

Abstract
Understanding the root causes of autoimmune diseases is hampered by the inability to access relevant human tissues and identify the time of disease onset. To examine the interaction of immune cells and their cellular targets in type 1 diabetes, we differentiated human induced pluripotent stem cells into pancreatic endocrine cells, including β cells. Here, we describe an in vitro platform that models features of human type 1 diabetes using stress-induced patient-derived endocrine cells and autologous immune cells. We demonstrate a cell-type-specific response by autologous immune cells against induced pluripotent stem cell-derived β cells, along with a reduced effect on α cells. This approach represents a path to developing disease models that use patient-derived cells to predict the outcome of an autoimmune response.
Keywords: type 1 diabetes, induced pluripotent stem cell-derived β cells, disease modeling, endoplasmic reticulum stress
Graphical Abstract
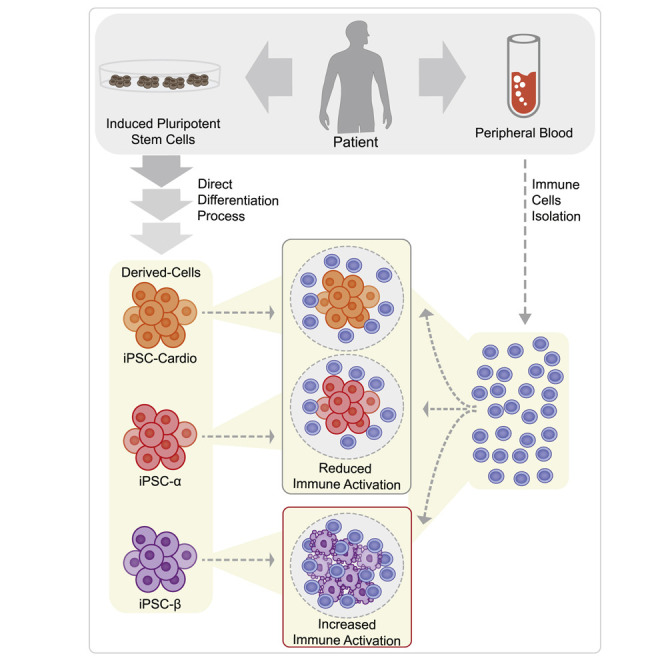
Highlights
-
•
β cell-specific response is achieved using iPSC-β cells and autologous immune cells
-
•
An in vitro immune response to iPSC-β cells requires ER stress
-
•
T cell activation is restricted to autologous iPSC-β cells
-
•
T cell activation is mediated by TCR engagement of peptide-HLA complexes on iPSC-β
Leite et al. describe an in vitro platform that models features of autoimmune type 1 diabetes using patient-derived endocrine cells and autologous immune cells. This represents a path to developing a model with patient-derived cells to predict and analyze the autoimmune response.
Introduction
Type 1 diabetes (T1D) results from an autoimmune destruction of insulin-secreting pancreatic β cells (World Health Organization, 2016). Genetic and environmental factors may render β cells susceptible to attack by the immune system and contribute to β cell dysfunction (van Belle et al., 2011; Pugliese, 2013); however, the exact triggers and progression to clinical onset are not fully understood.
Insulin accounts for up to 50% of protein production in β cells, and this increases in response to metabolic demand (Scheuner and Kaufman, 2008; Schuit et al., 1988). Consequently, β cells are prone to endoplasmic reticulum (ER) stress and protein misfolding that can lead to β cell failure (Engin, 2016). It has also been suggested that the onset of T1D is associated with viral infection (Hober and Sauter, 2010), β cell exposure to chemicals (Like and Rossini, 1976), reactive oxygen species (Delmastro and Piganelli, 2011), and inflammation (Eizirik et al., 2013). All of these environmental triggers can cause β cell ER stress, suggesting that ER stress may be a common factor in disease onset.
T1D was a lethal disease until the discovery of insulin in the 1920s, and since then, insulin injection remains the primary treatment for T1D patients (Banting et al., 1922). The transplantation of cadaveric islets along with immunosuppression has been successfully performed to treat T1D (Shapiro et al., 2000). The challenges associated with transplantation, including the paucity of available human islets and allogeneic immune responses, may be overcome through the use of patient induced pluripotent stem cell-derived β cells (iPSC-β). Human iPSC-β cells could have the potential to replace the function of the β cells lost in T1D (Pagliuca et al., 2014; Veres et al., 2019; Nair et al., 2019; Russ et al., 2015; Rezania et al., 2014), thereby reducing or eliminating the need for exogenous insulin. However, it is expected that autologous iPSC-β cells will still be vulnerable to recurrent autoimmunity following transplantation (Sutherland et al., 1984; Sibley et al., 1985). Immune evasion strategies, including encapsulation with biomaterials or genetic manipulation, represent ways forward in advancing iPSC-β cell transplant therapies. Developing such strategies would benefit from a model that can predict tissue rejection in T1D patients. Here, we describe an in vitro platform using iPSC-β and iPSC-α cells that recapitulates some aspects of the autologous immune interaction in human T1D. This platform makes it possible to more intently study human autoimmune interactions and may be used for immunogenic evaluation before cell replacement therapy.
Results
Immune Profiling of In Vitro-Generated Patient-Derived β and α Cells
iPSCs were derived from three T1D donor (T1D1–3) and one non-T1D (ND) donor (Figure 1 A; Table S1). After iPSC characterization (Figures S1A and 1B), cells were differentiated in vitro using two different protocols to produce islet-like clusters that are enriched in either insulin-producing iPSC-β cells (Veres et al., 2019) or glucagon-producing iPSC-α cells (Peterson et al., 2020) (β cell and α differentiation protocol, respectively) (Figure 1A). The efficiency of β or α cell generation varies between PSC lines, yet both the T1D and ND iPSC lines were able to differentiate into iPSC-β or iPSC-α clusters. The differentiation protocols produced ∼20% of the desired cell type (Figures 1B–1D), similar to previous reports using iPSC lines (Millman et al., 2016). While these designations indicate the predominant endocrine cell type in each protocol, iPSC-β clusters also have glucagon-producing α cells (10%) and vice versa (5%) (Figures 1B–1D, S1C, and S1D). These iPSC-β and -α cell-enriched preparations were used as autologous targets for immune studies, with peripheral blood mononuclear cells (PBMCs) isolated from the corresponding donors.
Figure 1.

Immune Profiling of In Vitro-Generated Patient-Derived β and α Cells
(A) Schematic of the differentiation workflow.
(B) Immunostaining of an iPSC-β cluster.
(C and D) Flow cytometry quantification of C-peptide- and glucagon-positive cells in the β-cell differentiation protocol (C) and α-cell differentiation protocol (C). n = 3 T1D and n = 1 ND donor, n = 3–7 differentiation batches per donor line.
(E and F) Flow cytometry quantification of HLA-A, -B, and -C iPSC-β clusters treated with thap (5 μM for 5 h) or IFNγ (100 U/mL, 48 h), gated on C-peptide-positive cells (E) and glucagon-positive cells (F). The values are represented as adjusted mean fluorescence intensity (MFI). n = 3 T1D and n = 1 ND donor, n = 3 differentiation batches per donor line. T1D1, T1D2, and T1D3 were pooled together.
(G) Relative mRNA expression of T1D-associated autoantigens in clusters untreated or treated with thap (5 μM for 5 h). Each column represents a donor and each row represents a gene. n = 3 T1D, n = 1 ND donor, n = 4 control human islets.
∗∗p < 0.01 and ∗∗∗p < 0.0005. Ordinary 1-way ANOVA. ns, non-significant. See also Figure S1.
Little is known about the immunogenicity of iPSC-β or iPSC-α cells (van der Torren et al., 2017). The human leukocyte antigen complex (HLA) gene locus is the top risk allele in T1D (Pociot, 2017), and HLA expression is required for a robust adaptive immune response (Clark et al., 2017). Since our goal was to recapitulate immune interactions that may occur in T1D pathogenesis, we investigated the expression of molecules relevant to T cell recognition in iPSC-β and iPSC-α from T1D and ND donors in stress conditions. The iPSC-α and -β cell preparations were treated with soluble interferon gamma (IFNγ) to mimic inflammatory stress and thapsigargin (thap) to induce ER stress (Kracht et al., 2017; Marre et al., 2018). Following IFNγ (Richardson et al., 2016) but not by thap treatment alone, iPSC-β and iPSC-α cells upregulated HLA class I (HLA-ABC) and other immunoregulatory molecules, including programmed death-ligand 1 (PD-L1) and Fas cell surface death receptor (FasR) (see Figures S1G–S1P, 1E, and 1F). The expression of molecules relevant to immune recognition was similar in both ND and T1D iPSC-α and -β cells. The IFNγ-induced upregulated HLA-ABC protein expression was the same between iPSC-β cells and human islets following cytokine treatment. The HLA-ABC expression was lower in resting iPSC-α and -β cells compared to control islets (Figure S1Q).
Several islet proteins have been identified as targets of autoimmunity in T1D, including proinsulin (INS), glutamic acid decarboxylase (GAD), tyrosine phosphatase-like insulinoma-associated antigen 2 (IA-2, PTPRN), islet-specific glucose-6-phosphatase catalytic subunit–related protein (G6PC2), the cation efflux transporter ZNT8 (SLC30A8), islet-amyloid polypeptide (IAPP), and others (Pugliese, 2017). CD8 T cell epitopes from these proteins have been identified in the PBMCs of recent-onset T1D patients, suggesting a role in β cell destruction (Velthuis et al., 2010). We determined the relative transcript expression encoding these protein targets in derived iPSC-β or -α cells. We found similar levels of mRNA in iPSC-β cells, albeit low in some cases, for G6PC2, SLC30A8, IAPP, INS, GAD, and IA-2 in both T1D and ND iPSC-β cells and in islets isolated from a control donor (Figure 1G).
T Cells Are Activated When Co-cultured with Autologous ER-Stressed iPSC-β Cells
To examine the interaction between iPSC-β and immune cells, we co-cultured autologous PBMCs with their corresponding iPSC-β cell clusters. Immune activation was evaluated by surface staining of T cell activation markers and by cytokine secretion after a 48-h co-culture.
We assessed the immune response of autologous PBMCs when co-cultured with iPSC-β clusters from ND and T1D donors (Figure 2 A). As a positive control, PBMCs were stimulated with anti-CD3/CD28 beads (Figures S2A–S2C). T cell activation was not observed when autologous PBMCs were co-cultured with the corresponding untreated iPSC-endocrine cells (Figure 2). Similarly, iPSC-β pre-stimulated with IFNγ to enhance antigen presentation did not elicit an immune response (Figures S2E and S2F). Previous studies have reported that β cell ER stress has implications for immunogenicity in T1D. ER stress has been shown to increase β cell immunogenicity and can lead to T1D (Eizirik et al., 2008). To mimic ER stress, iPSC-β cells were pre-treated with thapsigagin (thap) for 5 h before co-culturing with PBMCs for 48 h. Thap treatment of PBMCs alone did not induce T cell activation (Figure S2D).
Figure 2.

T Cells Are Activated When Co-cultured with Autologous ER-Stressed iPSC-β
(A) Experimental design: PBMCs co-cultured with autologous iPSC-β.
(B–D) Flow cytometry data of T cells after a 4-8h co-culture with iPSC-β cells (n = 3 T1D and n = 1 ND donor, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
(B) CD25+ and (C) CD69+ co-positive for CD3+, CD4+, or CD8+ cells, as indicated. The values are represented as adjusted MFI.
(D) Pro-inflammatory cytokine detection in supernatants collected after 48 h co-culture of PBMC with iPSC-β (n = 3 T1D and n = 1 ND donor, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
(E) Percentage of live iPSC-β after co-culture, gated for C-peptide+/glucagon− (n = 3 T1D and n = 1 ND donor, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0005, and ∗∗∗∗p < 0.0001. Ordinary 1-way ANOVA. ns, non-significant. See also Figure S2.
Co-culturing PBMCs with autologous T1D and ND iPSC-β, pre-treated with thap, resulted in upregulated immune cell activation markers CD25 and CD69 on T cell populations (Figures 2B, 2C, and S2G) and in increased levels of IFNγ, interleukin-2 (IL-2), IL-17, and C-X-C motif ligand 10 (CXCL10) (Figure 2D). Both CD69, a type II C-lectin receptor, and CD25, the high-affinity IL-2 receptor-α chain, are early activation markers expressed rapidly by human conventional CD8 and CD4 T cells following T cell receptor (TCR) engagement (Cibrián and Sánchez-Madrid, 2017; Caruso et al., 1997). We also observed a decrease in viable iPSC-β in the thap-treated co-culture experiments (Figures 2E and S4G). These results support a causal role of ER stress in eliciting an immune response, as previously reported by others. Autologous co-culture with iPSC-β with PBMCs induced an immune response in both T1D donors and ND donors. These observations are in line with reports describing that islet-reactive T cell frequencies in the blood do not distinguish T1D from ND individuals (Culina et al., 2018).
Next, we performed experiments to determine whether the observed T cell activation within the PBMCs is mediated by direct TCR engagement with the HLA complex on iPSC-β cells, rather than a T cell bystander activation caused by the inflammatory cytokine milieu or engagement of co-stimulatory molecules by other cells present in the PBMCs. First, the iPSC-β target cells were pre-treated with an HLA class I blocking antibody to prevent TCR binding to the peptide-HLA complex (Figures 3 A, 3B, S3A, and S3D). Second, we used a transwell co-culture system that precludes contact between the immune cells and iPSC-β (Figure 3C). Third, iPSCs were transduced with a B2M guide RNA and Cas9, leading to the decreased expression of HLA class I on differentiated iPSC-β cells (Figures 3D, S3B, and S3C). In all three experiments, reduced T cell activation was assessed by cytokine secretion and evaluation of the T cell activation markers CD25 and CD69 in CD8+ T cells present in the PBMCs. These results support the conclusion that T cell activation is mediated by direct TCR engagement of peptide-HLA complexes on iPSC-β.
Figure 3.

iPSC-β-Induced T Cell Activation and Killing Is Mediated by Direct T Cell-HLA Interaction
(A and B) PBMCs co-cultured for 48 h with autologous iPSC-β (n = 3 T1D donors, 3 differentiation batches per donor). iPSC-β were pre-treated with thap (5 μM for 5 h) and/or anti-HLA antibody for 30 min before co-culture.
(A) Flow cytometry after 48 h co-culture of T cell activation markers, CD25+ and CD69+, gated on CD8+ cells.
(B) Pro-inflammatory cytokine detection in supernatants collected after 48 h co-culture of PBMCs with autologous iPSC-β ± thap.
(C) Expression of CD25+ and CD69+, gated on CD8+ cells in a transwell system.
(D) Expression of the activation marker CD25+ and CD69+ on CD8+ cells after 48-h co-culture with autologous iPSC-β transduced with a lentivirus vector expressing a non-targeting (NT) or B2M guide RNA (gRNA) and Cas9 (n = 3 T1D donors, n = 3 differentiation batches per donor line). ∗p < 0.05 and ∗∗p < 0.01, Student’s t test. T1D1, T1D2, and T1D3 were pooled together. ns, non-significant.
(A–C) n = 3 T1D donors, n = 3 differentiation batches per donor line. T1D1, T1D2, and T1D3 were pooled together. ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.0005; 2-way ANOVA.
See also Figure S3.
Activation and Killing by T Cells Is Selective for iPSC-β
The main feature of T1D is selective destruction of pancreatic β, but not α, cells. To test whether the in vitro platform recapitulates that specificity, we generated additional iPSC-derived cell types—autologous iPSC-α cells and iPSC-cardiomyocytes—and performed co-culture experiments, as described above (Figure 4 A). All of the cell preparations (iPSC-β, iPSC-α, and cardiomyocytes) were pre-treated with thap before co-culturing with PBMCs.
Figure 4.

Activation and Killing by T Cells Is Selective for iPSC-β
(A) Experimental design: PBMCs co-cultured with autologous iPSC-derived cells.
(B, C, and F) CD25 or CD69 expression shown as MFI.
(B) PBMCs (gated on CD3+, CD4+, and CD8+ populations) co-cultured for 48 h with autologous iPSC-β (purple) or iPSC-α (red). (n = 3 T1D and n = 1 ND donor, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
(C) Donor-matched PBMCs (CD3+ gated) (n = 1 T1D, n = 3 differentiation batches per donor line) co-cultured for 48 h with autologous iPSC-β (purple) or iPSC-cardiomyocytes (orange).
(D) Percentage of live iPSC-β (C-peptide+/glucagon−) or iPSC-α (C-peptide−/glucagon+) from iPSC-β or iPSC-α differentiations, respectively (n = 3 T1D donors, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
(F) Representative flow cytometry histograms after 48 h of co-culture. Dashed histogram represents the control (untreated target cells).
(E) Percentage of apoptotic (apopxin+) iPSC-β (CD49a+/CD26−) or iPSC-α (CD49a−/CD26+) from iPSC-β or iPSC-α differentiations (n = 1 T1D iPSC donor, n = 3 differentiation batches per donor line).
(G) Unmatched PBMCs (CD3+ gated cells) co-cultured for 48 h with iPSC-α (n = 3 T1D donors, n = 3 differentiation batches per donor line). T1D1, T1D2, and T1D3 were pooled together.
(H) Donor-matched PBMCs (gated on CD3+, CD4+, and CD8+ populations) co-cultured for 48 h with autologous enriched iPSC-β or iPSC-α.
Data are means ± SEMs, 2-way ANOVA. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, and ∗∗∗∗p < 0.0001; ns, non-significant. See also Figure S4.
T cell activation was restricted to co-culture with autologous iPSC-β cells and not to other iPSC-derived cells (Figures 4B, 4C, 4F, S2G, and S4A–S4C). Since β cells are highly secretory cells and more likely to undergo ER stress, we set out to evaluate whether the T cell response induced by iPSC-β was due to the resistance of iPSC-α to ER stress. Both iPSC-β and -α cells showed an increase in ER stress-related proteins, PRKR-like ER kinase (PERK), protein disulfide isomerase (PDI), and binding immunoglobulin protein (BiP) after thap treatment, demonstrating that both cell types are sensitive to ER stress induction in vitro (Figures S1E and S1F).
To further explore iPSC-β cell specificity, we investigated whether iPSC-α cells are protected from T cell-mediated destruction compared to iPSC-β cells after autologous co-culture with PBMCs. We compared β and α cell numbers from their respective differentiation protocols (α versus β differentiation protocol) using two assays for survival after autologous co-culture with PBMCs: the percentage of live α and β cells by live cell count (Figures 4D, S4E, and S4F) and assessing apoptosis using the apopxin dye (Figures 4E and S4G). We found that fewer iPSC-β cells survive co-culture with autologous PBMCs compared to iPSC-α cells, and more iPSC-β cells are apoptotic compared to iPSC-α cells.
Since we observed a low activation of autologous immune cells in the presence of iPSC-α cells, we performed a co-culture experiment using unmatched PBMCs to test whether iPSC-α can induce an allogeneic T cell response. Allogeneic T cells upregulated the activation markers after co-culture with iPSC-α cells (Figure 4G). These results show that iPSC-α cells can be immunogenic and induce T cell reactivity in an allogeneic setting.
Our in vitro differentiation system generates a mixed population of endocrine cells. It is thus difficult to demonstrate whether the effects are cell specific or dependent on other cell types in the preparation. To address the question of β cell specificity, we used anti-CD49a staining to sort out iPSC-β cells. The negative fraction of the CD49a sorted cells was stained with CD26 to sort iPSC-α cells (Veres et al., 2019). This two-step method yields clusters of up to 80% β cells by CD49a sorting and up to 50% α cells by CD26 sorting (Figure S4D). After enriching both β and α cell populations, we performed co-culture experiments comparing both enriched populations and the original mixed population derived from the standard iPSC-β protocol. Co-culture with both enriched and standard iPSC-β cell clusters has similar levels of T cell activation while the α cell population induced minimal activation (Figure 4H). In summary, we show that an autoreactive, β cell-specific response can be achieved in vitro using iPSC-β cells and autologous immune cells, recapitulating key features of T1D.
Discussion
A deeper understanding of the immunopathogenesis of T1D, including how it differs among T1D individuals, is needed to develop rational strategies to find a cure or to prevent the disease. Recent advances in stem cell technologies and access to patient samples provide the means to explore T1D pathogenesis. We established an in vitro platform using iPSC-derived pancreatic endocrine cells and autologous immune cells that recapitulates key aspects of T1D. We demonstrate that the immune response to β cells requires ER stress, that a direct, physical interaction between the T cells and β cells is necessary, and the T cell-mediated activation and killing is preferential to β cells, but not α cells.
The findings of this research should be interpreted in light of two major limitations. First, these experiments required the use of blood provided from the same patient who supplied the iPSCs used to generate iPSC-derived cells. We had to synchronize the time of cell generation with donor availability to obtain blood to perform the experiments. Second, cell numbers are a limitation for the co-culture experiments, since we were only able to isolate on average 50 million PBMCs from each blood draw. The approach would be strengthened by improving techniques to select and culture circulating pancreatic β cell-reactive immune cells. A better source of pancreatic β cell-reactive immune cells may be the draining pancreatic lymph nodes in T1D patients, but these are not readily accessible.
We observed unexpected results, including the activation of T cells by ND iPSC-β cells, the inability of IFNγ to induce an immune response, apart from HLA class I upregulation, and the requirement of ER stress to induce autologous T cell activation. We believe that iPSC-β cells mimic healthy β cells after reprogramming from somatic cells, regardless of whether they were derived from a T1D or a ND donor, thus putting both T1D and ND iPSC-β cells on the same starting line. To mimic a β cell disease state, we had to induce ER stress in the iPSC-β cells. This supports the idea that the trigger of β cell destruction in T1D is enhanced β cell vulnerability caused by islet inflammation due to ER stress. We did not observe any difference in the ability of iPSC-β cells from T1D donors to stimulate an immune response relative to iPSC-β cells from ND donors, and in either case, the cells had to be subjected to ER stress to promote an immune response. It is perhaps relevant that the pancreata of individuals with T1D are significantly smaller than those studied from ND individuals (Regnell et al., 2016; Virostko et al., 2019), and even that first-degree relatives of an index case with T1D have smaller pancreata (Campbell-Thompson et al., 2019). While conjectural, it stands to reason that a metabolic load imposed on pancreatic β cells will be greater per β cell when there are fewer such cells to carry the load.
We observed that T cell activation was restricted to co-culture with the autologous iPSC-β cell preparation and not to the iPSC-α or cardiomyocyte preparation. iPSC-β cells expressed genes encoding peptides associated with T1D. Although we did not assess natural processing and presentation or verify peptide modification in the iPSC-derived cells, CD8+ T cells within PBMCs responded to stressed iPSC-β, suggesting that β cell-specific epitopes were generated. The importance of ER stress and the unfolded protein response in β cell pathophysiology and the autoimmune responses in T1D are not well understood (Marre et al., 2018; Eizirik et al., 2008). Our results add to this narrative by showing that ER stress in β cells is an important trigger for an immune response in vitro. Experiments using islet-specific T cell lines would take advantage of the system presented here and potentially strengthen the analysis and conclusions. It is unfortunate that the coronavirus disease 2019 (COVID-19) pandemic lockdown eliminated the possibility of obtaining the necessary patient blood samples for months, and we therefore could not perform these experiments. Our protocol for assessing antigen-specific T cell activation was the expression of both CD25 and CD69 on T cells (Caruso et al., 1997). A significant proportion of both CD8 and CD4 T cells are positive for CD69 expression, which may reflect the increased levels of inflammatory cytokines detected within the cultures. In contrast, expression of CD25 by human conventional CD8 and CD4 T cells is a better measure of antigen-specific stimulation of T cells and less likely to be significantly upregulated by cytokines alone (Shatrova et al., 2016). We postulate that the PBMC-iPSC-β cell co-cultures created an environment in which ER-stressed iPSC-β cells are presenting/releasing autoantigens and creating an inflammatory milieu that strongly stimulates autoreactive T cells to express CD25 and CD69 and that causes bystander T cells to upregulate CD69.
CD8+ T cells play a fundamental role in T1D pathogenesis, as they take center stage in the destruction of pancreatic β cells and contribute to sustained islet inflammation (Coppieters et al., 2012). Although we also observed CD4+ activation after co-culturing PBMCs and iPSC-β cells, we speculate that this activation is due to antigen presentation by professional antigen-presenting cells present in the PBMC preparation. However, we observed low levels of HLA-DR on iPSC-β that could be involved in HLA class II peptide presentation by β cells leading to CD4+ T cell activation. Further investigation is necessary to elucidate the mechanism by which CD4+s are activated in this system.
In summary, the in vitro platform described here can be used to better understand the cellular and molecular components relevant to immune cell interaction in human T1D. In addition, it opens the possibility of testing for the immune protection of tolerogenic engineered iPSC-β cells before cell replacement therapy. Genetically engineered iPSCs that evade immune destruction may ultimately reduce the need for harmful immunosuppression (Han et al., 2019; Deuse et al., 2019). Finally, our model can pave the way to the development of other disease models that take advantage of patient-derived cells to predict the outcome of an autoimmune response.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-C-peptide | Developmental Studies Hybridoma Bank (DHSB) | GN-ID4, RRID:AB_2255626 |
| Mouse anti-NKX6.1 | DHSB | F55A12, RRID:AB_532379 |
| Mouse anti-glucagon | Santa Cruz Biotech | Cat#SC-514592, RRID:AB_2629431 |
| Donkey anti-rat 594 | Life Technologies | Cat#A21209, RRID:AB_2535795 |
| Donkey anti-mouse Alexa 647 | Life Technologies | Cat#A31571, RRID:AB_162542 |
| Donkey anti-rabbit Alexa 488 | Life Technologies | Cat#A21206, RRID:AB_2535792 |
| Donkey anti-rabbit Alexa 594 | Life Technologies | Cat#A21209, RRID:AB_2535795 |
| Donkey anti-rabbit Alexa 647 | Life Technologies | Cat#A31573, RRID:AB_2536183 |
| Donkey anti-goat Alexa 647 | Life Technologies | Cat#A21447, RRID:AB_141844 |
| Donkey anti-sheep Alexa 488 | Life Technologies | Cat#A11015, RRID:AB_141362 |
| Donkey anti-rat 488 | Jackson Laboratories | Cat#712-546-153, RRID:AB_2340686 |
| Donkey anti-rat 405 | Abcam | Cat#ab175670, RRID:AB_11009056 |
| Mouse Anti-HLA-ABC PE-conjugated | Biolegend | W6/32, Cat#311406, RRID:AB_314875 |
| Mouse anti-HLA-E PE-conjugated | Biolegend | 3D12, Cat#342603, RRID:AB_1659250 |
| Mouse anti-HLA-DR APC-conjugated | Biolegend | L243, Cat#307610, RRID:AB_314688 |
| Mouse anti-PD-L1 APC-conjugated | Biolegend | 29E.2A3, Cat#329708, RRID:AB_940360 |
| Mouse anti-CD3 PB-conjugated | Biolegend | UCHT1, Cat#300417, RRID:AB_493094 |
| Mouse anti-CD8 PE-conjugated | Biolegend | T8-Leu2, Cat#344705, RRID:AB_1953243 |
| Mouse anti-CD4 PE/Cy7-conjugated | Biolegend | RPA-T4, Cat#300511, RRID:AB_314079 |
| Mouse anti-CD69 Alexa 647-conjugated | Biolegend | FN50, Cat#310918, RRID:AB_528871 |
| Mouse anti-CD25 Alexa 700-conjugated | Biolegend | M-A251, Cat#356118, RRID:AB_2562168 |
| Mouse anti-CD95/Fas APC-conjugated | eBioscience | DX2, Cat#17-0959-42, RRID:AB_10807091 |
| Mouse anti-CD49a PE-conjugated | BD Biosciences | Cat#559596, RRID:AB_397288 |
| Mouse anti-CD26 APC-conjugated | Miltenyi Biotech | Cat# 130-120-769, RRID:AB_2752189 |
| Mouse anti-Oct4 Alexa 488-conjugated | BD PharMingen | Cat#560253, RRID:AB_1645304 |
| Mouse anti-Sox2 PE-conjugated | BD PharMingen | Cat#560291, RRID:AB_1645334 |
| Mouse anti-SSEA4 V450-conjugated | BD PharMingen | Cat#561156, RRID:AB_10896140 |
| Mouse anti-TRA-1-60 (A647-conjugated | BD PharMingen | Cat#560850, RRID:AB_10565983 |
| Rabbit anti-PERK | Cell Signaling Tech. | D11A8, Cat#5683S, RRID:AB_10841299 |
| Rabbit anti-PDI | Cell Signaling Tech. | C81H6, Cat#3501S, RRID:AB_2156433 |
| Rabbit anti-BIP | Cell Signaling Tech. | C50B12, Cat#3177S, RRID:AB_2119845 |
| Rabbit anti-GAPDH | Abcam | Cat#ab181602, RRID:AB_2630358 |
| Bacterial and Virus Strains | ||
| lentiCRISPRv2 | Broad institute | RRID: Addgene_52961 |
| pHDM-vsvG, -tat, rev, gag/pol | Harvard Medical School DNA Resource Core | N/A |
| Sendai virus (SeV) Cytotune 2 kit | Life Technologies | Cat#A16517 |
| Biological Samples | ||
| Human peripheral blood | University of Massachusetts Medical School | N/A |
| Human Peripheral Blood Leukapheresis Pack | Stem Cell Technologies | Cat#70500.1 |
| Lenti-X 293T Cell Line | Takara Bio | Cat#632180 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Activin A | R&D Systems | Cat#338-AC |
| Rock Inhibitor Y-27632 | DNSK | Cat#DNSK-Kl15-02 |
| Chir99021 | Stemgent | Cat#04-0004-10 |
| KGF | Peprotech | Cat#100-19 |
| Retinoic acid | Sigma-Aldrich | Cat# R2625 |
| LDN193189 | Sigma-Aldrich | Cat#SML0559 |
| Sant1 | Sigma-Aldrich | Cat#S4572 |
| PBDU | EMD Millipore | Cat#524390 |
| XXI | EMD Millipore | Cat#565790 |
| Alk5i II | Axxora | Cat#ALX-270-445 |
| T3 | EMD Millipore | Cat#642511 |
| Betacellulin | ThermoFisher Scientific | Cat# 565790 |
| Human IFN gamma | R&D Systems | Cat#285IF |
| Thapsigargin | Sigma-Aldrich | Cat#T9033 |
| Critical Commercial Assays | ||
| MSD U-PLEX Metabolic group 1 kit | MesoScale Discovery | Cat# K151ACM |
| STEMdiff Cardiomyocyte Differentiation Kit | Stem Cell Technologies | Cat# #05010 |
| Experimental Models: Cell Lines | ||
| Human iPSC-line T1D1 | HSCI | iPSC 3107-001 |
| Human iPSC-line T1D2 | HSCI | iPSC 3107-002 |
| Human iPSC-line T1D3 | HSCI | iPSC 3107-003 |
| Human iPSC-line ND | HSCI | iPSC 3107-013 |
| Software and Algorithms | ||
| QuantStudio 6 Flex Real-Time PCR system | Applied Biosystems | N/A |
| FlowJo v10 | BD (Becton, Dickinson and Company) | N/A |
| GraphPad Prism8 | GraphPad Software | N/A |
| nCounter | NanoString technologies | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas A. Melton (dmelton@harvard.edu).
Materials Availability
This study did not generate any unique reagents. When appropriate, details and source information to synthesize non-commercially available intermediate chemicals are provided and original literature source cited. The corresponding author can be contacted for further details.
Data and Code Availability
The published report includes all data generated or analyzed during this study. No code was used or generated during this study.
Experimental Model and Subject Details
All procedures were performed in accordance with the IRB guidelines at Harvard University and University of Massachusetts Medical School under IRB and ESCRO Protocols E00024. All iPSC lines were generated at the iPSC Core at Harvard Department of Stem Cells and Regenerative Biology.
Method Details
iPSC and PBMCs Derivation from Human Donors
For iPSC derivation from blood, Sendai virus (SeV) Cytotune 2 kit (Life Technologies, Cat#A16517) was used to infect erythroblast-enriched populations following expansion. Briefly, 1 × 105 erythroblast-enriched populations were resuspended in 1mL of fresh StemSpan SFEM II with Erythroid expansion supplement. After that, Sendai virus vectors were reconstituted at MOI of 5 for KOS, MOI of 5 for Myc and MOI of 3 for Klf4. Subsequently, virus suspension was added to the cells. On the following day cells were collected in a 15 mL falcon tube and washed with StemSpan SFEM II with Erythroid expansion supplement by centrifugation at 300 × g for 10 min. Two days post infection, cells were transferred from a well of a 12-well plate to a 6-well plate culture dish containing a mouse embryonic fibroblast (MEF) feeder layer and cultured further for 2 days in hES Media (mTeSR™1 - Stem Cell Technologies; 85850); subsequently, media were changed daily. At 15 to 21 days post transduction, the transduced cells began to form colonies with iPSC morphology, and visible colonies were handpicked and transferred onto 12-well plates.
Cell Culture
Human induced pluripotent stem-cell maintenance and differentiation were carried out as previously described (Pagliuca et al., 2014). Induced pluripotent stem-cell lines were obtained from stocks maintained by the Melton laboratory. Induced pluripotent stem cell lines were maintained in cluster suspension culture format using mTeSR3D (Stem Cell Technologies, 03950) in 500 mL spinner flasks (Corning, VWR) spinning at 70 rpm in an incubator at 37°C, 5% CO2 and 100% humidity. Cells were passaged every 72h or 96h: induced human pluripotent stem-cell clusters were dissociated to single cells using gentle cell dissociation reagent (Stem Cell Technologies; 07174) and light mechanical disruption, counted and seeded at 0.7 M cells/ml in mTeSR3D + 10 μM Y27632 (DNSK International, DNSK-KI-15-02). Cell lines were authenticated by DNA fingerprinting, karyotyping (Cell Line Genetics) and all lines tested negative on routine mycoplasma contamination verifications. Differentiation flasks were started 72 h after passaging by replacing mTeSR3D medium with the appropriate differentiation medium including growth factors and small molecule supplements as previously described (Veres et al., 2019):
β Cell Differentiation Protocol
Stage 1: 24 hours in S1 medium supplemented with Activin A (100ng/ml), CHIR99021 (1.4 μg/ml) and Rock Inhibitor (10 μM), followed by 48 hours Activin A (100ng/ml) only.
Stage 2: 72 hours in S2 medium supplemented with KGF (50ng/ml) and Rock Inhibitor (10 μM).
Stage 3: 48 hours in S3 medium supplemented with KGF (50ng/ml), LDN193189 (200nM), Sant1 (0.25 μM), retinoic acid (2 μM), PBDU (500nM) and Rock Inhibitor (10 μM).
Stage 4: 5 days in S3 medium supplemented with KGF (50ng/ml), Sant1 (0.25 μM), retinoic acid (0.1 μM) and Rock Inhibitor (10 μM).
Stage 5: 7 days in BE5 medium supplemented with Betacellulin (20ng/ml), XXI (1 μM), Alk5i-II (10 μM) and T3 (1 μM). Sant1 (0.25 μM) was added in days 1 to 3, and retinoic acid was added at 0.1 μM in days 1 to 3, then at 0.025 μM.
Stage 6: 14-21 days in S3 medium, changed every 48h.
α Cell Differentiation Protocol
Stages 1-2: Same as β Cell Differentiation Protocol
Stage 3: 48 hours in S3 medium supplemented with LDN193189 (200nM), retinoic acid (2 μM) and Rock Inhibitor (10 μM).
Stage 4: 5 days in S3 medium supplemented with LDN193189 (200nM) and Rock Inhibitor (10 μM).
Stage 5: 7 days in S3 medium supplemented with Alk5i-II (10 μM).
Stage 6: 28 days in S3 medium supplemented with PBDU (500nM).
During feeds, the differentiating clusters were allowed to gravity-settle for 5–10 min, medium was aspirated and 300 mL of pre-warmed medium was added. All experiments involving human cells were approved by the Harvard University IRB and ESCRO committees.
For cardiomyocyte differentiations, cells were seeded as clumps on 48 well plates and differentiated using the STEMdiff Cardiomyocyte Differentiation Kit (Stem Cell Technologies, 05010).
Magnetic enrichment using CD49a and CD26
Stage 6 clusters were dissociated using TrypLE Express for 20 min at 37°C. Cells were then quenched with DMEM + 10% FBS and spun down. Remaining undissociated cell clusters were mechanically dissociated using a P1000 pipette. The dissociated single cells were resuspended in sorting buffer (PBS + 1% BSA + 2 mM EDTA) and filtered through a 37-μm mesh filter. Cells were counted and resuspended at a density of 10 million cells per 300 μL in 15 mL conical tubes. Cells were stained at room temperature for 20 min using a 1:100 dilution of anti-human CD49a PE-conjugated (BD 559596) antibody, covered from light and agitated every 3 min. Stained cells were washed twice with 15 mL of sorting buffer by spinning down (5 min, 300 g) and resuspended to their initial density of 10 million cells per 300 μl. To label with microbeads, 40 μL of anti-PE UltraPure MACS microbreads (Miltenyi 130-105-639) were added for each 10 million cells, and the cell solution was incubated for 15 min at 4°C, agitated every 5 min. The stained cells were washed twice as above, and resuspended to a target density of 25–30 million cells per 500 μl. Volumes of 500 μL (containing no more than 30 million cells) were then magnetically separated on LS columns (Miltenyi 130-042-401) in a QuadroMACS separator (Miltenyi 130-090-976) using the recommended protocol. Successful PE enrichment was verified by live-cell flow cytometry on an Attune NxT (Invitrogen) flow cytometer. The negative cell fraction was stained at room temperature for 20 min using a 1:100 dilution of anti-human CD26 APC-conjugated (Miltenyi Biotech 130-120-769) antibody, stained cells were washed as above and incubated with anti-APC MACS microbreads (130-090-855). The cells were then magnetically separated on LS columns as described above.
Human Primary Immune Cell Isolation and Co-Culture Experiments
We obtained blood from Type 1 Diabetic (T1D) and non-diabetic (ND), de-identified donors from University of Massachusetts Medical School. For unmatched blood, fresh Leukopaks from human donors were purchased (Stem Cell Technologies, 70500.1). Human primary peripheral mononuclear cells (PBMCs) were isolated using the density gradient medium, Ficoll-Paque Plus (GE health care life sciences, 17144002) and the SepMate tubes (Stem Cell Technologies, 85450); T cells were isolated using EasySep Human T Cell Isolation Kit (Stem Cell Technologies, 17951). Isolated T cells were cultured in X-VIVO 10 (Lonza, 04-380Q) media supplemented with 5% Human AB Serum (Valley Biomedical, HP1022HI), 5% Fetal Bovine Serum (ThermoFisher Scientific, A3840101), 1% Penicillin/Streptomycin (ThermoFisher Scientific, 15070063), GlutaMAX (ThermoFisher Scientific, 35050061), MEM Non-Essential Amino Acids (ThermoFisher Scientific, 11140050).
T Cell Activation Assay
iPSC-β and α were used as target cells. Fifty to 100,000 target cells were plated on 96-well round bottom plates and treated with IFNγ, 100 ng/ml (Peprotech, 300-02) for 48h or thapsigargin, 5uM (Sigma Aldrich, T9033) for 5h before the assay. Cells were washed to remove residual thapsigargin and IFNγ. The immune/target cell ratio for co-culture is indicated in the figure legend. After a 48 hour co-culture, CD4+ and CD8+ T cells were stained for T cell activation markers CD69 and CD25. T cells activated with Dynabeads Human T-Activator CD3/CD28 beads (ThermoFisher Scientific, 111.61) for 48 hours were used as positive control. The results are presented as adjusted mean MFI, with the mean MFI of unstimulated PBMCs subtracted from the mean MFI of activation.
Flow cytometry
Intracellular Marker Staining
Differentiated clusters, sampled from suspension cultures (1–2 ml), were dissociated using TrypLE Express (GIBCO, 12604013) at 37°C, mechanically disrupted into single cells, fixed using 4% PFA for 30 min at room temperature and stored in PBS at 4°C. For staining, fixed single cells were incubated in Perm/Wash Buffer (BD Biosciences, 554723) for 30 min at room temperature, then incubated in Perm/Wash Buffer with primary antibodies (1 h at room temperature), washed three times with Perm/Wash Buffer, incubated with secondary antibodies in Perm/Wash Buffer (1 h at room temperature), washed three times and resuspended in Perm/Wash Buffer. Stained cells were analyzed using the Attune NxT (ThermoFisher) flow cytometer. A sample gating strategy is shown in Figure S1. Results presented in this study are representative of more than ten differentiations.
Surface Marker Staining
PBS containing 4% Fetal Bovine Serum (FBS) was used as blocking and staining buffer. Immune cells or other dissociated single cells were washed and blocked with blocking buffer for 30 min at 4°C, then incubated in blocking buffer with conjugated antibodies (1h at 4°C), washed three times with blocking buffer, fixed using 4% PFA for 30 min at room temperature and stored in PBS at 4°C. Stained cells were analyzed using the Attune NxT (ThermoFisher) flow cytometer.
Immunofluorescence Microscopy
Differentiated clusters were fixed in 4% PFA for 1 h at room temperature, washed, frozen in OCT (Tissue-Tek) and sectioned. For staining, slides were incubated in CAS block (ThermoFisher, 008120) with primary antibody overnight at 4°C, washed three times, incubated in secondary antibody for 2 h at room temperature, washed, mounted in ProLong Diamond Antifade Mountant with DAPI, covered with coverslips and sealed with clear nail polish. Representative regions were imaged using Zeiss.Z2 with Apotome or Zeiss Cell Discoverer 7 microscopes. Images shown are representative of similar results in at least three biologically separate differentiations from matched or similar stages.
Quantitative Real-time PCR (qPCR)
Cells were treated with TRIzol (ThermoFisher Scientific) for RNA extraction following the manufacturer’s protocol. Purified RNA was reverse-transcribed into cDNA using the SuperScript IV first-strand synthesis kit (Invitrogen). GAD, PTPRN, G6PC2, SLC30A, IAPP and INS probes for TaqMan assays were purchased from ThermoFisher Scientific. All qPCR assays were performed using a QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems).
NanoString gene array
Stage 6 cells from the β Cell Differentiation Protocol were enriched as described in the section ‘Magnetic enrichment using CD49a and CD26.’ Prior to the nanostring assay, the enriched populations, CD49a+ (iPSC-β) and CD26+ (iPSC-α) fractions were lysed using the RLT buffer (RNeasy Lysis Buffer, QIAGEN). An nCounter gene expression assay was performed according to the manufacturer’s protocol. The assay utilized a custom-made NanoString codeset designed to measure 24 transcripts, including 3 putative housekeeping transcripts (see Table S2). The data was normalized to the average counts for all housekeeping genes in each sample and analyzed with nSolver software (NanoString Technologies).
Protein Extraction and Immunoblotting
Cells were homogenized in RIPA buffer (ThermoFiscer, 89901) supplemented with protease and phosphatase inhibitors (Abcam, 201119). Total protein content was determined by BCA assay (ThermoFisher, 23225). Equal protein amounts (˜10 mg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes (EMD Millipore). Membranes were immunoblotted with the indicated antibodies: PERK (Cell Signaling Technology, 5683S), PDI (Cell Signaling Technology, 3501S), BIP (Cell Signaling Technology, 3177S), GAPDH (Abcam, 181602).
Cytokines Analysis
Supernatant of co-culture experiments were assayed using the MSD proinflammatory custom panel, a highly sensitive multiplex enzyme-linked immunosorbent assay (ELISA) for quantitatively measuring cytokines including interferon γ (IFN-γ), interleukin (IL)-1β, IL-2, IL17, and C-X-C motif chemokine 10 (CXCL10) from the supernatants using an electrochemiluminescent detection method (MesoScale Discovery, Gaithersburg, MD, USA).
Viability and Apoptosis Assay
48h prior to co-culture with PBMCs, differentiated clusters were dissociated and reaggregated in V-bottom 96-well plates. PBMCs were then co-cultured to re-aggregated iPSC-islets, with and without 5h pre-treatment with 5uM thapsigargin (Sigma Aldrich, T9033). For live counts of iPSC-β or iPSC-α, clusters were dissociated, and single cells were fixed (1% paraformaldehyde) and stained with rat anti-C-peptide (DHSB, GN-ID4) and mouse anti-glucagon (Santa Cruz Biotech, SC-514592) antibodies for flow cytometry. For the apoptosis assay, clusters were dissociated, and single cells were stained at room temperature for 30 min using a 1:100 dilution of recently reported stem cell derived β-cell marker, anti-human CD49a (Veres et al., 2019) PE-conjugated (BD Biosciences, 559596), α-cell marker anti-human CD26 APC-conjugated (Miltenyi Biotech, 130-120-769), and Apopxin green indicator (Abcam, ab176750).
Lentivirus Preparation and Transduction
Lentiviral particles were produced by transfecting 293T cells (Takara Bio, Mountain View, CA, USA) with the packaging vectors pHDM-vsvg, pHDM-tat, pHDM-rev, and pHDM-gag/pol along with lentiviral backbone vectors using the TransIT-293 transfection reagent (Mirus, Madison, WI, USA). The following lentiviral vectors were used: non-targeting guide RNA (5′-TTTACGATCTAGCGGCGTAG-3′) or a B2M guide RNA (5′-GCTACTCTCTCTTTCTGGCC-3′) cloned into lentiCRISPRv2 [a gift from Feng Zhang (Addgene plasmid # 52961; http://addgene:52961; RRID:Addgene_52961 (Sanjana et al., 2014)]. Lentiviral particles were concentrated 48 h and 72 h post transfection using the PEG-IT virus precipitation reagent (Fisher Scientific, Waltham, MA, USA) overnight at 4°C followed by centrifugation at 1500 g for 30 min at 4°C and stored at −80°C. For transduction, cell clusters collected from spinner flask suspension cultures were dissociated in TrypLE Express (Life Technologies, Carlsbad, CA, USA) for 7 min, followed by mechanical dissociation and centrifugation at 300 g for 5 min at room temperature (RT). Cell pellets were resuspended at a density of 2.5 million cells/mL in the stage-matched medium with polybrene reagent (Santa Cruz, Dallas, TX, USA) at 8μg/mL. Single-cell suspensions were combined with concentrated lentiviral particles and plated on ultra-low attachment six-well plates on a rocker plate set at 70rpm in a humid 37°C incubator and 5% CO2.
Quantification and Statistical Analysis
Statistical analyses are described in detail where reported. Statistical analyses were carried out using Graphpad Prism software. Statistical assays were performed as described in each figure legend. n represents number of biological replicates in all cases where reported. Biological replicates refer to unique donor-derived batches of human islets or unique differentiations of iPSC-derived cells produced from unique suspension cultures.
Acknowledgments
We thank J. Babon, R. Pop, A.L. Faust, and K. Boulanger for discussions and feedback on the manuscript and the iPSC and Flow Cytometry Core Facilities at Harvard University for their support. D.A.M. is an Investigator of the Howard Hughes Medical Institute. N.C.L. is supported by American Diabetes Association grant #1-19-PMF-024. E.S. is supported by Juvenile Diabetes Research Foundation (JDRF) Post-Doctoral Fellowship (3-PDF-2018-590-A-N). This work was supported by grants from the Harvard Stem Cell Institute, the Helmsley Charitable Trust (2015PG-T1D044), JDRF (5-SRA-2014-284-Q-R), National Institutes of Health (NIH): NIH UO1DK104218, NIH R24OD0426640, and NIH UC4DK104218, and the JPB Foundation (award no. 1094).
Author Contributions
N.C.L. and E.S. conceived the study. The experiments were performed by N.C.L. and E.S. T.B.M. provided technical support and conceptual advice. M.A.B., D.L.G., and D.M.H. were involved in the experimental design and patient recruitment and advised on the project. N.C.L., E.S., and D.A.M. wrote the manuscript. D.A.M. designed and supervised the research.
Declaration of Interests
D.A.M. is a founder of Semma Therapeutics and an advisor to Vertex, who have licensed technologies developed in the Melton lab. All of the other authors declare no competing interests.
Published: July 14, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107894.
Supplemental Information
References
- Banting F.G., Best C.H., Collip J.B., Campbell W.R., Fletcher A.A. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can. Med. Assoc. J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- Campbell-Thompson M.L., Filipp S.L., Grajo J.R., Nambam B., Beegle R., Middlebrooks E.H., Gurka M.J., Atkinson M.A., Schatz D.A., Haller M.J. Relative Pancreas Volume Is Reduced in First-Degree Relatives of Patients With Type 1 Diabetes. Diabetes Care. 2019;42:281–287. doi: 10.2337/dc18-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso A., Licenziati S., Corulli M., Canaris A.D., De Francesco M.A., Fiorentini S., Peroni L., Fallacara F., Dima F., Balsari A., Turano A. Flow cytometric analysis of activation markers on stimulated T cells and their correlation with cell proliferation. Cytometry. 1997;27:71–76. doi: 10.1002/(sici)1097-0320(19970101)27:1<71::aid-cyto9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Cibrián D., Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M., Kroger C.J., Tisch R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017;8:1898. doi: 10.3389/fimmu.2017.01898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters K.T., Dotta F., Amirian N., Campbell P.D., Kay T.W., Atkinson M.A., Roep B.O., von Herrath M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012;209:51–60. doi: 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culina S., Lalanne A.I., Afonso G., Cerosaletti K., Pinto S., Sebastiani G., Kuranda K., Nigi L., Eugster A., Østerbye T., ImMaDiab Study Group Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci. Immunol. 2018;3:eaao4013. doi: 10.1126/sciimmunol.aao4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmastro M.M., Piganelli J.D. Oxidative stress and redox modulation potential in type 1 diabetes. Clin. Dev. Immunol. 2011;2011:593863. doi: 10.1155/2011/593863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuse T., Hu X., Gravina A., Wang D., Tediashvili G., De C., Thayer W.O., Wahl A., Garcia J.V., Reichenspurner H. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019;37:252–258. doi: 10.1038/s41587-019-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik D.L., Cardozo A.K., Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- Eizirik D.L., Miani M., Cardozo A.K. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013;56:234–241. doi: 10.1007/s00125-012-2762-3. [DOI] [PubMed] [Google Scholar]
- Engin F. ER stress and development of type 1 diabetes. J. Investig. Med. 2016;64:2–6. doi: 10.1097/JIM.0000000000000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Wang M., Duan S., Franco P.J., Kenty J.H., Hedrick P., Xia Y., Allen A., Ferreira L.M.R., Strominger J.L. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2019;116:10441–10446. doi: 10.1073/pnas.1902566116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hober D., Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat. Rev. Endocrinol. 2010;6:279–289. doi: 10.1038/nrendo.2010.27. [DOI] [PubMed] [Google Scholar]
- Kracht M.J., van Lummel M., Nikolic T., Joosten A.M., Laban S., van der Slik A.R., van Veelen P.A., Carlotti F., de Koning E.J., Hoeben R.C. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat. Med. 2017;23:501–507. doi: 10.1038/nm.4289. [DOI] [PubMed] [Google Scholar]
- Like A.A., Rossini A.A. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- Marre M.L., McGinty J.W., Chow I.T., DeNicola M.E., Beck N.W., Kent S.C., Powers A.C., Bottino R., Harlan D.M., Greenbaum C.J. Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4+ T Cells in Patients With Type 1 Diabetes. Diabetes. 2018;67:1356–1368. doi: 10.2337/db17-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millman J.R., Xie C., Van Dervort A., Gürtler M., Pagliuca F.W., Melton D.A. Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat. Commun. 2016;7:11463. doi: 10.1038/ncomms11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair G.G., Liu J.S., Russ H.A., Tran S., Saxton M.S., Chen R., Juang C., Li M.L., Nguyen V.Q., Giacometti S. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019;21:263–274. doi: 10.1038/s41556-018-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca F.W., Millman J.R., Gürtler M., Segel M., Van Dervort A., Ryu J.H., Peterson Q.P., Greiner D., Melton D.A. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson Q.P., Veres A., Chen L., Slama M.Q., Kenty J.H.R., Hassoun S., Brown M.R., Dou H., Duffy C.D., Zhou Q. A method for the generation of human stem cell-derived alpha cells. Nat. Commun. 2020;11:2241. doi: 10.1038/s41467-020-16049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pociot F. Type 1 diabetes genome-wide association studies: not to be lost in translation. Clin. Transl. Immunology. 2017;6:e162. doi: 10.1038/cti.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A. The multiple origins of Type 1 diabetes. Diabet. Med. 2013;30:135–146. doi: 10.1111/dme.12081. [DOI] [PubMed] [Google Scholar]
- Pugliese A. Autoreactive T cells in type 1 diabetes. J. Clin. Invest. 2017;127:2881–2891. doi: 10.1172/JCI94549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnell S.E., Peterson P., Trinh L., Broberg P., Leander P., Lernmark Å., Månsson S., Elding Larsson H. Pancreas volume and fat fraction in children with Type 1 diabetes. Diabet. Med. 2016;33:1374–1379. doi: 10.1111/dme.13115. [DOI] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Arora P., Rubin A., Batushansky I., Asadi A., O’Dwyer S., Quiskamp N., Mojibian M., Albrecht T. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Richardson S.J., Rodriguez-Calvo T., Gerling I.C., Mathews C.E., Kaddis J.S., Russell M.A., Zeissler M., Leete P., Krogvold L., Dahl-Jørgensen K. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. 2016;59:2448–2458. doi: 10.1007/s00125-016-4067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ H.A., Parent A.V., Ringler J.J., Hennings T.G., Nair G.G., Shveygert M., Guo T., Puri S., Haataja L., Cirulli V. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D., Kaufman R.J. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit F.C., In’t Veld P.A., Pipeleers D.G. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 1988;85:3865–3869. doi: 10.1073/pnas.85.11.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A.M., Lakey J.R., Ryan E.A., Korbutt G.S., Toth E., Warnock G.L., Kneteman N.M., Rajotte R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- Shatrova A.N., Mityushova E.V., Vassilieva I.O., Aksenov N.D., Zenin V.V., Nikolsky N.N., Marakhova I.I. Time-Dependent Regulation of IL-2R α-Chain (CD25) Expression by TCR Signal Strength and IL-2-Induced STAT5 Signaling in Activated Human Blood T Lymphocytes. PLOS ONE. 2016;11:e0167215. doi: 10.1371/journal.pone.0167215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley R.K., Sutherland D.E., Goetz F., Michael A.F. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Lab. Invest. 1985;53:132–144. [PubMed] [Google Scholar]
- Sutherland D.E., Sibley R., Xu X.Z., Michael A., Srikanta A.M., Taub F., Najarian J., Goetz F.C. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans. Assoc. Am. Physicians. 1984;97:80–87. [PubMed] [Google Scholar]
- van Belle T.L., Coppieters K.T., von Herrath M.G. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- van der Torren C.R., Zaldumbide A., Duinkerken G., Brand-Schaaf S.H., Peakman M., Stangé G., Martinson L., Kroon E., Brandon E.P., Pipeleers D., Roep B.O. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia. 2017;60:126–133. doi: 10.1007/s00125-016-4125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velthuis J.H., Unger W.W., Abreu J.R., Duinkerken G., Franken K., Peakman M., Bakker A.H., Reker-Hadrup S., Keymeulen B., Drijfhout J.W. Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes. 2010;59:1721–1730. doi: 10.2337/db09-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres A., Faust A.L., Bushnell H.L., Engquist E.N., Kenty J.H., Harb G., Poh Y.C., Sintov E., Gürtler M., Pagliuca F.W. Charting cellular identity during human in vitro β-cell differentiation. Nature. 2019;569:368–373. doi: 10.1038/s41586-019-1168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virostko J., Williams J., Hilmes M., Bowman C., Wright J.J., Du L., Kang H., Russell W.E., Powers A.C., Moore D.J. Pancreas Volume Declines During the First Year After Diagnosis of Type 1 Diabetes and Exhibits Altered Diffusion at Disease Onset. Diabetes Care. 2019;42:248–257. doi: 10.2337/dc18-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . 2016. Global Report on Diabetes.https://www.who.int/diabetes/global-report/en/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published report includes all data generated or analyzed during this study. No code was used or generated during this study.