallelization demonstrated by MCED[ 52 ] and a variable voxel size shown with the FluidFM[ 56 ] are strategies that have increased the volumetric throughput and enable deposition of 3D geometries more than hundred microns in length, width and height with submicron resolution[ 56 ]. Third, the electrochemical strategies are currently limited to metals only, while ink‐, laser‐transfer‐, and ion/electron‐beam‐based techniques allow for the deposition of all materials classes.[ 5 ] Consequently, electrochemical methods are—for the time being—restricted to applications that demand highest materials properties in metals but can adjust to the mentioned limitations.
4.3.4. Synthesis Techniques: Electron/Ion‐Induced CVD
It is generally accepted that FEBID and FIBID deposits obtained from MeCpPt(Me)3 as used in this study, feature a composite structure of metal nanoparticles embedded in an amorphous hydrogenated carbon (a‐C:H) matrix.[ 29 ] Typical metal contents achieved with MeCpPt(Me)3 in commercial setups at room temperature are approximately 10 – 15 at% for FEBID[ 28 , 29 ] and around 45 at% for FIBID,[ 29 ] although other volatile metal compounds that achieve much higher metal content have also been demonstrated.[ 29 , 99 ] As a result of the low metal content, the mechanical behavior is dominated by the carbonaceous matrix. The properties of a‐C:H are highly dependent on its sp2/sp3 ratio with the stiffness and strength increasing with increasing sp3 content.[ 100 , 101 ] In FEBID and FIBID, this ratio as well as the density of the matrix are variable with the primary electron or ion energy, dose, and on the balance of precursor flux versus electron/ion flux.[ 22 , 60 , 102 ] As the structure of the matrix is sensitive to the deposition parameters, our measurements (E: 60 – 140 GPa, H: 6 – 9.4 GPa, σ0.07: 2.6 GPa) and reported mechanical properties of FIBID and FEBID deposits (E: 10 – 100 GPa[ 22 , 59 , 60 , 61 ] [up to 150 GPa for core shell structures[ 103 ]], H: 3.6 – 7.6 GPa,[ 62 , 104 ] tensile strength σtensile: 1 – 2 GPa[ 59 ] [up to 6.6 GPa for core‐shell structures[ 103 ]] cover a large range. Interestingly, the measured elastic modulus of the FEBID deposits is higher than previously reported moduli of as‐deposited Pt‐C structures[ 22 , 60 , 61 ] and similar to that of e‐beam‐annealed FEBID deposits.[ 22 ] Possibly, the here‐used deposition conditions imparted an in‐situ e‐beam curing, which is suggested to lower the hydrogen content of the matrix and improve the reticulation of the carbon network.[ 22 ] Nevertheless, the mechanical properties are considerably lower than data of a‐C:H synthesized by conventional plasma deposition techniques (E: 40 – 760 GPa,[ 100 , 101 , 105 ] H: 5 – 30 GPa,[ 100 , 101 ] σtensile: 7 GPa[ 105 ]), presumably due to the softer Pt phase and the fact the FEBID and FIBID carbon matrix is still hydrogenated and features a high sp2/sp3 ratio (typically, sp2/sp3=1 – 9[ 102 , 106 ]).
The low volume fraction of metal is not a disadvantage for mechanical performance—the stiffness compares well to that of metals synthesized with the other small‐scale AM techniques, and the strength outperforms the electrochemically deposited metals by a factor of 2 – 3. Further, variation of the sp2/sp3 ratio enables optimization of the stiffness and strength for a given application.[ 22 , 23 ] A drawback of amorphous carbon is its brittleness compared to metals. However, as long as fracture toughness is not an issue, there is no need to increase the metal loading from a mechanical perspective. Of course, an increase of the metal content is necessary to achieve an electrical resistance that can compete with that of metals: the room‐temperature electrical resistivity of as‐deposited Pt‐carbon material is around 1 × 10−6 Ωm for FIBID (46 at% Pt) and 1–10 × 10−2 Ωm for FEBID (10–15 at% Pt), corresponding to approximately 10× (FIBID) and 105 × (FEBID) the resistivity of bulk metals.[ 28 ]
The pronouncedly lower mechanical properties measured for cryo‐FEBID samples are likely owed to the porosity of the structures (especially of the pillars). This porosity results from evaporation of non‐irradiated precursor‐gas upon heating the deposits from cryogenic to room temperature.[ 64 ] As the amount of non‐irradiated precursor is a function of total fluence,[ 64 ] optimization of the exposure parameters could probably result in an increased density and thus improved mechanical properties.
5. Conclusion and Outlook
We have surveyed the microstructure and resulting mechanical properties of metals fabricated by almost all contemporary small‐scale AM methods. This study provides a comprehensive overview of metals accessible using these techniques today, and thus provides groundwork for optimization of the materials required for tomorrow. Both commonalities and differences between the synthesized metals were found. Metals deposited by transfer techniques (from colloidal inks or metal melts) are typically nano‐ to microporous. In contrast, materials produced by synthesis techniques are usually dense, and in the case of electrochemical methods, often nanocrystalline. Thus, modern microscale AM techniques synthesize metallic materials with a wide range of microstructures and can supply materials for various potential applications: dense and nanocrystalline metals for mechanical applications, dense and microcrystalline metals for high electrical or thermal conductivity, or highly porous materials for the use in catalysis or optical metamaterials.
Two main consequences are drawn for the future development of the techniques and their applications. First, not all 3D‐printed metals are equal—the characteristic microstructure associated with each different technique varies considerably. Consequently, users that request specific materials properties need to select an appropriate AM method. For applications requiring materials of high stiffness or strength, electrochemical techniques or FEBID and FIBID are recommended. These techniques deliver materials with dense microstructure and excellent mechanical properties, equal to those of metals used in state‐of‐the‐art microfabrication. For applications that require low defect density, electrochemical methods are the first choice (potentially in combination with thermal annealing for grain coarsening). If porous microstructures can be tolerated, or are desirable, and high throughput is required, printing of inks or LIFT is typically the simplest and most efficient choice.
Second, the material challenges faced by ink‐based methods and LIFT need to be tackled—relying on electrochemical and electron/ion‐beam techniques only is not an option. Although electrochemical and electron/ion‐beam approaches offer high‐performance metals, they suffer from other drawbacks, such as comparably low volumetric growth rates and a limited range of materials. As high throughput (ideally in combination with high materials quality) is essential for many industrial applications, many microscale AM techniques strive toward higher deposition rates, parallel printing or deposition with variable voxel sizes. While parallelization as well as on‐the‐fly adjustments of the voxel size have been demonstrated with both synthesis[ 52 , 56 , 107 ] and transfer techniques,[ 17 , 108 , 109 , 110 ] transfer‐techniques typically offer deposition rates normalized by resolution that can be orders of magnitude higher than that of synthesis methods[ 5 ] (partial exceptions are EHD‐RP and cryo‐FEBID). Hence, transfer techniques generally have an inherent advantage for high throughput. Additionally, transfer methods offer a much wider range of accessible materials that also include non‐metals, which are not yet available with electrochemical techniques. For these two reasons, transfer methods are and will remain important for many applications of small‐scale printing, despite their shortcomings in materials quality. Consequently, there is a strong need to improve the materials synthesized by these methods in order to guarantee levels of performance required in typical applications. Hence, we advocate more detailed studies of printed materials' microstructure and, most importantly, optimization of thermal post‐print processing. To this end, the large body of established sintering strategies employed in powder metallurgy could be tapped to guide attempts toward an improved densification of printed inks. On the other hand, electrochemical and electron/ion‐beam‐based methods need to expand the available materials, their compatibility with non‐conductive substrates, and their volumetric growth rates. In general, an understanding of the processing‐structure‐property relationships for all techniques, including more detailed studies of the synthesized microstructure and its relation to deposition parameters, is required to enable future adjustment of materials properties to needs of potential applications.
In summary, some small‐scale AM methods can already provide device‐grade metals, but the narrow range of accessible chemistries and the comparably small build volumes of these methods still limit the application of additive techniques in modern microfabrication. However, if the porosity of ink‐derived metals and the limited applicability of electrochemical and electron‐beam‐based methods are addressed, microscale AM of inorganic materials has the potential to offer a large range of device‐grade materials and to become a versatile and powerful alternative to lithography‐based 3D manufacturing at small scales.
6. Experimental Section
Sample Preparation—Direct Ink Writing of Shear‐Thinning Ag Inks
The concept and setup for DIW of Ag structures, as well as the synthesis of the shear‐thinning Ag inks, have been described earlier.[ 15 , 19 ] In brief, the coagulated ink consisted of 75 – 85 wt% polyacrylic acid (PAA)‐coated Ag particles in DI water. Pads and pillars were printed on a Regenovo Bio‐Architect WS printer at speeds of 30 – 80 µm s−1 and extrusion pressures of 17 – 24 bar. Pads were printed with a layer‐by‐layer approach, while pillars were deposited by continuously retracting the nozzle from the substrate. All samples were printed directly onto soda lime glass substrates. After printing, samples were heated on a hotplate in air. As‐deposited samples were dried at 100 °C (30 min), and annealed samples were sintered at a final temperature of 300 °C after a controlled temperature ramp (100 °C (10 min), 150 °C (10 min), 200 °C (10 min), 250 °C (10 min), and finally, 300 °C for 30 min or 2 h). All samples were sputter‐coated with a conductive layer of Pt–Pd (8 nm, CCU‐010 safematic) prior to SEM imaging and mechanical testing to avoid charging in the electron microscope.
Direct Ink Writing of Newtonian Ag Inks
The basic process and setup for printing, as well as the synthesis of the Ag inks, was previously described by Lee et al.[ 38 ] The synthesized ink (PAA‐coated Ag particles, 25 wt% in DI water) shows Newtonian fluid characteristics at shear rates of 0 – 103 s−1, with a viscosity of 6.8 × 10−3 Pa s. Glass nozzles with an opening of 10 µm were fabricated with a P‐97 pipette puller (Sutter Instruments). The ink was filled from the back of the nozzle and drawn to the nozzle tip by capillary forces without applied pressure. Pads were printed with a layer‐by‐layer strategy (hatch distance 10 µm, layer height 10 µm) at a speed of 10 µm −1. Pillars were printed by retracting the nozzle at 10 µm s−1. All samples were printed on (111) Si wafers coated with a 1 µm thick Pt film. Annealing was performed in an ambient‐atmosphere furnace (ov‐11, JEIO‐Tech), either at 150 °C for 1 h or 450 °C for 12 h.
Electrohydrodynamic Printing of Au Inks
The printing procedure was previously described by Galliker et al.[ 40 ] Similarly, the ink has been reported elsewhere.[ 80 ] In brief, Au nanoparticles with Decanethiol ligands, ≈5 nm in diameter, were synthesized with the Stucky method[ 111 ] and dispersed in Tetradecane. The maximum ink concentration was ≈10 mg mL−1. All samples were printed on diced SiO2 wafers (UniversityWafer, USA). The printing parameters are similar to the process presented by Schneider et al.[ 18 ] Yet, the printing process was optimized for high mass flow for printing the large structures presented herein. Therefore, the 250 Hz AC actuation voltage was increased to 260 – 360 Vp and larger nozzles with an aperture diameter of 1.8 – 2 µm were used. These parameters resulted in an ejection frequency of ≈1 kHz of droplets ≈200 nm in diameter. In general, the samples were printed in a layer‐by‐layer fashion, where each layer was deposited in a serpentine‐like printing path. With a translation speed of 5 µm s−1 and a hatch distance (line pitch) of 300 nm, approximately 30 layers are required to deposit a pad of 5 µm in height. In contrast, pillars (especially small pillars) were printed with lower mass flow, using lower actuation voltages and smaller nozzles. As‐printed samples were annealed for 10 min at 400 °C in a constant gas flow of O2 at ambient pressure in a rapid thermal processing furnace (As‐One 150, Annealsys, France).[ 18 ]
Laser‐Assisted Electrophoretic Deposition of Au Nanoparticles
The principle, setup, and typical printing parameters for laser‐assisted electrophoretic printing have been described in detail in literature.[ 43 , 112 ] For deposition of Au structures, an aqueuous solution of Au nanoparticles (0.25 wt%, 3 nm dia., Tanaka Kikinzoku Kogyo K. K.) was dispensed between an ITO‐coated cover glass and an ITO‐coated glass substrate (200 nm ITO on soda lime glass, Geomatec inc.). An electric field of 12 kV m−1 was applied between these electrodes for electrophoresis (electrode distance: 160 µm). A CW Nd:YVO4 laser (532 nm, 5 mW, Spectra physics, Millenia Pro) was used for optical trapping of the Au particles (Objective lens: ×60, NA = 1.2, Olympus, UPLSAPO 60XW). For printing of pillars, the stage was lowered at 0.67 µm s−1. Pads were printed by depositing an array of overlapping pillars. Printed structures were annealed for 1 h at 300 °C in atmosphere in a furnace (AFM‐10, ASWAN).
Laser‐Induced Forward Transfer of Ag Nanoparticle Inks
The details of the method have been described in previous publications.[ 17 , 44 , 113 ] Donor substrates were prepared via doctor‐blading a commercial Ag ink (80 wt%, particle dia.: 7 – 12 nm, viscosity ≈90 Pa s, NPS Nanopaste, Harima Chemicals Group) onto lithographically defined wells (4 mm × 2 cm) in glass substrates. Laser printing was performed using a Nd:YVO4 (JDSU, Q301‐HD) pulsed laser (λ = 355 nm, 30 ns FWHM) with a laser fluence of ≈30 – 100 mJ cm−2. Pads were printed by laser‐transferring individual voxels of Ag ink in the shape of circular disks, and pillars were deposited by stacking multiple individual voxel disks.[ 113 ] All samples were printed onto diced (100), p‐type Si wafers (University Wafer, Inc.). Thermal annealing was performed in a furnace (ambient atmosphere) for one hour at temperatures in the range of 150 – 230 °C.
Laser‐Induced Forward Transfer of Melts of Au and Cu Thin Films
The printing setup for LIFT and the general principle have been reported in literature.[ 48 , 114 ] A 3 W laser with a pulse duration of 0.8 ns and a wavelength of 532 nm (Picospark, Teem Photonics) was used for the LIFT process. The laser was deflected by a scanning mirror, and its spot size was (28.0 ± 0.5) µm (4‐sigma) at the donor interface. Soda lime glass slides coated with 500 nm of either Cu or Au were used as donors. Substrates were soda lime glass coated with 10 nm Ti/100 nm Au. Printing was performed in ambient atmosphere with a donor‐substrate gap of 300 µm. All samples were printed with droplet overlaps of 3 µm[ 114 ] and a total of five layers. The pulse energy used was 5.5 – 7.5 µJ for Cu and 4 – 6.5 µJ for Au.
Meniscus‐Confined Electrodeposition of Cu
The method and setup were described by Seol et al.[ 50 ] In short, Cu was deposited from an electrolyte of 1.05 M CuSO4·5H2O (99 %, Samchun Chem.) in an aqueous solution of 0.8 M H2SO4 (95 %, Daejung Chem.) using a two‐electrode setup with a Pt anode. All samples were grown by pulsed electrodeposition (pulse profile: –1.7 V (1 s), 1 V (0.5 s)) at growth rates of 0.02 µm s−1 and 0.4 µm s−1 for pads and pillars respectively. Both, pads and pillars, were printed by retracting the nozzles (diameter: 25 µm for pads, 10 µm for pillars) from the substrate in steps of 10 µm. All samples were deposited on a 1 µm‐thick film of Pt on cut (111) Si wafers. No thermal annealing was performed.
FluidFM Electrodeposition of Cu
The FluidFM principle and general printing parameters have been reported elsewhere.[ 54 , 56 ] All structures were printed with FluidFM Nanopipette probes (300 nm opening, Cytosurge AG) mounted on either a FluidFM BOT (Cytosurge AG and Exaddon AG) in the case of pads, or on a FluidFM ADD‐ON (Cytosurge AG) for classical AFM systems (Nanowizard I, JPK) in the case of pillars. For printing, the probe was filled with a Cu electrolyte solution (1 M CuSO4 (Sigma Aldrich) in aq. 0.8 M H2SO4 (Sigma Aldrich), filtered using a Millex‐VV Syringe filter (0.1 µm, PVDF, Sigma Aldrich)) and immersed in a supporting electrolyte bath (aq. H2SO4, pH 3). The electrochemical cell was equipped with a graphite (BOT) or a Pt (ADD‐ON) counter electrode and a Ag/AgCl reference electrode. Samples were deposited onto Ti(3 nm)/Au(25 nm) films coated either on soda lime glass (Menzel Gläser, Thermoscientific) in the case of pillars, or Si substrates ((100), Microchemicals) in the case of pads. Pads were printed with a layer‐by‐layer strategy at a potential of –0.45 –0.6 V vs. Ag/AgCl, an applied pressure of 4 – 8 mbar and a layer height of 0.4 – 0.8 µm. Pillars were printed in a layer‐by‐layer fashion, at a potential of –0.67 V vs. Ag/AgCl, an applied pressure of 7 mbar and a voxel height of 0.25 µm. No thermal annealing was performed.
Electrohydrodynamic Redox Printing of Cu
The basic concept and setup for EHD‐RP have been published earlier.[ 57 ] In brief, a sacrificial Cu anode (0.1 mm dia. wire, 99.9985%, Alfa Aesar) was immersed in a printing nozzle filled with acetonitrile (Optima, Fisher Chemical). The nozzles were pulled (P‐2000 micropipette puller system, Sutter Instrument) from quartz capillaries (QF100‐70‐15, Sutter Instrument) to an aperture diameter of 135 – 145 nm. Cu wires were etched in pure nitric acid (Sigma Aldrich) for 15 s prior to use. Printing was conducted in Ar‐atmosphere (<40 ppm O2, as measured by a Module ISM‐3 oxygen sensor (PBI Dansensor)). Samples were printed onto Si substrates ((100), SiMat) with a Ti(3 nm)/Au(20 nm) coating deposited in our laboratory sputter facility by DC magnetron sputtering (PVD Products Inc.). 15 × 15 µm pads were printed layer by layer, with a serpentine‐like print path, a hatch distance (line pitch) of 100 nm and a rotation of the hatch direction by 90° in every subsequent layer. The voltage applied to the sacrificial anode was 110 – 150 V. The nozzle‐substrate separation was kept constant at 7.5 µm by retracting the nozzle after every printed layer by a distance equal to an estimated layer height. Different combinations of in‐plane speed, layer height and number of layers were used. Typical values were: in‐plane speed: 10 – 40 µm s−1, layer height: 25 – 100 nm, layers: 10 – 80. Nominally 1 × 1 µm wide, square pillars were printed with the same protocol as the pads. Printing parameters were: hatch distance: 100 nm, voltage: 100 V, nozzle‐substrate separation: 7.5 µm, in‐plane speed: 5 μ s−1, layer height: 200 nm, layers: 14. Two pillars 160 and 170 nm in diameter were printed with the same electric field but with no relative motion between substrate and nozzle.
Focused Ion and Electron Beam Induced Deposition of Pt
A Tescan Lyra FIB‐SEM (background pressure 9 × 10−6 mbar, operating pressure 1 × 10−5 mbar) with electron field emission gun, Orsay Physics Ga‐ion source, and Orsay Physics gas injection system (GIS) was used for FIBID and FEBID of Pt deposits. The GIS nozzle exit (inner diameter: 350 µm) was placed at a distance of approximately 200 µm to the area of deposition. Methylcyclopentadienyl platinum (IV) trimethyl (MeCpPt(Me)3) was used as a precursor gas, heated to 80 °C. The local precursor pressure above the substrate was calculated by Empa's freeware GIS simulator to be 0.4 and 0.8 Pa (2 × 103 and 4 × 103 ML s−1) for FIB and FEB deposition, respectively. All samples were deposited onto diced (100) Si wafers (Semiwafer). The deposition parameters for FIBID and FEBID pillars and pads are given as (beam current, beam energy, beam size, scan pattern, dwell time, pixel distance, refresh time, total time, and deposition rate). FIBID pillars: (5 pA, 10 kV, 32 nm, fly back, 80 ns, 25.6 nm, 122 µs, 10 min 28 s, 0.7 µm3 nA−1 s−1). FIBID pads: (285 pA, 10 kV, 32 nm, fly back, 80 ns, 25.6 nm, 12 ms, 19 min 34 s, 0.3 µm3 nA−1 s−1). FEBID pillars: (1 nA, 5 kV, 29 nm, rotating leading edge (RLE), 80 ns, 29 nm, 92.5 µs, 16 min 38 s, 0.0026 µm3 nA−1 s−1). FEBID pads: (3.9 nA, 5 kV, 180 nm, RLE, 1 µs, 180 nm, 6.9 ms, 70 min, 0.0064 µm3 nA−1 s−1).
Cryo‐FEBID of Pt
The setup has been described in detail by Bresin et al.,[ 63 , 64 ] including the important processes governing morphology, size, microstructure, and composition. All experiments were conducted in a FEI Nova 600 Nanolab dual beam system (base pressure 6.7 × 10−5 Pa). For deposition, the MeCpPt(Me)3 precursor gas is condensed on the substrate at ‐190 °C (custom‐built cryogenic stage cooled by liquid nitrogen in a heat exchanger) and subsequently patterned with the electron beam. Square pads were printed by injecting the precursor gas for 30 s (crucible temperature: 27 °C) and scanning the electron beam (0.62 nA, 20 kV) in a normal imaging raster with a pixel dwell time of 100 ns. Pillars were deposited by three cycles of condensation and e‐beam patterning. In contrast to the pads, the precursor was injected for 60 s, and beam parameters were 0.6 nA, 18 kV. Further, the individual layers were exposed with a stationary rather than a scanning beam, exposing a nominal area of 10 nm for 30 s. All structures were deposited onto p‐type (100) silicon with 100 nm of thermal oxide (plasma cleaned prior to deposition (20% O2 in Ar, Fischione Model 1020)).
Analysis
The morphology of the printed samples was studied with a FEG‐SEM (Magellan 400, FEI, USA). Cross‐sections of samples for microstructure analysis were cut and imaged with a dual‐beam FIB‐SEM (NVision40, Zeiss, Germany) using the InLens detection mode and an acceleration voltage of 5 kV and final polishing currents of 40 – 80 pA. The chemical composition was qualitatively analyzed by SEM EDX spectroscopy (Quanta 200F, FEI, equipped with an Octane Super EDX system, EDAX, software: Genesis, EDAX). All spectra were recorded with identical acquisition conditions: acceleration voltage: 10 kV, amplification time: 7.68 µs, live time: 60 s, dead time: 25 – 30%, scan size: 5 × 5 µm2 for small pads, 15 × 15 µm2 for large pads, no specimen tilt. In general, all samples were stored either under vacuum in a desiccator or at atmospheric pressures in a low‐humidity Ar cabinet.
The mechanical properties of the printed materials were measured by nanoindentation of pads and microcompression of micropillars. Nanoindentation was performed with three different testing setups: an Ultra Nanoindentation Tester (Anton Paar Tritec SA, Switzerland) or an iNano Nanoindenter (Nanomechanics, Inc., USA) were used to test samples of low surface roughness. Both systems were fitted with a diamond Berkovich indenter (Synton‐MDP, Switzerland). An Alemnis indenter (Alemnis GmbH, Thun, Switzerland)[ 115 ] fitted with either a Berkovich or a cube‐corner indenter (Synton‐MDP, Switzerland) was used for indentation of LIFT (melt)‐printed pads, as these samples showed higher surface roughness and thus required higher indentation depths (and thus forces). Typical linear loading rates were 5000 – 10000 µN min−1 and 10 mN min−1 for the Anton Paar Tritec and the Alemnis systems, respectively. The Nanomechanics system was used in constant strain rate mode, with a target strain rate of 0.1. Hardness and elastic modulus were recorded as a function of depth, either by performing progressive load–unload cycles (Anton Paar Tritec and Nanomechanics system) or a continuous measurement by superimposing a sinus oscillation on the load profile (110 Hz, Nanomechanics system). Reported values are as‐measured hardness and modulus values averaged over a depth range adjusted to the thickness, width and roughness of the tested pads. No model to account for substrate effects was applied. Non‐symmetrical or cut indents (caused by large‐scale roughness) were excluded from analysis. The thickness of all tested pads was assessed with a confocal optical profilometer (PLu neox, Sensofar). If pads vary in thickness, the lowest thickness is reported. RMS roughness of the roughest of all tested samples of a given technique, as judged from SEM images, was measured by AFM (Cypher, Oxford Instruments, scan size adjusted to pad width, max. 20 µm) or the optical profilometer in the case of LIFT (melt). The following Poisson's ratios were used for the conversion of indentation modulus to Young's modulus: Ag: 0.37, Au: 0.42, Cu: 0.36.[ 116 ] For all Pt FIBID and FEBID samples, we calculated with the Poisson's ratio of the matrix material (glassy carbon, 0.2) instead of the value for Pt. A constraint factor of 2.8[ 117 ] was used for conversion of hardness to strength.
Microcompression testing was performed using an in‐situ SEM Indenter (Alemnis GmbH, Thun, Switzerland)[ 115 ] mounted in a Vega3 SEM (Tescan, Brno, Czech Republic). This facilitated both sample positioning and direct observation of the deformation process. The indenter was fitted with diamond flat punches of 1.5 – 50 µm in diameter, depending on the width of the tested pillars. Pillars were compressed in progressive loading‐unloading cycles to a total strain of 10 – 25%. Compression was performed under displacement control at a rate proportional to the pillar's height to produce a strain rate on the order of 1 × 10−3 s−1. For determination of the yield stress, load–displacement curves were converted to stress–strain curves based on the average diameter of the deformed portion of the pillar. Due to the non‐uniform pillar diameter and the blurred onset of yield, no classic yield criterion was applied. Instead, we report the flow stress at 7% strain, which corresponds directly to the representative strain under a Berkovich indenter.[ 118 ] For the calculation of the Young's modulus, stress–strain curves based on the average load‐bearing cross‐section of the whole pillar were used. The Young's modulus was usually measured from the first unloading segment after the plastic yield point. No volume conservation was applied. Sneddon's model[ 119 ] was used to correct for pillar sink‐in, using the following elastic moduli of the different substrates: soda lime glass: 72 GPa, fused silica: 73 GPa, Si <100>: 130 GPa, Si <111>: 186 GPa. Metallic coating layers between pillar and substrate were ignored in the model, as the elastic field was expected to extend far beyond those layers. Prior to testing, the top portions of most pillars were cut using a dual‐beam FIB‐SEM (NVision40, Zeiss, Germany) to produce flat pillar tops and to trim the pillars to an aspect ratio 2 – 3. This helped to avoid buckling and to achieve a clearer plastic yield point which is less influenced by the rounded top of the pillars. Final Ga+‐ion milling currents of 80 pA – 3 nA were used, depending on the diameter of the pillars.
The measured mechanical data were normalized to literature values of polycrystalline thin films deposited by PVD or electrochemical deposition to decouple microstructural from chemical influences on the mechanical properties (to allow a comparison between the different chemical elements used by different AM methods) and to enable a facile comparison of the quality of printed metals to that of thin‐film materials synthesized by established deposition techniques. The following values were used (Table S3, Supporting Information): E Ag: 80.5 GPa, H Ag: 1.2 GPa, E Au: 80.2 GPa, H Au: 1.2 GPa, E Cu: 125 GPa, H Cu: 2 GPa, E Pt: 174 GPa, H Pt: 4 GPa. Literature data for E of Co and W FEBID deposits were normalized to E Co: 204 GPa and E W: 369 GPa, while the hardness values were not normalized. The measured flow stress σ0.07 was normalized by literature values for H/2.8. All FEBID and FIBID values were normalized to literature data of Pt to highlight the difference in mechanical behavior compared to metals, although we acknowledge that FEBID and FIBID deposits are mostly composed of amorphous carbon.
Conflict of Interest
D.P. is the cofounder of SCRONA. The other authors declare no conflict of interest.
Author Contributions
A.R. contrived and coordinated the study. R.S. supervised the project. A.R. provided SEM and FIB and nanoindentation analysis. J.M.W. and A.R. performed microcompression analysis. The following authors were responsible for sample fabrication with the different techniques: L.K. (EHD‐RP), K.A.D. (cryo‐FEBID), T.M. and F.I. (LAEDP), O.F. and Z.K. (LIFT (melt)), N.Z. and J.L. (DIW (s.t.)), K.C. and A.P. (LIFT (ink)), P.R. and D.P. (EHDP), S.L. and S.K.S. (DIW (N.) and MCED), I.U. (FEBID and FIBID), C.v.N. and T.Z. (FluidFM). A.R., J.M.W. and R.S. interpreted the results, while all authors discussed the results. A.R. wrote the manuscript and visualized the data. All authors reviewed and commented the manuscript.
Supporting information
Supporting Information
Acknowledgements
A.R., J.W., and R.S. thank S. Ganzeboom (ETH Zürich) for experimental support and gratefully thank D. Momotenko (Laboratory of Biosensors and Bioelectronics, ETH Zürich) for access to his nozzle‐puller. Electron‐microscopy analysis was performed at ScopeM, the microscopy platform of ETH Zürich. A.R., J.W., and R.S. acknowledge the financial support by Grant no. ETH 47 14‐2. The work by C.v.N. and T.Z. was supported by Swiss Agency for Technology and Innovation Innosuisse (Project Nr: PNFM‐NM 18511.1). C.v.N. would like to thank L. Hirt (formerly ETH Zürich) for experimental support. P.R. post‐treated his samples at the Binnig and Rohrer Nanotechnology Center (BRNC) at IBM Zurich and analyzed them at the ETH Center for Mirco‐ and Nanoscience (FIRST). P.R. acknowledges funding by the SFA Advanced Manufacturing program under the Powder Focusing project. K.C. and A.P. acknowledge that this work was funded by the Office of Naval Research (ONR) through the Naval Research Laboratory Basic Research Program. The contribution of S.L. and S.K.S. was supported in part by Korea Electrotechnology Research Institute (KERI) Primary research program (No. 20‐12‐N0101‐27) through the National Research Council of Science & Technology (NST) funded by Ministry of Science and ICT. I.U. would like to thank G. Bürki (Empa) and P. L. Gal (Tescan/Orsay Physics) for experimental support. I.U. further acknowledges the financial support of EU Horizon 2020 Marie Curie‐Sklodowska Innovative Training Network “ELENA”, grant agreement no 722149.
Reiser A., Koch L., Dunn K. A., Matsuura T., Iwata F., Fogel O., Kotler Z., Zhou N., Charipar K., Piqué A., Rohner P., Poulikakos D., Lee S., Seol S. K., Utke I., van C. Nisselroy, Zambelli T., Wheeler J. M., Spolenak R., Metals by Micro‐Scale Additive Manufacturing: Comparison of Microstructure and Mechanical Properties. Adv. Funct. Mater. 2020, 30, 1910491 10.1002/adfm.201910491
References
- 1. Onses M. S., Sutanto E., Ferreira P. M., Alleyne A. G., Rogers J. A., Small 2015, 11, 4237. [DOI] [PubMed] [Google Scholar]
- 2. Hohmann J. K., Renner M., Waller E. H., von Freymann G., Adv. Opt. Mater. 2015, 3, 1488. [Google Scholar]
- 3. Rogers J., Huang Y., Schmidt O. G., Gracias D. H., MRS Bull. 2016, 41, 123. [Google Scholar]
- 4. Zhang Y., Zhang F., Yan Z., Ma Q., Li X., Huang Y., Rogers J. A., Nat. Rev. Mater. 2017, 2, 17019. [Google Scholar]
- 5. Hirt L., Reiser A., Spolenak R., Zambelli T., Adv. Mater. 2017, 29, 1604211. [DOI] [PubMed] [Google Scholar]
- 6. Montemayor L., Chernow V., Greer J. R., MRS Bull. 2015, 40, 1122. [Google Scholar]
- 7. Soukoulis C. M., Wegener M., Nat. Photonics 2011, 5, 523. [Google Scholar]
- 8. Lee J.‐H., Singer J. P., Thomas E. L., Adv. Mater. 2012, 24, 4782. [DOI] [PubMed] [Google Scholar]
- 9. Lewis J. A., Adv. Funct. Mater. 2006, 16, 2193. [Google Scholar]
- 10. Gansel J. K., Thiel M., Rill M. S., Decker M., Bade K., Saile V., von Freymann G., Linden S., Wegener M., Science (80‐.). 2009, 325, 1513. [DOI] [PubMed] [Google Scholar]
- 11. Ergin T., Stenger N., Brenner P., Pendry J. B., Wegener M., Science (80‐.). 2010, 328, 337. [DOI] [PubMed] [Google Scholar]
- 12. Schaedler T. A., Jacobsen A. J., Torrents A., Sorensen A. E., Lian J., Greer J. R., Valdevit L., Carter W. B., Science (80‐.). 2011, 334, 962. [DOI] [PubMed] [Google Scholar]
- 13. Jang D., Meza L. R., Greer F., Greer J. R., Nat. Mater. 2013, 12, 893. [DOI] [PubMed] [Google Scholar]
- 14. Xia X., Afshar A., Yang H., Portela C. M., Kochmann D. M., Di Leo C. V., Greer J. R., Nature 2019, 573, 205. [DOI] [PubMed] [Google Scholar]
- 15. Ahn B. Y., Duoss E. B., Motala M. J., Guo X., Park S.‐I., Xiong Y., Yoon J., Nuzzo R. G., Rogers J. A., Lewis J. A., Science (80‐.). 2009, 323, 1590. [DOI] [PubMed] [Google Scholar]
- 16. Hu J., Yu M.‐F., Science (80‐.). 2010, 329, 313. [DOI] [PubMed] [Google Scholar]
- 17. Wang J., Auyeung R. C. Y., Kim H., Charipar N. A., Piqué A., Adv. Mater. 2010, 22, 4462. [DOI] [PubMed] [Google Scholar]
- 18. Schneider J., Rohner P., Thureja D., Schmid M., Galliker P., Poulikakos D., Adv. Funct. Mater. 2016, 26, 833. [Google Scholar]
- 19. Zhou N., Liu C., Lewis J. A., Ham D., Adv. Mater. 2017, 29, 1605198. [DOI] [PubMed] [Google Scholar]
- 20. Gavagnin M., Wanzenboeck H. D., Wachter S., Shawrav M. M., Persson A., Gunnarsson K., Svedlindh P., Stöger‐Pollach M., Bertagnolli E., ACS Appl. Mater. Interfaces 2014, 6, 20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo J., Pohl R., Qi L., Römer G.‐W., Sun C., Lohse D., Visser C. W., Small 2017, 13, 1602553. [DOI] [PubMed] [Google Scholar]
- 22. Arnold G., Winkler R., Stermitz M., Orthacker A., Noh J., Fowlkes J. D., Kothleitner G., Huth M., Rack P. D., Plank H., Adv. Funct. Mater. 2018, 28, 1707387. [Google Scholar]
- 23. Sattelkow J., Fröch J. E., Winkler R., Hummel S., Schwalb C., Plank H., ACS Appl. Mater. Interfaces 2019, 11, 22655. [DOI] [PubMed] [Google Scholar]
- 24. Schürch P., Pethö L., Schwiedrzik J., Michler J., Philippe L., Adv. Mater. Technol. 2018, 1800274, 1800274. [Google Scholar]
- 25. Winter S., Zenou M., Kotler Z., J. Phys. D. Appl. Phys. 2016, 49, 165310. [Google Scholar]
- 26. Daryadel S., Behroozfar A., Minary‐Jolandan M., Adv. Eng. Mater. 2019, 21, 1800946. [Google Scholar]
- 27. Skylar‐Scott M. A., Gunasekaran S., Lewis J. A., Proc. Natl. Acad. Sci. 2016, 113, 6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Botman A., Mulders J. J. L., Hagen C. W., Nanotechnology 2009, 20, 372001. [DOI] [PubMed] [Google Scholar]
- 29. Utke I., Hoffmann P., Melngailis J., J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2008, 26, 1197. [Google Scholar]
- 30. Tanaka T., Ishikawa A., Kawata S., Appl. Phys. Lett. 2006, 88, 081107. [Google Scholar]
- 31. Maruo S., Saeki T., Opt. Express 2008, 16, 1174. [DOI] [PubMed] [Google Scholar]
- 32. van Spengen W. Merlijn, Microelectron. Reliab. 2003, 43, 1049. [Google Scholar]
- 33. Yi Z., Guo J., Chen Y., Zhang H., Zhang S., Xu G., Yu M., Cui P., Microsystems Nanoeng. 2016, 2, 16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farahani R. D., Chizari K., Therriault D., Nanoscale 2014, 6, 10470. [DOI] [PubMed] [Google Scholar]
- 35. Fogel O., Winter S., Benjamin E., Krylov S., Kotler Z., Zalevsky Z., Addit. Manuf. 2018, 21, 307. [Google Scholar]
- 36. Vyatskikh A., Delalande S., Kudo A., Zhang X., Portela C. M., Greer J. R., Nat. Commun. 2018, 9, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oran D., Rodriques S. G., Gao R., Asano S., Skylar‐Scott M. A., Chen F., Tillberg P. W., Marblestone A. H., Boyden E. S., Science (80‐. ). 2018, 362, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee S., Kim J. H., Wajahat M., Jeong H., Chang W. S., Cho S. H., Kim J. T., Seol S. K., ACS Appl. Mater. Interfaces 2017, 9, 18918. [DOI] [PubMed] [Google Scholar]
- 39. Park J.‐U., Hardy M., Kang S. J., Barton K., Adair K., Mukhopadhyay D. kishore, Lee C. Y., Strano M. S., Alleyne A. G., Georgiadis J. G., Ferreira P. M., Rogers J. A., Nat. Mater. 2007, 6, 782. [DOI] [PubMed] [Google Scholar]
- 40. Galliker P., Schneider J., Eghlidi H., Kress S., Sandoghdar V., Poulikakos D., Nat. Commun. 2012, 3, 890. [DOI] [PubMed] [Google Scholar]
- 41. Galliker P., Schneider J., Poulikakos D., Appl. Phys. Lett. 2014, 104, 073105. [Google Scholar]
- 42. Schneider J., Bachmann T., Franco D., Richner P., Galliker P., Tiwari M. K., Ferrari A., Poulikakos D., Macromol. Biosci. 2013, 13, 973. [DOI] [PubMed] [Google Scholar]
- 43. Takai T., Nakao H., Iwata F., Opt. Express 2014, 22, 28109. [DOI] [PubMed] [Google Scholar]
- 44. Piqué A., Auyeung R. C., Kim H., Metkus K., Mathews S. A., J. Laser Micro/Nanoengineering 2008, 3, 163. [Google Scholar]
- 45. Mathews S. A., Auyeung R. C. Y., Kim H., Charipar N. A., Piqué A., J. Appl. Phys. 2013, 114, 064910. [Google Scholar]
- 46. Piqué A., Kim H., Auyeung R. C., Beniam I., Breckenfeld E., Appl. Surf. Sci. 2016, 374, 42. [Google Scholar]
- 47. Birnbaum A. J., Wahl K. J., Auyeung R. C. Y., Piqué A., J. Micromechanics Microengineering 2010, 20, 077002. [Google Scholar]
- 48. Zenou M., Sa'ar A., Kotler Z., Sci. Rep. 2015, 5, 17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fogel O., Cohen S., Kotler Z., Zalevsky Z., Procedia CIRP 2018, 74, 285. [Google Scholar]
- 50. Seol S. K., Kim D., Lee S., Kim J. H., Chang W. S., Kim J. T., Small 2015, 11, 3896. [DOI] [PubMed] [Google Scholar]
- 51. Behroozfar A., Daryadel S., Morsali S. R., Moreno S., Baniasadi M., Bernal R. A., Minary‐Jolandan M., Adv. Mater. 2018, 30, 1705107. [DOI] [PubMed] [Google Scholar]
- 52. Lin Y.‐p., Zhang Y., Yu M.‐f., Adv. Mater. Technol. 2019, 4, 1800393. [Google Scholar]
- 53. Daryadel S., Behroozfar A., Morsali S. R., Moreno S., Baniasadi M., Bykova J., Bernal R. A., Minary‐Jolandan M., Nano Lett. 2018, 18, 208. [DOI] [PubMed] [Google Scholar]
- 54. Hirt L., Ihle S., Pan Z., Dorwling‐Carter L., Reiser A., Wheeler J. M., Spolenak R., Vörös J., Zambelli T., Adv. Mater. 2016, 28, 2311. [DOI] [PubMed] [Google Scholar]
- 55. Momotenko D., Page A., Adobes‐Vidal M., Unwin P. R., ACS Nano 2016, 10, 8871. [DOI] [PubMed] [Google Scholar]
- 56. Ercolano G., Zambelli T., van Nisselroy C., Momotenko D., Vörös J., Merle T., Koelmans W. W., Adv. Eng. Mater. 2019, 1900961, 1900961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reiser A., Lindén M., Rohner P., Marchand A., Galinski H., Sologubenko A. S., Wheeler J. M., Zenobi R., Poulikakos D., Spolenak R., Nat. Commun. 2019, 10, 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fowlkes J. D., Winkler R., Lewis B. B., Stanford M. G., Plank H., Rack P. D., ACS Nano 2016, 10, 6163. [DOI] [PubMed] [Google Scholar]
- 59. Utke I., Friedli V., Fahlbusch S., Hoffmann S., Hoffmann P., Michler J., Adv. Eng. Mater. 2006, 8, 155. [Google Scholar]
- 60. Friedli V., Utke I., Mølhave K., Michler J., Nanotechnology 2009, 20, 385304. [DOI] [PubMed] [Google Scholar]
- 61. Lewis B. B., Mound B. A., Srijanto B., Fowlkes J. D., Pharr G. M., Rack P. D., Nanoscale 2017, 9, 16349. [DOI] [PubMed] [Google Scholar]
- 62. Wich T., in Autom. Nanohandling by Microrobots (Ed: Fatikow S.), Springer London, London: 2008, pp. 295–340. [Google Scholar]
- 63. Bresin M., Toth M., Dunn K. A., Nanotechnology 2013, 24, 035301. [DOI] [PubMed] [Google Scholar]
- 64. Bresin M., Thiel B., Toth M., Dunn K., J. Mater. Res. 2011, 26, 357. [Google Scholar]
- 65. Shugurov A., Panin A., Hui‐Gon C., Oskomov K., Sci. Technol. 2004. KORUS 2004. Proceedings. 8th Russ. Int. Symp. 2004, 3, 168. [Google Scholar]
- 66. Panin A., Shugurov A., Oskomov K., Phys. Solid State 2005, 47, 2055. [Google Scholar]
- 67. Okuda S., Kobiyama M., Inami T., Mater. Trans. JIM 1999, 40, 412. [Google Scholar]
- 68. Volinsky A. A., Moody N. R., Gerberich W. W., J. Mater. Res. 2004, 19, 2650. [Google Scholar]
- 69. Beegan D., Chowdhury S., Laugier M. T., Surf. Coatings Technol. 2003, 176, 124. [Google Scholar]
- 70. Lu L., Schwaiger R., Shan Z. W., Dao M., Lu K., Suresh S., Acta Mater. 2005, 53, 2169. [Google Scholar]
- 71. Beegan D., Chowdhury S., Laugier M. T., Surf. Coatings Technol. 2007, 201, 5804. [Google Scholar]
- 72. Chang S.‐Y., Chang T.‐K., J. Appl. Phys. 2007, 101, 033507. [Google Scholar]
- 73. Mall S., Lee H., Leedy K. D., Coutu R. A., in Part B Magn. Storage Tribol. Manuf. Tribol. Nanotribology; Eng. Surfaces; Biotribology; Emerg. Technol. Spec. Symp. Contact Mech. Spec. Symp. Nanotribology, San Antonio, Texas, USA: 2006, pp. 1377–1378. [Google Scholar]
- 74. Nili H., Walia S., Bhaskaran M., Sriram S., J. Appl. Phys. 2014, 116, 163504. [Google Scholar]
- 75. Mencik J., Swain M. V., MRS Proc. 1994, 356, 729. [Google Scholar]
- 76. Zhang Z., Chen D., Han W., Kimura A., Fusion Eng. Des. 2015, 98‐99, 2103. [Google Scholar]
- 77. Abadias G., Dub S., Shmegera R., Surf. Coatings Technol. 2006, 200, 6538. [Google Scholar]
- 78. Birnbaum A. J., Auyeung R. C., Wahl K. J., Zalalutidnov M., Laracuente A. R., Piqué A., J. Appl. Phys. 2010, 108, 1. [Google Scholar]
- 79. Birnbaum A. J., Zalalutdinov M. K., Wahl K. J., Pique A., J. Microelectromechanical Syst. 2011, 20, 436. [Google Scholar]
- 80. Richner P., Eghlidi H., Kress S. J. P., Schmid M., Norris D. J., Poulikakos D., ACS Appl. Mater. Interfaces 2016, 8, 11690. [DOI] [PubMed] [Google Scholar]
- 81. Podsiadlo P., Krylova G., Lee B., Critchley K., Gosztola D. J., Talapin D. V., Ashby P. D., Shevchenko E. V., J. Am. Chem. Soc. 2010, 132, 8953. [DOI] [PubMed] [Google Scholar]
- 82. Yan C., Arfaoui I., Goubet N., Pileni M. P., Adv. Funct. Mater. 2013, 23, 2315. [Google Scholar]
- 83. Greer J. R., Street R. A., J. Appl. Phys. 2007, 101. [Google Scholar]
- 84. Lee D. J., Oh J. H., Thin Solid Films 2010, 518, 6352. [Google Scholar]
- 85. Dou R., Xu B., Derby B., Scr. Mater. 2010, 63, 308. [Google Scholar]
- 86. Hashin Z., J. Appl. Mech. 1983, 50, 481. [Google Scholar]
- 87. Roberts A. P., Garboczi E. J., J. Am. Ceram. Soc. 2000, 83, 3041. [Google Scholar]
- 88. Hodge A. M., Biener J., Hayes J. R., Bythrow P. M., Volkert C. A., Hamza A. V., Acta Mater. 2007, 55, 1343. [Google Scholar]
- 89. Jakus A. E., Taylor S. L., Geisendorfer N. R., Dunand D. C., Shah R. N., Adv. Funct. Mater. 2015, 25, 6985. [Google Scholar]
- 90. Taylor S. L., Jakus A. E., Shah R. N., Dunand D. C., Adv. Eng. Mater. 2016. [Google Scholar]
- 91. Röthlisberger A., Häberli S., Spolenak R., Dunand D. C., J. Mater. Res. 2016, 31, 753. [Google Scholar]
- 92. Calvo M., Jakus A. E., Shah R. N., Spolenak R., Dunand D. C., Adv. Eng. Mater. 2018, 20, 1800354. [Google Scholar]
- 93. Kenel C., Casati N. P. M., Dunand D. C., Nat. Commun. 2019, 10, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zenou M., Sáar A., Kotler Z., Small 2015, 11, 4082. [DOI] [PubMed] [Google Scholar]
- 95. Feinaeugle M., Pohl R., Bor T., Vaneker T., Römer G.‐w., Addit. Manuf. 2018, 24, 391. [Google Scholar]
- 96. Fogel O., Toker G. B., Cohen‐Taguri G., Gergaud P., Gaillard F., Kotler Z., Zalevsky Z., J. Phys. D. Appl. Phys. 2019, 52, 285303. [Google Scholar]
- 97. Suryavanshi A. P., Yu M.‐F., Appl. Phys. Lett. 2006, 88, 083103. [Google Scholar]
- 98. Suryavanshi A. P., Yu M.‐F., Nanotechnology 2007, 18, 105305. [Google Scholar]
- 99. Huth M., Porrati F., Dobrovolskiy O., Microelectron. Eng. 2018, 185‐186, 9. [Google Scholar]
- 100. Jiang X., Reichelt K., Stritzker B., J. Appl. Phys. 1989, 66, 5805. [Google Scholar]
- 101. Weiler M., Sattel S., Giessen T., Jung K., Ehrhardt H., Veerasamy V. S., Robertson J., Phys. Rev. B 1996, 53, 1594. [DOI] [PubMed] [Google Scholar]
- 102. Li J., Bresin M., Dunn K., Thiel B., Microsc. Microanal. 2007, 13, 1472. [Google Scholar]
- 103. Córdoba R., Lorenzoni M., Pablo‐Navarro J., Magén C., Pérez‐Murano F., De Teresa J. M., Nanotechnology 2017, 28, 445301. [DOI] [PubMed] [Google Scholar]
- 104. Ding W., Dikin D. A., Chen X., Piner R. D., Ruoff R. S., Zussman E., Wang X., Li X., J. Appl. Phys. 2005, 98, 014905. [Google Scholar]
- 105. Cho S., Chasiotis I., Friedmann T. A., Sullivan J. P., J. Micromechanics Microengineering 2005, 15, 728. [Google Scholar]
- 106. Bret T., Mauron S., Utke I., Hoffmann P., Microelectron. Eng. 2005, 78–79, 300. [Google Scholar]
- 107. Post P. C., Mohammadi‐Gheidari A., Hagen C. W., Kruit P., J. Vac. Sci. Technol. B, Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 2011, 29, 06F310. [Google Scholar]
- 108. Lee J.‐S., Kim S.‐Y., Kim Y.‐J., Park J., Kim Y., Hwang J., Kim Y.‐J., Appl. Phys. Lett. 2008, 93, 243114. [Google Scholar]
- 109. Pan Y., Chen X., Zeng L., Huang Y., Yin Z., J. Micromechanics Microengineering 2017, 27, 125004. [Google Scholar]
- 110. Auyeung R. C. Y., Kim H., Mathews S., Piqué A., Appl. Opt. 2015, 54, F70. [DOI] [PubMed] [Google Scholar]
- 111. Zheng N., Fan J., Stucky G. D., J. Am. Chem. Soc. 2006, 128, 6550. [DOI] [PubMed] [Google Scholar]
- 112. Iwata F., Kaji M., Suzuki A., Ito S., Nakao H., Nanotechnology 2009, 20, 235303. [DOI] [PubMed] [Google Scholar]
- 113. Charipar K. M., Diaz‐Rivera R. E., Charipar N. A., Piqué A., in Laser 3D Manuf. V (Eds: Helvajian H., Piqué A., Gu B.), SPIE, February: 2018, p. 29. [Google Scholar]
- 114. Zenou M., Kotler Z., Opt. Express 2016, 24, 1431. [DOI] [PubMed] [Google Scholar]
- 115. Wheeler J. M., Michler J., Rev. Sci. Instrum. 2013, 84, 045103. [DOI] [PubMed] [Google Scholar]
- 116. Köster W., Franz H., Metall. Rev. 1961, 6, 1. [Google Scholar]
- 117. Maier V., Durst K., Mueller J., Backes B., Höppel H. W., Göken M., J. Mater. Res. 2011, 26, 1421. [Google Scholar]
- 118. Leitner A., Maier‐Kiener V., Kiener D., Adv. Eng. Mater. 2017, 19, 1600669. [Google Scholar]
- 119. Sneddon I. N., Int. J. Eng. Sci. 1965, 3, 47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information