

Abstract
Approximately 95% of human genes are alternatively spliced, and aberrant splicing events can cause disease. One pre-mRNA that is alternatively spliced and linked to neurodegenerative diseases is tau (microtubule-associated protein tau), which can cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) and can contribute to Alzheimer’s disease. Here, we describe the design of structure-specific lead small molecules that directly target tau pre-mRNA from sequence. This was followed by hit expansion and analogue synthesis to further improve upon these initial lead molecules. The emergent compounds were assessed for functional activity in a battery of assays, including binding assays and an assay that mimics molecular recognition of tau pre-mRNA by a U1 small nuclear ribonucleoprotein (snRNP) splicing factor. Compounds that emerged from these studies had enhanced potency and selectivity for the target RNA relative to the initial hits, while also having significantly improved drug-like properties. The compounds are shown to directly target tau pre-mRNA in cells, via chemical cross-linking and isolation by pull-down target profiling, and to rescue disease-relevant splicing of tau pre-mRNA in a variety of cellular systems, including primary neurons. More broadly, this study shows that lead, structure-specific compounds can be designed from sequence and then further optimized for their physicochemical properties while at the same time enhancing their activity.
Graphical Abstract
INTRODUCTION
RNA plays important roles in a variety of biological processes, including protein synthesis, gene regulation, and RNA transport.1 Mutations in an RNA sequence can lead to cancers, viral infections, and neurodegenerative diseases. Alternative pre-mRNA splicing is carried out by the spliceosome and is regulated via structured regions at intron–exon junctions that are recognized by various splicing factors. Mutations to the pre-mRNA sequence can lead to structural changes that influence the ability of the pre-mRNA to be recognized by the spliceosome. While targeting the spliceosome has been shown to be a potential treatment for cancers, such compounds affect pre-mRNA splicing globally, which may adversely impact their safety profile.2 Directly targeting a specific pre-mRNA could influence specific splicing events by enhancing or preventing the binding of proteins comprising the spliceosome machinery, which thereby afford an improved selectivity and safety profile.2
Current RNA-targeting modalities include antisense oligonucleotides (ASOs) and small molecules. Recently, an ASO has been approved by the U.S. Food and Drug Administration (FDA) to treat spinal muscular atrophy (SMA) and provided a transformative medicine (Spinraza, Biogen).3 However, ASOs have limited tissue distribution and can also stimulate the immune system.4 An alternative approach to target RNA is to use small molecules to target three-dimensionally structured regions. Molecular recognition of structured regions of an RNA with a small molecule is different than targeting an RNA with an oligonucleotide, as the oligonucleotide targets an unstructured region in an RNA.5 Interestingly, the small molecule Risdiplam, currently in clinical trials for SMA, was identified from a phenotypic screen and affects survival motor neuron (SMN) pre-mRNA splicing by binding to a complex between RNA and protein.6–8 In contrast, the ASO, nusinersen (Spinraza) affects SMN pre-mRNA splicing by targeting an intronic splicing silencer downstream of the 5′ splice site of exon 7.9,10
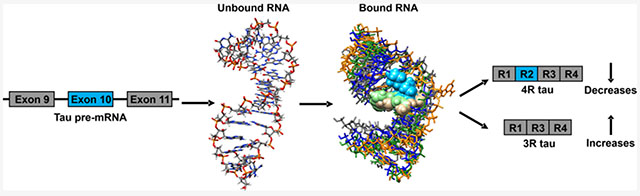
In frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), a mutation in a regulatory RNA structure in the pre-mRNA that encodes for the microtubule-associated protein tau (MAPT, commonly referred to as tau) causes aberrant alternative splicing and generation of a protein isoform prone to forming fibrils.11 In particular, this aberrant splicing event causes the overproduction of an isoform of tau that contains four microtubule binding domains, referred to as 4R, that aggregates. An increase in the formation of the 4R isoform of tau is also known to play a major role in other neurodegenerative diseases such as Alzheimer’s. In this work, we demonstrate that small molecules can be designed to target a structured region of tau pre-mRNA to modulate splicing. These small molecules were designed by our lead identification strategy, Inforna, that is built on experimentally identified and characterized RNA motif–small-molecule interactions.12,13 The 3D folds within a target RNA are compared to this database of privileged interactions, with overlap affording lead small molecule(s). This strategy has been used to identify small molecules that target structured RNAs such as micro-RNAs,14–16 CUG repeats,17 and an iron responsive element (IRE) of α-synuclein mRNA.18 The initial hits reported herein for tau mRNA subsequently can be optimized to serve as useful chemical probes of RNA function to affect disease processes.
The MAPT gene contains 16 exons, among which exons 2, 3, and 10 are alternatively spliced to yield six isoforms containing eight constitutive exons.11 Overproduction of 4R tau is due to mis-regulation of alternative splicing at exon 10 (Figure 1).19 The resulting 4R protein also has additional phosphorylation sites that changes protein solubility and ultimately leads to insoluble aggregates.19 The tau pre-mRNA contains various structures that regulate its biology.20 The FTDP-17 disease is caused by, for example, a C-to-U intronic mutation located 14 nucleotides (nt) downstream of the 5′ splice site of tau exon 10 (C[+14]U; commonly referred to as disinhibition–dementia–parkinsonism–amyotrophy complex (DDPAC), Figure 1).21 The mutation destabilizes the pre-mRNA hairpin structure on the exon 10–intron 10 junction, a splicing regulatory element (SRE), thereby increasing U1 small nuclear ribonucleoprotein (snRNP) binding and exon 10 inclusion.22 The proportion of 4R to 3R tau is therefore above a normal ratio in FTDP-17 patients.21 The introduction of mutations that thermodynamically stabilize the SRE hairpin have been shown to reduce the 4R/3R ratio.21 This suggests a therapeutic strategy in which the stabilization of the hairpin by a small molecule could be capable of inducing exon 10 skipping, thereby reducing the overproduction of the 4R version of tau.21
Figure 1.
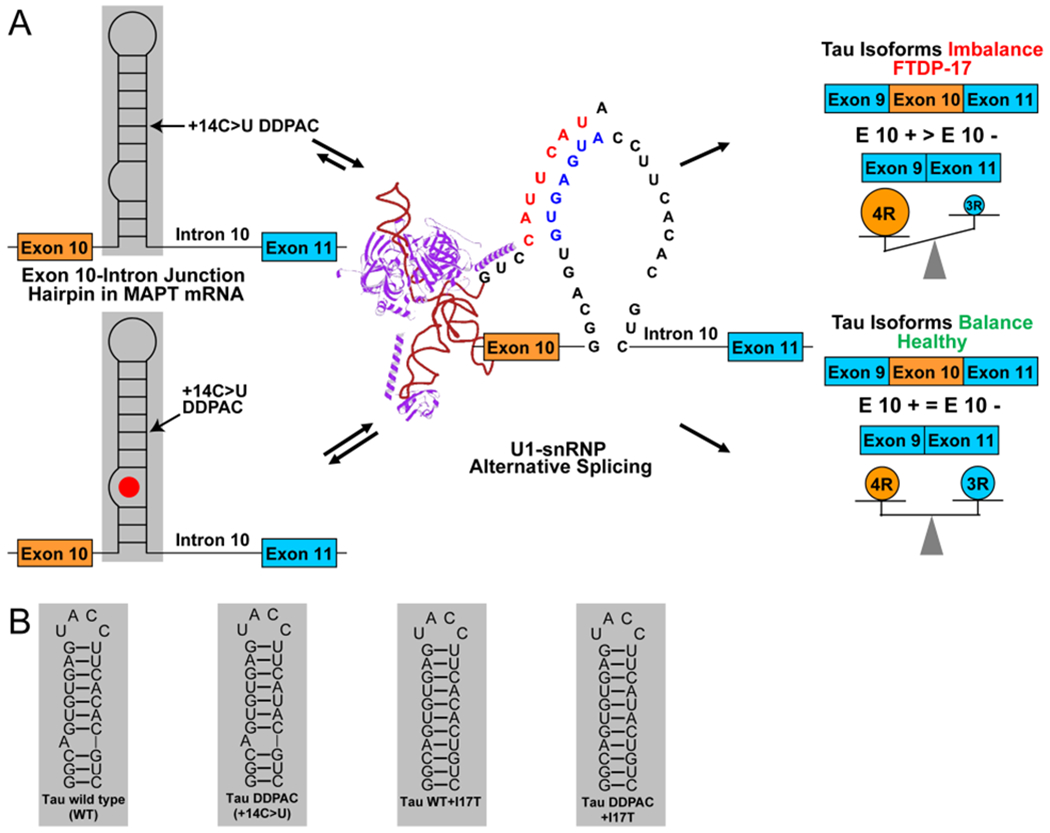
Outcome of alternative splicing of exons 9 to 11 of tau pre-mRNA is regulated by a hairpin at the exon 10–intron 10 junction. (A) Schematic of alternative splicing of exons 9 to 11 of tau pre-mRNA. A +14 C to U mutation destabilizes a hairpin at the exon 10–intron 10 junction, promoting U1 snRNP binding to the pre-mRNA, increasing exon 10 inclusion, and producing 4R tau. Binding of a ligand to the A-bulge site stabilizes the hairpin, increasing exon 10 skipping and production of 3R tau. (B) RNA structures used in this study. Constructs consist of the tau WT hairpin, the tau DDPAC hairpin consisting of a + 14C > U mutation, and mutants of both constructs in which a uridine is inserted across the strand from the A-bulge to form an AU pair that serve as negative controls.
A small molecule that binds to the three-dimensionally folded adenine-containing bulge (A-bulge) region of the SRE and induces exon 10 skipping in cell-based assays was previously identified23 by sequence-based design using the Inforna approach.12,13 This lead chemical probe has sub-optimal physicochemical properties. One of the major challenges in the fast-evolving area of targeting RNA with small molecules is to develop effective strategies for lead generation and subsequent optimization.13 Inforna provides a facile and potentially general route to identify small molecules that target folded RNA structures. Herein, we describe and implement a variety of synergistic approaches to characterize and subsequently optimize compounds that target the 3D structure of the tau pre-mRNA SRE. In the first approach, we identified all compounds that can bind to the tau SRE using Inforna. Analysis of these compounds defined a pharmacophore model for ligands that bind this structure. Querying these models for related compounds in improved physicochemical property space led to the identification of new chemical scaffolds for further optimization. In addition, we solved the solution structures of a variety of compounds bound to the SRE using NMR spectroscopy and restrained molecular dynamics (MD), thus enabling structure-based design methods to facilitate optimization. Each of these compounds were assessed for affecting the SRE using a suite of NMR, biochemical, and cell-based assays. Key observations were made with respect to (1) site-specific ligand binding to the SRE using a compound displacement assay, (2) effect on the dynamics of the SRE upon interaction with the compound as determined by changes in line width in NMR spectra, and (3) changes in the molecular recognition of U1 snRNP for tau pre-mRNA in the presence of the ligand. Armed with these approaches, we defined the structure–activity relationships (SAR) for molecular recognition and also identified new compounds with a variety of improved properties (Figure 2). Ultimately, new compounds were found that target the tau pre-mRNA A-bulge and exhibit robust functional activity in a multitude of cell lines. Compound 1 was the previously reported bioactive compound.23 Compounds 2 to 4 were found in a chemical similarity search using 1 as a query molecule, and compounds 5 to 7 were identified through hit expansion/optimization using a pharmacophore model devised from 1 to 4. Compound 8 was found from docking to an NMR structure of the 4–tau RNA complex, and a hit expansion using 8 as a query molecule resulted in the identification of compound 9 (details of compounds 1 to 9 are provided below). In particular, the modulation of the tau 4R/3R ratio was demonstrated in mouse neurons that were harvested from a mouse model that expressed human tau pre-mRNA. To the best of our knowledge, this is the first small molecule identified that promotes exon skipping by directly targeting RNA in a neuronal cell line.
Figure 2.
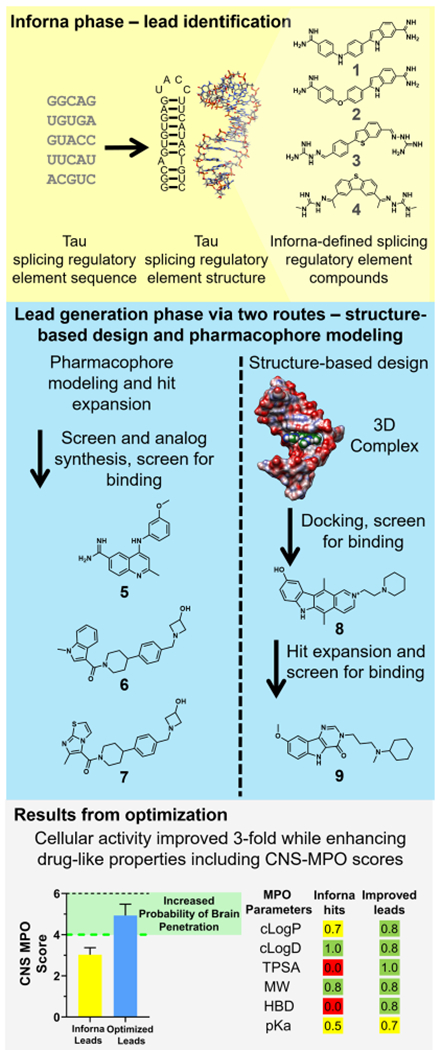
Identification of lead compounds that target the tau exon 10–intron 10 hairpin. Secondary and 3D structures of a splicing regulatory element (SRE) at the exon 10–intron 10 junction were determined by mutational analysis and NMR spectroscopy, respectively.19,66,78,79 An Inforna-based search of a database of RNA–small-molecule interactions that target the SRE led to the identification of compounds 1 to 4. Compounds 1 to 4 were then used as starting points to identify compounds with better physiochemical properties and potency. Pharmacophore modeling of compounds 1 to 4, followed by a chemical similarity search and subsequent analogue synthesis, identified compound 5 and a piperidine amide hit that served as the starting point for the synthesis of analogues 6 and 7. Separately, NMR spectroscopy and restrained molecular dynamics studies were used to determine the structure of 4 in complex with the A-bulge of the hairpin. Docking of the NCI diversity set to the tau RNA–4 complex led to 8, and hit expansion via similarity searching around 8 afforded 9. Hit expansions around compounds 1 to 4 resulted in compounds with improved cellular activities and drug-like properties, as indicated by central nervous system multiparameter optimization (CNS MPO) scores. The higher the CNS MPO score, the higher the probability is of achieving good ADME (absorption, distribution, metabolism, and excretion) proper ties and brain penetration. A majority of CNS drugs have CNS MPO > 4. Values of properties for Inforna compounds are averages of those of compounds 1 to 4. Values of properties for optimized compounds are averages of those of compounds 6, 7, and 9. Error bars indicate SD.
RESULTS
Using Chemical Similarity Searching to Identify Compounds That Affect Tau Pre-mRNA Splicing.
Previously, 1 was identified to target mutant tau exon 10–intron 10 hairpin by querying the tau exon 10–intron 10 hairpin secondary structure against an RNA motif–small molecule database via Inforna (Figure 2).23 This identified a series of small molecules that targeted the A-bulge in the exon 10 pre-mRNA hairpin. The binding of 1 to the pre-mRNA thermodynamically stabilized its folding and modestly induced exon 10 exclusion in the mature mRNA in cells.23 In order to identify more potent compounds that affect pre-mRNA splicing by directly targeting the pre-mRNA, 1 was used as a query molecule in chemical similarity searching campaigns to identify six compounds with a Tanimoto score, or a chemical similarity score, of greater than 90%.24 A chemotype analysis was completed on these six compounds, which was used to requery compound collections, affording an additional 14 compounds (Figure S1). The 20 compounds (Figure S1) were then tested in a luciferase reporter assay for targeting tau pre-mRNA splicing in cells (Figure S2).25 The reporter genes express luciferase in frame with exon 10 such that the alternative splicing outcomes can be measured by luciferase activity: the inclusion of exon 10 (4R) results in increased luciferase activity, while its exclusion (3R) leads to decreased luciferase activity (Figure 3). That is, when exon 10 is included, luciferase is produced and mimics the production of the tau 4R protein that causes FTDP-17. The effect of a compound on exon 10 splicing was studied using three constructs: (i) wild-type (WT) tau; (ii) the DDPAC mutation, which causes FTDP-17 (C to U mutation at position 14); and (iii) the double mutant, known as I17T, containing a C14U mutation and a U insertion opposite the bulged A that served as a cellular control because it lacks the A-bulge binding site (Figure 1).
Figure 3.
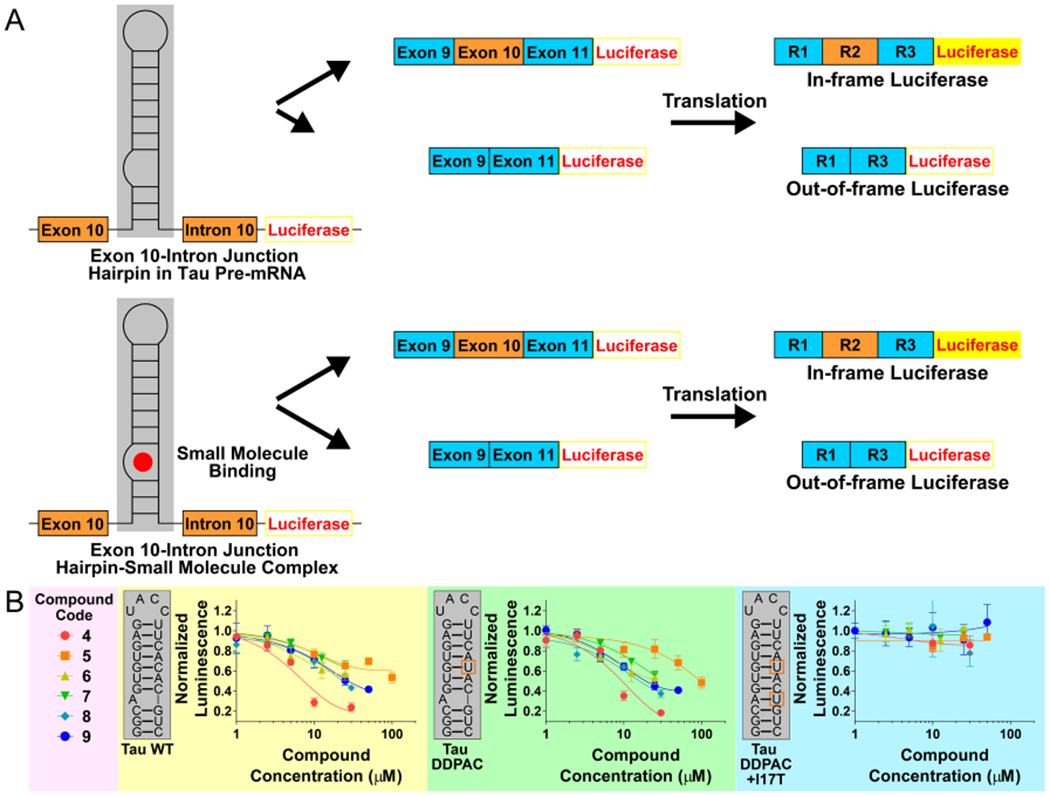
The effects of hit compounds on tau exon 10 splicing, as assessed by a cell-based luciferase reporter assay. (A) Schematic diagram of a mini-gene used in cell-based assays with HeLa cells transfected with a DDPAC or WT construct and treated with compounds. The hairpin at the exon 10–intron 10 junction controls access of U1 small nuclear (sn)RNA to the splice site. In the absence of ligand at the A-bulge, the C(+14)U mutation destabilizes the hairpin, promoting U1 snRNP loading on to the pre-mRNA and exon 10 inclusion. Ligand binding at the A-bulge site stabilizes the hairpin and shifts the equilibrium away from unfolded RNA. The firefly luciferase reporter gene is in frame with exon 10 and expressed when exon 10 is included. (B) Results of cell-based assays with HeLa cells transfected with a WT, a DDPAC, or a fully base-paired DDPAC+I17T construct and treated with various compounds. Mutations in the DDPAC and DDPAC+I17T constructs are indicated with orange boxes. Error bars indicate SD.
Compounds were screened at 20 μM in the cell-based luciferase assay with the DDPAC mutant reporter (Figure 3A). Compounds 2 to 4 reduced luciferase activity more than 1 at 20 μM concentration, and thus a full dose response was completed for them (Figure S3). Cells treated with 20 μM of 4 displayed a significant reduction of luciferase (~60% reduction, p < 0.01), with a statistically significant effect observed at 10 μM. Cells transfected with the WT reporter also exhibited a similar dose-dependence reduction in luciferase activity when treated with 4. No effect was observed when the compounds were tested for affecting the splicing of a tau mini gene pre-mRNA when the A-bulge binding site was replaced with an AU base pair, importantly showing that the effect of the compounds is selective for the A-bulge (Figure 3B).
Compound 4 was then studied in a human neuroblast cell line, BE(2)-M17, that stably expresses a DDPAC tau exon 10 mini-gene reporter. The mini-gene contains tau exons 9, 10, and 11 fused to NanoLuc-PEST. In contrast to the transfected tau mini genes discussed above, exon 10 skipping (3R) stimulates luciferase production. Compounds that simply inhibit the translation of luciferase have no activity in this assay. In agreement with studies in transfected HeLa cells, a dose-dependent effect was observed for 4 (Figure S4). Compound 4 also had improved solubility and, therefore, was amenable to NMR spectroscopy studies (vide infra).
The goal of the subsequent studies was to optimize the activity of compounds that bind the exon 10–intron 10 hairpin and rescue tau pre-mRNA splicing associated with disease. For example, we sought to enhance drug-like properties and therefore improve the physicochemical properties of the compounds, enhance the window between activity and toxicity in various cell lines, demonstrate that molecular recognition of this RNA target can affect pre-mRNA splicing in various cell lines and settings, and broadly define tools to identify and optimize compounds that affect splicing outcomes of not only tau but other pre-mRNAs.
Overview of NMR Spectroscopy of 4-Bound Tau RNA.
To provide insight into ligand binding and its effect on the stabilization of RNA structures, NMR spectra were collected on the A-bulge site in complex with 4. Distance restraints calculated from these spectra were used to model the structure of 4 bound to the tau RNA stem mimic duplex. Stacking interactions between 4 and the A-bulge suggest the compound may affect the emission of a fluorescent analogue of adenine, 2-aminopurine (2-AP), when 2-AP is substituted for the A-bulge. Thus, an assay using 2-AP as a reporter to assess binding was developed to identify other compounds that target the tau SRE (Figure 4A). The NMR spectra and structures of 4-bound tau RNA are discussed below in further detail.
Figure 4.
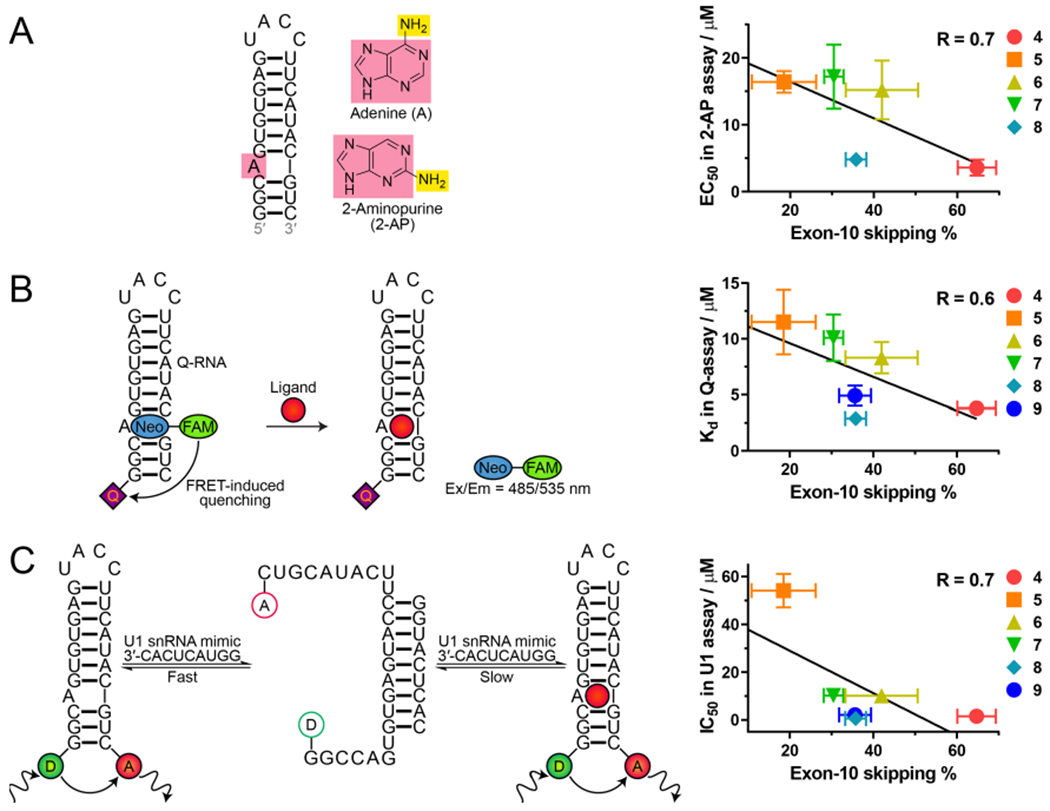
In vitro assays to assess compound binding to tau RNA. (A) 2-AP assay. Addition of compound quenches emission. No data are available for 9 due to spectral overlap with 2-AP. (B) Fluorescence resonance energy transfer (FRET) FRET-based quenching (Q-)assay. (C) FRET-based U1 snRNA assay. To the right of the schemes are plots showing correlation between each in vitro binding assay and exon 10 skipping determined at 10 μM for all compounds except 6 and 7, which were determined at 12.5 μM. Exon skipping assays were carried out with HeLa cells transfected with a DDPAC mini-gene. Error bars indicate SD.
Development of a High-Throughput Assay Using 2-AP to Identify Small Molecules That Perturb Tau A-Bulge Dynamics.
Our biophysical studies indicate that 4 binding perturbs the dynamics of the tau A-bulge, suggesting that a high-throughput binding assay could be developed by substituting the A-bulge with the fluorescent adenine mimic 2-aminopurine (2-AP). The fluorescence of 2-AP is dependent upon its microenvironment (stacked vs unstacked in a helix, for example). Indeed, 2-AP has been used previously to study the binding of aminoglycosides to a model of the bacterial A-site, particularly changes in the RNA’s structural dynamics.26 The optimal position for 2-AP to monitor compound binding was first determined in models of the WT and DDPAC hairpins. Replacing the A-bulge with 2-AP was optimal as compared to replacing an adenine in base pairs adjacent to the bulge, as assessed by the change induced by the binding of 4 (Figure S5).
As expected, 4 decreases the fluorescence intensity of both WT and DDPAC RNAs labeled with 2-AP in a dose-dependent fashion (Figure S6). Fitting the resulting curves afforded EC50 values of 6.2 ± 0.8 and 3.6 ± 1.2 μM for WT and DDPAC RNAs, respectively. Importantly, the 2-AP label does not have a very significant effect on small-molecule binding since Kd’s for unlabeled WT and DDPAC RNAs are 9.8 and 1.3 μM, respectively. Compound 4 was then tested with minimal WT and DDPAC duplex constructs used for NMR studies, but with 2-AP substitutions at the A-bulge site (Figure S6). For the WT and DDPAC duplexes, 4 decreased the fluorescence intensity in a dose-dependent manner, with EC50’s of 0.7 ± 0.1 and 2.1 ± 0.4 μM, respectively (Figure S6). The data confirmed that these duplexes were good model constructs for NMR studies of ligand—tau RNA complexes. Using 4 as a positive control, the 2-AP binding assay was investigated for suitability for higher throughput screening by calculating its Z-factor, which is a statistical parameter that quantifies the dynamic range and variation in a high-throughput screen and range from 0 to 1 (ideal).27 A score of >0.5 is considered suitable for high-throughput screens. The Z-factor for the 2-AP assay is 0.7, which indicates its suitability for completing a screen for RNA binding and affecting dynamics.
Development of a Competitive Binding Assay That Uses Fluorescent Quenching.
An in vitro competitive binding assay was developed to assess the binding of compounds to the exon 10–intron 10 hairpin. In the assay, the Q-RNA labeled at the 5’ end with a black hole quencher is bound to a Q-probe, which consists of a fluorescein amidite (FAM)-labeled neomycin (Figure 4B).29 Upon complex formation, the FAM emission intensity decreases as the quencher and the fluorophore are in close proximity.28 Displacement of the Q-probe from the A-bulge by a competitor molecule leads to the recovery of FAM fluorescence. Neomycin B was used in this binding assay because of its favorable aqueous solubility, and additionally, an NMR structure of the neomycin–tau RNA complex showed that the neomycin B 5′-OH group does not form contacts with the RNA and therefore can be functionalized to attach a fluorescent probe.29,30 The quenching assay allows for compounds to be screened for binding that had spectral overlap with 2-AP. The Z-factor for the quenching assay is 0.6, which also indicates that it may be adapted as a high throughput screening (HTS) method.
Development of a U1 snRNA Assay That Mimics Recognition of Tau Pre-mRNA by Spliceosomal Components.
In the U1 snRNA assay, cyanine Cy3 and Cy5 dyes are attached to the 5′ and 3′ ends, respectively, of the exon 10–intron 10 hairpin construct (Figure 4C). The addition of an oligonucleotide mimic of U1 snRNA causes the hairpin to unfold and form a complex, resulting in decreased FRET between Cy3 and Cy5. When a small molecule is added that binds to the tau A-bulge, the hairpin is thermally stabilized, which decreases the rate of complex formation between U1 snRNA and exon 10–intron 10 hairpin, as read out by a FRET decreasing rate. The U1 snRNA mimic assay is a kinetic assay that allowed us to investigate the ability of ligands to stabilize the exon 10–intron 10 hairpin and slow the unfolding rate, which mimics U1 snRNA unfolding of the same hairpin in cells and subsequent inclusion of exon 10. After the addition of ligand, the rate of increase of Cy3 emission was measured and compared to that measured for the free-form RNA.
The addition of 4 resulted in the lowest rate of Cy3 emission recovery. This result is consistent with the 2-AP binding affinity assays and demonstrates that the FRET-based assay may be used as an alternative assay for identifying compounds that bind to a target RNA. The Z-factor for the U1 snRNA assay is 0.6, indicating it may be adapted as a HTS method.
Primary Hit Expansion of Lead Compounds from the Initial Inforna Hits.
A hit expansion was performed based on compounds 1 to 4 with the goal of identifying improved leads from computational mining of small molecule collections. Several approaches were pursued in parallel: (1) Pharmacophore models were constructed using LigandScout,32 where the training set included the four active compounds along with 16 inactive structures (See Figure S1 for structures). Shared feature pharmacophore models were created to differentiate the active from inactive compounds. The selected pharmacophore model used in the virtual screen incorporates one hydrophobic interaction, two H-bond donors (HBDs), two H-bond acceptors (HBAs), one aromatic ring, and positive ionizable areas. Positive ionizable area features were excluded for the virtual screen to avoid retrieving highly polar molecules. The model was screened against the Pfizer corporate compound collection of approximately 3 million compounds, and the top-ranked 1000 hits were selected by the following process. A GPU-based implementation of the ROCS algorithm, FastROCS,33 was used to identify compounds from the Pfizer collection with high shape-based similarity to each of the four active compounds. A TanimotoCombo34 score was used to rank the compounds based on the shape and chemical similarity between the query (four active compounds) and compounds in the collection. As a result, 113 hits with a TanimotoCombo score >1.2 were selected.34 A 2D similarity search was also performed around the four active compounds using various types of fingerprints, including ECFP_4, FCFP_4, and MOE 2D.35 The similarity scores calculated using each of these fingerprint methods were normalized and combined using a mathematical rule.35 The resulting hits were then ranked using the combo score, and approximately 1000 compounds were selected for further triaging. Compound sets from the methods outlined above were combined and prioritized based on overall chemical attractiveness and physicochemical properties.
Using this approach, an initial set of 305 compounds in total were selected from the Pfizer compound collection and screened in the 2-AP assay and cell-based luciferase assay with the DDPAC mutant reporter. This resulted in 33 putative hits that were retested in triplicates in the 2-AP assay to inform the selection of a subset that was further profiled in both in vitro binding and luciferase assays in BE(2)-M17 cells. Among the most interesting chemotypes to emerge from these efforts was a piperidine amide that served as the starting point for a set of analogues, resulting in 6 and 7 as the top hits (Figure 2, Table S5). The nature of the amide heterocycle in 6 and 7 is clearly important for both RNA-binding affinity and functional activity, as the corresponding benzamide 10 was devoid of activity in the in vitro assays (Table S5). In addition to the piperidine amide series, amidine 5 was also identified from the hit expansion, and it was also selected for detailed profiling. The activities in the 2-AP assay were confirmed to be 16.4 μM for 5 and 15.2 and 16.9 μM for 6 and 7, respectively. These results were further corroborated by demonstrating the activity in the Q-assay and U1 mimic assay (Table S5). The translation from in vitro binding to tau splice modulation in cell-based assays was subsequently established by showing a dose-dependent effect in the luciferase assay with the DDPAC mutant reporter. Compounds 5 to 7 reduced the luciferase activity by 30% at 50, 7, and 12 μM concentrations, respectively (Figure 3). Further confirmations were also completed in the BE(2)-M17 luciferase assay (Figure S4).
Compared with compound 4, analogues 6 and 7 have significantly improved central nervous system multiparameter optimization (CNS MPO) scores (5.2 for 6, and 5.5 for 7, compared with 2.7 for 4, on a scale from 0 to 6). The higher the CNS MPO score is, the more likely it is that a compound will have good brain penetration (BBP) and overall favorable ADME (absorption, distribution, metabolism, and elimination) and safety attributes.36 The CNS MPO score is calculated from six physiochemical parameters: (1) calculated lipophilicity (cLogP); (2) calculated distribution coefficient at pH 7.4 (cLogD); (3) molecular weight (MW); (4) topological polar surface area (TPSA); (5) number of HBDs; and (6) pKa.36 In particular, the presence of multiple HBDs can adversely impact brain penetration. It is therefore noteworthy that compounds 6 and 7 only contain one HBD, which is a significant improvement relative to compounds 1 to 4. The identification of compounds 6 and 7 indicates that Inforna hits can serve as valuable starting points to identify RNA modulators with robust functional activity in cell-based assays and with favorable drug-like properties suitable for further lead optimization using standard medicinal chemistry approaches.
Using the 4–WT Tau RNA Structure and 2-AP Assay to Identify Novel Small Molecule Binders That Inform a Complementary Hit Expansion.
As the in vitro 2-AP assay is suitable for screening small molecules for binding to tau RNAs, small molecules from the NCI diversity set37 that were first triaged by docking were studied. Briefly, small molecules from the diversity set were docked into the structure of the 4–WT tau RNA complex and scored for binding. Glide docking scores were computed for each docked structure from free energy terms that favor binding, such as hydrogen-bonding and hydrophobic interactions, and terms that hinder binding, such as ligand strain and water desolvation.38 Compounds with a Glide docking score <–9 were considered hits. A subset of these compounds was commercially available (n = 15) and carried forward in binding assays with tau RNAs. Of the compounds (10 out of 15), 67% can be categorized into three chemotypes: ellipticines, bis-quinoliniums, and phenothiazines, which are known RNA binders. In particular, an ellipticine binds to r(CGG)exp and inhibits its association with DGCR8Δ (Kd = 76 ± 4 nM); bis-quinoliniums bind to and stabilize RNA G-quadruplexes, and a phenothiazine, acetopromazine, binds to the A-bulge loop in HIV-1 TAR RNA (Kd= 270 nM).39–41 The 15 compounds were screened for binding to 2-AP-labeled WT and DDPAC RNAs. Within these compounds, the most active compound, 8, decreased 2-AP emission with an EC50 of 4.8 μM and was active in the cell-based luciferase assay with an IC50 of around 10 μM (Figure 3 and Table S5).
To verify that these compounds were selective for the A-bulge, as compared to other bulges, competitive binding assays between 2-AP-labeled DDPAC RNA and unlabeled RNAs with each single nucleotide bulge were completed.42 These data are summarized in Table S6. For 5 to 7, saturable binding is only observed for the RNA containing the A-bulge, with IC50s ranging from 12 to 15 μM. Likewise, 4 showed a ~10-fold selectivity for the RNA with an A-bulge over a C-bulge, with no saturable binding observed for RNAs with G- or U-bulges (up to 200 μM concentration). Compound 8 was ~7-fold selective for the A-bulge (IC50 = 3.2 ± 0.6) over the other bulges (IC50’s ranging from 19 to 26 μM). Thus, all five compounds show selectivity for the A-bulge, as compared to other single nucleotide bulges.
Using 8 as a starting point, the Pfizer corporate compound collection of approximately 3 million compounds was again searched using a combination of the 2D similarity and FastROCS33 methods described above along with consideration for CNS physicochemical properties. Ultimately, about 200 compounds were selected for screening, leading to the identification of 9 as a promising lead (Figure 2). Compound 9 exhibits single-digit micromolar activity in the quenching and U1 assays with Kd and IC50 values of 4.9 and 2.1 μM, respectively, and it resides in a good CNS property space, as indicated by the CNS MPO score of 4.4. The activity in the 2-AP assay could not be established due to compound emission interfering with the 2-AP signal.
The improved biochemical potency of 9 translated into robust and dose-dependent functional activity in the cell-based luciferase assay, with an IC50 of approximately 10 μM. More importantly, 4 and 8 have significant cellular toxicity at 50 μM, whereas 9 has no toxicity even at 100 μM (Figure S7). The larger window between toxicity and activity indicates greater biocompatibility of 9. Finally, 5 to 9 were also tested and had no effect on the splicing of the A-bulge-mutated tau mini-gene in transfected HeLa cells (Figure 3B).
Interestingly, there is a good correlation between the EC50, Kd, or IC50 values of all compounds determined by the different assays and percentage of exon 10 skipping, with correlation coefficients of 0.6 to 0.7 for each assay (Figure 4). However, factors such as cellular uptake and solubility can affect a compound’s biological activity, resulting in a modest difference in in vitro Kd or EC50 values relative to cellular IC50 values.31 Among the three in vitro assays, the U1 snRNA mimic assay gives the best correlation with biological activity (R = 0.7), demonstrating this assay best reflects events that happen in the biological environment.
Thermodynamic Stability of Tau RNAs in Complex with Compounds 4–9.
Collectively, the biochemical and cellular assays suggest that compounds 4 to 9 thermally stabilize the tau RNA. To provide experimental evidence that this is the case, we completed optical melting experiments for the DDPAC, WT, and DDPAC+I17T constructs in the absence or presence of compounds (Figure S17). Among the unbound constructs, the DDPAC RNA had the lowest melting temperature (Tm), and the WT RNA had the highest Tm. The Tm of WT and DDPAC increased the most in the presence of compounds 4, 8, and 9, with greater effects on the DDPAC RNA than the WT RNA. These results are consistent with binding affinities determined from the three in vitro binding assays and activities in the cell-based assays. In agreement with in vitro binding assays, compounds 5, 6, and 7 increased the Tm of the WT and DDPAC RNAs less than the higher affinity binders 4, 8, and 9. Piperidine amides 6 and 7 increased the Tm of WT and DDPAC RNAs similarly to each other. In the DDPAC+I17T construct, only compound 8 significantly increased Tm and stabilized the RNA, in agreement with the lower selectivity observed in cell-based luciferase assays and RT-qPCR. Taken together, each compound binds to the WT and DDPAC tau RNA constructs and stabilizes the DDPAC construct to a greater degree than the WT construct.
Testing Compounds for Affecting Tau Pre-mRNA Alternative Splicing in Cells by RT-qPCR and Western Blot.
Next, we verified that the compounds with activity in vitro and in cell-based luciferase assays are functioning at the RNA level by measuring levels of 3R and 4R mRNA isoforms by RT-qPCR.21 In these experiments, a Vivo-Morpholino ASO complementary to the exon 10–intron 10 junction was used as a positive control (4R-to-3R ASO) and was expected to inhibit exon 10 inclusion.21 Compounds 4 to 9 were tested in HeLa cells expressing a WT or a DDPAC mini-gene as well as for their effects on endogenous tau in LAN5 neuroblastoma cells. Indeed, all six compounds reduced the 4R/3R ratio (4R tau mRNA expression divided by 3R tau mRNA expression) (Figure S8). Given that 9 is one of the most active compounds in all assays, we measured the change in endogenous tau 4R and 3R protein levels in LAN5 cells by Western blot. Compound 9 and the 4R-to-3R ASO both demonstrated a significant reduction of the 4R/3R ratio (4R tau protein abundance divided by 3R tau protein abundance), without changing total tau expression (Figure S9). To summarize the results of the hit expansions compared with initial Inforna hit compounds 1 to 4, the follow-up leads 6, 7 and 9 have dramatically increased CNS MPO scores, from an average of 3.0 to 4.8 (Figure 2), as well as modestly enhanced cellular activity. Considering that CNS drug candidates usually have CNS MPO scores over 4,36 the identification of 6, 7, and 9 starting from Inforna hits followed by subsequent hit expansions and analogue synthesis indicates that this approach can be a valuable lead generation strategy to identify drug-like leads for directly targeting RNA.
Demonstrating Binding of Compound 9 to Tau pre-mRNA in Cells Using Chem-CLIP.
Given that 9 is one of the most active compounds in all assays, we sought to profile its binding to tau pre-mRNA in cells. By studying the complex of 9 with the WT tau RNA, we designed compound 11 to further assess the in vitro and cellular target engagement of 9 by using Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) (Figure 5A) to study the compound occupancy.14,43–47 Competitive Chem-CLIP (C-Chem-CLIP), a complementary approach, was also used to evaluate the binding of parent compound 9 directly by competing for the binding of RNA targets with the Chem-CLIP probe 11. On the basis of the binding mode of 9 as established by NMR spectroscopy studies (vide infra), no stacking interaction was contributed by the cyclohexane of 9. Therefore, it provides a site that can be modified with cross-linking modules. We introduced a diazirine module for photo-cross-linking with biomolecules in proximity and an alkyne purification module for pull-down with streptavidin beads after click-chemistry-mediated conjugation to a biotin azide reporter (Figure 5B).48 A control compound 12 lacking the RNA-binding module was also synthesized (Figure 5B). Chem-CLIP probe 11 had a similar activity to 9 in in vitro and biological assays, indicating that the introduction of the diazirine alkyne module has a limited effect on compound binding to the target RNA (Figures S10 and S11, Table S5). The ability of 11 and 12 to react with the tau hairpin RNAs was assessed in vitro (Figure S12 and S13). Photoprobe 11 only reacted with tau WT and DDPAC RNA with an IC50 value of about 10 μM, while not capturing the I17T mutant. In contrast, 12, due to lacking an RNA-binding module, did not label any RNAs in this experiment (Figure S12). C-Chem-CLIP experiments were completed with 9 to compete with 11 for the RNA binding sites and thereby reduce the labeling of tau pre-mRNA with photoprobe 11. The IC50 of 9 in C-Chem-CLIP experiments was close to 10 μM, further bolstering 11 and 9 binding to RNA targets with a similar affinity.
Figure 5.
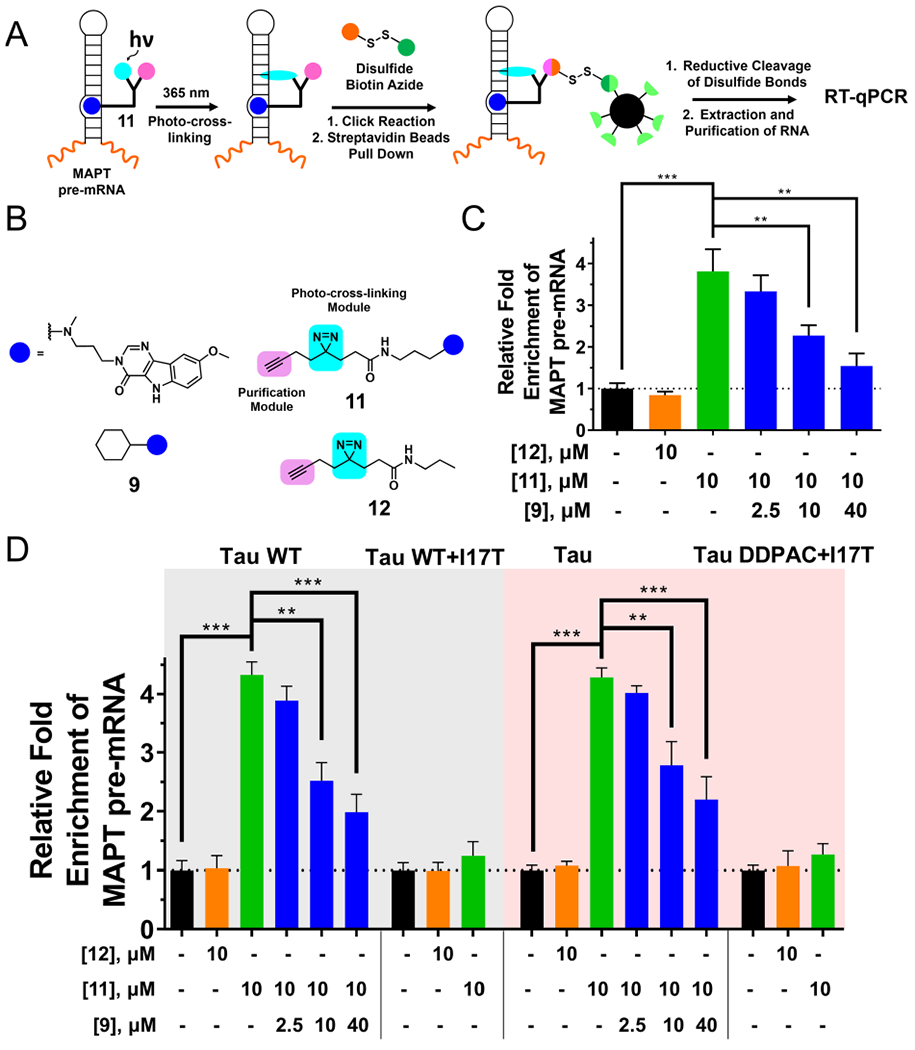
Target profiling in cells using Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP). (A) Scheme of Chem-CLIP to react with an RNA target in a cell to validate target engagement of small molecules. (B) The compounds used in Chem-CLIP and competitive Chem-CLIP (C-Chem-CLIP). (C) The Chem-CLIP probe of bioactive ligand 9 (compound 11) reacts with tau pre-mRNA in LAN5 cells, whereas control probe 12 does not. C-Chem-CLIP shows that when increasing concentrations of 9 are preincubated with LAN5 cells, the pull-down of tau pre-mRNA with a constant concentration of 11 is diminished in a dose-dependent manner. (D) Compound 11 reacts with tau pre-mRNA in WT and DDPAC mini-gene-transfected HeLa cells, whereas 12 does not. Preincubation of 9 diminished pull-down of 11 in C-Chem-CLIP. Compound 11 does not pull-down I17T mutant tau pre-mRNA in transfected cells. Collectively, Chem-CLIP pull-down of 11 and C-Chem-CLIP studies show that target engagement is dependent on the presence of the 3D structure of the RNA target, as mutations abate pull-down. Error bars indicate SD. ** P < 0.01 and *** P < 0.001, as determined by a two-tailed Student t-test.
We then used Chem-CLIP and C-Chem-CLIP to study target engagement in LAN5 neuroblastoma cells (Figure 5C) and transfected HeLa cells (Figure 5D). A 4-fold enrichment of tau pre-mRNA was observed with 10 μM of 11 but not 12 in the pulled-down fraction, as measured by RT-qPCR compared with the amount of tau pre-mRNA before pull-down (Figure 5C and 5D). The C-Chem-CLIP experiment was performed by preincubating cells with an increasing concentration of parent compound 9, followed by treatment with a constant concentration of 11. Similar to in vitro C-Chem-CLIP studies, the pull-down of tau pre-mRNA by 11 was inhibited in a dose-dependent manner when RNA targets were preoccupied by 9 (significantly at 10 μM; Figure 5C and 5D). Again, 12 did not pull down any of the RNA targets. Importantly, there was no significant enrichment by 11 in A-bulge-mutated tau-mini-gene-transfected HeLa cells, which confirms that the binding site of 11 is also the A-bulge, the same as parent compound 9 (Figure 5D). We then measured the enrichment of mature tau mRNAs, which do not contain the A-bulge, mRNAs containing iron responsive elements (IREs) with single nucleotide bulges, GAPDH, and β-actin mRNAs by 11 to assess its selectivity for the tau A-bulge (Figure S14). GAPDH and β-actin mRNAs were chosen as they represent highly abundant cellular RNAs. Compound 11 did not enrich these mRNAs, confirming it is selective for the tau exon 10–intron 10 junction in tau pre-mRNA.
Effect of Compounds on Other RNAs Containing the Tau A-Bulge.
As a further test of selectivity, we studied the effect of 7 and 9 on other RNAs that contain the same A-bulge at tau exon 10-intron 10 junction. We identified ten such RNAs in a database of all secondary structural elements found in human microRNA precursor hairpins (Figure S15).49 Treatment of LAN5 cells with 7 or 9 had no effect on the expression level of any of the RNAs studied (Figure S15). This could be due to a variety of factors, including: (i) the next nearest neighbor base pairs of the A-bulge in these sequences differ from those in the tau exon 10 hairpin, which may affect small molecule recognition; (ii) expression level; and (iii) in nine of the ten miRNAs, the A-bulge is located in a nonfunctional sites, i.e., not in a Drosha or Dicer processing site.
Compound 9 Affects Tau Pre-mRNA Splicing in hTau Mice Primary Neurons.
In addition to transfected and neuroblastoma cell lines, we evaluated 9 in cultured primary neurons extracted from htau transgenic mice. Brains from htau mice pups were isolated and, after removal of the meninges, the neurons from the cortex were isolated and cultured in precoated plastic plates (Figure 6).50 After 15 days in vitro (DIV), the cultured neurons were treated with 9, and the 4R/3R tau ratio was measured by RT-qPCR after 2 days, similar to studies in LAN5 cells. [Note that gentamicin was added to the culture medium at a final concentration of 4.2 nM. To verify that gentamicin did not affect tau exon 10 splicing, LAN5 cells were separately treated with gentamicin at 0.4, 4, and 40 nM, and the 4R/3R tau ratio was measured after 2 days (Figure S16). No effect on tau splicing was observed at any of the concentrations tested.]
Figure 6.
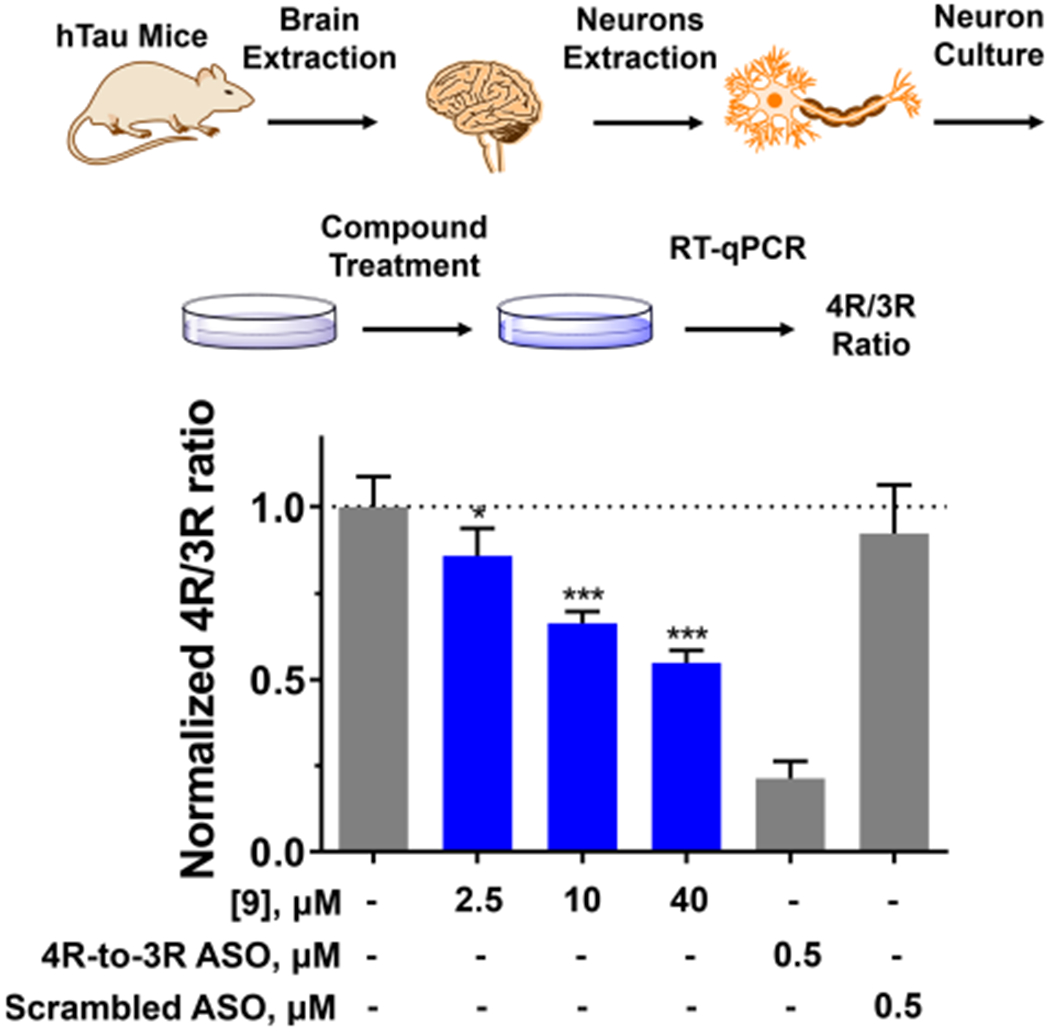
Effect of 9 on the pre-mRNA splicing outcome of tau exon 10 in primary neurons harvested from human tau transgenic mice (htau mice). (Top) Primary neurons were extracted and cultured from htau mice brains as described in Experimental Section. Total RNA was extracted after 48 h with treatment of DMSO (vehicle), 4R- to-3R ASO, or a Scrambled ASO control (“Scrambled”). The 4R/3R ratio, which was calculated as 4R tau mRNA expression divided by 3R tau mRNA expression, was measured by RT-qPCR. (Bottom) Compound 9 downregulated 4R/3R ratio dose-dependently, up to 50%. The 4R-to-3R ASO decreased 4R/3R ratio by 80%, whereas the scrambled ASO had no effect. Error bars indicate SD.
Compound 9 demonstrated dose-dependent downregulation of the 4R/3R ratio, up to 50%, showing that the bioactivity of 9 is not limited to transfected or neuroblastoma cells but is also observed in primary cortical neurons (Figure 6). Compound 9 is an initial lead molecule that emerged following a single hit expansion around 8. Future SAR studies are likely to give rise to second generation compounds with improved potency that may enable testing directly in htau transgenic mice.
Studies of RNA-Small Molecule Complexes.
Our NMR studies of 4 bound to a model of the tau exon 10-intron 10 junction enabled us to develop models of other compounds bound to the RNA, providing important insight into molecular recognition. Below, we discuss additional details about the 4-tau RNA complex as well a summary of our NMR studies of complexes with 5–9. In brief, NMR studies were commenced by acquiring ligand-based WaterLOGSY spectra51 on the RNA-small molecule complexes to confirm the binding of the small molecule to the RNA and the solubility of the complex (Figure 7). 1D 1H NMR spectra were then collected to identify the chemical shift and line width changes to the imino proton resonances as an additional indication of small-molecule binding. If these spectra indicate binding, then 2D 1H NMR spectra were acquired to assign the RNA and small molecule resonances and calculate distance restraints from intramolecular and RNA–small molecule NOEs. Finally, these distance restraints were applied to restrained molecular dynamics simulations to generate a structure of the complex: The results of 1D and 2D NMR spectra of unbound tau RNAs and 4-bound WT tau RNA are described in Supporting Information.
Figure 7.
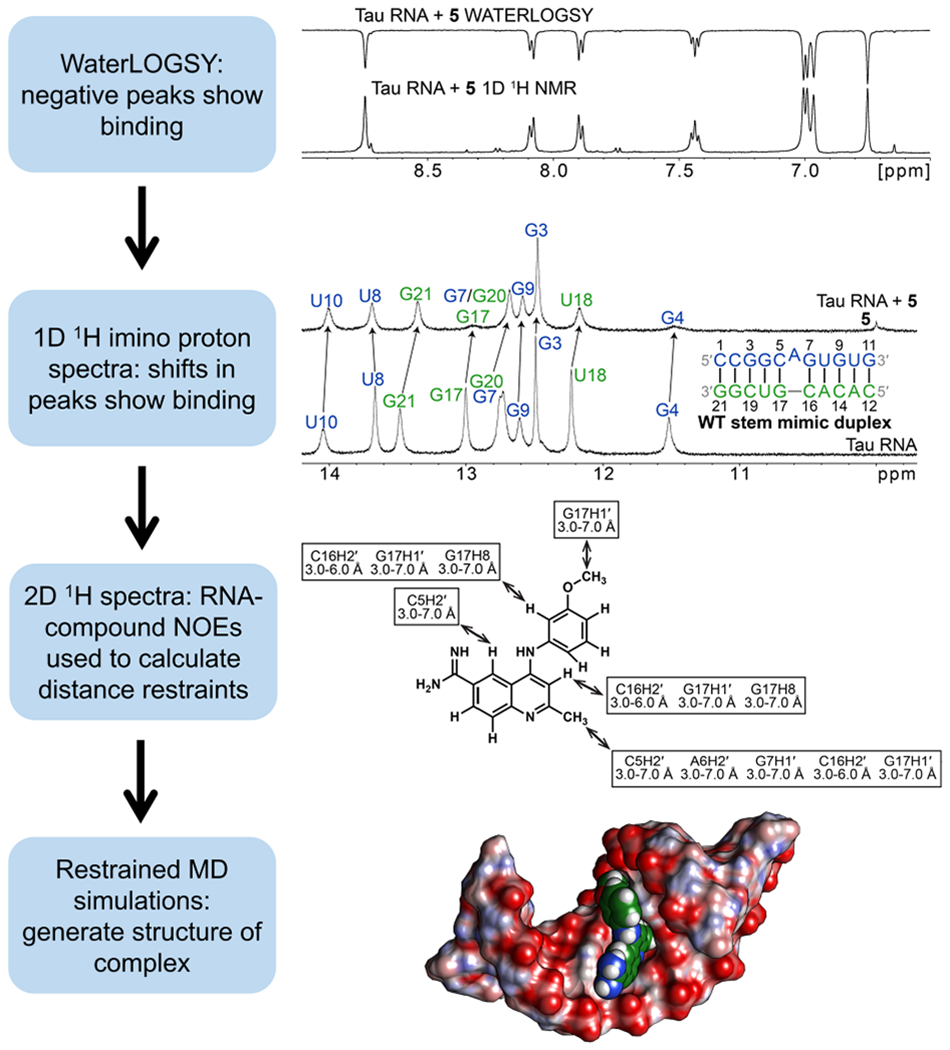
Scheme for NMR-based determination of structures of RNA–small molecule complexes. First, a WaterLOGSY spectrum was acquired on the RNA-small molecule complex to assess binding and solubility of the complex. Next, 1D 1H spectra were acquired to identify shifts in resonances that are expected to occur upon small-molecule binding. 2D NMR spectra were then acquired to assign resonances and calculate distance restraints, which were used to model the RNA-small molecule complex via restrained MD simulations. Compound 5 was presented as an example in this scheme.
Overall Structures of the Unbound RNA and WT Tau RNA–4 Complex.
The overall structure of the unbound WT tau RNA stem mimic, as determined by NMR spectroscopy, is shown in Figure 8A and described in Supporting Information. An ensemble of 20 structures of the WT tau RNA–4 complex was generated by using restrained molecular dynamics and 182 NOE distance restraints (Figure S27 and Table S10). The average root-mean-square deviation (RMSD) for the structures is 1.92 Å (Table 1), indicating good convergence. The bound RNA structure consists of an A-form helix (Figure 8B). All of the canonical base pairs on either side of the A-bulge form, consistent with the NMR data (Figure S22). The average C1′–C1′ distances among all base pairs range from 10.3 to 10.8 Å. The A-bulge is positioned in the minor groove, consistent with the break in the NOESY walk at residue A6 and the loss of NOEs from C5H2′ to A6H8 and A6H2′ to G7H8 (Figure 8B). The closing GC pairs are stabilized by stacking interactions with 4, and their formation is consistent with observed C-amino to G-imino NOEs (Figures 8, 9, and S20). However, the C16H1′ to G17H8 NOE for the bound structure was not observed, most likely due to the broadening, spectral overlap, or increased distances due to the stacking of 4 between these nucleotides. The helix is overwound at the 5′G4C5/3′U18G17 step. The G4-U18 pair has a relatively large propeller twist, base pair opening, and shear (see Supporting Information). As expected from the NOEs, the compound stacks between the neighboring bulge base pairs. Relatively large buckle values were observed for the G3–C19 and G7–C16 pairs (see Supporting Information). A hydrogen-bonded network helps to stabilize the structure. Specifically, hydrogen bonds occur in the major groove between the C5 amino protons and N3 of 4 and the C5 amino nitrogen and H02 of 4. These interactions may provide sequence specificity for the binding of 4 to the A-bulge loop flanked by the GC pairs. Considerable stacking among the fused aromatic rings of G7, 4, and G17 suggests that these stacking interactions stabilize the complex. Altogether, the binding of 4 to the RNA results in the formation of a complex largely stabilized by the hydrogen-bonding and stacking interactions.
Figure 8.
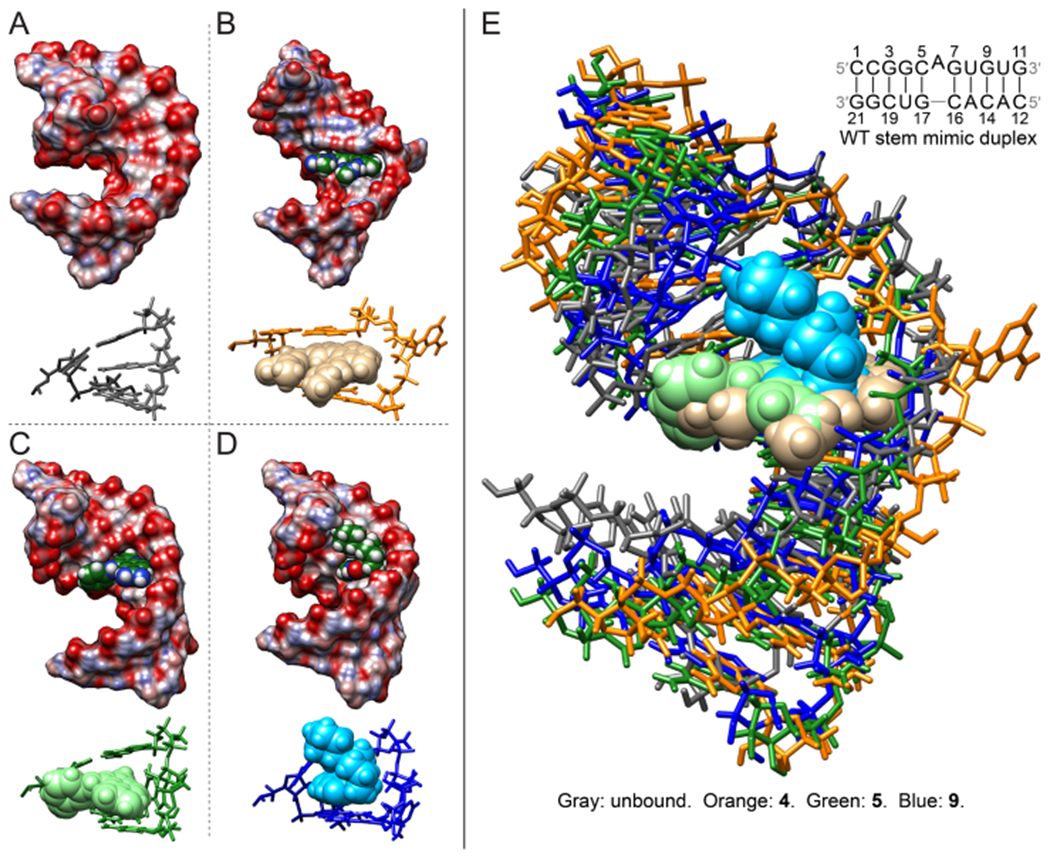
NMR solution structures of unbound and bound WT tau duplex model. (A) Structure of the free-form WT tau duplex model. (B) Structure of the 4-bound WT tau RNA. (C) Structure of the 5-bound WT tau RNA. (D) Structure of the 9-bound WT tau RNA. Structures were calculated by restrained simulated annealing. In panels (A) to (D), views of the binding site residues are depicted with stick representations for the RNA and sphere representations for compounds. (E) Overlay of unbound and 4-, 5-, and 9-bound structures. Compounds 4, 5, and 9 stack between the C5–G17 and G7–C16 closing base pairs. The helices in the 5- and 9-bound structures appear to be more tightly wound to form interactions with the compounds.
Table 1.
Structural Refinement Statistics of Unbound and Bound WT Tau Duplexes
| unbound RNA | 4-bound RNA | 5-bound RNA | 9-bound RNA | |
|---|---|---|---|---|
| no. of restraints | ||||
| all distance restraints, including hydrogen bonds | 191 | 182 | 258 | 173 |
| all NOE restraints | 150 | 141 | 221 | 146 |
| intraresidue | 69 | 68 | 121 | 75 |
| sequential residues | 79 | 62 | 79 | 57 |
| long range RNA (excluding 4–, 5–, or 9–RNA) | 2 | 2 | 2 | 2 |
| intramolecular 4, 5, or 9 | N/A | 2 | 6 | 1 |
| intermolecular 4–, 5–, or 9–RNA | N/A | 7 | 13 | 11 |
| hydrogen bonds | 41 | 41 | 37 | 27 |
| dihedral | 0 | 0 | 0 | 105 |
| RMSD of experimental restraints | ||||
| distance (Å) | 3.8 × 10−3 | 2.8 × 10−3 | 4.2 × 10−3 | 2.1 × 10−3 |
| dihedral (deg) | N/A | N/A | N/A | 2.1 × 10−3 |
| RMSD of structures for heavy atoms (Å) | ||||
| all residues, excluding 4, 5, or 9 | 1.79 ± 0.31 | 1.92 ± 0.49 | 1.11 ± 0.25 | 1.37 ± 0.36 |
| all residues and 4, 5, or 9 | N/A | 1.91 ± 0.47 | 1.12 ± 0.25 | 1.41 ± 0.34 |
| outer helices (residues 1–3, 9–14, and 19–21) | 2.01 ± 0.40 | 1.92 ± 0.61 | 1.02 ± 0.31 | 1.40 ± 0.47 |
| 4, 5, or 9 | N/A | 1.06 ± 0.38 | 0.50 ± 0.33 | 1.08 ± 0.60 |
| Bulge loop and inner base pairs, including 4, 5, or 9 for bound structure (residues 4–8 and 15–18) | 0.84 ± 0.19 | 1.53 ± 0.37 | 0.85 ± 0.17 | 1.14 ± 0.34 |
| Bulge loop and closing pairs, including 4, 5, or 9 for bound structure (residues 5–7 and 16–17) | 0.63 ± 0.19 | 1.43 ± 0.39 | 0.97 ± 0.22 | 1.23 ± 0.40 |
Figure 9.
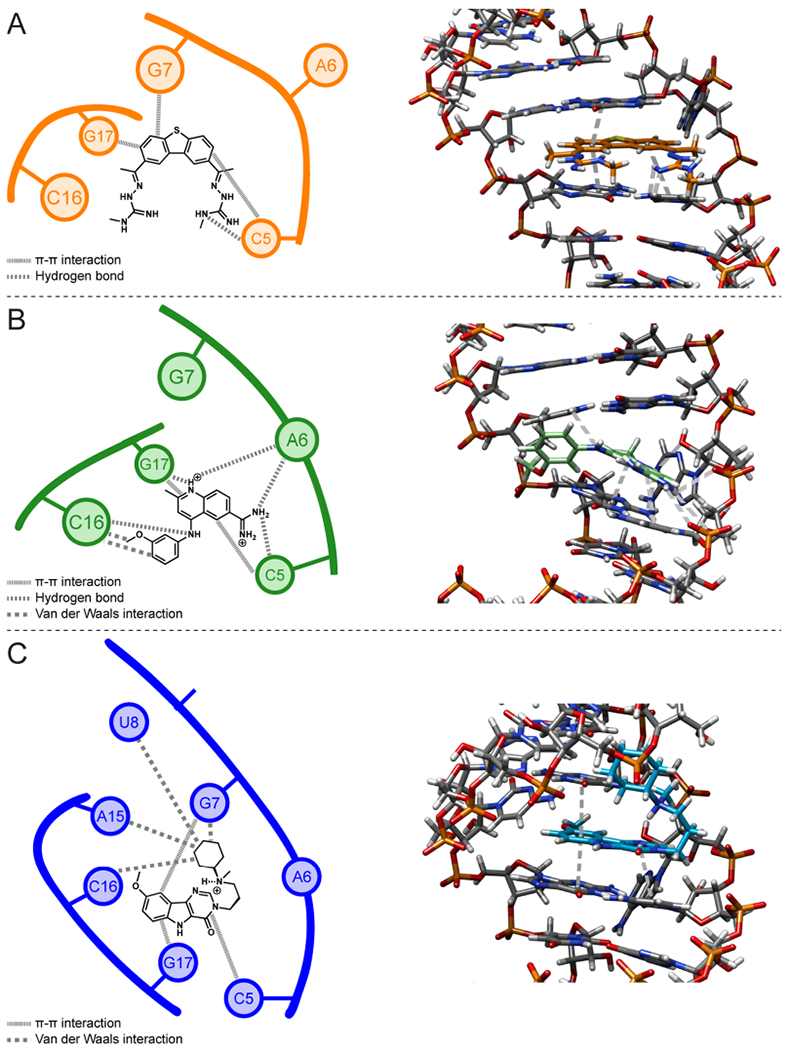
Schematic diagrams and 3D structures of intermolecular interactions between the WT tau duplex model and compounds 4, 5, and 9 in NMR structures. (A) Interactions in the tau RNA–4 complex. (B) Interactions in the tau RNA–5 complex. (C) Interactions in the tau RNA–9 complex. In the 3D structures, 4, 5, and 9 are colored orange, green, and blue, respectively, while the RNA is colored gray. Gray dashed lines in the 3D structures represent hydrogen-bonding and stacking interactions; van der Waals interactions are not shown.
1D NMR Spectra of RNAs Bound to Compounds 5-9.
An analysis of the bioactive compounds 5 to 9 revealed three different scaffolds. Among these scaffolds, RNA-ligand NOEs were not obtained for compounds with a 4-phenylpiperidine core (compounds 6 and 7). To investigate the differential binding of these scaffolds to the RNAs, NMR spectra were acquired on each compound in complex with tau RNAs. WaterLOGSY spectra were acquired on 5 to 9 in complex with the WT and DDPAC duplex constructs (Figures S29 to S33 and Tables S1A and S1B) and, as a control, on the compounds alone to determine whether indications of binding may result from aggregation. The results of these experiments, in addition to the 2D NMR spectra of 5 and 9 bound to WT tau RNAs, are summarized in Supporting Information.
Overall Structures of the WT Tau RNA Complexed with Compounds 5 and 9.
Ensembles of 20 structures were generated using 258 and 173 distance restraints for the tau RNA–5 and tau RNA–9 complexes, respectively, including 13 intermolecular restraints between the tau RNA and 5 and 11 intermolecular restraints between the tau RNA and 9 (Figure S28 and Tables 1, S11, and S12). The average RMSD for the WT tau RNA–5 and –9 complexes is 1.11 and 1.37 Å, respectively (Table 1), indicating good convergence. In each complex, all of the base pairs are consistent with the 1D and 2D imino proton data (Figure S40 and S41). The average helical rise and twist are similar for both WT tau RNA–5 and –9 complexes and consistent with an A-form helical structure of RNA, similar to that observed for the 4-bound complex (see Supporting Information). The average C1′–C1′ distances for the complexes range from 10.5 to 10.8 Å for all residues, which is also consistent with A-form helices. In each complex, the A bulge is displaced into the minor groove (Figure 8). In the 5-bound structure, the displaced A-bulge appears to stack within the minor groove, and the 9-bound structure has the A-bulge partially displaced, with the hydrogen-bonding surface of the adenine pointing out of the minor groove. In the 5-bound structure, the C7 methyl group is oriented toward the minor groove and is supported by observed NOEs between the methyl protons and the sugar protons of the adjacent base pairs to the A6-bulge (Figures 8 and S38). The 9-bound structure, with the 9-C7 methoxy group oriented toward the minor groove and the 9-C16 methyl group in the major groove, is also supported by NOEs between the methyl groups and the adjacent base pairs of the A-bulge (Figures 8 and S39). The methoxy groups of 5 and the tertiary amine of 9 lie in the major groove and generally do not form hydrogen-bonding interactions with the RNA. In the 5-bound structure, overlaps between the quinolone ring of 5 and C5 and G17 bases suggest that the complex is primarily stabilized by stacking interactions (Figure 9). Additionally, van der Waals interactions form between the methoxybenzene group and the RNA. In the 9-bound structure, overlaps between the aromatic rings of 9 and C5, G7, and G17 bases suggest that stacking appears to primarily occur between 9 and these bases (Figure 9). The N-methylcyclohexylamine group of 9 appears to form van der Waals interactions with the wall of the major groove. The buckle values for the C5–G17 and G7–C16 base pairs are 13.8 ± 8.0 and −25.1 ± 9.7°, respectively. In the 5-bound structure, relatively large buckle values of 20.5 ± 8.5 and −12.2 ± 10.5° are observed for the C5-G17 and G7-C16 base pairs, respectively. The helix is overwound at the 5′G4C5/3′U18G17 steps for both the 5- and 9-bound structures, with similar helical twist values for both structures. From the results of the NMR data and structural calculations, both 5 and 9 displace the A-bulge and stabilize the RNA by stacking between the adjacent base pairs.
Comparison of the Unbound and Bound Structures.
The bound and unbound structures consist of A-form helices with the same canonical base pairs. The average C1′–C1′ distances among all base pairs in the bound and unbound spectra are similar, but the 4-, 5-, and 9-bound structures are slightly less compressed than the unbound structure and have average helical rise values that are closer to typical A-form RNA (2.8 Å) than the unbound structure.52 The average helical twist values of the 5- and 9-bound structures are approximately that of the unbound structure (33.7°), while that of the 4-bound structure is slightly lower (31.0°). The major groove at the 5′G4C5/3′U18G17 and 5′G7U8/3′C16A15 steps widened upon the binding of 4 (see Supporting Information). In comparison, less widening of the major groove occurs at these steps upon the binding of 5 (see Supporting Information). The binding of 9 also resulted in less widening of the major groove width at these steps than the binding of 4 (see Supporting Information). Minor conformational changes of the G4–U18 pair occurred upon the binding of 4, as evident from the relatively small changes in propeller twist and base pair opening; however, the magnitude of the buckle increased by 6.9°. At the same time, greater changes occurred in the conformation of the closing G7–C16 pair, with a considerable decrease in the propeller twist, while the buckle increased in both that pair and the G3–C19 pair (see Supporting Information). In the 5-bound structures, the propeller twist and base pair opening of the G4-U18 pair changed by 11.3 and 9°, respectively, while the magnitude of the buckle increased by 8.3°. In the 9-bound ensemble of structures, the values for the propeller twist, base pair opening, and magnitude of the buckle of the G4–U18 pair changed by 10.7, 8.4, and 7.8°, respectively. Relative to the 4-bound structures, smaller changes in the propeller twist and buckle occurred at the G7–C16 pair of the 5-bound structures upon compound binding (see Supporting Information). A larger change in the buckle occurred at this base pair in the 9-bound structures (see Supporting Information). These data indicate that the binding of 4, 5, or 9 to the helix induces helical structure changes, especially in the closing base pairs of the bulge loop, that facilitate stacking, van der Waals, and/or hydrogen-bonding interactions between the ligand and the RNA.53 These interactions are facilitated by a balance of the hydrophobic and hydrophilic groups that maintains the solubility and membrane permeability.54 In particular, the number of HBDs and HBAs in 5 and 9 are outside of the range for which poor absorption is expected (over five and 10, respectively), while the molecular weights of 4, 5, and 9 are less than 500 g/mol.55 These interactions may contribute to the increased selectivity of the drugs for the target and thus reduce the toxicity associated with off-target binding.
DISCUSSION
Previous studies have rescued aberrant tau pre-mRNA splicing using ASOs, demonstrating that the pre-mRNA hairpin can be targeted for therapeutic benefit.21 The success of this ASO led to the investigation of small-molecule interactions with the tau pre-mRNA hairpin. In 2000, G. Varani’s group reported the NMR solution structure of a complex between neomycin and the tau SRE29 and then later the structure of the tau SRE bound to mitoxantrone (MTX), a more selective compound identified from a high-throughput screen.53 Using dynamic combinatorial chemistry, López-Senin et al. identified additional ligands that stabilize the hairpin, likely by binding around the adenine bulge.56 While these molecules have been shown to bind to the tau exon 10–intron 10 junction hairpin, the compounds did not rescue aberrant splicing.
The first compound, 1, that improved aberrant tau pre-mRNA splicing was identified from a structure-based search of compounds via Inforna that targets the 5′ splice site. An Inforna search using this compound as a query led to compound 4, which demonstrated improved binding affinity, bioactivity, and solubility, allowing for NMR structure determination of the complex. On the basis of this structure, an in vitro fluorescence-based binding assay was developed and then employed in two separate strategies to screen for compounds with improved physiochemical properties and activity. One strategy consisted of constructing a pharmacophore model using the Inforna hits, followed by hit expansion and analogue synthesis, which led to 6 and 7. The second strategy consisted of molecular docking of the NCI diversity set, testing of the top scoring compounds, and then hit expansion, which led to 9. Accounting for these and other parameters, the average CNS MPO score increased to 4.9 for lead compounds 6, 7, and 9 from 3.2 for the initial Inforna hits, 1 to 4. Among the optimized compounds, 9 was the most potent in cellulis and bound to the target RNA with the greatest affinity. Furthermore, 9 was more selective for the A-bulge site and less toxic than its parent compound 8. Thus, it was carried forward in Chem-CLIP studies to assess for target engagement. The C-Chem-CLIP studies using 11 showed that 9 selectively inhibited pull-down of the tau pre-mRNA in a dose-dependent manner in LAN5 cells expressing endogenous tau and in transfected HeLa cells expressing the A-bulge-containing hairpin. Separately, 9 decreased levels of 4R tau mRNA relative to 3R tau mRNA in humanized mouse primary neurons in a dose-dependent manner.
Compound selectivity was demonstrated via thermal melting experiments, miRNA expression profiling, and Chem-CLIP. In the thermal melting experiments, 4–9 stabilized the tau hairpin structures, with melting temperatures correlating with the binding affinities and bioactivities of the small molecules. The 2-AP competitive assays and thermal melting experiments also showed that the compounds selectivity bound to and stabilized constructs containing the A-bulge over RNAs lacking the A-bulge or other bulges. MiRNA expression profiling with compounds 7 and 9 showed that these compounds were selective against microRNAs containing A-bulge motifs in the same sequence context as in the tau exon 10 hairpin. In Chem-CLIP studies, photoprobe 11 selectively enriched pull-down of tau pre-mRNA over other mRNAs. Taken together, this study highlights two complementary approaches, both rooted in the evaluation of an RNA structure, that have successfully resulted in the identification of superior chemical matter, as evident by improvements in bioactivity, selectivity, physicochemical properties, and cell viability.
The compounds identified in this study generally consisted of fused aromatic rings that stabilize the exon 10 hairpin by stacking interactions. Compound 4 is a derivative of dibenzothiopene, itself an analogue of carbazole, while 8 and 9 contain ellipticine cores, and 5 contains a quinoline core. Indeed, DNA-targeting drugs based on these cores primarily act via DNA intercalation.57–59 Although intercalators may bind nonspecifically to RNA,60,61 cross-peaks between the tau WT NMR construct and compounds 4, 5, and 9, in particular asymmetric compounds 5 and 9, in NMR spectra support a limited number of binding modes. This indicates structure-specific recognition of the A-bulge site by these compounds, which are discussed in further detail below. However, the lack of strong evidence that compounds 6 and 7 bind to the tau RNA constructs under NMR conditions indicates that, for some compounds, no direct correlation may be observed between the binding interactions to the NMR constructs and in vitro or in cellulis activity.
The NMR structures of 4, 5, and 9 in complex with WT tau RNA show that these compounds share features with the pharmacophore model. The hydrogen bonds that the two guanidines of 4 form with the closing C5 cytosine of the A-bulge are consistent with the two HBDs and HBAs of the amidines in the pharmacophore model. Similarly, the amidine of 5 forms hydrogen bonds with the phosphates of the A-bulge and adjacent cytosine. In contrast, 9 does not form hydrogen bonds with the RNA. All three compounds contain fused ring systems that form stacking interactions with the closing base pairs of the A-bulge, consistent with the predicted stacking interactions by the aromatic heterocyclic indole of the pharmacophore model. Taken together, these results indicate that stacking interactions are necessary to stabilize the tau RNA-small molecule complex, while hydrogen-bonding interactions may enhance selectivity.
The recognition of the A-bulge site by 4 appears to be driven by hydrogen-bonding interactions between the guanidinyl groups of 4 and the closing nucleobases, while the aromatic portion of 4 forms stacking interactions with the closing base pairs. These hydrogen bonds lead to a specificity in drug binding over intercalation alone. Compound 5 forms hydrogen-bonding interactions with the phosphate backbone of C5, 2′-OH group of A6, and nucleobases of C16 and G17, while its methoxybenzene group appears to fill the region of the binding cavity across the strand from the A-bulge and is positioned to form van der Waals interactions with the C16 nucleotide opposite of the A-bulge (Figure 9B).62 In the 9–RNA complex, the N-methylcyclohexylamine of 9 fills the major groove of the RNA helix and forms van der Waals interactions with the G7, U8, A15, and C16 nucleotides. In addition to the enthalpic gains from the van der Waals interactions, filling a binding cavity with substituents that form such interactions can bring about entropic gains from desolvation that displaces water molecules from the binding cavity and allows them to join bulk water.62,63 In comparison to the 4-bound complex, the RNA in the 5- and 9-bound complexes appears to be more tightly wound to form more interactions with each compound, as evident from their greater helical twist values relative to that of the 4–RNA complex (vide supra). The shape complementarity between 5 and 9 and the RNA confers structural specificity, which has been known to lead to less toxicity associated with off-target binding.64
Selectivity may be gained from the substituents that fill the binding cavity if they do not fit into smaller binding pockets of off-target molecules, decreasing the off-target binding affinity.65 While adding charged groups that form electrostatic interactions may increase the selectivity at the target site, it can lead to poor oral bioavailability and membrane permeability.54 In contrast, adding hydrophobic moieties to drugs may lead to solubility problems.54 Therefore, a balance between the hydrophobic and hydrophilic nature of a drug must be achieved for the drug to be transported across membranes.55 Among these compounds, 4 has the most HBDs and HBAs (6 and 8, respectively), while 9 has the fewest (2 HBDs and 6 HBAs, respectively) and 5 is in between (4 HBDs and 5 HBAs). Thus, target recognition by 4 is primarily achieved by hydrogen-bonding interactions and stacking interactions between the closing base pairs of the A-bulge. In contrast, the absence of target-specific hydrogen-bonding interactions by 9 may be compensated by stacking interactions and van der Waals interactions formed by its N-methylcyclohexylamine group in the major groove. Intermediate to these compounds is 5, which forms up to six hydrogen bonds with the target RNA and van der Waals interactions with its methoxybenzene group in the major groove but to a lesser degree than the N-methylcyclohexylamine group of 9. However, its smaller fused ring system compared to 4 or 9 results in fewer stacking interactions with the RNA. Taken together, compounds 5 and 9 contain a balance of hydrophilic and hydrophobic groups to maintain drug-likeness while achieving target selectivity via different binding modes based on their available functional groups.
The NMR structure of the unbound stem mimic (Figure 8) exhibits similar structural features as those of the 25-nt WT tau hairpin previously reported by Varani et al.66 In these structures the adenine is either partially stacked on the C4-G14 base pair or slightly displaced into the minor groove. The bound stem mimic complexes have similar features to the MTX-bound WT hairpin reported by Zheng et al.53 MTX, 4, 5, and 9 form stacking interactions with the GC pairs adjacent to the A-bulge. In all structures, the A-bulge is displaced from the helix, retaining the anti conformation. For the MTX- and 4-bound structures, the adenine adopts a more external position, while in the 5- and 9-bound structures, the adenine is within the minor groove. NMR data supporting the degree to which the adenine is displaced include the absence of intramolecular NOEs to the A-bulge in the 4–RNA spectrum and presence of H6/H8 to HT connectivities through the A-bulge in the 5– and 9–RNA spectra. The structures of the bound 4, 5, and 9 have fewer hydrogen bonds than those observed for the bound MTX. A second set of NOEs corresponding to the unbound conformation are observed among RNA residues around the binding site in both the MTX and 4-bound complexes, suggesting that the RNAs exist in an equilibrium of bound and unbound conformations. Such NOEs were not observed in the 5- or 9-bound complexes, which may suggest a faster exchange between the bound and unbound conformations. The 4- and MTX-bound complexes share similar structural features, with predominant stacking between the adjacent base pairs, while the 5- and 9-bound complexes exhibit less stacking but have more interactions within the major groove. The greater amount of stacking of 4 relative to 5 or 9 may result in slower dissociation, resulting in both bound and unbound conformation NOEs in the 4-bound complex.
The NMR structures of the compounds bound to the tau SRE, together with the binding assays, show that compounds that target the tau SRE can have different binding modes. While the 2-AP assay is a measure of the displacement of the A-bulge from the helix, the U1 assay is a measure of the stabilization of the hairpin by small molecules. These differences in the binding modes may explain why 9 has a similar Kd value to 4 in the U1 assay yet does not significantly displace the A-bulge from the helix. The different binding modes for stabilizing the tau SRE are evident when comparing the MTX- and neomycin-bound tau SRE structures reported by Varani’s group, where MTX binds by the displacement of the A-bulge from the helix, while neomycin binds to the major groove with a minor displacement of the A-bulge.29,53,65. Understanding these aspects of binding, intercalation, and presence in the major groove and how they control splicing are key to developing therapeutics against the tau SRE.
Comprehensive studies of these compounds can promote a better understanding of how to chemically control a pre-mRNA splicing outcome. Small molecules that affect SMN pre-mRNA splicing for treatment of SMA facilitate exon inclusion by stabilizing a SMN2 pre-mRNA-protein complex. Wang et al. showed that one of these compounds, SMN-C2, binds to SMN2 splicing activators FUBP1 and KSHRP and increases their affinity with the exon.47 In contrast, Zhang et al. identified a small molecule that inhibited G-quadruplex-dependent exon inclusion by disrupting RNA G-quadruplex structures near the splice site and inhibiting their association with the splicing factor hnRNPF.67 Together, these studies demonstrate that small molecules can be used to target pre-mRNAs and influence alternative splicing outcomes. A deeper understanding of how these molecular recognition events affect this process and an understanding how to target these structures to affect exon inclusion or exclusion could therefore be broadly important.
CONCLUSIONS
In this work, 4 was identified from a chemical similarity search of lead compound 1 and has improved activity for in inducing the exclusion of tau exon 10. Compound 4 and other initial hits from chemical similiarty searching were subsequently used as starting points to identify leads with improved drug-like characteristics via two complementary strategies. In the first approach, pharmacophore modeling of 1 to 4 and substructure and similarity searching followed by analogue synthesis led to piperidine amides 6 and 7. Amidine 5 was also identified from the hit expansion approach and selected for profiling. The 2-AP, Q-, and U1 snRNA mimic assays provided complementary means to identify screening hits and confirmed that these compounds indeed bind to the tau hairpin construct. Translation of in vitro biochemical activity to a functional effect on splice modulation was demonstrated using luciferase-based cell assays and RT-qPCR experiments. The second hit generation approach relied on the docking of ligands from the NCI diversity set to the NMR-derived structure of 4 bound to the tau pre-mRNA. A select set of compounds were subsequently screened in the in vitro assays, resulting in the identification of 8.
Hit expansion around 8 afforded compound 9, which demonstrated a dose-dependent reduction of the ratio of 4R- to-3R tau mRNA levels in mouse primary neurons. Furthermore, compound 9 reduced the pull-down of tau pre-mRNA in cells in Chem-CLIP experiments using photoprobe 11, thereby demonstrating target engagement in cells. Detailed NMR experiments were performed to characterize the binding of 4, 5, and 9 to the tau hairpin. NMR spectroscopy and restrained simulated annealing of these compounds in complex with a model of the WT tau RNA stem showed that they stabilize the tau hairpin by stacking and hydrogen-bonding interactions with base pairs adjacent to the A-bulge. This work demonstrated that a combination of structure-based drug design, in vitro screening, thermal melting, and cell-based assays can be used to identify compounds with drug-like properties that target tau pre-mRNA and reduce levels of 4R tau in FTDP-17. This strategy may be used to identify and optimize compounds that target other incurable diseases caused by aberrant pre-mRNA splicing. Furthermore, the ability to improve activity and drug-like properties of hits obtained from the Inforna platform bodes well for designing structure-specific ligans from sequence, affording lead compounds that target RNA. It is conceivable that drug-like hits identified using the strategies described herein can subsequently be optimized using traditional medicinal chemistry approaches.
EXPERIMENTAL SECTION
Compounds.
Compounds 1 to 4 and 8 (Figure 2) were obtained from the National Cancer Institute (NCI). Compounds 5 to 7 and 9 and 10 (Figure 2) were obtained from Pfizer. A second batch of compound 9, in addition to compounds 11 and 12, were synthesized as described in the Supporting Information.
Cell Culture.
HeLa cells were maintained in DMEM with 10% fetal bovine serum (FBS) and 1% GlutaMax at 37 °C with 5% CO2. Cells were discarded after 40 passages. The BE(2)-M17 cells were stably transfected with a Nanoluc-PEST reporter in frame with tau exons 9–11, resulting in an increase in the signal when exon 10 is excluded. Stably transfected cells were selected for by using Zeocin. Cells were grown in DMEM/F12 50:50 mix with 10% FBS, 1% Glutamax, and 100 μg/mL Zeocin at 37 °C in a 5% CO2 atmosphere and were passaged no more than 10 times according to the suggested protocol from ATCC. LAN5 cells were grown in RPMI 1640 medium supplemented with a 1× penicillin/streptomycin solution (Corning) and 20% FBS at 37 °C with 5% CO2. The protocol for neuron cell culture is described below (htau Mice Primary Neuron Experiments).
Luciferase-Reporter Assay to Study Exon 10 Alternative Splicing.
The exon 10 mini-genes, both WT and DDPAC, fused to firefly luciferase, have been previously described.23 HeLa cells were transfected with WT, WT+I17T, DDPAC, or DDPAC+I17T tau minigenes with Lipofectamine 2000 (Life Technologies) or JetPrime (Polyplus Transfection) per the manufacturers’ recommended procedures. For large-scale compound screening, HeLa cells (75 mm dish) were batch transfected and then trypsinized from the surface. The cells were resuspended in growth medium, transferred to 384-well plates using a WellMate dispenser (Thermo Scientific), and then treated with compound (transferred using a Beckman pintool in 100 nL volumes) for 18 h at 37 °C and 5% CO2. Compound treatment was completed in the same fashion for both transfected and stably expressing cell lines. The activity of hit compounds was assessed by completing a dose–response analysis in 96-well plates.
After the treatment period, the cells were washed with 1× DPBS, and then lysed with 10 mM sodium phosphate buffer (pH 7.8) supplemented with 0.2% Triton 100-X (1× TLysis Buffer). The lysed cells were incubated with a buffered solution of firefly luciferase substrate (200 mM Tris-HCl, pH 8, 15 mM MgSO4, 0.1 mM EDTA, 25 mM dithiothreitol (DTT), 1 mM ATP, 0.2 mM coenzyme A, and 200 μM luciferin) for 10 min, and then luminescence was measured with a Biomek FLx800 plate reader (1.0 s integration time).68
Cell Viability.
Cells were treated with compound as described above. After the removal of the compound-containing growth medium, the cells were washed with 1× DPBS and incubated in growth medium containing 10% WST-1 cell proliferation reagent (Roche) or CellTiter Fluor (Promega) for 40 min. The absorbance of the solution was measured at 450 and 650 nm with a SpectraMax M5 plate reader (Molecular Devices) for cells treated with WST-1. The background signal at 650 nm was subtracted from the WST-1 signal at 450 nm for analysis. For cells treated with CellTiter Fluor, the fluorescence was measured with 390 nm excitation and 505 nm emission wavelengths. Cells treated with a compound were compared to untreated cells to determine the effect of the compound on cell viability.
Quantitative Reverse Transcription PCR (RT-qPCR).
HeLa cells were seeded in 60 mm dishes and incubated for 12–16 h at 37 °C. The cells were transfected with WT, WT+I17T, DDPAC, or DDPAC+I17T luciferase reporter minigenes using JetPrime (Polyplus Transfection) per the manufacturer’s recommended protocol and incubated for 4 h. After transfection, HeLa cells were collected; reseeded in 24-well plates at a 0.6 × 105 cell/well density; treated with compounds, a Vivo-Morpholino 4R-to-3R ASO (Gene Tools, LLC) against the exon 10–intron 10 junction with the sequence 5′-TGAAGGTACTCACACTGCCGC-3′, or a scrambled control; and incubated for 48 h. LAN5 cells were seeded in 24-well plates at a 1 × 105 cell/well density, treated with the same compounds or oligonucleotides, and incubated for 48 h. Total RNA was extracted from the cells and analyzed via RT-qPCR to determine the levels of 3R and 4R tau mRNA. Cells were washed with 1× DPBS buffer and then lysed and extracted using the Quick-RNA MiniPrep kit (Zymo Research) following the manufacturer s protocol. The concentration of the extracted RNA was measured with a Nano Drop 2000 (Thermo Fisher Scientific). The cDNA was generated from 150 ng of the total RNA using qScript (Quanta Bio) following the manufacturers recommended protocol. The qPCR reactions consisted of SYBR Green 5× PCR mix (invitrogen), 200 nM primers, and 10 ng of RT product. Each biological replicate was tested in three technical replicates. The levels of 18S rRNA were monitored as the endogenous control for relative quantification (see Table S14 for primers). The primers for RT-qPCR in transfected HeLa cells and LANS cells are provide in Table S14.
Western Blot.
The protein expression of 4R, 3R, and total tau was analyzed in LANS cells after treatment of 4R-to-3R ASO, compound 9, or DMSO vehicle for 48 h. The cells were gently washed twice with ice-cold 1× DPBS and lysed in 2% SDS lysis buffer containing protease (539137, Millipore) and phosphatase inhibitor cocktails (P5726, Sigma), followed by brief sonication. After quantifying the protein concentration using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA), an equal amount of protein from each sample was separated in 15% SDS-PAGE (GenScript, Piscataway, NJ) and transferred onto polyvinylidene difluoride (PVDF) membranes. Nonspecific binding was blocked with 5% (w/v) nonfat dry milk for 1 h at room temperature, and the membranes were probed overnight with primary antibodies specific for 4R tau (1:2000, 05-804 from MilliporeSigma), 3R tau (1:5000, 05-803 from MilliporeSigma), and total tau (1:5000, 46687S from Cell Signaling). β-Actin (1:10000, 3700S from Cell Signaling) was used as a protein loading control. After three 10 min washes with 1× TBST (Tris-buffered saline, 0.5% Tween 20), the membranes were incubated in horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10000) for 1 h at room temperature and washed three times with TBST. Immunocomplex signals of each protein were then detected using an enhanced chemiluminescence detection system (ECL, PerkinElmer, Waltham, MA). The abundance of protein in each sample was determined based on the band intensity from an ImageJ analysis and then normalized to β-actin bands.
2-AP Assay.
Sequences corresponding to the WT and DDPAC hairpins that contain a 2-AP substitution at the A-bulge site (2-AP-labeled WT/DDPAC hairpin) were folded by heating the complex to 70 °C for 3 min in a 1× Assay Buffer (0.185 M NaCl, 8 mM NaH2PO4, pH 6.3) and slowly cooling to room temperature. Bovine serum albumin (BSA) was added to the RNA solution to a final concentration of 40 μg/mL. All binding assays were carried out in triplicate in 96- or 384-well half area plates at room temperature with a Tecan plate reader (gain, 100; integration time, 40 μs). Fluorescence excitation and emission wavelengths were set at 310 and 375 nm, respectively. Dissociation constants were fit to eq 1:15
| (1) |
where I and I0 are the observed fluorescence intensities in the presence and absence of RNA, respectively, Δε is the difference between the fluorescence intensity in the presence of an infinite concentration of RNA and the absence of RNA, [FL]0 and [RNA]0 are the concentrations of small molecule and RNA, respectively, and Kt is the dissociation constant.
Competitive binding assays were completed using a previously published protocol.18 Briefly, compounds 4–8 were incubated with 2 μM 2-AP-labeled tau DDPAC hairpin RNA and with increasing concentrations of competitive DDPAC RNAs containing a C-bulge, a G-bulge, or a U-bulge substitution. The concentration of each compound corresponded to their EC50 determined by 2-AP binding assays (Table S5). The change of the fluorescence intensity was fit to eq 2:
| (2) |
where θ is the percentage of 2-AP tau RNA bound, [RNA] is the concentration of 2-AP-labeled tau RNA, EC50 is the EC50 of 2-AP-labeled tau RNA and compound, [compound] is the concentration of compound, Ct is the concentration of competing, unlabeled RNA, Kd is the competitive dissociation constant of competing, unlabled RNA, and A is a constant.
2-AP Assay on Hit Expansion Library.
An aliquot of 10 μM 2-AP-labeled WT or DDPAC hairpin was folded in 1× Assay Buffer. BSA was added to the RNA solution to a final concentration of 40 μg/mL. Aliquots of 16μ/L containing 0.5% DMSO and 50 μM of 4 in a 1× Assay Buffer were separately dispensed into empty wells of assay-ready plates as negative and positive controls, respectively. Then, an equal volume of 1× Assay Buffer was dispensed into the other wells of the 384-well plates using an Aurora liquid handler. Plates were incubated for 15 min at room temperature and preread on a Tecan plate reader using 310 nm excitation and 380 nm fluorescence emission wavelengths to investigate the compound internal fluorescence. Next, 4 μL of an RNA solution was then dispensed into the plates. Plates were incubated for 30 min at room temperature and read on the Tecan plate reader using the same wavelengths described in 2-AP Assay.
Quenching Assay.
An aliquot of 2 μM quencher-labeled RNA (QjRNA.) with a sequence corresponding to the DDPAC hairpin was folded in a 1× Assay Buffer. BSA and Qjprobe were added to the RNA solution to final concentrations of 40 μg/mL and 10 nM, respectively. The Q;RNA and Q-probe solution was incubated at room temperature for 15 min. Compound was added to the solutions, and serial dilutions were performed on the RNA–compound solutions. After 30 min of incubation at room temperature, the fluorescence was measured using a BioTek FLx800 plate reader with 485 nm excitation and 535 nm emission wavelengths.
U1 snRNA Mimic Assay.
A 200 nM aliquot of dual-labeled RNA with a sequence corresponding to the DDPAC hairpin was folded in a 1× Assay Buffer at 65 °C for 5 min and then slowly cooled to room temperature. BSA was added to the solution to a final concentration of 40 μg/mL. Compounds were then incubated with the RNA solutions at room temperature for 15 min. A U1 snRNA mimic with the sequence 5′-GGUACUCAC-3′ was added to the solution to a final concentration of 20 μM. The fluorescence intensity of Cy3 was measured as a function of time (in kinetics mode) for 20 min using a Cary Eclips fluorescence spectrophotometer, with excitation and emission wavelengths of 532 and 568 nm, respectively.
Molecular Docking.
The NCI diversity set comprising 1974 compounds was prepared in LigPrep (Schrodinger, Inc.). Briefly, the ionization in the pH range of 5 to 9, tautomers, and stereoisomers options were used to calculate all the possible chemical structures. The 3947 compounds generated from these calculations were minimized using an OPLS 2005 force field. An NMR model of the tau A-bulge structure bound to 4 was prepared by removing the bound ligand, assigning a bond order, and adding the missing hydrogen atoms. Glide (Schrodinger, Inc.) docking experiments were completed in the HTVS mode with no constraint. The grid size for the docking was set to 20 × 20 × 20 Å3 around the center of the 4 position. The final ligand poses with a Glide docking score of –9 or better were selected for further analysis (n = 32).
In Vitro Chem-CLIP and C-Chem-CLIP.
Approximately 10000 counts of 32P 5′-end-labeled tau WT, tau DDPAC, tau WT+I17T, or tau DDPAC+I17T RNA were folded in 100 μL of a 20 mM HEPES buffer at 60 °C for 5 min. After cooling to room temperature, dilutions of 11 or 12 were added and incubated at 37 °C for 30 min. Alternatively, for C-Chem-CLIP, dilutions of competing parent compounds, 9, were incubated with RNA for 30 min before adding 11 or 12. The reaction mixture was then irradiated with 365 nm UV light in a UV stratalinker for 10 min. A click mixture (0.5 μL of disulfide biotin azide (Click Chemistry Tools), 10 mM; 1 μL of CuSO4, 10 mM; 1 μL of THPTA, 50 mM; and 1 μL of sodium ascorbate, 250 mM) was then added to each reaction and incubated at 37 °C for 2 h. Next, 10 μL of streptavidin beads (Dynabeads MyOne streptavidin C1 beads (Thermo Scientific)) was added to the reaction for pull-down of cross-linked RNA for 10 min. Unreacted RNA was removed by placing reaction tubes on a magnetic separation rack. The RNAs cross-linked to a small molecule remain attached to the beads via a biotin–streptavidin interaction. The click mixture buffer was then collected, and the beads were washed three times with wash buffer (10 mM Tris-HCl, pH 7.0, 1 mM EDTA, 4 M NaCl, 0.2% Tween-20). The wash buffer was collected and mixed with the click mixture buffer. The amounts of radioactivity in the buffer and on the beads were quantified with a Beckman Coulter LS6500 liquid scintillation counter. The percentage of RNA captured was determined by using the radioactivity on the beads divided by the total radioactivity in the buffer and on the beads.
Cellular Chem-CLIP and C-Chem-CLIP.
LAN5 cells were grown to ~80% confluency as monolayers in 100 mm dishes. The HeLa cells were transfected, as described above in Luciferase-Reporter Assay to Study Exon 10 Alternative Splicing, and grown to ~80% confluency as monolayers in 100 mm dishes. The cells were then treated with 10 μM 11 or 12 for 6 h. For C-Chem-CLIP, dilutions of competing parent compound, 9, were incubated with cells for 12 h before adding 11 or 12. After treatment, the cells were washed with 1× DPBS, and then 10 mL of ice-cold 1× DPBS was added before irradiation with 365 nm UV light in a UV stratalinker for 10 min. The cells were scraped, and the total RNA was extracted using TRIzol LS reagent (Invitrogen) per the manufacturer’s protocol. Approximately 20 μg of the total RNA was then incubated with a click mixture in 25 mM HEPES buffer at 37 °C for 2 h, as described in In Vitro Chem-CLIP and C-Chem-CLIP. The total RNA was cleaned up by RNA Clean XP beads (Beckman Coulter) according to the manufacturer’s protocol. Pull-down by streptavidin beads and the washing steps were performed as described in In Vitro Chem-CLIP and C-Chem-CLIP. After washing, the RNA on the beads was cleaved using a 1:1 mixture of TCEP (200 mM) and K2CO3 (600 mM) with shaking at 37 °C for 30 min. The reaction mixture was quenched with one volume of iodoacetamide (400 mM) with shaking at room temperature for 30 min. The RNA in the buffer was collected by using a magnetic separation rack and was cleaned up by RNA Clean XP beads. RT-qPCR, as described above (Quantitative Reverse Transcription PCR), was performed on RNA before and after pull-down using the forward primer –5′-GGAAGTGGTGTGAGTGCGTACAC-3′ and the reverse primer –5′-CACCTTCAGCCAACTTCCAATG-3′. The relative fold enrichment of tau pre-mRNA was measured using eq 3:
| (3) |
where is the difference between the Ct values for the RNA of interest and a housekeeping gene (18S) in total RNA from cells and is the difference between the Ct values for the RNA of interest and the same housekeeping gene in RNA after pull-down.
hTau Mice Primary Neuron Experiments.
The details pertaining to media preparation are presented in the following.
Dissection Medium.
The dissection medium consisted of the following: 430 mL of culture-grade H2O (Fisher Scientific, SH3052902), 50 mL of 10× HBSS without Ca2+ and Mg2+ (Invitrogen, 14185052), 10 mL of HEPES (Invitrogen, 15630080), 5 mL of pyruvate (Invitrogen, 11360070), 5 mL of glucose solution (Thermo, A2494001), and 100 μL of gentamicin (Invitrogen, 15710064).
Plating Medium.
Plating medium is comprised the following: 465 mL of Neurobasal (Invitrogen, 21103049); 25 mL of FBS, heat inactivated (Invitrogen, 10082139); 10 mL of Glutamax-I (Invitrogen, 35050061); and 100 μL of gentamicin (Invitrogen, 15710064).
Feeding Medium.
Feeding medium contains the following: 490 mL of Neurobasal-A (Invitrogen, 10888022), 10 mL of Glutamax-I (Invitrogen, 35050061), and 100 μL of gentamicin (Invitrogen, 15710064). Note, add 2% B27 just before feeding (Invitrogen, 17504044).
Miscellaneous Reagents.
Miscellaneous reagents include the following: poly-D-lysine (PDL; Sigma, P7280), Trizma Tris base (Sigma, T1194), and Papain suspension (Worthington, LS003124).
Culture plates (Corning CellBIND surface microplates) were precoated with 0.1 mg/mL PDL, which was dissolved in Tris buffer overnight or for 2 h at 37 °C. Next, 1 mL per pup of dissection medium was placed on ice. Then, 5 mL per pup of plating medium for the disassociation step, with the required volume of feeding medium for plating, and 1 mL per pup of dissection medium with 1% Papain were prewarmed at 37 °C. The cortices from P0 htau mouse pups (B6.Cg-Mapttm1-(EGFP)Klt Tg(MAPT)8cPdav/J, Jackson Laboratories) were removed and placed in ice cold dissection medium for dissection and removal of meninges. Then, the dissected cortices were placed into ice-cold dissection medium. The tails of P0 htau mouse pups were collected for further genotyping. After dissection, cold dissection medium was aspirated, and the warmed dissection medium/Papain solution was added. The solution was placed at 37 °C for 20 min, and then, one volume of plating medium was added to stop Papain activity. Meanwhile, the PDL solution in precoated plates was aspirated. The plates were washed with 1× DPBS with added feeding medium and incubated at 37 °C. The medium in dissection tubes was carefully aspirated. The remaining tissue was carefully washed three times with plating medium. After washing, 2 mL of fresh plating medium was added. The tissue was dissociated by gently triturating through a 5 mL serological pipet three times and then through a P1000 pipet tip three times. Then, 1 mL of the supernatant was transferred to a new 15 mL tube, and then, 1 mL of fresh plating medium was added. Trituration was repeated three to six times through the P1000 tip until all chunks of tissue were dissociated, and 1 mL of the supernatant was transferred every time. The cell mixture in new tube was diluted to 10 mL with plating medium and then run through a 40 μM cell strainer. Then, 10 mL of cells was spun down at 1900 rpm for 4 min. The cell pellet was resuspended in 1 mL of fresh plating medium and then diluted to the required density in feeding medium +2% B-27. The neuron cells were plated in prewarmed plates. Approximately 50% of the medium was changed every 3–4 days with feeding medium +2% B-27. P0 htau mouse pups were genotyped before compound treatment. After 15 days, the neuron cells were treated with 9 and Vivo-Morpholino ASO and were scrambled control and incubated at 37 °C for 48 h. The total RNA was extracted and analyzed via RT-qPCR, as described above (Quantitative Reverse Transcription PCR). Primers used for the evaluation of mouse neuron cells are provided in Table S14.
Optical Melting Experiments.
The thermal stabilities of WT, DDPAC, and DDPAC+I17T RNA constructs were analyzed by optical melting experiments with and without compounds. The RNAs were heated to 95 °C and slowly cooled to room temperature. The samples were then cooled to 15 °C upon the addition of or in the absence of compound and then heated to 85 °C at a rate of 1 °C/min. The absorbance was monitored at 260 nm. The melting curves were fitted with MeltWin 3.5.69
Design and Preparation of RNA Samples for NMR.
All RNA constructs were purchased from GE Healthcare Dharmacon, Inc. Oligoribonucleotides that form the duplex, including r(5′-CCGGCAGUGUG-3′) + r(5′CACACGUCGG-3′) to mimic the stem of the WT tau SRE and r(5′-CCGGCAGUGUG-3′) + r(5′CAUACGUCGG-3′) to mimic the stem of the DDPAC tau SRE, were deprotected and desalted with PD-10 columns according to the manufacturer’s recommended protocol. Equimolar amounts of the sequences were combined in 400 μL of NMR buffer (10 mM KH2PO4/K2HPO4 and 0.05 mM EDTA (pH 6.0)) to make three separate samples. The final concentrations of RNA in the samples were 250, 400, 500, or 700, including 20 μL of D2O in each sample to provide a lock signal. RNA samples were reannealed by heating to 80 °C for 3 min and were slowly cooled to room temperature to form duplexes.
NMR Spectroscopy.
Shigemi NMR tubes (Shigemi, Inc.) were used for all NMR experiments. NMR spectra were acquired on Bruker Avance III 600, 700, and 850 MHz spectrometers equipped with cryoprobes. The 1D spectra and 2D NOESY and DQF-COSY spectra with different mixing times were acquired at 281.5 and 298 K on samples in 95% H2O/5% D2O and 100% D2O alone or in the presence of 4–9. 1D LH NMR spectra were acquired on either WT or DDPAC tau hairpin stem mimic duplexes alone. Each compound was then titrated into the final compound/RNA molar ratios of 0.0, 0.5, 1.0, 1.5, and 2.0. WaterLOGSY (water-ligand observed via gradient spectroscopy) experiments were carried out on samples of the WT tau stem mimic duplex mixed with each compound to determine the formation of the RNA-small molecule complexes.70–72 Samples for WaterLOGSY experiments contained 15 μM RNA and 300 μM compound for a 1/20 RNA/compound ratio. WaterLOGSY spectra were phased to give negative signals for negative NOEs with water.71,72 Samples for 2D NMR spectroscopy consisted of 700 μM free-form WT tau RNA, to which 4 was added to a final concentration of 700 μM for studies of the 4–RNA complex. Additionally, 2D spectra were acquired on a separate sample containing 400 μM WT tau RNA and 600 μM 5 or 9. For 1D and 2D spectra of samples in 95% H2O/5% D2O, an excitation sculpting sequence73 was applied during acquisition to suppress the water signal. Proton chemical shifts were referenced internally to the frequency of water. Then, 2D NMR spectra were processed with NMRPipe,74 and the resonances were assigned with SPARKY.75
Methods for Obtaining NOE Restraints and Structure Calculations.
NOESY cross peaks were integrated assuming a Gaussian distribution, and the volumes were converted to distances using H5–H6 pyrimidine peaks as the reference. Distance restraints were created by setting the lower and upper bounds to ±0.5 Å the calculated distance for each peak. Peaks with potential spin diffusion were given upper bounds of + 1.0 Å. Interbase (H6/8-H6/8) restraints were assigned between 3.5 and 5.0 Å and 5.0 and 6.5 Å based on their relative intensity at a mixing time of 400 ms. All imino protons involved in GC and AU base pairs exhibited cross peak patterns indicative of WC base pairing, and therefore, standard WC base pair restraints were applied for these pairs. The G4–U18 mismatch was not restrained in the calculations. Structures were calculated by restrained molecular dynamics using AMBER14 or AMBER16.76 The system was heated to 2000 K over 10 ps and then cooled to 0 K, totaling 35 ps in implicit solvent using the 99SB and GAFF force fields. Charges were assigned to 4, 5, and 9 using the AM1-BCC method model. NMR restraints were applied at the peak temperature. From a single initial structure of free-form or 4-bound WT tau RNA, 100 calculations were performed with randomized initial trajectories. For the 5- and 9-bound WT tau RNA duplex, 60 calculations were performed with randomized initial trajectories. The top 20 structures were selected by their adherence to the NMR data. Helical and base pair properties were extracted with X3DNA.77
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Samuel Hasson (Pfizer) and Lyn Jones (Pfizer) for initiating the project and Xiangming Kong (The Scripps Research Institute) and Chunhua Yuan (The Ohio State University) for assistance with NMR experiments. Minimization by simulated annealing and molecular dynamics simulations were performed with computational resources provided by The Scripps Research Institute, State University of New York at Fredonia, and Florida Atlantic University. This study made use of the Campus Chemical Instrument Center NMR facility at The Ohio State University.
Funding
This work was supported by Pfizer, the National Institutes of Health (R01-GM097455, DP1-NS096898, and P01-NS099114 to M.D.D, and S10-OD021550 to The Scripps Research Institute), the Tau Consortium and Rainwater Charitable Foundation (to M.D.D.), and the Huntington’s Disease Society of America (J.L.C.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00768.
Analysis of 1D and 2D NMR spectra of unbound and bound RNAs; set of 20 compounds identified by Inforna and screened in cell-based luciferase assay; the effect of compounds on expression of luciferase in cell-based assays and on relative amounts of 4R and 3R Tau mRNA assessed by RT-qPCR; binding assays of 4 with 2-AP substituted RNA constructs; cell toxicity data for compounds assessed by the cell viability reagent; protein modulation with compound 9 and 4R-to-3R ASO treatment; results of Chem-CLIP and C-Chem-CLIP experiments with compounds 9, 11, and 12; the effects of compounds 7 and 9 on microRNA expression; the effect of gentamicin on relative amounts of 4R and 3R Tau mRNA in LAN5 cells assessed by RT-qPCR; the effect of compounds on the thermodynamic stability of Tau RNA constructs; 1H titration and WaterLOGSY NMR spectra with the compounds; 2D 1H NOESY NMR spectra of unbound and bound WT stem mimic duplexes; synthesis of compounds 5–7 and 9–11; summary of chemical shifts changes of WT and DDPAC stem mimic duplexes induced by compounds; 1H NMR chemical shifts of unbound and bound stem mimic duplexes; Kd EC50, or IC50 (μM) values for compounds determined from in vitro binding assays for compounds binding to RNA constructs; NOE restraints used for modeling of unbound and bound stem mimic duplexes; primers of RT-qPCR, Chem-CLIP, and microRNAs (PDF)
Unbound RNA (PDB)
4-Bound tau WT RNA contruct (PDB)
5-Bound tau WT RNA contruct (PDB)
9-Bound tau WT RNA contruct (PDB)
(XLSX)
Accession Codes
NMR chemical shifts for the unbound and 4-, 5-, and 9-bound tau WT RNA construct were deposited in the Biological Magnetic Resonance Data Bank as entries 30697, 30698, 30699, and 30700, respectively. Atomic coordinates for the unbound and bound tau WT RNA constructs were deposited in the RCSB Protein Data Bank as entries 6VA1, 6VA2, 6VA3, and 6VA4, respectively.
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c00768
Contributor Information
Jonathan L. Chen, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Peiyuan Zhang, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Masahito Abe, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Haruo Aikawa, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Liying Zhang, Pfizer Worldwide Research and Development, Cambridge, Massachusetts 02139, United States.
Alexander J. Frank, Department of Chemistry & Biochemistry, State University of New York at Fredonia, Fredonia, New York 14063, United States
Timothy Zembryski, Department of Chemistry & Biochemistry, State University of New York at Fredonia, Fredonia, New York 14063, United States.
Christopher Hubbs, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
HaJeung Park, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Jane Withka, Pfizer Worldwide Research and Development, Cambridge, Massachusetts 02139, United States.
Claire Steppan, Pfizer Worldwide Research and Development, Groton, Connecticut 06340, United States.
Lucy Rogers, Pfizer Worldwide Research and Development, Groton, Connecticut 06340, United States.
Shawn Cabral, Pfizer Worldwide Research and Development, Groton, Connecticut 06340, United States.
Martin Pettersson, Pfizer Worldwide Research and Development, Cambridge, Massachusetts 02139, United States.
Travis T. Wager, Pfizer Worldwide Research and Development, Cambridge, Massachusetts 02139, United States
Matthew A. Fountain, Department of Chemistry & Biochemistry, State University of New York at Fredonia, Fredonia, New York 14063, United States
Gavin Rumbaugh, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Jessica L. Childs-Disney, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter, Florida 33458, United States
Matthew D. Disney, Department of Chemistry and Neuroscience, The Scripps Research Institute, Jupiter.
REFERENCES
- (1).Mattick JS; Makunin IV Non-coding RNA. Hum. Mol. Genet 2006, 15 (suppl_1), R17–R29. [DOI] [PubMed] [Google Scholar]
- (2).Tazi J; Durand S; Jeanteur P The spliceosome: A novel multi faceted target for therapy. Trends Biochem. Sci 2005, 30 (8), 469–478. [DOI] [PubMed] [Google Scholar]
- (3).Chiriboga CA; Swoboda KJ; Darras BT; Iannaccone ST; Montes J; De Vivo DC; Norris DA; Bennett CF; Bishop KM Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86 (10), 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gallego J; Varani G Targeting RNA with small-molecule drugs: Therapeutic promise and chemical challenges. Acc. Chem. Res 2001, 34 (10), 836–843. [DOI] [PubMed] [Google Scholar]
- (5).Disney MD Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 2019, 141 (17), 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Naryshkin NA; Weetall M; Dakka A; Narasimhan J; Zhao X; Feng Z; Ling KK; Karp GM; Qi H; Woll MG; Chen G; Zhang N; Gabbeta V; Vazirani P; Bhattacharyya A; Furia B; Risher N; Sheedy J; Kong R; Ma J; Turpoff A; Lee CS; Zhang X; Moon YC; Trifillis P; Welch EM; Colacino JM; Babiak J; Almstead NG; Peltz SW; Eng LA; Chen KS; Mull JL; Lynes MS; Rubin LL; Fontoura P; Santarelli L; Haehnke D; McCarthy KD; Schmucki R; Ebeling M; Sivaramakrishnan M; Ko CP; Paushkin SV; Ratni H; Gerlach I; Ghosh A; Metzger F Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345 (6197), 688–693. [DOI] [PubMed] [Google Scholar]
- (7).Palacino J; Swalley SE; Song C; Cheung AK; Shu L; Zhang X; Van Hoosear M; Shin Y; Chin DN; Keller CG; Beibel M; Renaud NA; Smith TM; Salcius M; Shi X; Hild M; Servais R; Jain M; Deng L; Bullock C; McLellan M; Schuierer S; Murphy L; Blommers MJ; Blaustein C; Berenshteyn F; Lacoste A; Thomas JR; Roma G; Michaud GA; Tseng BS; Porter JA; Myer VE; Tallarico JA; Hamann LG; Curtis D; Fishman MC; Dietrich WF; Dales NA; Sivasankaran R SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 2015, 11 (7), 511–517. [DOI] [PubMed] [Google Scholar]
- (8).Ratni H; Ebeling M; Baird J; Bendels S; Bylund J; Chen KS; Denk N; Feng Z; Green L; Guerard M; Jablonski P; Jacobsen B; Khwaja O; Kletzl H; Ko CP; Kustermann S; Marquet A; Metzger F; Mueller B; Naryshkin NA; Paushkin SV; Pinard E; Poirier A; Reutlinger M; Weetall M; Zeller A; Zhao X; Mueller L Discovery of Risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem 2018, 61 (15), 6501–6517. [DOI] [PubMed] [Google Scholar]
- (9).Ottesen EW ISS-N1 makes the First FDA-approved drug for spinal muscular atrophy. Transl. Neurosci 2017, 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Singh NN; Howell MD; Androphy EJ; Singh RN How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017, 24 (9), 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brunden KR; Trojanowski JQ; Lee VMY Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discovery 2009, 8 (10), 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Disney MD; Winkelsas AM; Velagapudi SP; Southern M; Fallahi M; Childs-Disney JL Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol 2016, 11 (6), 1720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Velagapudi SP; Gallo SM; Disney MD Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 2014, 10 (4), 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Costales MG; Hoch DG; Abegg D; Childs-Disney JL; Velagapudi SP; Adibekian A; Disney MD A designed small molecule inhibitor of a non-coding RNA sensitizes HER2 negative cancers to Herceptin. J. Am. Chem. Soc 2019, 141 (7), 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Velagapudi SP; Cameron MD; Haga CL; Rosenberg LH; Lafitte M; Duckett DR; Phinney DG; Disney MD Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (21), 5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Costales MG; Aikawa H; Li Y; Childs-Disney JL; Abegg D; Hoch DG; Pradeep Velagapudi S; Nakai Y; Khan T; Wang KW; Yildirim I; Adibekian A; Wang ET; Disney MD Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (5), 2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rzuczek SG; Colgan LA; Nakai Y; Cameron MD; Furling D; Yasuda R; Disney MD Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 2017, 13 (2), 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang P; Park HJ; Zhang J; Junn E; Andrews RJ; Velagapudi SP; Abegg D; Vishnu K; Costales MG; Childs-Disney JL; Adibekian A; Moss WN; Mouradian MM; Disney MD Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (3), 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hong M; Zhukareva V; Vogelsberg-Ragaglia V; Wszolek Z; Reed L; Miller BI; Geschwind DH; Bird TD; McKeel D; Goate A; Morris JC; Wilhelmsen KC; Schellenberg GD; Trojanowski JQ; Lee VMY Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282 (5395), 1914–1917. [DOI] [PubMed] [Google Scholar]
- (20).Chen JL; Moss WN; Spencer A; Zhang P; Childs-Disney JL; Disney MD The RNA encoding the microtubule-associated protein tau has extensive structure that affects its biology. PLoS One 2019, 14 (7), No. e0219210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Donahue CP; Muratore C; Wu JY; Kosik KS; Wolfe MS Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J. Biol. Chem 2006, 281 (33), 23302–23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Grover A; Houlden H; Baker M; Adamson J; Lewis J; Prihar G; Pickering-Brown S; Duff K; Hutton M 5’ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure That regulates alternative splicing of exon 10. J. Biol. Chem 1999, 274 (21), 15134–15143. [DOI] [PubMed] [Google Scholar]
- (23).Luo Y; Disney MD Bottom-up design of small molecules that stimulate exon 10 skipping in mutant MAPT pre-mRNA. ChemBioChem 2014, 15 (14), 2041–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rogers DJ; Tanimoto TT A computer program for classifying plants. Science 1960, 132 (3434), 1115–1118. [DOI] [PubMed] [Google Scholar]
- (25).Peacey E; Rodriguez L; Liu Y; Wolfe MS Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012, 40 (19), 9836–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jean JM; Hall KB 2-Aminopurine fluorescence quenching and lifetimes: Role of base stacking. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (1), 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang JH; Chung TD; Oldenburg KR A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 1999, 4 (2), 67–73. [DOI] [PubMed] [Google Scholar]
- (28).Johansson MK; Fidder H; Dick D; Cook RM Intramolecular dimers: A new strategy to fluorescence quenching in dual-labeled oligonucleotide probes. J. Am. Chem. Soc 2002, 124 (24), 6950–6956. [DOI] [PubMed] [Google Scholar]
- (29).Varani L; Spillantini MG; Goedert M; Varani G Structural basis for recognition of the RNA major groove in the tau exon 10 splicing regulatory element by aminoglycoside antibiotics. Nucleic Acids Res. 2000, 28 (3), 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Donahue CP; Ni J; Rozners E; Glicksman MA; Wolfe MS Identification of tau stem loop RNA stabilizers. J. Biomol. Screening 2007, 12 (6), 789–799. [DOI] [PubMed] [Google Scholar]
- (31).Teuscher KB; Zhang M; Ji H A versatile method to determine the cellular bioavailability of small-molecule inhibitors. J. Med. Chem 2017, 60 (1), 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wolber G; Langer T LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model 2005, 45 (1), 160–169. [DOI] [PubMed] [Google Scholar]
- (33).Grant JA; Gallardo MA; Pickup BT A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem 1996, 17 (14), 1653–1666. [Google Scholar]
- (34).Kearnes S; Pande V ROCS-derived features for virtual screening. J. Comput.-Aided Mol. Des 2016, 30 (8), 609–617. [DOI] [PubMed] [Google Scholar]
- (35).Sastry GM; Inakollu VS; Sherman W Boosting virtual screening enrichments with data fusion: coalescing hits from two-dimensional fingerprints, shape, and docking. J. Chem. Inf. Model 2013, 53 (7), 1531–1542. [DOI] [PubMed] [Google Scholar]
- (36).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1 (6), 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Holbeck SL Update on NCI in vitro drug screen utilities. Eur. J. Cancer 2004, 40 (6), 785–793. [DOI] [PubMed] [Google Scholar]
- (38).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- (39).Disney MD; Liu B; Yang WY; Sellier C; Tran T; Charlet-Berguerand N; Childs-Disney JL A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem. Biol 2012, 7 (10), 1711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Halder K; Largy E; Benzler M; Teulade-Fichou MP; Hartig JS Efficient suppression of gene expression by targeting 5′-UTR-based RNA quadruplexes with bisquinolinium compounds. ChemBioChem 2011, 12 (11), 1663–8. [DOI] [PubMed] [Google Scholar]
- (41).Mayer M; James TL NMR-based characterization of phenothiazines as a RNA binding scaffold. J. Am. Chem. Soc 2004, 126 (13), 4453–4460. [DOI] [PubMed] [Google Scholar]
- (42).Ke Y; Wu J; Leibold EA; Walden WE; Theil EC Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. Fine-tuning of mRNA regulation? J. Biol. Chem 1998, 273 (37), 23637–23640. [DOI] [PubMed] [Google Scholar]
- (43).Costales MG; Haga CL; Velagapudi SP; Childs-Disney JL; Phinney DG; Disney MD Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc 2017, 139 (9), 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Childs-Disney JL; Disney MD Approaches to validate and manipulate RNA targets with small molecules in cells. Annu. Rev. Pharmacol. Toxicol 2016, 56, 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yang WY; Wilson HD; Velagapudi SP; Disney MD Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J. Am. Chem. Soc 2015, 137 (16), 5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Guan L; Disney MD Covalent small-molecule-RNA complex formation enables cellular profiling of small-molecule-RNA interactions. Angew. Chem. Int. Ed 2013, 52 (38), 10010–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang J; Schultz PG; Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (20), E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168 (3), 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu B; Childs-Disney JL; Znosko BM; Wang D; Fallahi M; Gallo SM; Disney MD Analysis of secondary structural elements in human microRNA hairpin precursors. BMC Bioinformatics 2016, 17, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Rumbaugh G; Sillivan SE; Ozkan ED; Rojas CS; Hubbs CR; Aceti M; Kilgore M; Kudugunti S; Puthanveettil SV; Sweatt JD; Rusche J; Miller CA Pharmacological selectivity within class I histone deacetylases predicts effects on synaptic function and memory rescue. Neuropsychopharmacology 2015, 40 (10), 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dalvit C; Pevarello P; Tato M; Veronesi M; Vulpetti A; Sundstrom M Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol NMR 2000, 18 (1), 65–8. [DOI] [PubMed] [Google Scholar]
- (52).Fessl T; Lilley DMJ Measurement of the change in twist at a helical junction in RNA using the orientation dependence of FRET. Biophys. J 2013, 105 (9), 2175–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zheng S; Chen Y; Donahue CP; Wolfe MS; Varani G Structural basis for stabilization of the tau pre-mRNA splicing regulatory element by novantrone (mitoxantrone). Chem. Biol 2009, 16 (5), 557– 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Aboul-Ela F Strategies for the design of RNA-binding small molecules. Future Med. Chem 2010, 2 (1), 93–119. [DOI] [PubMed] [Google Scholar]
- (55).Sarkar A; Kellogg GE Hydrophobicity - Shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem 2010, 10 (1), 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).López-Senin P; Gómez-Pinto I; Grandas A; Marchán V Identification of ligands for the tau exon 10 splicing regulatory element RNA by using dynamic combinatorial chemistry. Chem. - Eur. J 2011, 17 (6), 1946–1953. [DOI] [PubMed] [Google Scholar]
- (57).Canals A; Purciolas M; Aymami J; Coll M The anticancer agent ellipticine unwinds DNA by intercalative binding in an orientation parallel to base pairs. Acta Crystallogr., Sect. D: Biol. Crystallogr 2005, 61 (7), 1009–1012. [DOI] [PubMed] [Google Scholar]
- (58).Ryckebusch A; Garcin D; Lansiaux A; Goossens J-F; Baldeyrou B; Houssin R; Bailly C; Henichart J-P Synthesis, cytotoxicity, DNA interaction, and topoisomerase II inhibition properties of novel indeno[2,1-c]quinolin-7-one and indeno[1,2-c]isoquinolin-5,11-dione derivatives. J. Med. Chem 2008, 51 (12), 3617–3629. [DOI] [PubMed] [Google Scholar]
- (59).Shaikh MS; Karpoormath R; Thapliyal N; Rane RA; Palkar MB; Faya AM; Patel HM; Alwan WS; Jain K; Hampannavar GA Current perspective of natural alkaloid carbazole and its derivatives as antitumor agents. Anti-Cancer Agents Med. Chem 2015, 15 (8), 1049–1065. [DOI] [PubMed] [Google Scholar]
- (60).Rehman SU; Sarwar T; Husain MA; Ishqi HM; Tabish M Studying non-covalent drug-DNA interactions. Arch. Biochem. Biophys 2015, 576, 49–60. [DOI] [PubMed] [Google Scholar]
- (61).Han GW; Kopka ML; Langs D; Sawaya MR; Dickerson RE Crystal structure of an RNA.DNA hybrid reveals intermolecular intercalation: dimer formation by base-pair swapping. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (16), 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kawasaki Y; Chufan EE; Lafont V; Hidaka K; Kiso Y; Mario Amzel L; Freire E How much binding affinity can be gained by filling a cavity? Chem. Biol. Drug Des 2010, 75 (2), 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ferenczy GG; Keseru GM On the enthalpic preference of fragment binding. MedChemComm 2016, 7 (2), 332–337. [Google Scholar]
- (64).Guengerich FP Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet 2011, 26 (1), 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kawasaki Y; Freire E Finding a better path to drug selectivity. Drug Discov. Today 2011, 16 (21–22), 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Varani L; Hasegawa M; Spillantini MG; Smith MJ; Murrell JR; Ghetti B; Klug A; Goedert M; Varani G Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc. Natl. Acad. Sci. U. S. A 1999, 96 (14), 8229–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zhang J; Harvey SE; Cheng C A high-throughput screen identifies small molecule modulators of alternative splicing by targeting RNA G-quadruplexes. Nucleic Acids Res. 2019, 47 (7), 3667–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hampf M; Gossen M A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem 2006, 356 (1), 94–99. [DOI] [PubMed] [Google Scholar]
- (69).McDowell JA; Turner DH Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: Solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry 1996, 35 (45), 14077–14089. [DOI] [PubMed] [Google Scholar]
- (70).Dalvit C; Fogliatto G; Stewart A; Veronesi M; Stockman B WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 2001, 21 (4), 349–359. [DOI] [PubMed] [Google Scholar]
- (71).Chu S; Zhou G; Gochin M Evaluation ofligand-based NMR screening methods to characterize small molecule binding to HIV-1 glycoprotein-41. Org. Biomol. Chem 2017, 15 (24), 5210–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Zega A NMR methods for identification of false positives in biochemical screens. J. Med. Chem 2017, 60 (23), 9437–9447. [DOI] [PubMed] [Google Scholar]
- (73).Hwang TL; Shaka AJ Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112 (2), 275–279. [Google Scholar]
- (74).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- (75).Goddard TD; Kneller DG SPARKY, NMR Assignment and Integration Software, 3; University of California, San Francisco: San Francisco, CA, 2004. [Google Scholar]
- (76).Case DA; Babin V; Berryman JT; Betz RM; Cai Q; Cerutti DS; Cheatham TEI; Darden TA; Duke RE; Gohlke H; Goetz AW; Gusarov S; Homeyer N; Janowski P; Kaus J; Kolossvary I; Kovalenko A; Lee TS; LeGrand S; Luchko T; Luo R; Madej B; Merz KM; Paesani F; Roe DR; Roitberg A; Sagui C; Salomon-Ferrer R; Seabra G; Simmerling CL; Smith W; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Kollman PA AMBER 14; University of California, San Francisco: San Francisco, CA, 2014. [Google Scholar]
- (77).Lu XJ; Olson WK 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31 (17), 5108–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Hutton M; Lendon CL; Rizzu P; Baker M; Froelich S; Houlden H; Pickering-Brown S; Chakraverty S; Isaacs A; Grover A; Hackett J; Adamson J; Lincoln S; Dickson D; Davies P; Petersen RC; Stevens M; de Graaff E; Wauters E; van Baren J; Hillebrand M; Joosse M; Kwon JM; Nowotny P; Che LK; Norton J; Morris JC; Reed LA; Trojanowski J; Basun H; Lannfelt L; Neystat M; Fahn S; Dark F; Tannenberg T; Dodd PR; Hayward N; Kwok JBJ; Schofield PR; Andreadis A; Snowden J; Craufurd D; Neary D; Owen F; Oostra BA; Hardy J; Goate A; van Swieten J; Mann D; Lynch T; Heutink P Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393 (6686), 702–705. [DOI] [PubMed] [Google Scholar]
- (79).Spillantini MG; Murrell JR; Goedert M; Farlow MR; Klug A; Ghetti B Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (13), 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.