

Abstract
Objective
To determine the inflammatory analytes that predict clinical progression and evaluate their performance against biomarkers of neurodegeneration.
Methods
A longitudinal study of MCI‐AD patients in a Discovery cohort over 15 months, with replication in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) MCI cohort over 36 months. Fifty‐three inflammatory analytes were measured in the CSF and plasma with a RBM multiplex analyte platform. Inflammatory analytes that predict clinical progression on Clinical Dementia Rating Scale‐Sum of Boxes (CDR‐SB) and Mini Mental State Exam scores were assessed in multivariate regression models. To provide context, key analyte results in ADNI were compared against biomarkers of neurodegeneration, hippocampal volume, and CSF neurofilament light (NfL), in receiver operating characteristic (ROC) analyses evaluating highest quartile of CDR‐SB change over two years (≥3 points).
Results
Cerebrospinal fluid inflammatory analytes in relation to cognitive decline were best described by gene ontology terms, natural killer cell chemotaxis, and endothelial cell apoptotic process and in plasma, extracellular matrix organization, blood coagulation, and fibrin clot formation described the analytes. CSF CCL2 was most robust in predicting rate of cognitive change and analytes that correlated to CCL2 suggest IL‐10 pathway dysregulation. The ROC curves for ≥3 points change in CDR‐SB over 2 years when comparing baseline hippocampal volume, CSF NfL, and CCL2 were not significantly different.
Interpretation
Baseline levels of immune cell chemotactic cytokine CCL2 in the CSF and IL‐10 pathway dysregulation impact longitudinal cognitive and functional decline in MCI‐AD. CCL2’s utility appears comparable to biomarkers of neurodegeneration in predicting rapid decline.
Introduction
Alzheimer’s disease (AD), which often presents early in its course with episodic memory loss, is the most common cause of dementia. It is increasingly recognized that there is considerable heterogeneity in AD phenotype and clinical trajectories. 1 , 2 Molecular factors that underpin this heterogeneity, however, remain ill‐defined. Increasing evidence suggests that inflammatory pathways may regulate AD progression. 3 , 4 , 5 , 6 There has been a significant increase in interest for evaluating inflammatory changes in clinical AD with several studies reporting inflammation related changes in AD clinical cohorts over the last 5 years. 7 , 8 , 9 , 10
There are also some key gaps in clarifying the role of inflammation across multiple clinical studies. These gaps include determining which specific peripheral and central immunological analytes and related pathways impact rate of cognitive decline in human AD. There have been challenges in interpreting these results against longitudinal rate of disease progression. Often, the directionality and magnitude of these associations on clinical outcomes also often differ, likely due to the use of a small number of measured inflammatory analytes, varied methodologies, and different stages of disease. 6 , 11 , 12 , 13 , 14 , 15 , 16 In addition, it is unclear how any of these analytes compare with other widely used biomarkers of neurodegeneration, MRI hippocampal volume, and the novel biomarker neurofilament light (NfL) on disease progression.
To help identify inflammatory pathways or networks of inflammatory analytes pertinent to cognitive decline in AD, it is crucial to develop approaches that evaluate multiple analytes concomitantly and interrogate their clinical significance when expressed together. We therefore have taken a systematic approach to answer these gaps in mild cognitive impairment (MCI) with AD consistent CSF biomarkers, (MCI‐AD) in whom the nature of inflammatory changes were characterized by the same multiplex panel of inflammatory analytes in the both the CSF and plasma. After evaluation in our Discovery cohort, we validated the results among MCI patients in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. Using bioinformatics and classical statistical tools, we determined (1) the key inflammatory analytes at baseline that best predict future cognitive decline, (2) the biological pathways most likely disrupted in relation to the above analytes and (3) how they compare to neurodegeneration biomarkers, MRI hippocampal volume, and cerebrospinal fluid (CSF) NfL. We tested the hypotheses that (1) a proinflammatory analyte profile at baseline would relate to a faster rate of longitudinal clinical progression in the MCI‐AD and (2) key inflammatory analytes have at least comparable utility to neurodegeneration biomarkers in predicting future cognitive decline.
Materials and Methods
Discovery cohort
Forty‐eight MCI‐AD patients at baseline in whom the diagnosis of MCI‐AD with CSF Aβ 42 and p‐tau181 levels consistent with a diagnosis of AD and consensus evaluation of two neurologists and a neuropsychologist the details of which have been published previously. 17 , 18 The study was approved by the Cleveland Clinic Institutional Review Board. Eight patients did not complete the longitudinal evaluations due to nonmedical reasons by their personal choice. The ADmark® Alzheimer's evaluation uses sandwich Enzyme Linked Immunosorbant Assay(ELISA) kits [Innotest β‐amyloid[1‐42], Innotest hTAU‐Ag, Innotest Phospho‐Tau[181P], Innogenetics, Ghent, Belgium]. APOE status was determined by blood samples(10 ng per subject) dispensed into 96‐well plates for TaqMan allelic discrimination detection of single nucleotide polymorphisms that discriminate the APOE alleles (rs429358, rs7412) (Life Technologies). Table 1 provides data on the Discovery cohort demographics. Inclusion, exclusion criteria are specified in Data S1. Additional clinical and environmental factors have been documented in Table S1 (Fig. 1).
Table 1.
Demographics of Discovery and ADNI cohorts.
| Discovery (N = 48) | ADNI ( N = 134) | P‐value | |||
|---|---|---|---|---|---|
| N | Statistics | N | Statistics | ||
| Age at enrollment | 48 | 68.1 ± 7.3 | 134 | 74.9 ± 7.2 | <0.001a1 |
| Gender | 48 | 134 | 0.23c | ||
| Male | 28 (58.3) | 91 (67.9) | |||
| Female | 20 (41.7) | 43 (32.1) | |||
| Years of education | 48 | 16.0 [12.5, 18.0] | 134 | 16.0 [14.0, 18.0] | 0.27b |
| APOE ε4positive | 48 | 37 (77.1) | 134 | 72 (53.7) | 0.005c |
| MMSE ‐ baseline | 48 | 24.8 ± 3.1 | 134 | 26.9 ± 1.8 | <0.001a2 |
| CDR‐SB ‐ baseline | 48 | 2.1 ± 1.2 | 134 | 1.5 ± 0.89 | 0.002a2 |
| CSF AB42, pg/mL | 48 | 305.9 [216.1, 367.1] | 134 | 144.5 [129.0, 171.0] | |
| CSF t‐tau, pg/mL | 48 | 454.3 [335.2, 771.3] | 134 | 90.6 [67.8, 134.0] | |
| CSF p‐tau, pg/mL | 48 | 79.6 [59.3, 104.6] | 134 | 35.7 [23.0, 45.8] | |
Statistics presented as Mean ± SD, Median [P25, P75], N (column%).
P‐values: a1 = t‐test, a2 = Satterthwaite t‐test, b = Wilcoxon Rank Sum test, c = Pearson's chi‐square test, d = Fisher's Exact test.
P‐value < 0.05 is noted in bold.
CDR‐SB, Clinical dementia rating scale‐sum of boxes; MMSE, Mini mental state exam.
Figure 1.
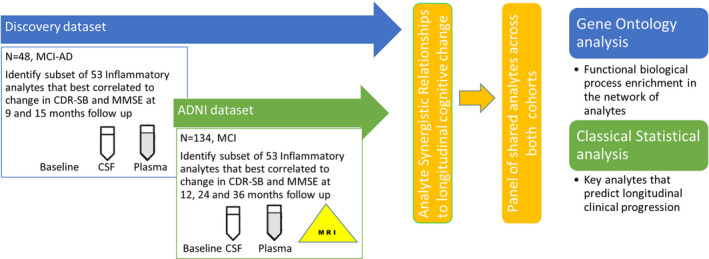
Methodological overview.
Cognitive and functional measures
Mini–Mental State Examination (MMSE), 19 and Clinical Dementia Rating scale (CDR‐SB) 20 were conducted to characterize the degree of their baseline cognitive and functional deficit. Mini–Mental State Examination and CDR‐SB scores were also evaluated longitudinally to evaluate cognitive change at 9 and 15 months from baseline evaluations.
Inflammatory biomarkers
The biomarker analysis protocol used in this study has been previously described (18). In brief, CSF and plasma were collected and analyzed by an independent laboratory via the validated RBM Multi‐Analyte Profile (MAP) platform from Myriad Genetics (Salt Lake City, UT). Samples were evaluated for levels of 86 analytes using a custom MAP:HumanMAP®v2.0 + IL1 and 16 using a Luminex platform. Validation has been performed as defined by the Clinical and Laboratory Standards Institute and is therefore replicable across multiple runs. Cerebrospinal fluid and plasma samples were collected contemporaneously. Only those analytes with at least 50% response rate above the limit of detection in the Discovery cohort were included for further analysis.
ADNI validation cohort
Alzheimer’s Disease Neuroimaging Initiative is a longitudinal multicenter study designed to develop clinical, imaging, genetic, and biochemical biomarkers for the early detection and tracking of AD. Alzheimer’s Disease Neuroimaging Initiative was launched by the National Institute of Aging and is a multicenter project with additional support from private pharmaceutical companies, and nonprofit organizations. ADNI 1 eligibility criteria are described in the ADNI 1 protocol http://adni.loni.usc.edu/methods/documents/.
The demographics at baseline among the subset of all 134 ADNI MCI participants who had CSF and plasma multiplex data were used in the validation analysis are shown in Table 1. Longitudinal cognitive evaluations in the ADNI cohort documented at 12, 24, and 36 months were included in the analysis. AD CSF biomarker data were downloaded from http://adni.loni.usc.edu/ in which the CSF Aβ1‐42, t‐tau, and p‐tau181 concentration data were generated using the Research Use Only (RUO) INNOBIA AlzBio3 immunoassay (Fujirebio, Belgium). Median values for each subject were used in the analysis. Amyloid‐positivity based on a published, autopsy‐confirmed cutoff value (<192 pg/mL) were used in a subgroup analysis to define MCI‐AD 21 , 22 of which there were 106 participants (see Data S1). Cerebrospinal fluid samples were measured for levels of 159 analytes using the RBM DiscoveryMAP® v.1.0 panel. The RBM HumanMAP® v.2.0 used in the Discovery cohort is a subset of the RBM DiscoveryMAP® v.1.0 and uses a Luminex platform with the same quality control and thresholding process used in ADNI dataset and are comparable. CSF NFL was measured using a sandwich ELISA method and provided as pg/ml (NF‐light ELISA kit,UmanDiagnostics AB, Umeå, Sweden), as described previously. The lower limit of quantification for this assay was 50 ng/L. 23 Subject data quality was checked.
Bioinformatics and statistical analysis
Ontology analysis and network analysis to identify inflammatory analytes
Our final comprehensive list of 53 candidate inflammatory analytes (from 86 analytes in RBM MAP platform in the Discovery cohort) is provided in Table 2 and has been previously described 18 and rationale describing selection is in Data S1.
Table 2.
List of inflammatory analytes analyzed in relation to longitudinal cognitive change.
| RBM Name | Gene | RBM Name | Gene |
|---|---|---|---|
| 1. Alpha‐1‐Antitrypsin (AAT) | SERPINA1 | 27. Interleukin‐12 Subunit p40 | IL12B |
| 2. Alpha‐2‐Macroglobulin | A2M | 28. Interleukin‐12 Subunit p70 | IL12P70 |
| 3. Apolipoprotein A‐I | APOA1 | 29. Interleukin‐15 | IL15 |
| 4. Beta‐2‐Microglobulin | B2M | 30. Interleukin‐17 | IL17A |
| 5. Brain‐Derived Neurotrophic Factor | BDNF | 31. Interleukin‐18 | IL18 |
| 6. Complement C3 | C3 | 32. Interleukin‐8 | CXCL8 |
| 7. C‐Reactive Protein | CRP | 33. Interleukin‐23 | IL23A |
| 8. Eotaxin‐1 | CCL11 | 34. Macrophage Inflammatory Protein‐1 alpha | CCL3 |
| 9. Fibrinogen | FGA | 35. Macrophage Inflammatory Protein‐1 beta | CCL4 |
| 10. Factor VII | F7 | 36. Matrix Metalloproteinase‐3 | MMP3 |
| 11. Ferritin | FTH1 | 37. Matrix Metalloproteinase‐9 | MMP9 |
| 12. Granulocyte‐Macrophage Colony‐Stimulating Factor | CSF2 | 38. Monocyte Chemotactic Protein 1 | CCL2 |
| 13. Granulocyte Colony‐Stimulating Factor | CSF3 | 39. Matrix Metalloproteinase‐2 | MMP2 |
| 14. Haptoglobin | HP | 40. Myeloperoxidase | MPO |
| 15. Intercellular Adhesion Molecule 1 | ICAM1 | 41. Neuron‐Specific Enolase (NSE) | ENO2 |
| 16. Interferon gamma | IFNG | 42. Plasminogen Activator Inhibitor 1 (PAI‐1) | SERPINE1 |
| 17. Interleukin‐1 alpha | IL1A | 43. Serotransferrin | TF |
| 18. Interleukin‐1 beta | IL1B | 44. Stem Cell Factor | SCF |
| 19. Interleukin‐1 receptor antagonist | IL1RN | 45. T‐Cell‐Specific Protein RANTES | CCL5 |
| 20. Interleukin‐2 | IL2 | 46. Tissue Inhibitor of Metalloproteinases 1 | TIMP1 |
| 21. Interleukin‐3 | IL3 | 47. Tumor Necrosis Factor alpha | TNF |
| 22. Interleukin‐4 | IL4 | 48. Tumor Necrosis Factor beta | LTA |
| 23. Interleukin‐5 | IL5 | 49. Tumor necrosis Factor Receptor 2 | TNFRSF1B |
| 24. Interleukin‐6 | IL6 | 50. Vascular Cell Adhesion Molecule‐1 | VCAM1 |
| 25. Interleukin‐7 | IL7 | 51. Vascular Endothelial Growth Factor | VEGFA |
| 26. Interleukin‐10 | IL10 | 52. Vitamin D‐Binding Protein | GC |
| 53. von Willebrand Factor | VWF |
Subgroup searching for analyte synergistic relationships
Univariate analyses often fail in validation as they are trained on the specific patient dataset of the discovery project, and average the heterogeneities present, while the validation cohort may have differences in the levels of individual analytes from multiple environmental factors. Network biology methods, specifically network based biomarker models, can effectively integrate heterogeneities at the patient level and provide robust validation across cohorts. In order to evaluate analyte levels that show higher correlation when considered together (synergistic relationship) rather than individual analyte correlation by univariate analysis alone, we performed an exhaustive search to find analyte subgroups whose aggregate levels maximally correlated with cognitive change measures. 18 , 24 , 25 Hypothesis 1 (H1) tested how likely we were to see greater or equal correlation with random analyte subgroups by sampling 10,000 random analyte subgroups from among all analytes that met the 50% detection threshold and computing the correlation values. Hypothesis 2 (H2) tested how likely we were to randomly observe greater or equal correlation between the aggregate activity of an analyte subgroup and a response marker by permuting the values of each response marker 10,000 times and computing the correlation values to the aggregate analyte levels. P‐values were estimated as the proportion of randomized responses with equal or greater correlation to aggregate analyte levels than the actual response.
Functional pathway analysis on analytes of interest
The analytes of significance identified as being shared across both the Discovery and ADNI cohorts in at least one time point in the analyte subgroup search above were entered into STRING: functional protein association networks for pathway enrichment analysis. 26 The most significant GO terms related to biological process identified in STRING (P < 0.05) were summarized into nonredundant hierarchical terms by their semantic similarity via ReviGO, 27 using SimRel 28 as the clustering algorithm with a similarity measure of 0.70, with the whole Uniprot database providing GO term sizes and reported in appropriate plots. The Reactome pathway database was used in enrichment analysis to identify targeted analyte pathways of interest in any secondary analysis. 29
Structural MRI acquisition and processing
ADNI 1.5‐T MRI scans from MCI subjects performed at baseline were processed using cross‐sectional FreeSurfer (version 5.1.0, default parameters). 30 The processing included conformation to isotropic cubic mm resolution, bias field correction, segmentation of the hippocampi from which the bilateral hippocampal volume was computed, and estimation of total intracranial volume. 31 The baseline hippocampal volume was corrected for head size by division by the intra cranial volume.
Clustering analysis
To further provide biological context to the key shared analyte robustly identified across both ADNI and Discovery cohorts it was subjected to consensus clustering, an unsupervised class discovery approach to identify co‐occurring analytes. 32 Using the quantitative values of the analytes, the number and possible memberships to clusters were found for each cluster. Cluster size for each group was fixed where relative increase in consensus was observed to have no appreciable increase. The analyte groups were tested for enrichment in pathways using ClusterProfiler tool using the Reactome Pathway Database 33 and the top five enriched Reactome pathways were interpreted.
Statistical analysis
Given the limited number of conversion events (dementia onset or highest quartile of CDR‐SB change) during intermediate follow‐ups (9 and 12 months) in both the Discovery and ADNI MCI cohorts, neither Cox proportional hazards model nor a time‐dependent receiver operating characteristic (ROC) were found to be adequate for the data. With the Discovery sample size, and conservatively choosing a Bonferroni corrected significance level of 0.005 to allow for up to 10 key analytes to be compared, there would be 80% power to detect correlations between analytes and changes in cognitive measures that exceed 0.5. Key inflammatory analytes shared between both cohorts, curated from the synergistic subgroup analysis described previously and used for functional pathway analysis, were next evaluated in predicting cognitive change after adjusting for age, sex, baseline MMSE, and APOEε4 status (present vs. absent). With five adjustment factors, the Discovery cohort models included no more than three biomarkers concurrently to maintain a 5 to 1 ratio of observations to variables as noted in prior statistical literature. 34 Two statistical approaches for variable selection were used. First, a best subset regression analysis was used to identify two to three biomarkers that best predicted outcome. In this approach, the third biomarker solution was used only if including the third biomarker increased the R2 by at least 1%. As an alternative, penalized regression using LASSO regression was performed, using Akaike information criterion (AIC) as the stopping rule. In general, as the LASSO approach was very conservative the best subset results are presented.
A log (base 2) transformation allowed Pearson correlations to be fit for exploratory univariate analysis. Along with estimates of correlation, 95% confidence intervals and p‐values with false discovery rate (FDR) adjustment were calculated. Normality of biomarkers was evaluated using Shapiro–Wilk tests and graphical methods. Sensitivity analyses were also performed to evaluate the robustness of the key analytes of significance. All analyses were performed at each time point using the patients with available cognitive change measures at that point. Separate analyses were performed at each time point because we hypothesized that the biomarkers related to cognitive change may change over time and the sample size did not allow for modeling of interactions to capture these complex relationships. We chose not to impute change measures for missing responses as doing so would not improve the information available. 35
Classical ROC analyses were performed for subjects only in the ADNI cohort, to explore if key CSF inflammatory analytes were comparable to hippocampal volume and CSF NfL in their clinical utility as all three data were available. Analyses were performed for a base model with above three variables alone and for an adjusted model that included age, sex, education years, APOEε4 status, and CSF Aβ/ptau ratio. The area under the ROC curve (AUC) was maximized in these analyses. In this setting, the AUC measures the intrinsic ability of the analytes to discriminate between subjects who progress ≥ 3 CDR‐SB points, the highest quartile CDR‐SB change in the ADNI cohorts for two year follow‐up. Analysis was performed using SAS software (version 9.4), R software (version 3.x; Vienna, Austria), and SPSS (Version 22.0. Armonk, NY: IBM Corp) an overall significance level of 0.05 was assumed.
Results
Subject demographics of the Discovery and ADNI cohorts are presented in Table 1.
Analyte subgroup analysis for synergistic relationships in discovery and ADNI cohorts
Analyte levels that show higher correlation when considered together rather than the individual component analytes (synergistic analyte analysis) are reported in Tables S3 and S4 and Figures 2A and 3A. The Discovery cohort had a similar number of analytes compared with the ADNI cohort that relate to cognitive changes in the CSF and in plasma. The analytes noted in relation to CDR‐SB change in a consistent direction among both cohorts in the CSF included CCL2, CCL4, and FGA, while in plasma the shared analytes in a consistent direction included APOA1, BDNF, and vWF. Among both cohorts, the CSF analytes noted in relation to MMSE change in a consistent direction across both cohorts included AAT, MMP3, and CRP, while in plasma MMP2, CXCL8, A2M, and Factor VII were best correlated to MMSE change consistently across both cohorts (Figs. 2A and 3A).
Figure 2.
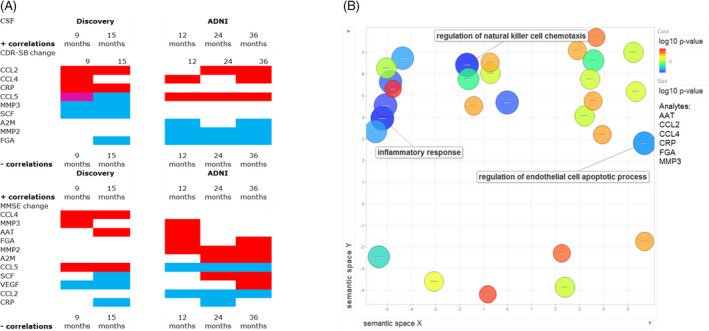
(A) Heat map denoting significant analytes from ADNI and Discovery datasets in the CSF from the subgroup synergistic analysis correlated to cognitive change (CDR‐SB or MMSE) for 9,12,15,24, and 36 months. Red: positive correlation, Blue: negative correlation, Magenta: has representation in both positive and negative correlation network of analytes. Given that higher CDR‐SB is worse cognition and function while lower MMSE is worse cognition, a positive correlation in CDR‐SB relates to worsening cognition while the negative MMSE correlations relates to worsening cognition. (B) Abundance of Gene Ontology (GO) terms related to biological processes that enrich for key shared analytes between ADNI and Discovery datasets in the CSF in relation to cognitive change (CDR‐SB or MMSE). Analytes included: AAT, CCl2, CCL4, CRP, FGA, and MMP3. GO term most representative of each cluster is noted.
Figure 3.
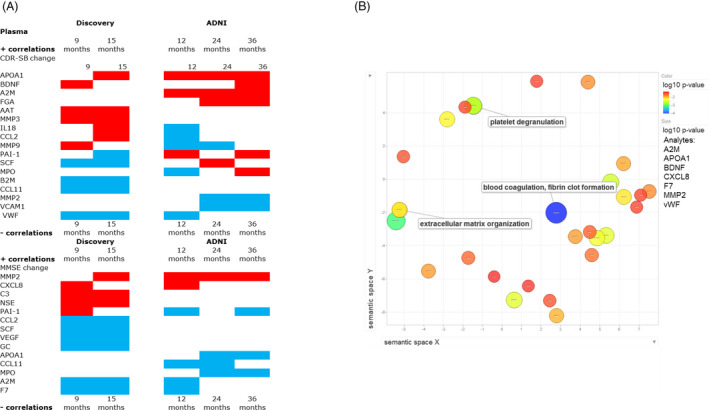
(A) Heat map denoting significant analytes from ADNI and Discovery datasets in the plasma from the subgroup synergistic analysis correlated to cognitive change (CDR‐SB or MMSE) for 9,12,15,24, and 36 months. Red: positive correlation, Blue: negative correlation, Magenta: has representation in both positive and negative correlation network of analytes. 3B: Abundance of Gene Ontology (GO) terms related to biological processes that enrich for key shared analytes between ADNI and Discovery datasets in the plasma in relation to cognitive change. Analytes chosen for their consistency of response included: A2M, APOA1, BDNF, CXCL8, F7, MMP2, and vWF. GO term most representative of each cluster is noted.
Functional pathway analysis
The analytes that significantly correlated to at least one time point of longitudinal follow‐up in both Discovery and ADNI cohorts in a consistent direction within the subgroup synergistic analysis were ranked as the most robust for functional pathway analysis. These analytes (for CSF: AAT, CCl2, CCL4, CRP, FGA, and MMP3 and for plasma: A2M, APOA1, BDNF, CXCL8, F7 MMP2, and vWF) were entered into STRING for functional pathway enrichment analysis for CSF and plasma in different runs. In the CSF, the largest clusters were most representative of natural killer cell chemotaxis, and regulation of endothelial cell apoptotic process. While in the plasma, clusters were representative of extracellular matrix organization, blood coagulation and fibrin clot formation, and platelet degranulation (Figs. 2B and 3B).
Statistical analysis
Univariable analysis in CSF and plasma
In a complementary analysis, univariable Pearson correlations after FDR correction in the Discovery cohort, only CSF levels of CCL2 positively correlated to change in CDR‐SB scores at 15 months, and after adjusting for covariates (age, sex, baseline cognitive score, APOE ε4 status), the association of CCL2 was still significant (Table S4A).
Within the ADNI cohort, there were no significant CSF analytes on univariable analysis that related to change in CDR‐SB or MMSE scores after adjusting for covariates. In the plasma, only levels of MMP2 was negatively correlated to change in MMSE scores at 24 months, and the association was still significant after adjusting for covariates (age, sex, baseline cognitive score, APOE ε4 status) while applying the FDR correction (Table S5).
Multivariable models CSF and plasma
Across both cohorts, CSF CCL2 was the most robustly selected parameter being chosen in 9 out of 10 models, and was significant at the 0.05 level in five models. MMP3 was chosen in four models and met significance threshold in three models. CSF CCL4 selected in four models, was significant in only two models. In the Discovery cohort, the multivariable models explained 27–42% of the variability in cognitive change, while in ADNI, only 13–19% of the variability in cognitive change was explained with these models (Table 3). Additional sensitivity analysis to evaluate the role of extreme measures using a rank based analysis noted CSF CCL2 as significant in all prior models, MMP3 was no longer significantly related to CDR‐SB change at 15 months.
Table 3.
CSF biomarkers of CDR‐SB and MMSE change are shown for both cohorts at specified time points, after adjustment for age, sex, APOE status and education.
| Time | CDR‐SB change | MMSE change | ||||
|---|---|---|---|---|---|---|
| Factor | Estimate (95% CI) | P‐value | Factor | Estimate (95% CI) | P‐value | |
| Discovery cohort | ||||||
| 9 months | CCL2 | 1.23 (0.24, 2.22) | 0.016 | AAT | 1.50 (−0.85, 3.85) | 0.20 |
| MMP3 | −0.97 (−1.64, −0.29) | 0.006 | CCL2 | −1.54 (−4.09, 1.02) | 0.23 | |
| MMP3 | 2.05 (0.32, 3.78) | 0.022 | ||||
| 15 months | FGA | −0.27 (−0.73, 0.20) | 0.25 | AAT | 1.53 (−1.24, 4.30) | 0.27 |
| CCL2 | 2.82 (1.30, 4.34) | <0.001 | CCL2 | −2.36 (−5.47, 0.76) | 0.13 | |
| MMP3 | −0.94 (−1.82, −0.05) | 0.039 | CCL4 | 1.48 (−0.15, 3.12) | 0.073 | |
| ADNI Cohort | ||||||
| 12 months | AAT | 0.37 (−0.10, 0.85) | 0.12 | CCL2 | −1.45 (−2.54, −0.36) | 0.009 |
| CCL4 | 0.30 (−0.03, 0.63) | 0.071 | FGA | 0.49 (0.13, 0.86) | 0.008 | |
| FGA | −0.23 (−0.40, −0.05) | 0.011 | ||||
| 24 months | CCL2 | 0.86 (−0.04, 1.75) | 0.062 | CCL2 | −1.46 (−3.19, 0.26) | 0.096 |
| CCL5 | 0.41 (0.02, 0.79) | 0.041 | CCL5 | −0.43 (−1.18, 0.31) | 0.25 | |
| CRP | −0.26 (−0.65, 0.13) | 0.20 | ||||
| 36 Months | CCL2 | 1.43 (0.13, 2.73) | 0.031 | CCL2 | −2.87 (−5.16, −0.58) | 0.015 |
| CCL4 | 0.76 (0.01, 1.52) | 0.047 | CCL4 | −1.36 (−2.71, −0.00) | 0.050 | |
| MMP3 | −0.53 (−1.25, 0.19) | 0.15 | CCL5 | 0.74 (−0.50, 1.98) | 0.24 | |
P‐value < 0.05 is noted in bold
In the plasma, a clear pattern replicating the findings between Discovery and ADNI data does not appear to be present in the multivariable models (Table 4). Across both cohorts, there were no analytes with correlations in opposing directions to the same cognitive change scores when correlations exceeded +/‐ 0.2 in the univariable models in CSF and plasma. In the multivariable models with MMSE, plasma BDNF and MMP2 differ in correlation direction between cohorts. Plasma MMP2 changes correlation direction within the ADNI cohort over time, indicating the instability of this effect when cognitive change is characterized by MMSE.
Table 4.
Plasma biomarkers of CDR‐SB and MMSE change are shown for both cohorts at specified time points, after adjustment for age, sex, APOE status and education.
| Time | CDR‐SB Change | MMSE Change | ||||
|---|---|---|---|---|---|---|
| Factor | Estimate (95% CI) | P‐value | Factor | Estimate (95% CI) | P‐value | |
| Discovery cohort | ||||||
| 9 months | CXCL8 | 0.37 (−0.39, 1.12) | 0.33 | A2M | −4.24 (−7.67, −0.81) | 0.017 |
| F7 | −1.19 (−2.04, −0.34) | 0.007 | F7 | −1.97 (−3.81, −0.13) | 0.037 | |
| MMP2 | −0.70 (−1.80, 0.41) | 0.21 | BDNF | 0.28 (−0.23, 0.79) | 0.27 | |
| 15 months | CXCL8 | 1.75 (0.66, 2.84) | 0.003 | A2M | −4.37 (−8.62, −0.12) | 0.044 |
| APOA1 | −1.41 (−3.75, 0.93) | 0.23 | F7 | −1.83 (−4.26, 0.60) | 0.13 | |
| F7 | −0.85 (−2.14, 0.44) | 0.19 | MMP2 | −1.28(−4.47, 1.92) | 0.42 | |
| ADNI Cohort | ||||||
| 12 months | A2M | 0.66 (0.06, 1.27) | 0.032 | CXCL8 | 1.03 (0.34, 1.71) | 0.004 |
| APOA1 | 0.28 (−0.13, 0.69) | 0.18 | APOA1 | −0.61 (−1.50, 0.28) | 0.18 | |
| BDNF | −0.24 (−0.63, 0.15) | 0.23 | ||||
| 24 months | APOA1 | 0.57 (−0.08, 1.23) | 0.086 | APOA1 | −1.16 (−2.35, 0.03) | 0.057 |
| BDNF | 0.23 (−0.05, 0.51) | 0.10 | MMP2 | 1.48 (0.88, 2.08) | < 0.001 | |
| MMP2 | −0.56 (−0.89, −0.22) | 0.001 | ||||
| 36 months | A2M | 1.73 (−0.28, 3.74) | 0.090 | A2M | −1.29 (−3.91, 1.33) | 0.33 |
| MMP2 | 1.12 (−0.19, 2.42) | 0.093 | APOA1 | −1.26 (−3.13, 0.61) | 0.18 | |
P‐value < 0.05 is noted in bold.
Inflammatory diseases and NSAID intake
No difference was noted in the analytes of significance when adjusted for current NSAID intake or when considering inflammatory diseases in clinical history within the Discovery cohort. Adjusting for individual CSF/plasma albumin ratio again noted no impact on key analytes of significance (analyses not presented).
Comparing inflammatory analytes in ADNI versus the Discovery cohort
The inflammatory analyte correlations to cognitive change within the Discovery cohort were larger than in the ADNI dataset (Tables S4 and S5). The analytes that were identified as correlating to at least one longitudinal cognitive measure across both datasets could be further validated after accounting for multiple covariates in the multivariable models (Tables 3 and 4). CSF CCL2 was identified as a key analyte shared between cohorts in relation to CDR‐SB change. In a subgroup analyses of ADNI MCI participants with CSF Aβ42 < 192 pg/mL (and additionally CSF t‐tau/Aβ42> 0.4), the direction of correlation between CCL2 and CDR‐SB change on univariable analysis was consistent between 24 and 36 months and met significance threshold at 36 months (Table S6). From the multivariable analysis, a doubling of baseline CSF CCL2 levels in the Discovery cohort predicted a 2.8 point estimated increase in CDR‐SB change at 15 months. A similar doubling in the ADNI MCI cohort predicted a 0.9 point increase of CDR‐SB over 24 months and 1.4 point over 36 months, after accounting for age, sex, education, APOE ε4 status, and baseline cognitive scores (Table 3, Fig. 4).
Figure 4.
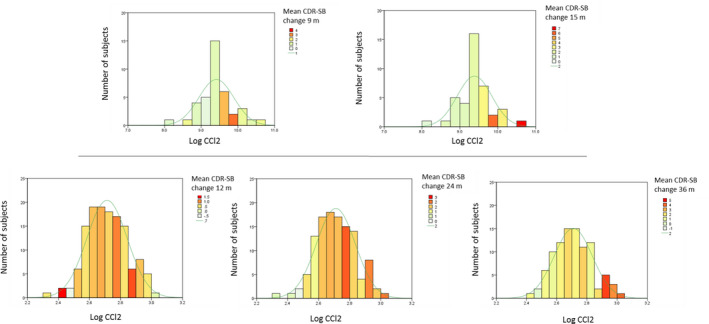
Histogram with normal distribution score of baseline CSF CCL2 levels, in relation to longitudinal CDR‐SB change (in heat colors) and average CDR‐SB change of cohort at each time point (as a line in legend). Data from Discovery cohort (9 and 15 months), ADNI (12, 24 and 36 months).
To provide a biological context to the role for CCL2, we next explored the clustering of CCL2 to other inflammatory analytes. We note that in the Discovery cohort CCL2 closely clusters with B2M, CXCL8, FGA, MMP2, TIMP1, and VCAM1 in the CSF (Fig. S1) and plasma CCL2 with BDNF, CCL4, CCL11, IL‐18, PAI‐1, and VEGFA (Fig. S2). These analytes in the CSF and plasma taken together are enriched in interleukin‐10 (IL‐10), interleukin‐4 (IL‐4), and interleukin‐13 (IL‐13) signaling pathways within the Reactome database (Tables S7 and S8). In the ADNI cohort, CSF CCL2 did not significantly cluster with other CSF analytes, while in the plasma, CCL2 and correlated inflammatory plasma analytes were enriched for the IL‐10 signaling pathway (Fig. S3, Tables S9 ). Summary statistics of these analytes are provided in Data S1.
CSF versus plasma inflammatory analytes in predicting future cognitive decline
Receiver operating characteristics based on the logistic regression models determined the utility of CCL2 compared with baseline CSF NfL and hippocampal volume to predict the highest quartile of CDR‐SB change over two years in the ADNI cohort. The area under the curve (AUC) of the adjusted model (age, sex, years of education, APOE ɛ4 genotype, and CSF Aβ 1–42/p‐tau) when it included CSF CCL2 was 0.66, CSF NfL was 0.63, and hippocampal volume was 0.69 (Fig. 5). The resulting AUCs and overlapping 95% confidence intervals were statistically not different for these biomarkers (Table S10). A nonsignificant correlation between CSF CCL2 and baseline hippocampal volume, r = −0.15, P = 0.12 was noted, while the correlation between CSF CCL2 and CSF NfL was r = 0.19, P = 0.042.
Figure 5.
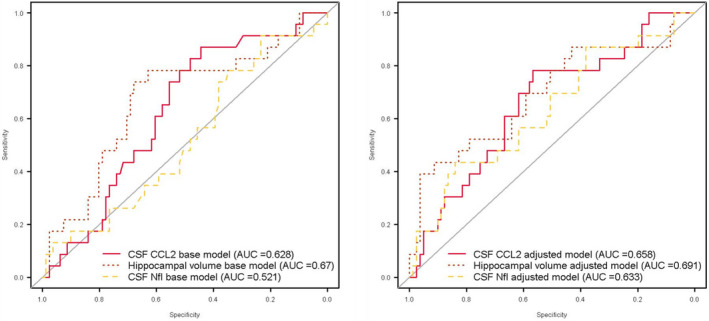
Receiver operating characteristic analysis curves denoting CSF CCL2, CSF NfL, and hippocampal volume for rapid cognitive decline (≥ 3 CDR SB change over two years, highest quartile among subjects) from ADNI cohort. Base models of the three biomarkers alone and adjusted models accounting for age, sex, years of education, APOE ɛ4 genotype, and CSF Aβ1–42/p‐tau.
Discussion
This study undertook an unbiased approach to evaluate the role for inflammatory analytes on clinical progression using a multi‐analyte panel with well characterized MCI patient cohorts and positive AD CSF biomarkers. Our results across two cohorts of baseline CSF CCL2 predicting rapid clinical decline lends credence to prior reports of CCL2 impacting clinical progression in AD. 13 , 36 In addition as a novel finding, by carefully characterizing chemokines that cluster closely with CCL2 in the CSF and plasma using bioinformatics tools, we are able to posit the relevance of IL‐10 inflammatory pathway dysregulation as a correlate of clinical progression in MCI. This result is consistent with our initial hypothesis that a proinflammatory analyte profile at baseline would relate to a faster rate of longitudinal clinical progression in the MCI stage. Furthermore providing clinical context to these results, we note that baseline measurements of the cytokine CSF CCL2 has comparable discriminatory power to neurodegeneration markers, CSF NfL and MRI hippocampal volume, in predicting the highest quartile of CDR‐SB change over two years (≥3 CDR‐SB) in MCI‐AD, but has limited correlation with either. Of note, 3 point CDR‐SB change is over twice the change among neuropathology confirmed AD patients in the National Alzheimer’s Coordinating Center (NACC) database (Avg 0.9, SD 0.7, per year), when they met criteria for MCI at initial visit. 2
CCL2 (also known as monocyte chemoattractant protein‐1, MCP‐1) is among the key cytokines that recruit monocytes to a site of inflammation. Infiltration of these blood derived immune cells toward Aβ plaque have been studied. 37 , 38 An increase in CSF CCL2 levels has been linked to the transition from MCI to AD and a faster rate of cognitive decline. 13 , 36 Plasma CCL2 levels have been observed to increase with the increasing severity of AD dementia and associated with a 2‐year rate of cognitive decline in AD and MCI. 36 , 39 Among asymptomatic normal aging individuals, longitudinal increases in plasma CCL2 levels were associated with decline in memory 40 and associated with increased long‐term risk of stroke in a meta‐analysis of population studies. 41
IL‐10 Reactome pathway was enriched among the CSF and plasma inflammatory analytes that CCL2 clustered with in both ADNI and the Discovery cohorts, while IL‐13 and IL‐4 pathways were enriched only in the Discovery cohort. IL‐10, IL‐13, and IL‐4 pathways are all associated with anti‐inflammatory changes noted in relation to inhibition of autoimmunity and infections. 42 , 43 The downstream proinflammatory factors in the IL‐10 Reactome pathway often suppressed by IL‐10 were elevated. Given the Lower Limit of Quantification (LLOQ) of the CSF IL‐10 assay only 3 of 48 patients in the Discovery cohort had measurable IL‐10 levels, limiting the assay’s utility in estimating IL‐10’s direct relevance in this context. In contrast to CCL2, the levels of MMPs were less consistently correlated to cognitive change in both cohorts.
CCL2 compared to neurodegeneration markers
To evaluate the utility of CSF CCL2 in predicting rapid cognitive decline, we compared its effectiveness against baseline CSF NfL and MRI hippocampal volume as they both provide complementary information. Imaging measures like hippocampal volume represent the magnitude of the neuropathologic damage accumulated over time, unlike CSF markers like NfL that reflect its production/clearance at one time point. 44 CSF NfL has been noted to correlate with rapid cognitive decline in MCI stage of AD and is thought to be related to degeneration of large‐caliber axons. 45 MRI measure of hippocampal volume is accepted as an indicator of neurodegeneration in the A/T/N classification. 44 In the ROC analysis, CSF CCL2 had nonsignificant differences from CSF NfL and hippocampal volumes in predicting rapid cognitive decline in the adjusted models that included covariates of clinical importance. In the base model when these biomarkers were considered without adjustment for covariates, CSF CCL2 performed slightly better than CSF NfL (0.63 vs. 0.52), but was still within the 95% CI. The lack of correlation between CCL2 and neurodegeneration biomarkers suggests that they likely capture different pathobiological signatures contributing to cognitive decline in MCI‐AD.
Inflammatory changes in the context of neurodegeneration markers and clinical progression
In our prior analysis, we had found TNFR2, SCF, and Ferritin strongly correlated to neurodegeneration markers and were expressed in the brain transcriptome. 18 In the current analysis, the above analytes were not significant in predicting longitudinal cognitive change within three years. The key inflammatory pathways are also likely to be distinct in different stages of clinical AD and needs further elucidation.
Even as migration of neutrophils toward amyloid plaques are noted in some mouse models of AD, 46 this is an area that needs future investigation in MCI‐AD patients to determine if CCL2 determined immune cell migration plays a role in impacting cognitive outcomes. The study data also point to a wide variance in the inflammatory analyte levels within patients at the same stage of MCI‐AD suggesting different degrees of inflammatory pathway dysregulation. Taken together these data suggests a promise for targeted therapies against key inflammatory pathways among patients in whom it is most dysregulated to have a significant clinical outcome within the time widows described in this study. The shared AUCs across CSF CCL2 and neurodegeneration biomarkers in predicting rapid cognitive change over two years but a lack of significant correlation between them, suggest they could play parallel roles in predicting disease progression.
Differences between Discovery and ADNI cohorts
The current study noted key shared inflammatory analyte correlations that were consistent between the Discovery and ADNI cohorts. The Discovery cohort analytes had larger correlation coefficients than ADNI in relation to degree of cognitive decline. The correlations of cognitive/functional change to CSF CCL2 are robust in the Discovery data at 15 months, and get stronger over the 3‐year time of follow‐up in the ADNI data. This may reflect a slower progressing population in ADNI, or an earlier stage of MCI in ADNI compared to Discovery cohort as noted in Figure 4 and Table 1. The common analytes meeting significance threshold in both ADNI and Discovery cohort in the CSF was CCL2 and in the plasma was CXCL8 (IL‐8). CXCL8 was also the only analyte that clustered with CCL2 in both the Discovery and ADNI cohort plasma pathway analysis (Table S11). Additional differences between the two cohorts include differences in duration of follow‐up, AD biomarker levels at baseline and patient recruitment characteristics: a memory clinic sample of MCI subjects with notable cognitive concerns in the Discovery cohort, versus a longitudinal MCI cohort in ADNI with likely different medical and environmental biases, as noted in the less robust correspondence in plasma analytes compared to CSF between the two cohorts. Despite these differences in data collection and patient variables, key shared analytes were still identified at baseline to have a longitudinally clinical impact across both cohorts.
Strengths and limitations
The study’s strengths include a) concomitant measurement of the same analytes in the CSF and plasma; b) well‐characterized patients including clinical variables (inflammation, vascular risk factors and medications), APOE ε4 status, and AD biomarkers, longitudinal assessment of cognition and validation across two different cohorts with potentially different recruitment biases; c) multiple internal and external validity checks to account for quality of data and measurements, and d) going beyond single analyte associations to meaningfully assess multiple analytes and narrow our focus to key activated biological processes related to inflammation in AD with high confidence.
Despite these strengths, this study is not comprehensive in determining the profile of inflammatory analytes as some (e.g., YKL‐40, sTREM2) were not analyzed. The Discovery cohort and ADNI longitudinal cognitive measurements do not have the same follow‐up duration. Additional inflammatory pathways could also be contributing to cognitive outcomes than those posited following our analysis. We also have limited insight based on baseline measures alone, as they themselves could be dynamic and change longitudinally, contributing to variation in the temporal window of strongest cognitive outcome in different stages of AD in both cohorts.
Future studies are needed to evaluate potential inflammatory analytes/pathways not covered in this analysis. Our results pass a stringent multiple comparisons cutoff, but it is possible that with weaker enrichment patterns other analytes of significance may become more salient with increased sample sizes. Lack of neuropathologic confirmation of diagnosis also limits our understanding of the role for mixed pathology in MCI‐AD.
Conclusion
We found that cognitive decline in MCI‐AD was best predicted by CSF CCL2 and likely related to IL‐10 pathway dysregulation in the CSF and plasma. Baseline CSF CCL2 has comparable utility to CSF NfL and hippocampal volume in predicting rapid cognitive decline. Exploring the triggers of this inflammatory response related to chemotaxis of immune cells and prospect of their modulation provides a potential therapeutic opportunity that is of clinical interest in MCI‐AD.
Author Contributions
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
J.A.P: 1) A, B, C 2) A, B 3) A
BTL: 1) A 2) C 3) B
JB: 2) A, B 3) B
KL: 2) B
GB: 2) A, B 3) B
AP: 2) C, 3) B
LS: 2) C, 3) B
MN: 2) C, 3) B
LMB: 1) B, 2) C, 3) B
SMR: 1) B, 2) C, 3) B
MC: 1) B, 2) C, 3) B
JBL: 1) B, 2) C, 3) B
Conflict of Interest
Jagan A Pillai has received research funding from the National Institutes of Health, Alzheimer’s Association and Keep Memory Alive foundation.
Bruce T Lamb has received honoraria or consulting fees from Eli Lilly, Amgen, and Eisai, and research funding from the National Institutes of Health, US Department of Defense, the Alzheimer’s Association, and the BrightFocus Foundation.
Stephen M. Rao has received honoraria, royalties, or consulting fees from: Biogen, Genzyme, Novartis, American Psychological Association, International Neuropsychological Society, and research funding from the National Institutes of Health, US Department of Defense, National Multiple Sclerosis Society, CHDI Foundation, Biogen, and Novartis.
James B. Leverenz has received a) Consulting fees from Acadia, Aptnyx, Biogen, Eisai, GE Healthcare, Sanofi, and Takeda b) Grant support from the Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, Biogen, Department of Defense, GE Healthcare, Genzyme/Sanofi, Lewy Body Dementia Association, Michael J Fox Foundation, National Institute of Health (NIA, NINDS).
Mads Neilsen has shares in Cerebriu A/S.
Akshay Pai has shares in Cerebriu A/S.
Mark Chance: None.
Lynn Bekris: None.
Gurkan Bebek: None.
James Bena: None.
Lei Kou: None.
Supporting information
Figure S1. Discovery cohort CSF levels were clustered to reveal correlated measurements using Consensus clustering.
Figure S2. Discovery cohort plasma levels were clustered to reveal correlated measurements using consensus clustering.
Figure S3. ADNI cohort plasma levels were clustered to reveal correlated measurements using consensus clustering.
Table S1. Additional clinical characteristics of the Discovery and ADNI cohorts and environmental characteristics of the Discovery cohort.
Table S2. Analyte subgroup analysis with most significant inflammatory analytes in the Discovery cohort.
Table S3. Analyte subgroup analysis with most significant inflammatory analytes in the ADNI cohort.
Table S4. Univariate correlations between inflammatory analyte and cognitive change in the Discovery cohort after controlling for baseline cognitive score, age, sex, and APOE ε4 status.
Table S5. Univariate correlations between inflammatory analyte and cognitive change in the ADNI cohort after controlling for baseline cognitive score, age, sex, and APOE ε4 status.
Table S6. CSF CCL2 correlation to ADNI CDR‐SB change after adjustment for baseline CDRSB score, age, sex, and APOE ε4 status with different AD biomarker thresholds.
Table S7. REACTOME pathways (Top 5) that enrich for CSF levels of CCL2 and its highly clustered analytes (B2M, CXCL8, FGA, MMP2, TIMP1, and VCAM1) in Discovery Cohort.
Table S8. REACTOME pathways (Top 5) that enrich for Plasma levels of CCL2 and its highly clustered analytes (AAT, BDNF, CCL4, CCL11, IL‐18, and VEGFA) in Discovery Cohort.
Table S9. REACTOME pathways (Top 5) that enrich for Plasma levels of CCL2 and its highly clustered analytes AAT, BDNF CCL2, CCL4, CCL5,CXCL8,KITLG (SCF), and MPO in ADNI Cohort.
Table S10. ROC analysis looks at how CDR‐SB progression ≥3 points over 2 years are predicted by CSF CCL2, CSF NfL, and hippocampal volume corrected for intracranial volume. Data from 88 patients in the ADNI dataset that had concomitant CSF CCl2, NfL, and hippocampal volume data at two years follow‐up.
Table S10. Comparison of significant analytes in the pathway analysis and multivariable models.
Data S1. Supplementary methods and summary statistics.
Acknowledgments
We thank the patients and families who took part in the Discovery cohort at Cleveland Clinic Lou Ruvo Center for Brain Health. Rosa Gonzalez, Jessica Lee, Maria Khrestian, Hilda Sosic, and Christopher Zalewski.
Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding Information
This study is funded by 2014‐NIRG‐305310 Alzheimer’s Association, NIA K23AG055685, 1P30 AG062428‐01, Keep Memory Alive Foundation and the Jane and Lee Seidman fund. “Research reported in this publication was supported by the National Institute On Aging of the National Institutes of Health under Award Number K23AG055685. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.” Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding Statement
This work was funded by NIA grants 1P30 AG062428‐01 and K23AG055685‐01; Keep Memory Alive Foundation grant ; Jane and Lee Seidman grant ; Alzheimer's Disease Neuroimaging Initiative grant ; National Institutes of Health grant U01 AG024904; Department of Defense grant W81XWH‐12‐2‐0012; National Institute of Biomedical Imaging and Bioengineering grant ; AbbVie grant ; Alzheimer’s Association grant 2014‐NIRG‐305310; Alzheimer’s Drug Discovery Foundation grant ; Araclon Biotech grant ; BioClinica, Inc. grant ; Biogen grant ; Bristol‐Myers Squibb Company grant ; CereSpir, Inc. grant ; Cogstate grant ; Eisai Inc. grant ; Elan Pharmaceuticals, Inc. grant ; Eli Lilly and Company grant ; EuroImmun grant ; F. Hoffmann‐La Roche Ltd grant ; Genentech, Inc. grant ; Fujirebio grant ; GE Healthcare grant ; IXICO Ltd. grant ; Janssen Alzheimer Immunotherapy Research & Development, LLC. grant ; Johnson & Johnson Pharmaceutical Research & Development LLC. grant ; Lumosity grant ; Lundbeck grant ; Merck & Co., Inc. grant ; Meso Scale Diagnostics, LLC. grant ; NeuroRx Research grant ; Neurotrack Technologies grant ; Novartis Pharmaceuticals Corporation grant ; Pfizer Inc. grant ; Piramal Imaging grant ; Servier grant ; Takeda Pharmaceutical Company grant ; Transition Therapeutics grant ; Canadian Institutes of Health Research grant ; Northern California Institute for Research and Education grant ; Alzheimer’s Therapeutic Research Institute grant .
References
- 1. Cummings JL. Cognitive and behavioral heterogeneity in Alzheimer's disease: seeking the neurobiological basis. Neurobiol Aging 2000;21:845–861. [DOI] [PubMed] [Google Scholar]
- 2. Pillai JA, Appleby BS, Safar J, Leverenz JB. Rapidly progressive alzheimer's disease in two distinct autopsy cohorts. J Alzheimers Dis 2018;64:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holtzman DM, Mandelkow E, Selkoe DJ. Alzheimer disease in 2020. Cold Spring Harb Perspect Med 2012;2:a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 2007;7:161–167. [DOI] [PubMed] [Google Scholar]
- 5. Bettcher BM, Kramer JH. Longitudinal inflammation, cognitive decline, and Alzheimer's disease: a mini‐review. Clin Pharmacol Ther 2014;96:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wyss‐Coray T, Rogers J. Inflammation in Alzheimer disease‐a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2012;2:a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 2018;28:e867–e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brosseron F, Traschütz A, Widmann CN, et al. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alz Res Ther 2018;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whelan CD, Mattsson N, Nagle MW, et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol Commun 2019;7:169 10.1186/s40478-019-0795-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suárez‐Calvet M, Araque Caballero MÁ, Kleinberger G, et al.; Dominantly Inherited Alzheimer Network . Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 2016;8:369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song F, Poljak A, Smythe GA, Sachdev P. Plasma biomarkers for mild cognitive impairment and Alzheimer's disease. Brain Res Rev 2009;61:69–80. [DOI] [PubMed] [Google Scholar]
- 12. Swardfager W, Lanctot K, Rothenburg L, et al. A meta‐analysis of cytokines in Alzheimer's disease. Biol Psychiatry 2010;68:930–941. [DOI] [PubMed] [Google Scholar]
- 13. Westin K, Buchhave P, Nielsen H, et al. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS One 2012;7:e30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reaux‐Le Goazigo A, Van Steenwinckel J, Rostene W, Melik PS. Current status of chemokines in the adult CNS. Prog Neurogibol 2013;104:67–92. [DOI] [PubMed] [Google Scholar]
- 15. Saleem M, Herrmann N, Swardfager W, et al. Inflammatory markers in mild cognitive impairment: a meta‐analysis. J Alzheimers Dis 2015;47:669–679. [DOI] [PubMed] [Google Scholar]
- 16. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pillai JA, Maxwell S, Bena J, et al. Key inflammatory pathway activations in the MCI stage of Alzheimer's disease. Ann Clin Transl Neurol 2019;6:1248–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Folstein MF, Folstein SE, McHugh PR. "Mini‐mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 20. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 21. Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta‐amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 2005;51:336–345. [DOI] [PubMed] [Google Scholar]
- 22. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nordlund A, Rolstad S, Hellstrom P, et al. The Goteborg MCI study: mild cognitive impairment is a heterogeneous condition. J Neurol Neurosurg Psychiatry 2005;76:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nibbe RK, Koyuturk M, Chance MR. An integrative ‐omics approach to identify functional sub‐networks in human colorectal cancer. PLoS Comput Biol 2010;6:e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maxwell S, Chance MR, Koyuturk M. Linearity of network proximity measures: implications for set‐based queries and significance testing. Bioinformatics 2017;33:1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res 2017;45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlicker A, Domingues FS, Rahnenfuhrer J, Lengauer T. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics 2006;7:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011;6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fabregat A, Sidiropoulos K, Viteri G, et al. Reactome pathway analysis: a high‐performance in‐memory approach. BMC Bioinformatics 2017;18:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. FreeSurfer . FreeSurfer Software Suite. 2020.http://www.freesurfer.net/. Accessed April 15, 2020 .
- 31. Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 32. Wilkerson MD, Hayes DN. Consensus ClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu GC, Wang LG, Han YY, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vittinghoff E, McCulloch CE. Relaxing the rule of ten events per variable in logistic and Cox regression. Am J Epidemiol 2007;165:710–718. [DOI] [PubMed] [Google Scholar]
- 35. Von Hippel PT. Regression with missing Ys: an improved strategy for analyzing multiply imputed data. Sociol Methodol 2007;37:83–117. [Google Scholar]
- 36. Lee WJ, Liao YC, Wang YF, et al. Plasma MCP‐1 and cognitive decline in patients with Alzheimer's disease and mild cognitive impairment: a two‐year follow‐up study. Sci Rep 2018;8:1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamamoto M, Horiba M, Buescher JL, et al. Overexpression of monocyte chemotactic protein‐1/CCL2 in beta‐amyloid precursor protein transgenic mice show accelerated diffuse beta‐amyloid deposition. Am J Pathol 2005;166:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hohsfield LA, Humpel C. Migration of blood cells to beta‐amyloid plaques in Alzheimer's disease. Exp Gerontol 2015;65:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galimberti D, Schoonenboom N, Scheltens P, et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol 2006;63:538–543. [DOI] [PubMed] [Google Scholar]
- 40. Bettcher BM, Neuhaus J, Wynn MJ, et al. Increases in a Pro‐inflammatory chemokine, MCP‐1, are related to decreases in memory over time. Front Aging Neurosci 2019;11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Georgakis MK, Malik R, Bjorkbacka H, et al. Circulating monocyte chemoattractant protein‐1 and risk of stroke: meta‐analysis of population‐based studies involving 17 180 individuals. Circ Res 2019;125:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelly‐Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin‐4 and interleukin‐13 signaling connections maps. Science 2003;300:1527–1528. [DOI] [PubMed] [Google Scholar]
- 43. Couper KN, Blount DG, Riley EM. IL‐10: the master regulator of immunity to infection. J Immunol 2008;180:5771–5777. [DOI] [PubMed] [Google Scholar]
- 44. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016;73:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baik SH, Cha MY, Hyun YM, et al. Migration of neutrophils targeting amyloid plaques in Alzheimer's disease mouse model. Neurobiol Aging 2014;35:1286–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Discovery cohort CSF levels were clustered to reveal correlated measurements using Consensus clustering.
Figure S2. Discovery cohort plasma levels were clustered to reveal correlated measurements using consensus clustering.
Figure S3. ADNI cohort plasma levels were clustered to reveal correlated measurements using consensus clustering.
Table S1. Additional clinical characteristics of the Discovery and ADNI cohorts and environmental characteristics of the Discovery cohort.
Table S2. Analyte subgroup analysis with most significant inflammatory analytes in the Discovery cohort.
Table S3. Analyte subgroup analysis with most significant inflammatory analytes in the ADNI cohort.
Table S4. Univariate correlations between inflammatory analyte and cognitive change in the Discovery cohort after controlling for baseline cognitive score, age, sex, and APOE ε4 status.
Table S5. Univariate correlations between inflammatory analyte and cognitive change in the ADNI cohort after controlling for baseline cognitive score, age, sex, and APOE ε4 status.
Table S6. CSF CCL2 correlation to ADNI CDR‐SB change after adjustment for baseline CDRSB score, age, sex, and APOE ε4 status with different AD biomarker thresholds.
Table S7. REACTOME pathways (Top 5) that enrich for CSF levels of CCL2 and its highly clustered analytes (B2M, CXCL8, FGA, MMP2, TIMP1, and VCAM1) in Discovery Cohort.
Table S8. REACTOME pathways (Top 5) that enrich for Plasma levels of CCL2 and its highly clustered analytes (AAT, BDNF, CCL4, CCL11, IL‐18, and VEGFA) in Discovery Cohort.
Table S9. REACTOME pathways (Top 5) that enrich for Plasma levels of CCL2 and its highly clustered analytes AAT, BDNF CCL2, CCL4, CCL5,CXCL8,KITLG (SCF), and MPO in ADNI Cohort.
Table S10. ROC analysis looks at how CDR‐SB progression ≥3 points over 2 years are predicted by CSF CCL2, CSF NfL, and hippocampal volume corrected for intracranial volume. Data from 88 patients in the ADNI dataset that had concomitant CSF CCl2, NfL, and hippocampal volume data at two years follow‐up.
Table S10. Comparison of significant analytes in the pathway analysis and multivariable models.
Data S1. Supplementary methods and summary statistics.