

Abstract
Purpose of Review:
Protein homeostasis (proteostasis) is maintained by an integrated network of physiological mechanisms and stress response pathways that regulate the content and quality of the proteome. The maintenance of cellular proteostasis is key to ensuring normal development, resistance to environmental stress, coping with infection, and promoting healthy aging and lifespan. However, recent studies have revealed that several proteostasis mechanisms can function in a cell-type specific manner, including within hematopoietic stem cells (HSCs). Here, we review recent studies demonstrating that the proteostasis network functions uniquely in HSCs to promote their maintenance and regenerative function.
Recent Findings:
Despite being highly-conserved cellular processes, several components of the proteostasis network, including protein synthesis, protein degradation, and the unfolded protein response, are regulated differently in HSCs as compared to restricted hematopoietic progenitors. Disruption of these proteostasis mechanisms are particularly detrimental to HSC maintenance and function. Together, these findings suggest that these aspects of cellular physiology are uniquely regulated in HSCs to maintain proteostasis, and that precise control of proteostasis is particularly important to support life-long HSC maintenance and regenerative function.
Summary:
Emerging data suggest that the proteostasis network is uniquely configured within HSCs to promote their longevity and hematopoietic function. Future work uncovering cell-type specific differences in proteostasis network configuration, integration, and function will be essential for understanding how HSCs function during homeostasis, in response to stress, and in disease.
Keywords: proteostasis, hematopoietic stem cell, protein synthesis, translation, protein degradation, proteasome, autophagy, unfolded protein response, aging, longevity
Introduction
Protein homeostasis (proteostasis) is maintained by an integrated network of physiological mechanisms and stress response pathways that dynamically coordinate protein synthesis, folding, trafficking and degradation to regulate proteome content and quality [1] (Fig. 1). Recent studies investigating various components of the proteostasis network have revealed that they play a critical role in regulating HSC maintenance and function during homeostasis and in response to stress. Strikingly, many of these pathways, despite being highly-conserved, exhibit exquisite celltype specificity, and often function differently in HSCs than in restricted hematopoietic progenitors. Components of the proteostasis network regulate multiple unique aspects of HSC biology, including their self-renewal, quiescence, regenerative potential and metabolism. However, we still have limited knowledge of how these proteostasis mechanisms directly impact the content and quality of the HSC proteome, and how their integrated configuration works in concert to support stem cell fate, function and longevity. In this review, we revisit seminal work on HSC proteostasis and discuss recent studies that are unraveling the intricacies of proteostasis network configuration and function required by HSCs.
Figure 1. Schematic of the proteostasis network.
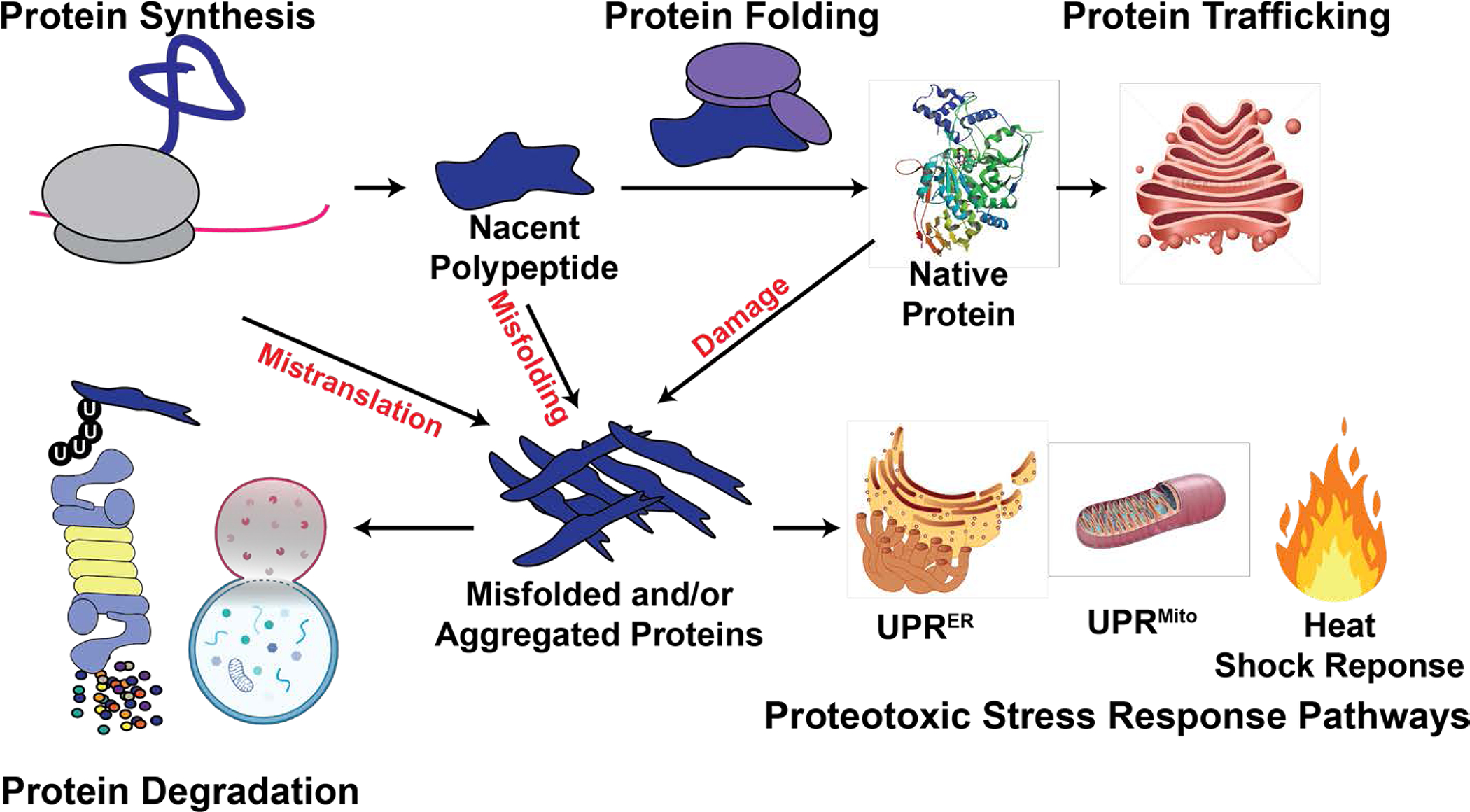
Proteostasis is maintained by an integrated network of physiological mechanisms and stress response pathways that regulate the content and quality of the proteome. Misfolded proteins can arise from errors in translation, protein misfolding, and protein damage. Stress response pathways sense and mitigate the accumulation of misfolded and/or aggregated proteins. Terminally misfolded and aggregated proteins can be eliminated through degradation, via the ubiquitin-proteasome system or autophagy-lysosome system.
Hematopoietic stem cells require low protein synthesis
Protein synthesis is a highly-conserved cellular process that is essential for life. As such, it has long been considered a housekeeping function performed similarly by most cells. However, recent advances that have enabled quantification of relative protein synthesis rates within single cells in vivo have changed this paradigm [2–5]. Distinct populations of hematopoietic stem, progenitor and differentiated cells each establish their own homeostatic rate of protein synthesis, and the amount of protein produced per hour can differ greatly (by an order of magnitude or more) between these closely related cell populations [5,6].
Murine hematopoietic stem cells (HSCs) in young adult bone marrow synthesize markedly less protein per hour than any restricted hematopoietic progenitor population yet analyzed (Fig. 2A). Importantly, this does not simply reflect HSC quiescence. Although dividing HSCs exhibit modest increases in protein synthesis, dividing HSC still have significantly lower rates of protein synthesis as compared to dividing restricted progenitors [5]. Interestingly, subsequent studies have demonstrated that stem cells in multiple other tissues also have relatively low rates of protein synthesis, indicating that that this is a broadly conserved feature of somatic stem cells [7–10].
Figure 2. HSCs depend on low protein synthesis.
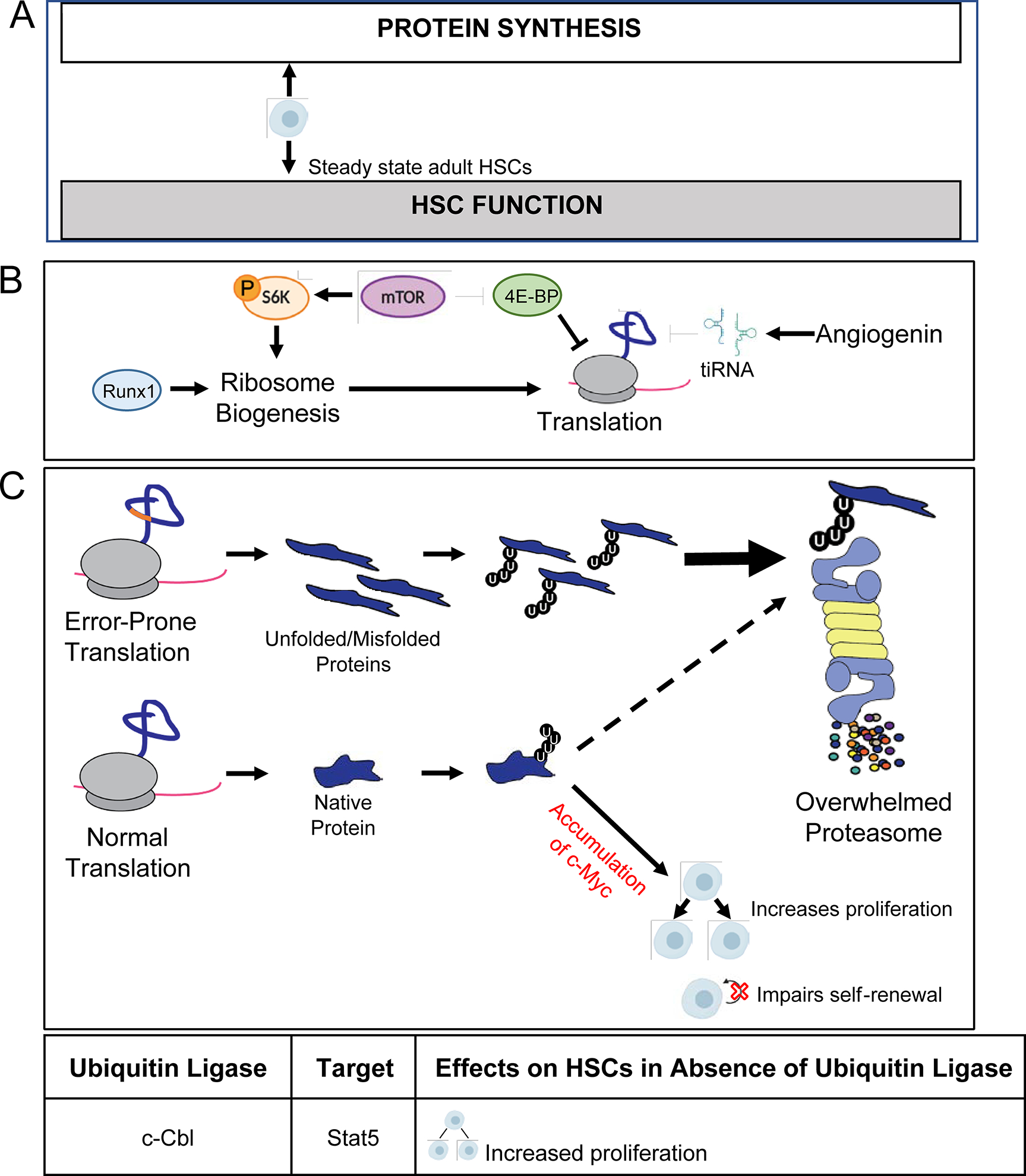
A) HSCs exhibit low rates of protein synthesis and require tight control of protein synthesis. A modest decrease in the rate of translation impairs regenerative activity, whereas a modest increase in the rate of protein synthesis impairs HSC function and maintenance. B) Reduced ribosome biogenesis, hypophosphorylation of 4E-BPs, and tiRNA production contribute to attenuation of protein synthesis in HSCs. Runx1 deficiency reduces ribosome biogenesis and global rates of protein translation. mTOR signaling inactivates 4E-BPs to promotoe the initiation o cap-dependent translation. Angiogenin produced in the bone marrow microenvironment promotes production of tiRNA, which can suppress translation within HSCS. C) Increasing protein synthesis increases promotes the accumulation of unfolded/misfolded proteins that can overload the proteasome, leading to a block in the degradation of proteins that are normally turned over by the proteasome (dashed line), including c-Myc. In HSCs, stabilization of c-Myc results in increased proliferation and loss of self-renewal.
Precise regulation of protein synthesis is critical for HSC maintenance and function (Fig. 2A). A mutation in the gene encoding the ribosomal protein L24 (Rpl24Bst/+) [11] reduces protein synthesis by ~30% within HSCs in vivo, and impairs their long-term multilineage reconstituting potential in competitive transplantation assays [6]. Conditional deletion of the tumor suppressor gene Pten, a negative regulator of the PI3K and mTOR signaling pathways, increases protein synthesis by ~30% within HSCs in vivo, and leads to a total loss of HSC maintenance and function [5,12,13]. Remarkably, when the Rpl24Bst/+ mutation and Pten-deficiency are present together, protein synthesis within HSCs is restored to near-normal levels, and HSC maintenance and function are largely rescued [5]. Together, these findings indicate that protein synthesis is tightly regulated in HSCs and that HSCs require low rates of protein synthesis.
Several studies have begun to uncover the mechanisms that contribute to attenuating protein synthesis within HSCs (Fig. 2B). Surprisingly, protein synthesis does not appear to be limited in HSCs by an absence of ribosomes. HSCs express similar amounts of ribosomal components as restricted progenitors that exhibit much higher rates of protein synthesis [5,14*]. Consistent with this observation, HSCs are dependent on maintaining normal amounts of ribosome production. Deletion of the transcription factor Runx1 reduces ribosome biogenesis and protein synthesis within HSCs, and impairs HSC maintenance [15]. One way that HSCs limit protein synthesis despite having abundant translational machinery, is through the cell intrinsic control of certain signaling pathways. For example, HSCs express higher levels of hypophosphorylated Eif4e-binding proteins (4E-BPs) than restricted progenitors. Hypophosphorylated 4E-BPs are translational repressors that are inactivated by the mTOR signaling pathway [16]. Compound deletion of 4E-BP1 and 4E-BP2 modestly and specifically increases protein synthesis within HSCs in vivo and impairs their serial reconstituting activity [6]. In addition to cell intrinsic mechanisms, protein synthesis within HSCs is also attenuated by cell extrinsic mechanisms. Angiogenin, expressed by cells in the bone marrow microenvironment, promotes the biogenesis of tRNA-derived stress-induced small RNAs (tiRNAs) that can reduce protein synthesis through multiple mechanisms [17]. Angiogenin deficiency preferentially increases protein synthesis within HSCs and is associated with increased proliferation and diminished reconstituting activity [18]. Notably, each of these mechanisms on its own only modestly contributes to restricting protein synthesis within HSCs. This suggests that there are several mechanisms of HSC translational control that have yet to be elucidated, and that several mechanisms may work in concert or redundantly to maintain low protein synthesis within HSCs.
Early studies on HSC protein synthesis raised a fundamental question of why low protein synthesis is necessary for HSC maintenance. Interestingly, genetic or environmental interventions that reduce protein synthesis can extend organismal lifespan in an evolutionarily conserved manner [19]. The effects of attenuating protein synthesis on lifespan are mediated in part by enhancing protein quality control and promoting improved maintenance of proteostasis [20,21]. Translational errors can lead to protein misfolding and the formation of toxic aggregates. High rates of protein synthesis can increase amino acid misincorporation [22], while reducing translation can decrease the synthesis of defective translational products and promote the clearance of misfolded proteins [23]. This raises the possibility that mechanisms that enhance proteostasis maintenance to extend organismal lifespan are conserved at the cellular level to maintain long-lived stem cells in vivo. Consistent with this idea, fetal HSCs, which exhibit higher rates of protein synthesis as compared to adult HSCs, depend upon bile acids produced in the fetal liver to prevent excessive protein aggregation and attenuate endoplasmic reticulum (ER) stress [24]. This mechanism for coping with increased protein synthesis load may contribute to supporting robust HSC expansion during fetal development.
A direct link between protein synthesis and protein quality was recently demonstrated in adult HSCs (Fig. 2C). HSCs contain significantly less unfolded and ubiquitinated protein (a surrogate for misfolded protein) than restricted myeloid progenitors [25*]. The reduced abundance of unfolded/misfolded protein cannot be explained by differences in proteasome activity, which is lower in HSCs. Increasing protein synthesis via conditional deletion of Pten, increases the abundance of unfolded/misfolded protein in HSCs. The accumulation of unfolded/misfolded protein in Pten-deficient HSCs was prevented by the Rpl24Bst/+ mutation. This finding indicates that HSCs depend upon low protein synthesis to prevent the biogenesis of unfolded/misfolded protein. Subsequent work demonstrated that the accumulation of misfolded protein can directly impair HSC maintenance and function. Aarssti/sti HSCs, which have defective tRNA editing activity [26], accumulate modest amounts of ubiquitinated proteins, and exhibit impaired maintenance and self-renewal activity in vivo. In contrast, accumulation of unfolded/misfolded protein has minimal effects on the function of restricted progenitors [25*]. HSC self-renewal and function are thus particularly dependent on the maintenance of proteostasis.
Although several studies have revealed a need to suppress overall protein synthesis for normal HSC maintenance, a key outstanding question is to determine the extent that translational control influences specific mRNAs and proteins [27*]. Future studies utilizing next generation sequencing of ribosome-bound mRNAs [28,29] will be necessary to elucidate the extent and impact of translational control on the HSC proteome.
Protein degradation promotes hematopoietic stem cell maintenance
Although HSCs restrict protein synthesis to reduce the biogenesis of defective translational products, unfolded, misfolded and aggregated proteins can still accumulate within HSCs due to residual errors in translation, protein misfolding, or protein damage (e.g. caused by reactive oxygen species) [30,31]. HSCs may be highly sensitive to disruptions in proteostasis because they are long-lived and mostly quiescent [32]. Long-lived cells can accumulate misfolded proteins throughout life, and quiescent cells less frequently dilute out misfolded proteins through cell division [33]. Thus, in addition to precise regulation of protein synthesis, HSCs may also depend upon cell-type specific mechanisms of protein degradation to regulate proteostasis.
There are two well characterized cellular pathways for protein degradation: the ubiquitin proteasome system and autophagy. These pathways oversee the turnover of normal proteins and also serve as the last line of defense against the accumulation of misfolded proteins [34]. Each of these pathways is essential for regulating HSC maintenance and function, although little is yet known about how these pathways contribute to proteostasis maintenance within HSCs (Fig. 3).
Figure 3. Protein degradation mechanisms promote HSC self-renewal.
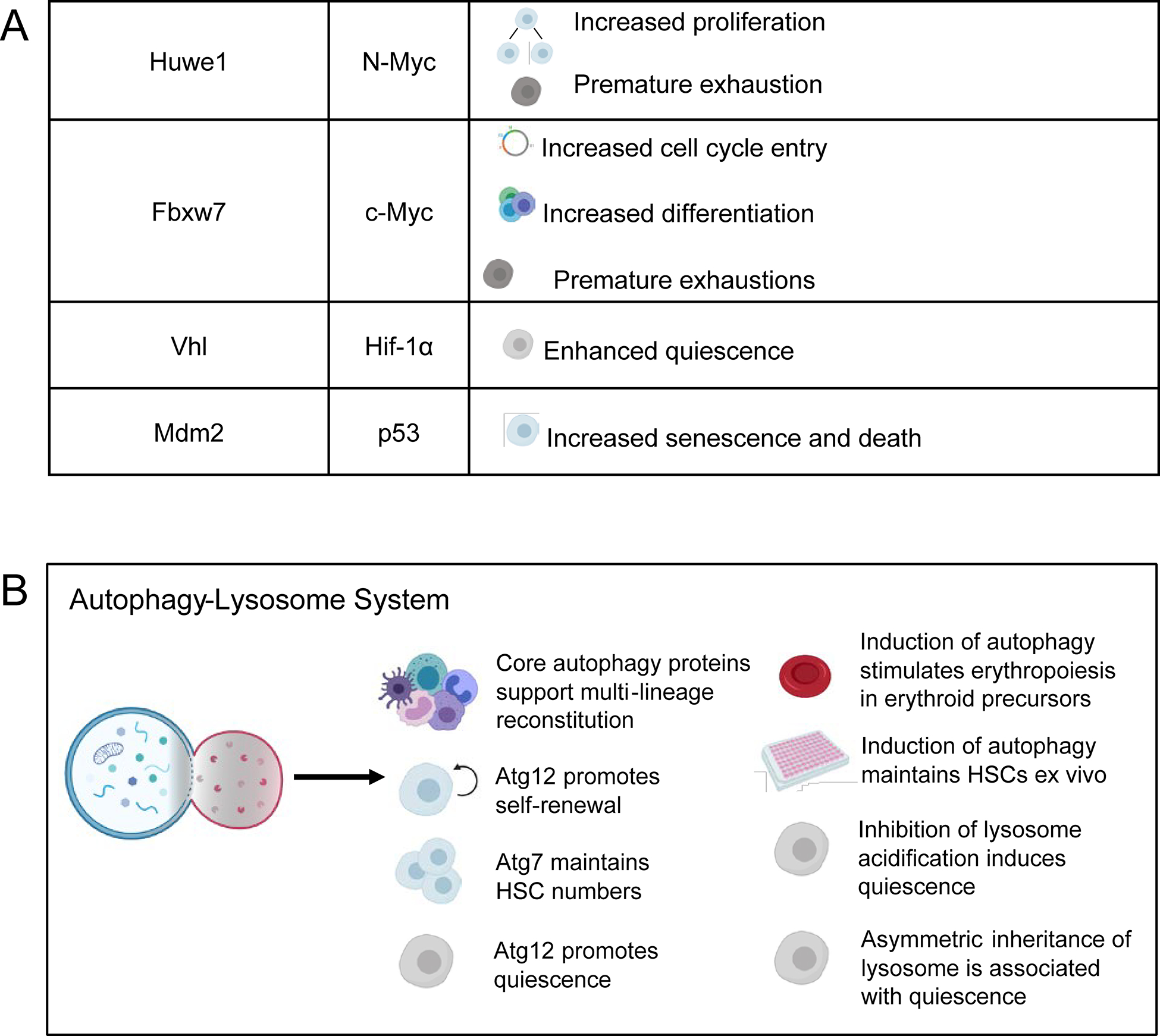
A) Ubiquitin ligases mark the key HSC regulators Stat5, N-myc, c-Myc, Hif-1α, and p53 for degradation by the proteasome. Stabilization or degradation of these target proteins in the absence of the ubiquitin ligases impairs HSC function. B) Core autophagy proteins support multi-lineage reconstitution, self-renewal, quiescence, and maintain HSC numbers. Pharmacological induction of autophagy also maintains HSCs ex vivo and stimulates erythropoiesis. Inhibition of lysosomal acidification induces quiescence, and the asymmetrical inheritance of lysosomes is associated with quiescence.
Both misfolded and normal proteins can be marked for proteasome degradation through the covalent attachment of ubiquitin. Protein ubiquitination is orchestrated by a cascade of ubiquitin ligases, beginning with ubiquitin activating enzyme (E1), which activates and transfers ubiquitin to the ubiquitin conjugating enzyme (E2). E2 then interacts with a ubiquitin ligase (E3) which recognizes specific target proteins for ubiquitination [35]. Much of what is known about how the proteasome regulates HSCs comes from genetic studies of specific ubiquitin ligases that posttranslationally control the abundance of key HSC regulators (Fig. 3A). Deletion of the ubiquitin ligase Fbxw7 leads to increased abundance of its target c-Myc, which drives HSC cell cycle entry, differentiation and exhaustion [36–38]. Similarly, deletion of Huwe1 leads to increased abundance of N-myc, which also promotes HSC proliferation and exhaustion [39]. Deletion of c-Cbl increases abundance and phosphorylation of Stat5, leading to increased HSC proliferation [40]. Hif-1α abundance is also regulated by the ubiquitin proteasome system via the E3 ligase Vhl [41] and can induce HSC quiescence when stabilized [42]. The ubiquitin ligase Mdm2, which targets p53 for degradation [43], promotes HSC survival by blunting the expression of p53 in response to stress [44]. The proteasome thus plays a key role in regulating HSC maintenance and function by controlling protein turnover and proteome content during homeostasis and in response to stress.
Although specific proteasome activity is critical for regulating proteome content and cell fate determination in HSCs, little is yet known about how the proteasome regulates protein quality within HSCs. However, recent findings revealed new insights into how proteostasis may influence proteasome-mediated control of protein turnover within HSCs. HSCs exhibit less proteasome capacity than restricted myeloid progenitors, and a modest accumulation of misfolded proteins in Aarssti/sti HSCs was recently shown to overwhelm proteasome capacity [25*] (Fig. 2C). Overwhelming of the proteasome was associated with stabilization and accumulation of the HSC proteasome target c-Myc, increased HSC proliferation, and impaired self-renewal. An intriguing possibility is that HSCs may “sense” proteostasis disruption through proteasome load, and resulting stabilization of c-Myc occurs to promote cell cycle entry which can restore proteostasis through division mediated dilution. In cases where protein stress is sustained within HSCs, c-Myc accumulation may ultimately promote differentiation and elimination of stressed HSCs from the stem cell pool.
In addition to the proteasome, proteins can be degraded through the autophagy-lysosome pathway. Autophagy is a highly-conserved pathway of self-degradation induced in response to metabolic [45], proteotoxic [46,47] and oxidative stress [48,49]. The process of autophagy begins with the formation of autophagosomes, which are specialized vesicles enclosing cytosolic contents destined for degradation. Autophagosomes then fuse with lysosomes to form autolysosomes, where degradation is completed by lysosomal enzymes. Organelles and macromolecules including normal, misfolded and aggregated proteins can be degraded via autophagy in a bulk or selective fashion [50].
Components of the core autophagy pathway support both fetal and adult HSCs (Fig. 3B). Deletion of Fip200, a member of the ULK complex, depletes fetal HSCs, and impairs reconstituting capacity and erythroid maturation [51]. Similarly, deletion of Atg7 reduces the numbers of adult HSCs and impairs short- and long-term reconstitution [52]. HSCs deficient for Atg12 also exhibit self-renewal defects, impaired reconstitution, and a loss of quiescence [53].
The ability to initiate a robust autophagic response is a key feature of HSCs. In contrast to myeloid progenitors, long-lived HSCs are able to mount an autophagic response to metabolic stress induced by cytokine withdrawal during ex vivo culture. Expression of Foxo3a within HSCs maintains a pro-autophagy gene expression program that can protect HSCs in response to stress [54]. Activation of autophagy may also promote ex vivo HSC maintenance. Combined inhibition of mTORC1 and GSK-3 promotes human HSC maintenance ex vivo, an effect that is associated with expression of a pro-autophagy gene signature [55,56]. Likewise, inhibition of sphingolipid enzyme DEGS1 enhances ex vivo HSC self-renewal in part by activating autophagy [57*]. Induction of autophagy can also stimulate erythropoiesis, suggesting a possible role for autophagy in early erythroid differentiation and development [58].
The role of autophagy in HSCs has largely been studied in the context of mitochondrial homeostasis. Deletion of genes encoding vital autophagy proteins impairs normal HSC function in part by increasing mitochondrial mass and reactive oxygen species, and induction of autophagy promotes mitochondrial clearance (mitophagy) and health [51–53]. Recently, lysosome retention has also been linked to HSC quiescence and mitochondrial health. Quiescent HSCs that maintain low mitochondrial membrane potential accumulate large lysosomes and degrade lysosomes less efficiently. Inhibition of lysosome acidification enhances HSC quiescence, increases the frequency of HSCs that can be recovered from ex vivo culture, and promotes balanced lineage output [59*]. The asymmetric distribution of lysosomes is one mechanism that promotes lysosome retention. Lysosomes and related vesicles (autophagosomes and mitophagosomes) can be asymmetrically distributed to daughter cells. Daughter cells receiving fewer lysosomes have elevated translational activity and are induced to differentiate [60*].
Despite autophagy being a major proteolytic pathway, its role in HSC regulation beyond mitophagy remains largely unknown. In particular, whether autophagy has a direct role in maintaining HSC proteostasis is unexplored. Since HSCs display lower levels of proteasome activity compared to restricted progenitors, an interesting possibility is that autophagy may play an outsized role in safeguarding the HSC proteome.
Protein stress response pathways regulate HSCs
The accumulation of unfolded/misfolded proteins can trigger the activation of cell compartment-specific stress response pathways that can modulate cellular behavior to rebalance proteostasis. Distinct unfolded protein responses are present in both the ER (UPRER) and the mitochondria (UPRMito), and cytosolic protein stress is sensed and resolved by the heat shock response [61–63]. Both the UPRMito and UPRER can influence HSC function (Fig. 4), but little is yet known about how the heat shock response contributes to the regulation of HSC maintenance and function in response to protein stress.
Figure 4. Unfolded protein responses promote HSC function during homeostasis and in response to stress.
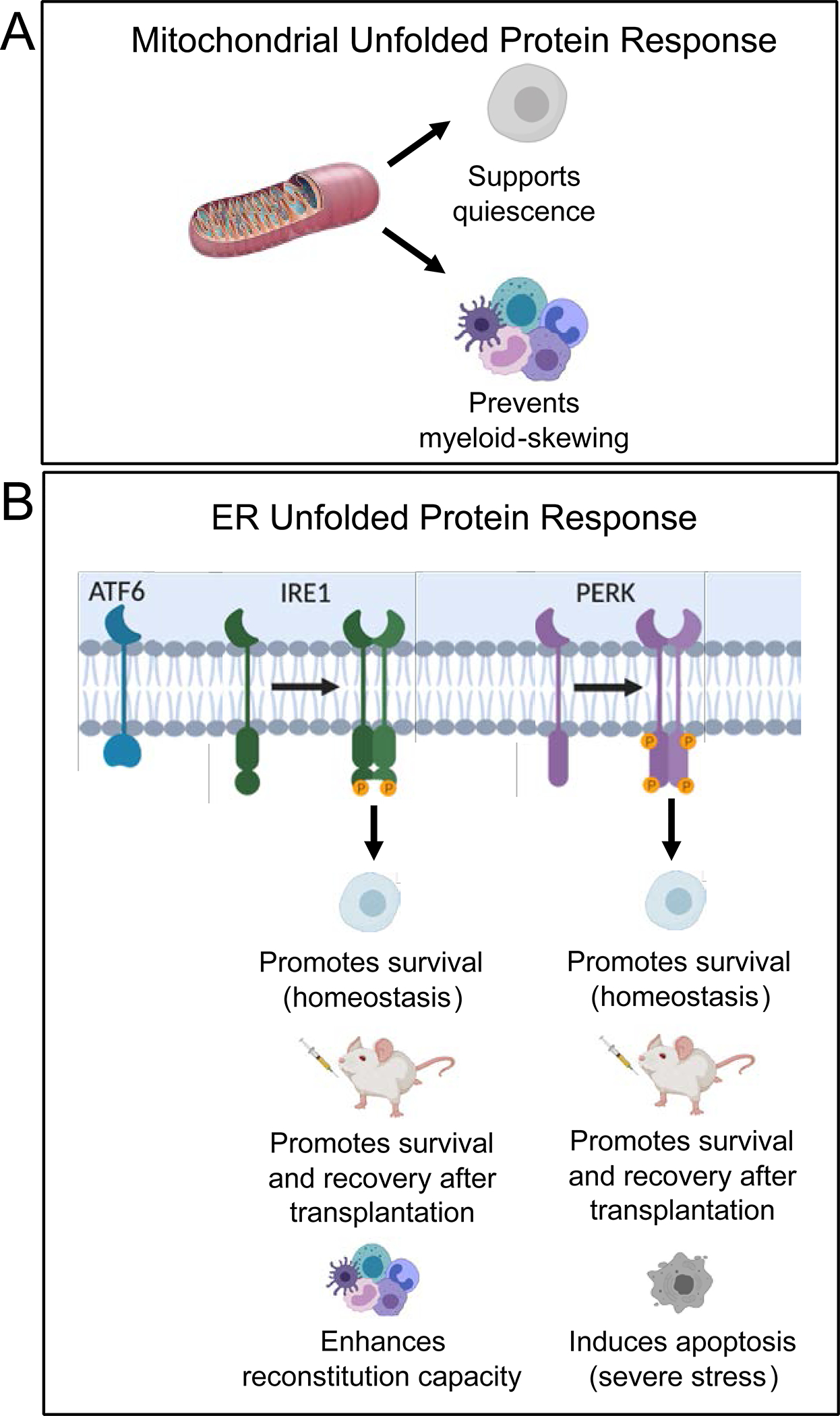
A) Induction of the UPRMito supports HSC quiescence and prevents myeloid skewing. B) The UPRER is induced under homeostatic and severe levels of stress. Activation of the Ire1 and Perk branches of the UPRER promote HSC survival during homeostasis whereas activation of Perk can also induce apoptosis during severe stress to uphold the integrity of the HSC pool. Activation of Ire1 and Perk support survival and recovery of HSCs after transplantation and Ire1 promotes reconstitution capacity.
The UPRMito is initiated to relieve protein stress in mitochondria [61]. In HSCs, Sirt7 relieves mitochondrial protein folding stress by repressing Nrf1 activity and mitochondrial translation, which promotes HSC maintenance and prevents myeloid-skewing during differentiation [64]. Induction of the UPRMito is especially important during the transition from quiescence to activation, which requires increased mitochondrial biogenesis. UPRMito activation during the exit from quiescence appears to act as a quality control checkpoint that ensures that HSCs are metabolically fit for proliferation [65] (Fig. 4A).
The UPRER mitigates the accumulation of unfolded proteins in the ER [63], The UPRER consists of three independent but parallel branches mediated by distinct effectors – Atf6, Perk, and Ire1 [66]. Each branch of the UPRER can sense unfolded proteins and initiate a cascade of events to restore proteostasis by attenuating protein synthesis, upregulating expression of folding chaperones, or if necessary inducing apoptosis [63].
Moderate activation of the UPRER can promote HSC maintenance and regenerative function under steady state and stress conditions (Fig. 4B). Activation of the Ire1 pathway preserves HSC reconstitution activity and protects HSCs from stress-induced apoptosis [67*]. Estradiol enhances HSC regenerative activity and improves bone marrow recovery following irradiation partly by activating the Ire1 pathway [68]. PERK induced activation of ATF4 promotes HSC maintenance under low, homeostatic levels of stress [69*]. However, sustained activation of PERK by more severe stressors, such as pharmacological induction of ER stress, preferentially induces apoptosis within HSCs [70]. Dppa5 can diminish ER stress, protect HSCs from proapoptotic signals triggered by the UPRER, and enhance reconstituting activity [71]. Much remains to be uncovered about how the UPRER functions under both physiological and stress conditions, but it is apparent that it functions uniquely within stem cells to maximize HSC maintenance and function while preserving the integrity of the HSC pool [70].
Recent work has begun to reveal the complexity and integrated dynamics of stress response pathways with other components of the proteostasis network in HSCs. The mechanism of ATF4 activation is particularly noteworthy, as it influences translational dynamics and autophagy. Induction of the Perk branch of the UPRER can suppress overall protein synthesis via phosphorylation of eIF2a [72]. Paradoxically, however, high levels of phosphorylated eIF2a promote translation of specific transcripts, including ATF4 [73]. ATF4 is a transcription factor that, depending on context, drives the expression of pro-survival or pro-apoptosis genes [74]. The eIF2a-mediated upregulation of ATF4 expression in HSCs promotes survival in response to stressors such as transplantation and valine depletion [69*]. Induction of ATF4 also occurs within ex vivo cultured HSCs following the inhibition of DEGS1. In this context, ATF4 induction promotes autophagy within HSCs and is associated with enhanced self-renewal of cultured cord blood derived hematopoietic stem and progenitor cells [57*]. Although ATF4 can induce expression of autophagy genes during stress [75], the integrated coordination of the UPRER, autophagy and other components of the proteostasis network within HSCs is not yet fully understood.
Proteostasis and the aging stem cell
The loss of proteostasis and stem cell exhaustion are two of the “hallmarks of aging” [76]. Aged HSCs exhibit diminished regenerative function, reduced lymphoid differentiation potential and clonal outgrowth that is associated with compromised immunity as well as an increased incidence of anemia, bone marrow failure and hematological malignancies in the elderly [32,77–81]. However, whether proteostasis is lost within aging HSCs, and whether disruptions in proteostasis contribute to age-related declines in HSC function is largely unknown.
In support of a potential role for proteostasis dysfunction contributing to age-related changes in HSCs, young adult HSCs deficient in autophagy experience a loss of quiescence and exhibit a bias towards myeloid differentiation, and old HSCs impaired in autophagy display exacerbated aging phenotypes [53]. Further, aged HSCs express increased levels of some heat shock proteins, raising the possibility that they are under proteotoxic stress and require stress response factors to maintain proteostasis [82].
Aged HSCs exhibit well-characterized features of aging, including an accumulation of DNA damage [83], telomere shortening [84], altered gene expression [85–87], mitochondrial dysfunction and elevated abundance of reactive oxygen species [88]. However, functional interventions within these pathways only modestly rescued HSC function in aged animals [89–93]. One intriguing possibility is that these age-related changes contribute to HSC dysfunction by impinging on proteostasis maintenance. DNA strand breaks in aged HSCs could lead to the production of altered transcripts that are translated into truncated or erroneous proteins susceptible to misfolding. Reactive oxygen species can oxidize and modify proteins making them prone to unfolding [94]. Aging HSCs exhibit increased transcription of ribosomal proteins and hypomethylation of ribosomal RNA, which could influence translation [85,87]. In addition, altered ribosome biogenesis and proteotoxic stress can induce the expression of tumor suppressor genes that impair HSCs and whose expression is increased during aging [2]. Moving forward, a pressing question is thus to examine whether the regulation of proteostasis is a key determinant of HSC aging.
Conclusion
Many components of the proteostasis network have emerged as fundamental regulators of HSC maintenance and function (Fig. 2–4). Surprisingly, despite being highly-conserved, many of these proteostasis mechanisms exhibit cell-type specific regulation and function, and stem cells appear to be particularly dependent on the precise control of proteostasis. Looking forward, there is still a great deal to uncover about how proteostasis mechanisms influence HSC function at steady state, during development and aging, in response to stress, and in disease. Furthermore, it is crucial to recognize that proteostasis mechanisms work in a coordinated network, and changes in one component often result in compensatory changes in one or more other components. The studies reviewed here suggest that the proteostasis network is uniquely configured in HSCs, and unraveling the functional and quantitative relationships that exist within the proteostasis network may be fundamental to harnessing their full regenerative and therapeutic potential.
Key Points.
Components of the proteostasis network function in a cell-type specific manner and fundamentally regulate HSC self-renewal, quiescence, regenerative potential, and metabolism.
HSCs are particularly dependent on the maintenance of proteostasis.
The direct role of the proteostasis network in maintaining proteome content and quality in HSCs is largely unexplored.
A collapse or disruption proteostasis may contribute to age-related and disease-associated changes in HSC function.
Acknowledgements
This research was supported by NIH/NIDDK (R01DK116951; R01DK124775), the American Society of Hematology (Scholar Award), the Sanford Stem Cell Clinical Center, and NIH/NCI 2P30CA023100-28. Figures were created with Biorender.com. The authors declare no competing financial interests.
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 2008; 319:916–919. [DOI] [PubMed] [Google Scholar]
- 2.Buszczak M, Signer RA, Morrison SJ. Cellular differences in protein synthesis regulate tissue homeostasis. Cell 2014; 159:242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hidalgo San Jose L, Signer RAJ. Cell-type-specific quantification of protein synthesis in vivo. Nat Protoc 2019; 14:441–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Xu Y, Stoleru D, Salic A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci U S A 2012; 109:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014; 509:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Signer RA, Qi L, Zhao Z, et al. The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev 2016; 30:1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanco S, Bandiera R, Popis M, et al. Stem cell function and stress response are controlled by protein synthesis. Nature 2016; 534:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llorens-Bobadilla E, Zhao S, Baser A, et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 2015; 17:329–340. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez CG, Teixeira FK, Czech B, et al. Regulation of Ribosome Biogenesis and Protein Synthesis Controls Germline Stem Cell Differentiation. Cell Stem Cell 2016; 18:276–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zismanov V, Chichkov V, Colangelo V, et al. Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 2016; 18:79–90. [DOI] [PubMed] [Google Scholar]
- 11.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 2004; 131:3907–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307–318. [DOI] [PubMed] [Google Scholar]
- *14.Jarzebowski L, Le Bouteiller M, Coqueran S, et al. Mouse adult hematopoietic stem cells actively synthesize ribosomal RNA. RNA 2018; 24:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that ribosomal RNA levels do not correlate with rates of protein synthesis, which suggests that ribosome biogenesis does not limit protein synthesis in HSCs.
- 15.Cai X, Gao L, Teng L, et al. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015; 17:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010; 328:1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanov P, Emara MM, Villen J, et al. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell 2011; 43:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goncalves KA, Silberstein L, Li S, et al. Angiogenin Promotes Hematopoietic Regeneration by Dichotomously Regulating Quiescence of Stem and Progenitor Cells. Cell 2016; 166:894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 2011; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen M, Taubert S, Crawford D, et al. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007; 6:95–110. [DOI] [PubMed] [Google Scholar]
- 21.Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 2007; 445:922–926. [DOI] [PubMed] [Google Scholar]
- 22.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 2009; 10:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherman MY, Qian SB. Less is more: improving proteostasis by translation slow down. Trends Biochem Sci 2013; 38:585–591. [DOI] [PubMed] [Google Scholar]
- 24.Sigurdsson V, Takei H, Soboleva S, et al. Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 2016; 18:522–532. [DOI] [PubMed] [Google Scholar]
- *25.Hidalgo San Jose L, Sunshine MJ, Dillingham CH, et al. Modest Declines in Proteome Quality Impair Hematopoietic Stem Cell Self-Renewal. Cell Rep 2020; 30:69–80.e66. [DOI] [PMC free article] [PubMed] [Google Scholar]; By showing that HSCs depend upon low rates of protein synthesis to minimize accumulation of unfolded and misfolded proteins, this work highlights a direct role between protein synthesis and protein quality.
- 26.Lee JW, Beebe K, Nangle LA, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006; 443:50–55. [DOI] [PubMed] [Google Scholar]
- *27.Chua BA, Van Der Werf I, Jamieson C, Signer RAJ. Post-Transcriptional Regulation of Homeostatic, Stressed, and Malignant Stem Cells. Cell Stem Cell 2020; 26:138–159. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review thoroughly discusses how the proteostasis network regulates the function and identity of embryonic and adult stem cells, including HSCs.
- 28.Ingolia NT, Brar GA, Rouskin S, et al. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 2012; 7:15341550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGlincy NJ, Ingolia NT. Transcriptome-wide measurement of translation by ribosome profiling. Methods 2017; 126:112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009; 16:574–581. [DOI] [PubMed] [Google Scholar]
- 31.Kirkwood TB, Holliday R, Rosenberger RF. Stability of the cellular translation process. Int Rev Cytol 1984; 92:93–132. [DOI] [PubMed] [Google Scholar]
- 32.Signer RA, Morrison SJ. Mechanisms that regulate stem cell aging and life span. Cell Stem Cell 2013; 12:152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010; 11:777–788. [DOI] [PubMed] [Google Scholar]
- 34.Proteasomal Dikic I. and Autophagic Degradation Systems. Annu Rev Biochem 2017; 86:193224. [DOI] [PubMed] [Google Scholar]
- 35.Zheng N, Shabek N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 2017; 86:129–157. [DOI] [PubMed] [Google Scholar]
- 36.Reavie L, Della Gatta G, Crusio K, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol 2010; 11:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson BJ, Jankovic V, Gao J, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med 2008; 205:1395–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson A, Murphy MJ, Oskarsson T, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 2004; 18:2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King B, Boccalatte F, Moran-Crusio K, et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat Immunol 2016; 17:13121321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathinam C, Thien CB, Langdon WY, et al. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev 2008; 22:992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 2008; 9:285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010; 7:391–402. [DOI] [PubMed] [Google Scholar]
- 43.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296–299. [DOI] [PubMed] [Google Scholar]
- 44.Abbas HA, Maccio DR, Coskun S, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 2010; 7:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol 2012; 4:a008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880–884. [DOI] [PubMed] [Google Scholar]
- 48.Rouschop KM, Ramaekers CH, Schaaf MB, et al. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother Oncol 2009; 92:411–416. [DOI] [PubMed] [Google Scholar]
- 49.Tal MC, Sasai M, Lee HK, et al. Absence of autophagy results in reactive oxygen speciesdependent amplification of RLR signaling. Proc Natl Acad Sci U S A 2009; 106:2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bento CF, Renna M, Ghislat G, et al. Mammalian Autophagy: How Does It Work? Annu Rev Biochem 2016; 85:685–713. [DOI] [PubMed] [Google Scholar]
- 51.Liu F, Lee JY, Wei H, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010; 116:4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mortensen M, Soilleux EJ, Djordjevic G, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011; 208:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho TT, Warr MR, Adelman ER, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017; 543:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warr MR, Binnewies M, Flach J, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494:323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang J, Nguyen-McCarty M, Hexner EO, et al. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med 2012; 18:1778–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen-McCarty M, Klein PS. Autophagy is a signature of a signaling network that maintains hematopoietic stem cells. PLoS One 2017; 12:e0177054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *57.Xie SZ, Garcia-Prat L, Voisin V, et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2019; 25:639653, e637. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that sphingolipid modulation can activate autophagy in HSCs and subsquently enhance self-renewal ex vivo.
- 58.Doulatov S, Vo LT, Macari ER, et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med 2017; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *59.Liang R, Arif T, Kalmykova S, et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020; 26:359–376.e357. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that quiescent HSCs accumulate enlarged lysosomes but exhibit lower lysosomal activity. Importantly, this paper also shows that curbing lysosome activity can enhance quiescence.
- *60.Loeffler D, Wehling A, Schneiter F, et al. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019; 573:426–429. [DOI] [PubMed] [Google Scholar]; This work shows that lysosomes can be asymmetrically inherited by daughter HSCs and that lysosome inheritance can predict translational activity and differentiation.
- 61.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 2010; 123:3849–3855. [DOI] [PubMed] [Google Scholar]
- 62.Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2010; 2:a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 64.Mohrin M, Shin J, Liu Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015; 347:1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mohrin M, Widjaja A, Liu Y, et al. The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell 2018; 17:e12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519–529. [DOI] [PubMed] [Google Scholar]
- *67.Liu L, Zhao M, Jin X, et al. Adaptive endoplasmic reticulum stress signalling via IRE1alphaXBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat Cell Biol 2019; 21:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that Ire1 protects HSCs from stress-induced apoptosis and preserves reconstitution capacity.
- 68.Chapple RH, Hu T, Tseng YJ, et al. ERalpha promotes murine hematopoietic regeneration through the Ire1alpha-mediated unfolded protein response. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *69.van Galen P, Mbong N, Kreso A, et al. Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Rep 2018; 25:1109–1117.e1105. [DOI] [PubMed] [Google Scholar]; This work shows that activation of Atf4 promotes HSC maintenance during homeostasis and stress conditions such as transplantation and valine depletion.
- 70.van Galen P, Kreso A, Mbong N, et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 2014; 510:268–272. [DOI] [PubMed] [Google Scholar]
- 71.Miharada K, Sigurdsson V, Karlsson S. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep 2014; 7:1381–1392. [DOI] [PubMed] [Google Scholar]
- 72.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmicreticulum-resident kinase. Nature 1999; 397:271–274. [DOI] [PubMed] [Google Scholar]
- 73.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- 74.Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab 2017; 28:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.B’Chir W, Maurin AC, Carraro V, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013; 153:11941217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beerman I, Bhattacharya D, Zandi S, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A 2010; 107:5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cho RH, Sieburg HB, Muller-Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood 2008; 111:5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol 2013; 13:376–389. [DOI] [PubMed] [Google Scholar]
- 80.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chambers SM, Shaw CA, Gatza C, et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 2007; 5:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beerman I, Seita J, Inlay MA, et al. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014; 15:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allsopp RC, Cheshier S, Weissman IL. Telomere shortening accompanies increased cell cycle activity during serial transplantation of hematopoietic stem cells. J Exp Med 2001; 193:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flach J, Bakker ST, Mohrin M, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014; 512:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Janzen V, Forkert R, Fleming HE, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006; 443:421–426. [DOI] [PubMed] [Google Scholar]
- 87.Sun D, Luo M, Jeong M, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014; 14:673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006; 12:446–451. [DOI] [PubMed] [Google Scholar]
- 89.Allsopp RC, Morin GB, Horner JW, et al. Effect of TERT over-expression on the long-term transplantation capacity of hematopoietic stem cells. Nat Med 2003; 9:369–371. [DOI] [PubMed] [Google Scholar]
- 90.Attema JL, Pronk CJ, Norddahl GL, et al. Hematopoietic stem cell ageing is uncoupled from p16 INK4A-mediated senescence. Oncogene 2009; 28:2238–2243. [DOI] [PubMed] [Google Scholar]
- 91.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity 1996; 5:207–216. [DOI] [PubMed] [Google Scholar]
- 92.Norddahl GL, Pronk CJ, Wahlestedt M, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 2011; 8:499–510. [DOI] [PubMed] [Google Scholar]
- 93.Wahlestedt M, Norddahl GL, Sten G, et al. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood 2013; 121:4257–4264. [DOI] [PubMed] [Google Scholar]
- 94.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012; 24:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]