

Abstract
Cardiomyopathies caused by double gene mutations are rare but conferred a remarkably increased risk of end‐stage progression, arrhythmias, and poor outcome. Compound genetic mutations leading to complex phenotype in the setting of cardiomyopathies represent an important challenge in clinical practice, and genetic tests allow risk stratification and personalized clinical management of patients. We report a case of a 50‐year‐old woman with congestive heart failure characterized by dilated cardiomyopathy, diffuse coronary disease, complete atrioventricular block, and missense mutations in cardiac myosin‐binding protein C (MYBPC3) and myopalladin (MYPN). We discuss the plausible role of genetic profile in phenotype determination.
Keywords: cardiac myosin‐binding protein C, complete atrioventricular block, diffuse coronary atherosclerosis, dilated cardiomyopathy, myopalladin
1. INTRODUCTION
Cardiomyopathies assessment represents a key role in the setting of heart failure and sudden death approach (McKenna, Maron, & Thiene, 2017). Compound genetic mutations leading to complex phenotype in the setting of cardiomyopathies represent an important challenge in clinical practice, and genetic tests are helpful in the clinical management of patients as they can provide important information on risk stratification, to achieve personalized therapy indicating differential surveillance strategies among individuals. Here, we report a case of a 50‐year‐old woman with congestive heart failure characterized by dilated cardiomyopathy, diffuse coronary disease and complete atrioventricular block, and genetic evidence of missense mutations in cardiac myosin‐binding protein C (MYBPC3) and myopalladin (MYPN).
2. CASE REPORT
A 50‐year‐old woman was referred to our cardiovascular department for ankles swelling and shortness of breath. An electrocardiogram showed sinus tachycardia and complete atrioventricular block with ventricular rhythm 40 bpm and diffuse ST depression (Figure 1). Then, the patient was admitted to the cardiology intensive care unit after a clear evidence of heart failure with peripheral congestion; she had a history of nontoxic multinodular goiter and no previous history of cardiovascular disease or cardiovascular risk factors. An echocardiogram demonstrated a dilated cardiomyopathy with end‐diastolic diameter of the left ventricle (LV) of 62 mm, severely depressed left ventricle ejection fraction (LVEF), 20%, mild hypertrophic septum (13.5 mm), trabecular aspect of lateral and inferior LV wall, thrombus in the apex, and severe tricuspid regurgitation with inferior vena cava reflow (Figure 2). Furthermore, at a cardiac magnetic resonance, the short‐term inversion recovery (STIR) detected subendocardial edema in the septum, anterior and lateral walls. Late sequences showed subendocardial necrosis at the level of the anterior‐middle and later‐middle segments, and full‐thickness necrosis in the lateral‐apical segment.
Figure 1.

Sinus tachycardia and complete atrioventricular block with ventricular rhythm 40 bpm and marked diffuse ST depression (red arrows indicate P wave)
Figure 2.
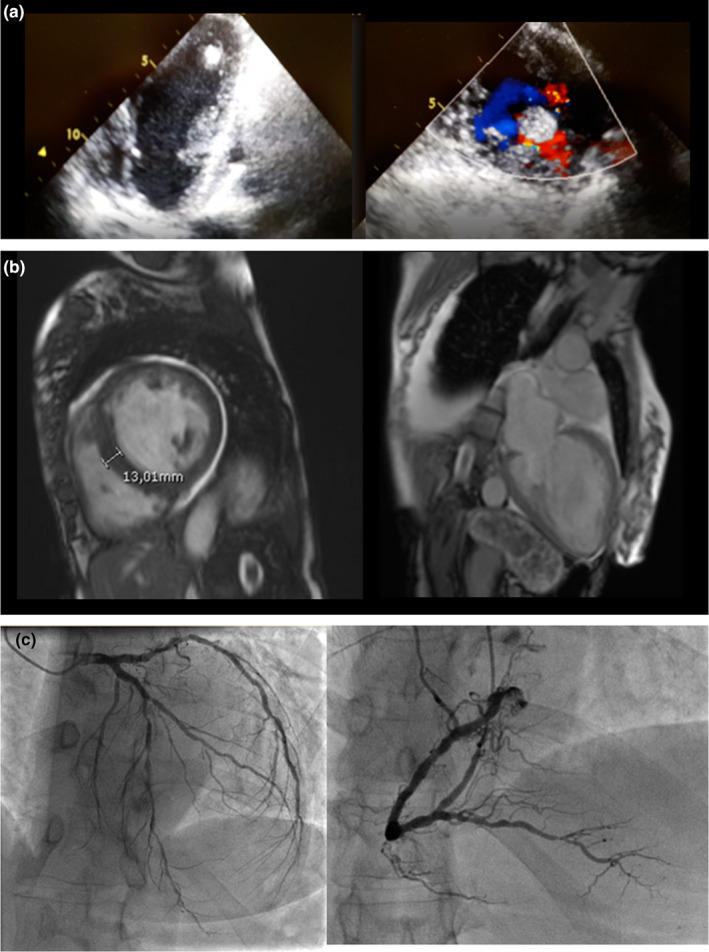
(a) Echocardiography evidence of left ventricular apical thrombosis and hypertrabeculation; (b) cardiac RMN showing septum hypertrophy and necrosis in the lateral‐apical segment; (c) coronary angiography showing diffuse atherosclerosis
A coronary angiography showed calcified critical stenosis of many proximal vessels and diffuse peripheral atheromasic disease with consequent recommendation to surgical revascularization. As the patient was asymptomatic for angina with negative troponin test and in presence of the complete atrioventricular (AV) block, severe LV dysfunction and apical thrombosis, he underwent cardiac resynchronization therapy defibrillator (CRT‐D) and started therapy with ACE‐inhibitor, mineralocorticoid receptor antagonist, beta‐blocker, warfarin, acetylsalicylic acid, and furosemide. After 1 month, we observed complete regression of apical thrombosis associated to clinical improvement with increased LVEF. Then, the patient underwent surgical myocardial revascularization with the mammary artery to anterior interventricular artery and venous by‐pass to the obtuse marginal and posterior interventricular artery. Five months later, the patient was in sinus rhythm with ventricular CRT‐D stimulation, NHYA class I, improved contractile function (LVEF 45%–48%) and mild tricuspidal insufficiency, still stable at 1 year follow‐up.
Genetic analysis by next‐generation sequencing (NGS), after informed consent of the patient, was performed for suspected genetically dilated cardiomyopathy and identified a double mutation in 11p11.2, cardiac myosin‐binding protein C (MYBPC3) gene, and in 10q21.3, myopalladin gene (MYPN). We verified that this patient was heterozygous for two missense mutations: the first in gene MYBPC3: c.2459G > A, p.(Arg820Gln) in exon 24, and the second in gene MYPN: c.3335C > T, p.(Pro1112Leu) in exon 18.
3. MATERIALS AND METHODS
3.1. Libraries preparation and Next‐generation sequencing (NGS)
Peripheral blood samples were taken from the patient, and genomic DNA was isolated by using Bio Robot EZ1 (Quiagen). The quality of DNA was tested on 1% electrophorese agarose gel, and the concentrations were quantified with Nanodrop 2000 C spectrophotometer (Thermo Fisher Scientific).
A library of all coding regions of the 76 genes, including genes related to hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic cardiomyopathy, and channelopathies, was obtained using the Haloplex target enrichment kit (Agilent Technologies) according to the manufacturer's instructions. The libraries were pooled, and NGS was performed on a MiSeq sequencer (Illumina) using a MiSeq Reagent kit V3 300 cycles flow cell. All putatively pathogenic variants were confirmed by Sanger sequencing. Polymerase chain reaction (PCR) products were sequenced using ABI Prism 3,100 Genetic Analyzer (Thermo Fisher Scientifi) and the BigDye Terminator v1.1 sequencing kit (Applied Biosystems, Foster City, CA, USA).The produced raw paired‐end reads underwent quality checking by using the FastQC tool [Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc] and then aligned to the hg19 reference genome sequence by means of Bowtie (Langmead & Salzberg, 2012). Depth of coverage statistics for the target regions was calculated by TEQC ver. 3.47 (Hummel, Bonnin, Lowy, & Roma, 2011). Variants were called by means of the HaplotypeCaller tool of GATK ver. 3.58 (McKenna et al., 2010), while functional annotation was carried out by ANNOVAR tool, using RefSeq gene and transcript annotations (updated to December 2016) (Wang, Li, & Hakonarson, 2010). Variants were found in dbSNP ver. 150 (Sherry et al., 2001), ExAC ver. 0.311 (Lek et al., 2016), and Exome Variant Server (http://evs.gs.washington.edu/EVS, accessed at December 2016), HRC (McCarthy, 2017), Kaviar (Glusman, Caballero, Mauldin, Hood, & Roach, 2011) and ClinVar (Landrum et al., 2017). Predictions of functional consequences for missense variants were further collected by querying the dbNSFP ver. 3.2 resource and retrieving precomputed pathogenicity predictions and evolutionary conservation measures (Liu, Jian, & Boerwinkle, 2011).
Different filtering strategies were implemented in order to determine the candidate causative variants for the patient. Interesting variants were those exhibiting relevant functional annotations (e.g., they were either missense, or splicing, or stopgain, or stoploss or frameshift mutations), clinical significance according ClinVar, low (≤0.05) or absent Minor Allele Frequency in public population databases, absence in healthy samples or presence in samples with same clinical traits.
4. RESULTS
Next‐generation sequencing revealed two missense variant in heterozygous state, one in the exon 17 of the MYPN gene (NM_032578) (OMIM 608,517) c.866G > C resulting in a p.(Pro1112Leu) substitution (GRCh37/hg19), and one in the exon 24 of the MYBPC3 gene (NM_000256) (OMIM 600,958) c.2459 G > A p.(Arg820Glu). Variants were detected with a depth of coverage >706× and 662×, respectively, with elevate quality scores (i.e., Phred quality > 3,000 and genotype quality = 99). The amino acid substitution MYBPC3 (Arg820Glu) was known (dbSNP ID: rs2856655) but very rare (0,00,002 estimated frequency by the ExAC and gnomAD database) and predicted as deleterious or probably damaging by several software tools, including SIFT, PolyPhen2, MutationTaster, CADD, and M‐CAP, and is reported to be likely pathogenic in ClinVAr. The amino acid substitution MYPN p.(Pro1112Leu) was less rare (dbSNP ID: rs71534278; 0,003 estimated frequency by the ExAC and gnomAD database), and it is predicted as deleterious or probably damaging by several software tools, including SIFT, PolyPhen2, CADD, and M‐CAP, but its contribution to the phenotype is not clear. The variants were confirmed by Sanger sequencing.
5. DISCUSSION
In this case report, the coexistence of advance heart failure, atrioventricular block, significative and diffuse coronary atherosclerosis, and dilated cardiomyopathy in a relatively young woman without apparent cardiovascular risk factors led us to perform genetic screening, and believing the particular clinical picture could have a deeper pathological explanation.
In literature, the MYBPC3 gene is responsible for 40%–50% of all hypertrophic cardiomyopathies (HCM) (Sabater‐Molina, Pérez‐Sánchez, Hernández Del Rincón, & Gimeno, 2018), but MYBPC3 mutations can be associated to overlapping phenotype leading several forms of cardiomyopathies, as dilated cardiomyopathy (DCM) and left ventricular noncompaction (Sedaghat‐Hamedani et al., 2017; Zhao et al., 2015). Most MYBPC3 mutations are heterozygous and carriers often have a late disease onset with a benign disease progression (Carrier, Mearini, Stathopoulou, & Cuello, 2015). Konno and collaborators confirmed that elderly patients with the p.(Arg820Gln) mutation in the MYBPC3 gene may show “burnt‐out” phase HCM, with DCM phenotype appearance, characterized by LV systolic dysfunction with a diffuse LV hypokinesis, slow LV ejection fraction, and mild fibrosis, without myocardial hypertrophy and myofibrillar disarray at myocardial biopsy (Konno et al., 2003). This underlines the difficult differential diagnosis between “burnt‐out” dilated HCM and DCM, as in our case, with a mild hypertrophic form evolved into dilatation.
The occurrence of complete atrioventricular block could have many explanations; among them, we could identify an ischemic‐related block at infrahissian level, explained by diffuse coronary atherosclerosis. On the other hand, MYBPC3 could be implicated by unknown mechanism, as already suggested by a previous case, a 16‐year‐old girl affected by missense mutation, with syncope and ICD implantation, and subsequent evidence of complete heart block (Walsh et al., 2012); furthermore, Chida and collaborators suggests a predictive role of MYBPC3 mutation for sudden death in patient affected by hypertrophic cardiomyopathy, maybe related to abnormal heart conduction over ventricular arrhythmias (Chida et al., 2017).
Myopalladin mutations cause various forms of cardiomyopathy via different protein–protein interactions as disturbed myofibrillogenesis and nuclear shuttling and lead to abnormal assembly of terminal Z‐disk within the cardiac transitional junction and intercalated disk (Purevjav et al., 2012). The p.(Pro1112Leu) residue of MYPN is well conserved among the orthologous proteins, the replacement of the polar proline for a hydrophobic leucine may not be pathogenic on its own, (Duboscq‐Bidot et al., 2008), but the coexistence of other mutations may modulate the phenotype (Ingles et al., 2005; Tsoutsman, Bagnall, & Semsarian, 2008).
Myopalladin is part of the family members of palladin that includes also myotilin, expressed in skeletal and cardiac muscle.
The ability to connect with several molecules, in particular with α‐actinin, localized along stress fibers and at the Z line of cardiac muscles, suggests that palladin family has the potential to serve as a cytoskeleton scaffold and signaling mediator (Jin et al., 2010). Palladin may also play a role in the pathology of myocardial infarction; several lines of evidence suggest that palladin is involved in vascular smooth muscle cell phenotypic switching and migration, contributing to formation and repair of lesion in response to vascular injury or atherosclerosis disease (Shiffman et al., 2005). In our patient suffering from diffuse coronary artery disease without the common cardiovascular risk factors, we hypothesized that the myopalladin mutation could favor the atherosclerosis process due to its similarity with the palladin, a critical paralog implicated in vascular plaque progression (Modos et al., 2016; Waard, Achterberg, Beauchamp, Pannekoek, & Vries, 2003). Whether confirmed in future studies, we would much more take in consideration genetic profiling in cardiac ischemic disease, ensuing strict cardiovascular risk assessment in patient offspring.
6. CONCLUSION
Atrioventricular block can induce chronic heart failure, and electrocardiogram represents the gold standard for detection of rhythm disturbance. Furthermore, a complete evaluation of the patient, from anamnesis to cardiac resonance and coronary angiography, can lead to a better definition of clinical puzzle, while genetic profile allows the identification of genetic variants possibly implicated in the clinical picture. The missense mutations in MYBPC3 gene (c.2459G > A, p.(Arg820Gln)) could be responsible for dilated cardiomyopathy and AV block, while the mutations in MYPN (c.3335C > T, p.(Pro1112Leu)) could be for the first time described in human as associated to diffuse and aggressive coronary atherosclerosis, and may contribute to phenotypic variability.
Cardiomyopathies caused by double‐gene mutations are rare but conferred a remarkably increased risk of end‐stage progression, arrhythmias, and poor outcome. Compound genetic mutations leading to overlapping phenotype in the setting of cardiomyopathies represent an important challenge in clinical practice, and genetic tests are helpful in the clinical management of patients as they can provide important information on risk stratification, to achieve personalized therapy indicating differential surveillance strategies among individuals.
CONFLICT OF INTEREST
Authors do not have conflict of interest.
ACKNOWLEDGMENTS
We thank Italian Ministry of Health for current research grant 2017.
Mastroianno S, Palumbo P, Castellana S, et al. Double missense mutations in cardiac myosin‐binding protein C and myopalladin genes: A case report with diffuse coronary disease, complete atrioventricular block, and progression to dilated cardiomyopathy. Ann Noninvasive Electrocardiol. 2020;25:e12687 10.1111/anec.12687
REFERENCES
- Carrier, L. , Mearini, G. , Stathopoulou, K. , & Cuello, F. (2015). Cardiac myosin‐binding protein C (MYBPC3) in cardiac pathophysiology. Gene, 573(2), 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chida, A. , Inai, K. , Sato, H. , Shimada, E. , Nishizawa, T. , Shimada, M. , … Nakanishi, T. (2017). Prognostic predictive value of gene mutations in Japanese patients with hypertrophic cardiomyopathy. Heart and Vessels, 32(6), 700–707. [DOI] [PubMed] [Google Scholar]
- Duboscq‐Bidot, L. , Xu, P. , Charron, P. , Neyroud, N. , Dilanian, G. , Millaire, A. , … Villard, E. (2008). Mutations in the Z‐band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovascular Research, 77(1), 118–125. [DOI] [PubMed] [Google Scholar]
- Glusman, G. , Caballero, J. , Mauldin, D. E. , Hood, L. , & Roach, J. C. (2011). Kaviar: An accessible system for testing SNV novelty. Bioinformatics, 27(22), 3216–3217. 10.1093/bioinformatics/btr540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel, M. , Bonnin, S. , Lowy, E. , & Roma, G. (2011). TEQC: An R package for quality control in target capture experiments. Bioinformatics, 27(9), 1316–1317. 10.1093/bioinformatics/btr122 [DOI] [PubMed] [Google Scholar]
- Ingles, J. , Doolan, A. , Chiu, C. , Seidman, J. , Seidman, C. , & Semsarian, C. (2005). Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. Journal of Medical Genetics, 42(10), e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L. , Gan, Q. , Zieba, B. J. , Goicoechea, S. M. , Owens, G. K. , Otey, C. A. , & Somlyo, A. V. (2010). The actin associated protein palladin is important for the early smooth muscle cell differentiation. PLoS ONE, 5(9), e12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno, T. , Shimizu, M. , Ino, H. , Matsuyama, T. , Yamaguchi, M. , Terai, H. , … Mabuchi, H. (2003). A novel missense mutation in the myosin binding protein‐C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. Journal of the American College of Cardiology, 41(5), 781–786. [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2017). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acid Research, 2018(46), 1062–1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Eric, V. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Jian, X. , & Boerwinkle, E. (2011). dbNSFP: A lightweight database of human non‐synonymous SNPs and their functional predictions. Human Mutation, 32(8), 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, S. (2017). A reference panel of 64,976 haplotypes for genotype imputation. Nature Genetics, 48(10), 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, W. J. , Maron, B. J. , & Thiene, G. (2017). Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circulation Research, 121(7), 722–730. 10.1161/CIRCRESAHA.117.309711 [DOI] [PubMed] [Google Scholar]
- Modos, D. , Brooks, J. , Fazekas, D. , Ari, E. , Vellai, T. , Csermely, P. , … Lenti, K. (2016). Identification of critical paralog groups with indispensable roles in the regulation of signaling flow. Scientific Reports, 6(6), 38588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purevjav, E. , Arimura, T. , Augustin, S. , Huby, A.‐C. , Takagi, K. , Nunoda, S. , … Towbin, J. A. (2012). Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Human Molecular Genetics, 21(9), 2039–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater‐Molina, M. , Pérez‐Sánchez, I. , Hernández Del Rincón, J. P. , & Gimeno, J. R. (2018). Genetics of hypertrophic cardiomyopathy: A review of current state. Clinical Genetics, 93(1), 3–14. [DOI] [PubMed] [Google Scholar]
- Sedaghat‐Hamedani, F. , Haas, J. , Zhu, F. , Geier, C. , Kayvanpour, E. , Liss, M. , … Meder, B. (2017). Clinical genetics and outcome of left ventricular non‐compaction cardiomyopathy. European Heart Journal, 38(46), 3449–3460. [DOI] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acid Research, 29(1), 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman, D. , Ellis, S. G. , Rowland, C. M. , Malloy, M. J. , Luke, M. M. , Iakoubova, O. A. , … Kane, J. P. (2005). Identification of four gene variants associated with myocardial infarction. American Journal of Human Genetics, 77(4), 596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoutsman, T. , Bagnall, R. D. , & Semsarian, C. (2008). Impact of multiple gene mutations in determining the severity of cardiomyopathy and heart failure. Clinical and Experimental Pharmacology and Physiology, 35(11), 1349–1357. [DOI] [PubMed] [Google Scholar]
- de Waard, V. , van Achterberg, T. A. E. , Beauchamp, N. J. , Pannekoek, H. , & de Vries, C. J. M. (2003) Cardiac Ankyrin Repeat Protein (CARP) expression in human and murine atherosclerotic lesions. Arteriosclerosis, Thrombosis, and Vascular Biology, 23, 64. [DOI] [PubMed] [Google Scholar]
- Walsh, M. A. , Grenier, M. A. , Jefferies, J. L. , Towbin, J. A. , Lorts, A. , & Czosek, R. J. (2012). Conduction abnormalities in pediatric patients with restrictive cardiomyopathy. Circulation: Heart Failure, 5(2), 267–273. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR : Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acid Research, 38(16), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Feng, Y. , Zhang, Y.‐M. , Ding, X.‐X. , Song, Y.‐Z. , Zhang, A.‐M. , … Xia, X.‐S. (2015). Targeted next‐generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. International Journal of Molecular Medicine, 36(6), 1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]