

Abstract
Identifying the molecular mechanisms that control differential gene expression (DE) is a major goal of basic and disease biology. We develop a systems biology model to predict DE, and mine the biological basis of the factors that influence predicted gene expression, in order to understand how it may be generated. This model, called DEcode, utilizes deep learning to predict DE based on genome-wide binding sites on RNAs and promoters. Ranking predictive factors from the DEcode indicates that clinically relevant expression changes between thousands of individuals can be predicted mainly through the joint action of post-transcriptional RNA-binding factors. We also show the broad potential applications of DEcode to generate biological insights, by predicting DE between tissues, differential transcript-usage, and drivers of aging throughout the human lifespan, of gene coexpression relationships on a genome-wide scale, and of frequently DE genes across diverse conditions. Researchers can freely utilize DEcode to identify influential molecular mechanisms for any human expression data - www.differentialexpression.org.
Introduction
While all human cells share DNA sequences, gene regulation differs among cell types and developmental stages, and in response to environmental cues and stimuli. Accordingly, when gene expression is not properly regulated, cellular homeostasis can be perturbed, often affecting cell function and leading to disease1. These distinctions between cell states are observed as differential expression (DE) of gene transcripts. DE have been catalogued for tens of thousands of gene expression datasets, in the context of distinctions between species, organs, and conditions. Despite the important and pervasive nature of DE, it has been challenging to shift from these observations towards a coherent understanding of the underlying generative processes that would essentially decode DE– a transition which is essential for progress in basic and disease biology. We address this gap by exploiting novel computational and systems biology approaches to develop a predictive model of DE based on genome-wide regulatory interaction data.
Diverse molecular interactions have been shown to generate DE, and jointly regulate gene expression at the transcriptional and post-transcriptional levels. Major classes of gene regulatory interactions have been catalogued at the genomic scale, including transcription factor (TF)-promoter interactions2, protein-RNA interactions3, RNA-RNA interactions4, chromatin interactions5, and epigenetic modifications on DNAs6, histones, and RNAs7. Statistical models of gene expression can help fulfil the purpose of these resources in describing the origins of gene regulation and DE1. However, such raw data resources have outpaced model development, likely due to the challenge of uniting diverse molecular data into a single accurate model.
Among many possible statistical approaches to predicting DE, deep learning (DL) blends diverse data sources in a way that approximates the convergence of regulatory interactions. Indeed, DL has been applied to genomic research8, 9 including RNA splicing10, genomic variant functions11, and RNA/DNA binding12. However, accurate prediction is only one component of understanding DE; additional genomic and systems biology analysis are helpful in understanding how predictions are fuelled by existing molecular concepts, mechanisms, and classes.
To decode the basis of DE in terms of molecular regulatory interactions, we first learn to predict it with a high degree of accuracy, using a DL model we call “DEcode”. This model combines several types of gene regulatory interactions and allows us to prioritize the main systems and molecules that influence DE on a tissue-specific basis. We further establish likely molecular mechanisms for this gene regulation and validate the influence of the predicted strongest regulators. In parallel, we predict the origin of person-to-person DE, which is the major component of experimental and clinical studies. These particularly challenging predictions are validated on a genome-wide scale, as we identify key drivers of coexpression, and also drivers for phenotype-associated DE. Utilizing diverse genomic datasets, we identify a complex, yet strikingly consistent set of principles that control DE. DEcode can be applied to the majority of current and future gene expression data, to accelerate basic and disease biology, by identifying the origins of DE in each experiment.
Results
Predicting differential expression across human tissues
The overarching goal of this study is to understand the major regulatory principles of DE signature by predicting gene expression as a function of molecular interactions. These results should be tissue-specific and ideally have sufficient accuracy to predict the relatively small expression changes observed between individual humans. To accomplish this, we utilized deep convolutional neural networks in a system called DEcode that can predict inter-tissue variations and inter-person variations in gene expression from promoter and mRNA features (Figure 1). The promoter features included: the genomic locations of experimentally-determined binding sites of 762 TFs13 and the mRNA features encompassed the locations of experimentally-determined binding sites of 171 RNA-binding proteins (RBPs)14 and predicted binding sites of 213 miRNAs15 (Supplementary Table 1). First, we applied the DEcode framework to tissue-specific human transcriptomes of 27,428 genes and 79,647 transcripts measured in the GTEx consortium16 to predict log-fold changes across 53 tissues against the median expression of all tissues, as well as the median expression of all tissues with a multi-task learning architecture. The predicted median expression levels showed high consistency with the actual observations for both gene-level (Spearman's rho = 0.81) and transcript-level (Spearman's rho = 0.62) (Figure 2a). Moreover, the model predicted the differential transcript usage within the same gene (Spearman's rho = 0.44). The DEcode models also predicted the DE profiles across 53 tissues for both gene (mean Spearman's rho = 0.34), transcript (mean Spearman's rho = 0.32), and transcript-usage levels (mean Spearman's rho = 0.16) (Figure 2b). The predicted gene expression for the testing genes was indeed tissue-specific, as they showed less correspondence with the expression profiles from alternate tissues (Figure 2c).
Figure 1.

Overview of building and evaluating the DEcode transcriptome prediction model. DEcode takes the promoter features and the mRNA features for each gene/transcript as inputs and outputs its expression levels under various conditions. We conducted a series of evaluations to show that DEcode defines major principles in gene regulation in arbitrary gene expression data. This capacity is strongly supported on a comparative basis to alternative methods, and on an absolute basis across diverse applications, which include, through predictions of transcript-usage, person-specific gene expression, frequently DE genes of multiple external disease-related gene sets.
Figure 2.

Performance of DEcode for predicting differential expression across 53 tissues. (a) Predictive performances on the median absolute expression levels across tissues. The predicted log2-TPM values for 2,705 genes and 7,631 transcripts, and the transcript usage for 1,485 genes that had multiple transcripts were compared with the actual expression using Spearman's rank correlation. (b) Predictive performances on the tissue-specific expression profiles. The predicted fold changes of 2,705 genes and 7,631 transcripts were compared with the actual data using Spearman's rank correlation. Predictive accuracies of the differences in the transcript usage across tissues were also computed for 1,485 genes. The colour of the bar indicated the tissue groups based on the similarity of gene expression profiles. (c) The heatmap showing pairwise correlations between the predicted and the actual tissue-specific expression profiles of 53 tissues for the testing genes. (d) Performance comparison of DEcode with ExPecto. The root-mean-square errors (RMSE) of DEcode models for expression-levels of 714 genes coded on chromosome 8 was compared with those of ExPecto. The relative improvement of DEcode over ExPecto in median RMSE of the 10 runs was displayed as a bar plot and the error bar represents median absolute deviation.
To provide context for the statistical benefit of the use of systems biology data for predicting DE, we contrast it to a raw-sequence-based method called ExPecto11, as it was designed to predict GTEx gene expression from epigenetic states, estimated from promoter sequences via DL. In this comparison, DEcode showed an average of 6.1% improvement in root mean square error over ExPecto (Figure 2d) which translates into an average correlation coefficient with actual gene expression of 0.42 - a 50% increase over 0.28 from ExPecto (Supplementary Figure 1).
Regulators for differential expression across tissues
Beyond the predictive performance of DEcode, we utilize the model to help define the biological processes regulating DE. Many studies have demonstrated that TFs-promoter interactions are critical determinants of transcriptional activity of promoters and thereby define gene expression levels2. However, it is unclear to what extent RNA features, which we define as each RNA’s binding sites of RBPs and miRNAs, contribute to gene expression levels. To answer this question, we re-trained the DEcode model, randomizing either RNA features, promoter features, or both. We found that RNA features alone explained the gene-level log-fold changes less accurately than the model with all features, but as well as the model trained with promoter features (Figure 3a). Interestingly, the RNA-based model performed better than the promoter-based model for the prediction of the transcript expression and differential transcript-usage.
Figure 3.

Identification and characterization of key predictors. (a) The predictive performances of the models trained with a different set of features. The performances for the fold-change across 53 tissues were compared with the paired sample t-test. (b) Comparison of predictive contribution among classes of regulators. The regulator’s predictive contribution to the fold-change across 53 tissues was estimated based on DeepLIFT score. The two-sample Wilcoxon test was used to assess the statistical significance of the differential contribution. (c) GO enrichment map for key predictors. GO enrichment analysis for key predictors for the fold-change across 53 tissues was conducted using the pre-ranked GSEA. The significant associations (FDR<0.05) were visualized with green/blue node colours representing certain classes of regulators. (d) Key predictors for the tissue-specific transcriptomes. DeepLIFT scores of the top 5 key predictors in each tissue were displayed as a heatmap. (e) Example relationships between the predictive contributions of a regulator and its expression levels across tissues. A line of best fit based on linear regression was depicted with 95 percent confidence intervals. (f) The overlap between the key predictors for the fold-change across 53 tissues and LoF intolerant genes, OMIM genes, lethal genes in mice, essential genes in cell lines, hub proteins in protein-protein interaction networks.
To further quantify the importance of each regulator weighted in the DEcode models, we calculated DeepLIFT scores, which are a measure of the additive contribution of its binding site to each prediction17, 18, for each task of predicting fold-change (Supplementary Table 2). Based on the DeepLIFT scores, TFs and RBPs showed comparable effects on the gene-level prediction (Figure 3b). However, at the transcript and transcript-usage-level, RBPs tended to influence the prediction more than TFs. We also found biological processes enriched for the influential predictors ranked by the DeepLIFT scores that are coherent with known gene regulation (Figure 3c and Supplementary Table 3). For instance, the predictors for gene and transcript expression were associated with histone modification, whereas splicing-related factors were only enriched for predictors of transcript and transcript-usage.
We also examined critical predictors for each of 53 tissues (Figure 3d). The DeepLIFT scores across tissues recapitulated the contribution of binding sites of known master regulators in each tissue such as REST for brain tissues19, SPI1 and RUNX1 for immune-related tissues20, TP63 and KLF4 for skin21, HNF4A for liver22, and PPARG for adipose-related tissues23, which suggested the differences in predictive contributions of binding sites of a given regulator reflect the differential activities of regulators across tissues. We hypothesized that the differential activities of a regulator could be in part explained by the relative abundance of a regulator across tissues. Based on this hypothesis, we contrasted DeepLIFT scores for the binding sites of each regulatory factor and its expression levels across tissues. We indeed found that 99 RBPs and 410 TFs showed significant correlations between DeepLIFT scores of their binding sites and their expression levels (FDR < 5%) (Supplementary Figure 2). These relationships were not based on differences in expression profiles between brain and non-brain tissues, as the relationships remained the same without brain tissues (Supplementary Figure 3). The sign of the correlation possibly reflects whether the binding of a regulator to RNA increased or decreased the abundance of the RNA. For instance, the model suggested that PPARG and PTBP1 are positive regulators of gene expression as DeepLIFT scores of PPARG or PTBP1 binding sites were higher in the tissues expressing PPARG or PTBP1 at higher levels (Figure 3e), which are consistent with their known functions23,24. Conversely, the expression levels of REST, a transcriptional repressor19, or METTL14, an RNA methyltransferase destabilizing RNAs25, showed inverse correlations with their DeepLIFT scores as expected. These results indicated that the DEcode model is interpretable and reflects biological mechanisms for controlling RNA abundance.
We hypothesized that if key predictors in DEcode models are truly impactful transcriptome regulators, then defects in such regulators would have significant impacts on cellular phenotypes and thereby lead to disease. To examine this hypothesis, first, we obtained genes whose loss-of-function (LoF) mutations are depleted through the process of natural selection, from the Exome Aggregation Consortium (ExAC)26 and thus that are considered to play important roles in individual fitness. We found that these LoF-mutation-intolerant regulators had greater DeepLIFT score magnitudes for the prediction of the fold-change across 53 tissues (Figure 3f and Supplementary Table 4). In particular, these associations are based on genes that are intolerant to LoF mutations only in a single allele (Supplementary Figure 4). Further, we confirmed that mutations in the predicted key regulators tend to cause genetic disorders based on the assessment with the Online Mendelian Inheritance in Man (OMIM). Interestingly, their roles on fitness are likely preserved across species, as dysfunctions of the predicted key regulators also lead pre-weaning lethality in mice. Moreover, we found the loss-of-function of the predicted key regulators of the transcriptome could also impair cellular viability. Intriguingly, the key predictors are more likely to be hubs in protein-protein interaction networks, which is the same property observed with disease-causing genes27. These results were robust, as they were also supported by the DeepLIFT scores for the transcript and transcript-usage prediction (Supplementary Figure 5). Together, the results indicated that the key predictors of transcriptome indeed play critical roles in maintaining vital cellular and body functions. Thus the DEcode model could be used to prioritize disease-causing genes, and this capability points toward the broader validity of predicted key regulators.
Predicting differential expression across individuals
Next, we asked whether the same input of promoter and RNA features could also predict relative expression differences across individuals within the same tissue. We extended the DEcode framework to model DE across individuals for 14 representative tissues with a sample size greater than 100 in GTEx. This was challenging, as the average variance in gene expression within tissues was less than 25% of that between tissues (Supplementary Figure 6).
The person-specific models successfully predicted fold changes across individuals with a mean Spearman’s correlation of ~0.28 (Figure 4a). The performance was further increased to 0.34 when we filtered out the models that worked poorly for the validation data (Supplementary Figure 7). The models were indeed person-specific, as they did not predict gene expression profiles of unrelated individuals. To examine if the model captured the person-specific expression shared across tissues28, we compared gene expression between tissues within the same individuals and between different individuals. The predicted expression showed better concordance between tissues from the same individuals, as is the case with actual expression data, which indicated the model captured the person-specific regulatory mechanisms, even though we did not use any direct information that could identify individuals (Figure 4b).
Figure 4.

Performance of the person-specific models. (a) The predictive performances of the person-specific models for the actual data from the same individuals and unrelated random individuals. (b) The person-specific models predicted person-specific expressions shared across tissues. (c) The performances of the models trained with a distinct feature set. (d) Per-person key predictive regulators for the hippocampus transcriptome. We selected the top 5 key predictors of the hippocampus transcriptome for each individual and their DeepLIFT scores were displayed as a heatmap. (e) Comparison of per-gene predictive accuracy between DEcode and PrediXcan. The number of genes that showed a positive Pearson’s correlation between predicted and actual gene expression levels at FDR 5% was calculated for each method. The testing genes on chromosome 1 were used in this comparison. (f) Per-gene prediction accuracy is associated with the number of features present in RNAs and promoters. The histogram represents Pearson’s correlations between the predicted and the actual expression for each gene. The line plot shows the average number of RNA and promoter features of genes in each bin of the histogram. Spearman’s correlation between the number of features and per-gene correlations is displayed in the line plot. The error bars indicate standard errors.
Next, to gauge the contribution of RNA and promoter features to the person-specific expression profiles, we re-trained models with randomized RNA features, promoter features, or both. The RNA-feature-based model performed on average 85% as well as the model trained with all features. This corresponded to an average 173% performance gain, compared to the promoter-feature-based model, which suggested that the post-transcriptional controls are the major determinants of the DE across individuals (Figure 4c). The model also allowed us to investigate the person-specific activities of regulators by calculating DeepLIFT scores (Figure 4d). At least 100 of regulators out of 933 regulators in each tissue showed a significant correlation between their DeepLIFT scores and expression levels across individuals (Extended Data Figure 1a). The signs of these correlations were consistent between tissues, and consistent with those of the cross-tissue model (Extended Data Figure 1bc). This suggested that DE between individuals and between tissues can be modelled by the universal relationships between regulators and their targets.
To examine whether specific genes contributed to the per-person accuracy of the predicted gene expression, we also assessed its accuracy on a per-gene basis. The predicted expression of a majority of the testing genes (78% on average) showed significant positive correlations with the actual gene expression (FDR<5%). To assess whether systems biology data improved per-gene performance over a raw-sequence-based method, we compared DEcode with PrediXcan29, which predicts person-specific gene expression from genetic variations in cis-regulatory regions of genes. The PrediXcan model predicted gene expression levels of only about 11% of the testing genes at FDR less than 5%, which was far less than that of DEcode (Figure 4e). This suggested that the differential activity of transcriptional and post-transcriptional regulators has a larger effect on gene expression than genetic variations in cis-regulatory regions.
The genes that DEcode could predict with the greatest accuracy were similar across tissues (Extended Data Figure 1d). This suggested that the predictability of gene expression is defined by gene characteristics rather than a target tissue. We, therefore, explored gene characteristics that were associated with the per-gene accuracy of the predicted expression. We found that the models showed higher performance for the genes that are registered in multiple gene annotation databases than those found only in the GENCODE database (Extended Data Figure 1e). Since the GENCODE-specific genes are novel or putative and thus their annotations might not be well established, it is reasonable that the performance of the model for those genes was lower than other well-established genes. Beyond the annotation reliability, we found that the number of known binding features for each gene had a larger effect on the predictability (Extended Data Figure 1f). This suggested that the more information on RNA and promoter interactions is available, the more the prediction becomes accurate. Interestingly, the number of binding features in RNAs was a stronger determinant of the predictive accuracy than that in promoter regions (Figure 4f). RNA-protein interactions are largely missing as global RNA-binding profiles are available for only about 10% of known RBPs30. Thus, the incompleteness of RNA features is likely to be an origin of lower accuracy for a portion of genes.
Generative process of trait-related expression changes
Next, we asked whether the person-specific expression profiles predicted by the DEcode models also retained trait-associated DE changes. For this, we conducted DE analysis against the donor’s age and sex using the predicted gene expression data. Notably, test statistics of the predicted data showed significant positive correlations with those of the actual data in all tissues for both traits (Figure 5a). Especially, age- and sex-specific expression changes were well preserved in the predicted data in lung (Spearman's rho = 0.59, P < 2.2e-16) and hippocampus (Spearman's rho = 0.47, P < 2.2e-16), respectively. The predicted associations were tissue-specific as they were the closest to those of corresponding tissues in 9 and 11 out of 14 tissues for age and sex, respectively (Figure 5b). We also explored the regulators for the age- and sex-related gene expression changes by associating regulator's DeepLIFT scores with age and sex. We found that many regulators, for instance, 717 in the tibial artery and 904 in the breast mammary tissue, showed age- and sex-dependent changes at FDR 5%, respectively (Figure 5c and Supplementary Table 5), which showed the capability of DEcode to associate transcriptional regulators with phenotypes. Although there were more TFs associated with phenotypes than RBPs and miRNAs, overall collective impacts of RNA features on the generative process of DEs for age and sex were greater than those of promoter features in most tissues (Supplementary Figure 8).
Figure 5.

Application of the person-specific models to analyze phenotype-related gene signatures. (a) The scatter plots showing the relations between the associations of genes with age and sex using predicted expression and those using the actual expression. Spearman’s correlation between t-statistics based on the predicted and the actual gene expression is displayed in the scatter plot. (b) The pairwise Spearman’s correlations between the predicted and the actual associations of genes with age and sex in all tissues. The numbers in diagonal elements of the heatmap indicate the ranks of similarity of the predictions with the actual observations in the corresponding tissues. (c) The regulators whose DeepLIFT scores were associated with age or sex. The Benjamini–Hochberg procedure was used to control the false discovery rate at 5% for each phenotype.
Regulatory basis of gene co-expression relationships
Co-expression analysis is a frequent component of transcriptome studies as gene-to-gene co-expression relationships are regarded as functional units of the transcriptional system31. Therefore, we examined if the DEcode models could detect known gene co-expression relationships. These tests were both a potential validation of the person-specific DEcode predictions, and a means to explore the biological basis of co-expression. We found that the gene co-expression relationships in the predicted gene expression profiles separated gene pairs with positive and negative correlation in the actual gene expression data in each tissue (Figure 6a). Furthermore, the predicted gene expression profiles also detected inter-tissue co-expression relationships (Figure 6b). The accuracy of these results motivated us to investigate key factors driving co-expression, via the DEcode predictions. RNA features alone could explain co-expression relationships better than promoter features in most tissues (Extended Data Figure 2), which again suggested the significant contribution of RNA features to person-specific transcriptomes.
Figure 6.

Regulatory basis of gene co-expression relationships. (a) Co-expression relationships in the predicted gene expression. We defined the ground truth co-expression relationships as gene pairs with the absolute Spearman’s correlation greater than 0.7 in the actual testing data. The density of Spearman’s correlation between the co-expressed gene pairs in the predicted gene expression data was estimated based on the Gaussian kernel. (b) Inter-tissue gene co-expression relationships in the predicted gene expression. We defined the ground truth co-expression relationships as gene pairs with the absolute Spearman’s correlation greater than 0.5 in the actual test data. The violin plots show Spearman’s correlation of the inter-tissue co-expressed gene pairs based on the predictions from all models or the models whose performances on validation data were greater than 50% percentile in each tissue. (c) The effect of the binding site removal on co-expression between LAPTM5 and CD53. (d) The key regulators for the co-expression between LAPTM5 and CD53. (e) Percent of the co-expression relationship explained by the expression levels of the key regulators. The white diamond and the error bars in the bar indicated the average and 95% percentile of the percent of variance explained by randomly picked regulators, respectively. The scatter plots and linear regression lines show the effect of the key regulators on the co-expression.
To further assess the capability of DEcode to decipher the mechanisms leading a specific co-expression relationship, we focused on the co-expression of LAPTM5 and CD53, which were robustly co-expressed both in the simulated expression data and the actual data in all tissues except whole-blood. Using the trained model, we simulated the consequences of disruptions of promoter and mRNA features. The co-expression relationship was weakened when the features near the transcriptional start site (TSS) and 1,000 bp downstream of the TSS in LAPTM5 or near TSS and 500 bp upstream of TSS in CD53 were removed (Figure 6c). These observed effects were reasonable as many TFs bind to these regions. We further examined the specific regulators for the co-expression relationships by simulating knockout (KO) effects of regulators. The in-silico KO experiments revealed that immune-related TFs such as SPI1 and TBX21 potentiated the co-expression relationships consistently across multiple tissues (Figure 6d). To validate if these regulators indeed induced the co-expression relationships, we conducted a mediation analysis that is an orthogonal computational method to infer the effect of regulators on downstream targets. The set of the 10 regulators together mediated up to 94% of covariance, which was significantly greater than the same number of randomly picked regulators (Figure 6e).
Molecular regulations for frequently DE genes
A recent meta-analysis of over 600 human transcriptome data revealed that some genes are more likely to be detected as DE genes than others in diverse case-control studies32. From this observation, Megan et al. formulated the “DE prior”, a global ranking of gene’s generic likelihood of being DE. The genes with high DE prior rank were significantly more enriched with frequently DE genes, as compared to other functional gene sets, such as those contained in gene ontology. However, the regulatory-origin behind the ranking of these highly responsive genes has yet to be uncovered. Therefore, we used DEcode to examine whether the DE prior rank could be generated by gene regulatory interactions, and to identify critical regulators for frequently DE genes. The ability of DEcode to predict global DE prior ranks was highly significant (P < 2.2e-16) and practically relevant (Spearman's rho = 0.53) (Extended Data Figure 3a). Furthermore, DEcode was able to identify genes with high (90th percentile and greater) DE prior probability (AUCROC = 0.81, 95% confidence interval = 0.78 - 0.84) (Extended Data Figure 3b). Re-training the model with randomized inputs indicated that TF-promoter interactions were the major factors explaining the DE prior rank. To further characterize TFs that contributed to the prediction, we defined critical TFs based on the DeepLIFT score (Supplementary Table 6) and performed pathway analysis on them. We found that critical TFs were enriched for cancer or inflammatory-related pathways (FDR<5%) such as pathways in cancer (Fold = 3.1, P = 4.2e-5), JAK-STAT signalling pathway (Fold = 6.8, P = 4.8e-5), chemokine signalling pathway (Fold = 7.3, P = 1.4e-4), and acute myeloid leukemia (Fold = 4.5, P = 3.6e-4) (Supplementary Table 7). This result is consistent with the disease-related data context for DE prior, which is 62% cancer-related and 23% inflammatory-related. Supported by the ability to predict DE prior ranks, and by the consistency of these results, this application of DEcode illustrates how it goes beyond DE gene lists, to uncover major key drivers for generating DE.
Discussion
We introduced the DEcode framework, which integrates a wealth of genomic data into a unified computational model of transcriptome regulations to predict multiple transcriptional effects in tissue- and person-specific transcriptomes. Systems biology analysis of these results provided biological insights regarding the regulatory mechanisms of transcriptome. For instance, it suggested that significant contributions of post-transcriptional regulators for explaining tissue-specific DE, differential transcript-usage, and even individual differences in transcriptomes and thus post-transcriptional controls might be key mechanisms to fine-tune the transcriptome in response to environmental and genetic factors.
We designed the DEcode framework as multi-task learning that predicts transcriptomes of multiple samples simultaneously with shared feature extraction layers as opposed to single-task learning that builds a prediction model specific to each sample. The multi-task design not only reduced learning time, as feature extraction layers are optimized for all samples jointly and training data is generated only once, but also improved prediction accuracy for both tissue-specific expression and person-specific expression (Supplementary Figure 9). This improvement is likely because multi-task learning was able to extract RNA and promoter features that were generalizable across samples, which prevented overfitting to a specific sample. In addition, the multi-task design would benefit in comparing differential contributions of key predictive features between samples, as it shares the same feature extraction processes, which is a critical step to lead a biologically meaningful interpretation of the DEcode model.
Transcriptome analysis often identifies differentially expressed genes and then assesses the enrichment of functional genes such as TF-targets one by one. The person-specific DEcode model offers several comparative advantages over this traditional approach. First, DEcode can take into account the effects of multiple regulators simultaneously as opposed to one at a time. Second, DEcode can estimate the person-specific regulator’s activities that can be used to identify regulators associated with a phenotype of interest. Third, DEcode can simulate the consequence of KO perturbations for each gene. This step can reduce the number of candidate key drivers of gene expression changes by an order of magnitude or more, and facilitates the design of follow-up experiments. Indeed, key predictors nominated based on DEcode showed significant and robust overlaps with various diseases-causing/related gene sets. Therefore, DEcode can extract more actionable information from transcriptome data, which will benefit a variety of transcriptome studies.
Looking toward even more expansive applications, the DEcode framework has the flexibility to incorporate other types of genomic information such as raw sequences, DNA methylation, histone marks, and RNA modifications, and also can be extended to other organisms. Thus, DEcode framework provides a direct bridge between accumulating genomic big data and individual transcriptome studies, allowing researchers to predict molecules that control DE associated with any condition or disease.
Methods
Transcriptome data processing
To prepare gene expression data used for the model training, we downloaded the median gene TPM (transcripts per million) from 53 human tissues from the v7 release of GTEx portal (https://gtexportal.org). We kept 27,428 genes expressed greater than two TPM in at least one tissue and log2-transformed TPM with the addition of 0.25 to avoid a negative infinity. Then, we calculated the median log2-TPM across 53 tissues and log2-fold-changes relative to the median of all tissues. The processed gene-level expression data comprised 27,428 genes with 54 columns including relative fold-changes for 53 tissues and the median log2-TPM across 53 tissues. To compile transcript-level data, we downloaded the individual-level transcript TPM from the GTEx portal and computed the median transcript TPM by tissue. We processed the transcript data in the same way we did for the gene-level data. The resulted transcript-level data included 79,647 transcripts that corresponded to 23,813 genes. For building person-specific DEcode models, we obtained the gene-level TPM for each individual in 14 tissues from the GTEx portal. We filtered out lowly-expressed genes in each tissue and kept genes expressed greater than one TPM in at least 50% of samples. Then, we log2-transformed TPM with the addition of 0.25 and then quantile normalized the log2-TPM. Finally, we removed the effects of technical covariates including rRNA rate, intronic rate, and RIN number via linear regression for each gene followed by quantile normalization.
Promoter and RNA binding features.
To generate RNA and DNA feature matrices, we downloaded genomic locations of binding sites of 171 RBPs from POSTAR214 as of Oct 2018, 218 miRNAs from TargetScan Release 7.215, and 826 TFs from GTRD13 as of Oct 2018. Then, we mapped the binding sites of RBPs, miRNAs, and TFs to promoters and exons defined in the GTF file provided by the GTEx portal. A promoter region of each gene was defined as the region from 2,000 bp upstream of the TSS to 1,000bp downstream of the TSS. We only used interactors that bind to promoters or RNA-coding regions of at least 30 genes, or transcripts as the predictors in each model. To reduce the size of the input, an RNA-coding region and a promoter region of each gene was binned with 100 bp intervals and the number of bases bound to each RBP, miRNA, or TF was counted in each interval. This step generated RNA and DNA feature matrices for each gene described in Figure 1.
Training tissue-specific models
For training the gene-level model of tissue-specific expression, we reserved all 2,705 genes coded on chromosome 1 as the testing data and the rest of the genes was randomly split into training data (22,251 genes) and validation data (2,472 genes). In the case of the transcript model, we used all 7,631 transcripts coded on chromosome 1 as the testing data and the rest of the transcripts was randomly split into training data (64,978 transcripts) and validation data (7,038 transcripts). This cross-chromosome train-test split prevents information leaking from intra-chromosomal interactions and potential overlaps of regulatory regions. The individual binding features (TF, RBP, and miRNA) were normalized by their maximum value across genes. The relative fold-changes for 53 tissues were scaled together to set the standard deviation as one and the median log2-TPM was separately scaled to set the standard deviation as one. These steps were conducted for the training data first and then the same scaling factors were used for the validation and the testing data to avoid information leaking from those data. We constructed and trained DL models using Keras (version 2.1.3) with a TensorFlow (version 1.4.1) backend. Hyper-parameters were optimized using hyperopt (version 0.2)33 based on the mean squared error against the validation data. The detailed structure of the model was described in Supplementary Figure 10. The training was done using mini-batches of 128 training examples with a learning rate of 0.001 for Adam optimizer. The number of maximum training epochs was set to 100 with early-stopping of 10 based on validation loss. This training cycle was repeated 10 times and the best model for the validation data was selected as the final model (Supplementary Figure 11). All models were trained using TITAN X Pascal graphics processing units (Nvidia).
Comparison of DEcode with ExPecto
To perform a fair comparison between DEcode and ExPecto11, we used 18,550 genes that were commonly included in both studies and trained models with the same set of genes for training and evaluation. Since ExPecto model was originally built using genes on chromosome 8 as the testing data, we followed the same procedure as we reserved all 714 genes coded on chromosome 8 as the testing data and the rest of the genes was randomly split into training data (16,052 genes) and validation data (1784 genes). The epigenetic states estimated by ExPecto were downloaded from the ExPecto repository (https://github.com/FunctionLab/ExPecto) as of Nov 2019. Given the epigenetic states, we built a prediction model for tissue-specific gene expression for each tissue via XGBoost based on the training script downloaded from the ExPecto repository. We modified the original script so that the early stopping of the model optimization was decided based on the performance on the validation data instead of the testing data. This modification prevented the overfitting of the model to the testing data. We used the same hyper-parameters for XGBoost as in the script. We built 10 models for each method using the same genes for training, validation, and testing to predict gene expression in the 53 tissues.
DeepLIFT score calculation
To evaluate the importance of input features to the prediction, we calculated DeepLIFT (Deep SHAP) scores17 using DeepExplainer implementation (version 0.27.0)18. The DeepLIFT method estimates the contribution of each input compared to a reference input in a trained DL model. To compute the contribution of the presence of a binding site, we used a reference that does not have any binding sites in both promoters and RNAs with the median length of all genes in the testing data. DeepLIFT scores follow a summation-to-delta property where the summation of input contributions (DeepLIFT scores) is equal to the difference in the predicted value compared to the prediction from the reference input. We calculated DeepLIFT scores for each gene and transcript in testing data for each of 53 outputs, then summed up the scores over promoter or RNA regions for each feature, and finally averaged them over genes. To estimate the collective impact on differential expression across 53 tissues, we averaged absolute DeepLIFT scores of all tissues for each regulator. To evaluate the regulator’s contribution to the prediction of transcript-usage, we calculated the variance of DeepLIFT scores across transcript variants for each gene and then averaged them over genes. We performed pre-ranked gene set enrichment analysis (GSEA)34 to assess gene ontology terms in MSigDB v7.135 associated with key TFs and RBPs. The absolute DeepLIFT scores were log-transformed and standardized to make them close to a normal distribution and then applied to GSEA with 10,000 permutations. The Enrichment Map was utilized for visualization of the results36.
Disease genes
The probability that a gene is intolerant for a loss-of-function mutation was downloaded from the release 0.3.1 of the ExAC portal (http://exac.broadinstitute.org). We defined genes with probability greater than 99% as intolerant of homozygous or heterozygous LoF mutations. Disease genes were obtained from the OMIM portal as of June 2019 (https://www.omim.org/). We excluded provisional gene-to-phenotype associations and genes associated with non-disease phenotypes, multifactorial disorders, or infection. We obtained mouse-lethal genes from Gene Discovery Informatics Toolkit (v1.0.0)37 that provided pre-processed gene lists from the murine knock-out experiments registered in Mouse genome informatics (MGI)38 and the International Mouse Phenotyping Consortium (IMPC)39. The results of CRISPR screening for the genes essential for proliferation or viability conducted in the DepMap project40 were downloaded from Enrichr portal41, 42 as of June 2019 (https://amp.pharm.mssm.edu/Enrichr). Enrichr portal provided two CRISPR screening results conducted independently at Broad Institute and the Sanger Institute. To reduce the false positives in the CRISPR screening, we used essential genes that were identified in both of the two independent screenings. Protein-protein interaction networks were downloaded from eXpression2Kinases Web43 which consists of 210,221 interactions across 15,716 proteins.
Training person-specific models
To train person-specific models, we utilized the same model structure as the tissue-model, except that the number of model outputs was modified to match the sample size of the tissue. We re-used the parameters of convolutional layers in the tissue-model and only parameters in the fully-connected layers were tuned (Supplementary Figure 12). We used the same gene splits and the same procedure of normalization and scaling as the tissue-model for training and evaluating models. We evaluated the model prediction for each individual separately based on validation data and filtered out the individual models that performed less than 50% percentile of all individual models for some analyses.
Training PrediXcan models
To build a prediction model for gene expression from genotype data, we trained PrediXcan29 models with GTEx gene expression and genotype data. A QCed vcf file of GTEx genotype data called by whole-genome sequence was downloaded from dbGaP for 635 individuals. We filtered out variants with a missing rate greater than 1% and minor allele frequency less than 1% and kept 9,219,660 variants for PrediXcan. We followed the model building procedure employed in PredictDB (http://predictdb.org/), a repository of PrediXcan models, as of Nov 2019. Briefly, we randomly split the samples into 5 folds. Then for each fold, we removed the fold from the data and used the remaining data to train an elastic-net model using 10-fold cross-validation to tune the lambda parameter. With the trained model, we predicted gene expression values for the hold out samples. We applied the PrediXcan method to predict the same gene expression data used for the person-specific DEcode models. We built PrediXcan model for each gene using variants located within 1 Mbp upstream and downstream of its TSS. A missing value of the genotype data was replaced with an average dosage of non-missing samples.
Differential expression analysis for age and sex
Limma44 was used to identify genes associated with age using gender as a covariate. The log2-TPM values of genes in the testing data were used. We also tested the associations between DeepLIFT scores for predictors and age via limma to identify regulators for DE against ages and sex. The Benjamini–Hochberg procedure was used to control the false discovery rate at 5%.
in silico binding-site disruption experiment
To simulate the consequence of the removal of binding sites on the expression of LAPTM5 and CD53, we generated 10,000 synthetic inputs for each of LAPTM5 and CD53 where all binding sites in each interval of its promoter and RNA were randomly removed. From each of these synthetic inputs, we computed predicted expression values and correlated them with ones of another gene without any disruptions in its binding sites. Then, we used multiple linear regression to associate the location of disrupted regions with the correlation values between LAPTM5 and CD53 to estimate the effects of the disruption in each region on the co-expression relationship.
in silico knockout experiment
To simulate the effect of regulator knockout (KO) on the expression of LAPTM5 and CD53, we generated 10,000 synthetic inputs for each LAPTM5 and CD53 where each protein or miRNA bound to its promoter or RNA was randomly removed from their feature matrices. From each of these synthetic inputs, we computed predicted expression values and correlated them with ones of another gene without any removals in its feature matrices. Then, we used multiple linear regression to associate KOs of regulators with the correlation values between LAPTM5 and CD53 to estimate the effects of the KO of each regulator on the co-expression relationship. We applied the Bonferroni correction to control multiple testing and the regulators with the corrected p-value less than 0.05 in all tissues were chosen as the key drivers of the co-expression.
Conditional independence test
To validate the effect of the predicted drivers on co-expression, we conducted a mediation analysis using a conditional independence test. A mediation analysis evaluated the hypothesis where if LAPTM5 and CD53 are co-expressed due to the predicted regulators, normalizing expression levels of the two genes by the expression levels of the regulators would decrease the co-expression relationships. Specifically, we regressed the actual log2-TPM values of LAPTM5 and CD53 with the actual log2-TPM values of the predicted drivers and computed R2 (variance explained) between the residuals of two genes. The R2 based on the actual gene expression and one from the residuals were compared to quantify the covariance explained by the predicted drivers. To evaluate the significance of this effect, we repeated this process 1,000 times with an equal number of randomly picked genes that have a binding site in LAPTM5 or CD53 as regressors.
DEcode model for DE prior rank
DE prior rank was downloaded from https://github.com/maggiecrow/DEprior. In the DE prior rank, each gene has a probability-like value where zero is the minimum and one is the maximum. To convert this value to a non-bounded scale, we applied the logit transformation to the DE prior value. We assigned a value of 10 to a gene that had an infinite value after the logit transformation. We used the same gene splits as the GTEx-tissue-model, which resulted in 13,433 genes for training, 1,504 genes for validation, and 1,674 genes for testing. We trained the DEcode model for DE prior rank using the same procedure as with the GTEx-person-specific models. To evaluate the contribution of promoter and RNA features to the prediction, the model was also trained with randomized input features. Receiver operating characteristic (ROC) curve analysis was performed using pROC R package45 with a default setting. We performed pathway analysis of the TFs with a DeepLIFT score greater than 90th percentile using KEGG pathways46. KEGG pathway gene sets were downloaded from MSigDB v6.135. The enrichment significance was based on results of the hypergeometric test, with 757 unique TF genes as a background, against KEGG pathways composed of at least 5 background genes. FDR was controlled at 5%. We manually curated the 159 disease-related data sets used in the construction of the DE prior ranking, to determine the number of data sets related to cancer or inflammatory disease.
Definition of boxplot elements
We used R ggplot2 package to draw boxplots in this manuscript with the default setting. The upper, centre and lower line of the boxplot indicates 75%, 50%, and 25% quantile, respectively. The upper and lower whisker of the boxplot indicates 75% quantile + 1.5 × interquartile range (IQR) and 25% quantile - 1.5 × IQR.
Data availability
The data sources of GTEx data, TF and RBP binding peaks, miRNA binding locations, disease-related genes, protein-protein interaction data, pathways, gene ontology, and the DEprior rank that were used for model training and interpretation are available in Supplementary Table 8. Processed data are available through our Code Ocean capsule (https://doi.org/10.24433/CO.0084803.v1).
Code availability
DEcode software and pre-trained models for tissue- and person-specific transcriptomes are available at www.differentialexpression.org, https://github.com/stasaki/DEcode, and Code Ocean capsule (https://doi.org/10.24433/CO.0084803.v1). DEcode is licensed under the BSD 3-Clause license.
Extended Data
Extended Data Fig. 1. Characterizations of the person-specific models.

(a) The consistency between the predictive contributions of regulators and their expression. Spearman’s correlation was used to evaluate the relation between DeepLIFT scores vs log2-TPMs in each tissue. A relation with the absolute correlation > 0.3 and FDR < 5% was defined as significant. (b) The consistency of predictive contributions across tissues. We selected the regulators that showed the significant relationships between their DeepLIFT scores with log2-TPMs in multiple tissues. Then, the consistency of directions of the correlations was assessed and visualized as a histogram. (c) The actual correlation profile between DeepLIFT scores and log2-TPMs. We selected 99 regulators that showed consistent relationships in more than four tissues. (d) The pairwise similarity of per-gene prediction accuracy between tissues. Spearman’s correlation was used for this comparison. (e) The association of per-gene prediction accuracy with the gene status. The gene status indicates whether genes are registered in multiple databases (Known) or only in the GENCODE database (Novel or Putative). (f) Decomposition of variance in the per-gene prediction accuracy. To compare the variances in the per-gene prediction accuracy explained by the gene status, the number of promoter features, and RNA features, we used a variance decomposition method (lmg) implemented in relaimpo R package.
Extended Data Fig. 2. The major feature types contributed to the gene co-expression.

We defined the gene pairs with the absolute Spearman’s correlation greater than 0.3 and the sign of the correlation matched with one with the ground truth as the successfully predicted gene pairs. The successfully predicted gene pairs of the model trained with the full set of features were split into three groups based on the performance of the models trained with only RNA features or promoter features.
Extended Data Fig. 3. DEcode predicts the regulatory principles behind frequently DE genes across diverse conditions.
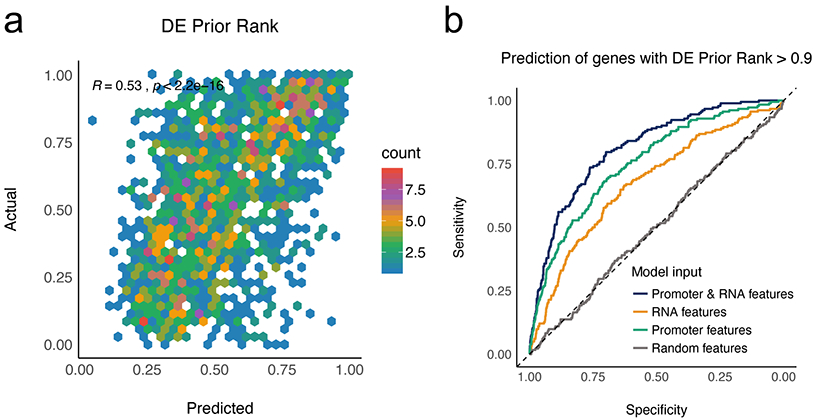
(a) The scatter plots showing the relations between predicted and actual DE prior rank. The predicted logit of DE prior rank was converted to probability and compared with actual DE prior rank with Spearman’s correlation. (b) The performances of the models trained with a distinct feature set. ROCs represent the performances of models in predicting genes with whose DE prior rank greater than 0.9.
Supplementary Material
Acknowledgments
We thank Dr. Lei Yu for managing access to GTEx data. The study was supported by NIH grants P30AG010161, R01AG061798, and R01AG057911. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 10/01/2018.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Lee T & Young R Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert SA et al. The Human Transcription Factors. Cell 172, 650–665 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glisovic T, Bachorik JL, Yong J & Dreyfuss G RNA-binding proteins and post-transcriptional gene regulation. FEBS letters 582, 1977–1986 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoenfelder S & Fraser P Long-range enhancer-promoter contacts in gene expression control. Nature reviews. Genetics 20, 437–455 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Smith ZD & Meissner A DNA methylation: roles in mammalian development. Nature reviews. Genetics 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Roundtree IA, Evans ME, Pan T & He C Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avsec Ž et al. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nature biotechnology 37, 592–600 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libbrecht MW & Noble WS Machine learning applications in genetics and genomics. Nature reviews. Genetics 16, 321–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaganathan K et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e24 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Zhou J et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature genetics 50, 1171–1179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alipanahi B, Delong A, Weirauch MT & Frey BJ Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature biotechnology 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Yevshin I, Sharipov R, Valeev T, Kel A & Kolpakov F GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic acids research 45, D61–D67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y et al. POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic acids research 47, D203–D211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal V, Bell GW, Nam J & Bartel DP Predicting effective microRNA target sites in mammalian mRNAs. eLife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melé M et al. Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shrikumar A, Greenside P & Kundaje A Learning important features through propagating activation differences. arXiv, 1704.02685 (2017).
- 18.Lundberg SM & Lee S A unified approach to interpreting model predictions. Advances in Neural Information Processing Systems 30, 4765–4774 (2017). [Google Scholar]
- 19.Chong JA et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80, 949–957 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Imperato MR, Cauchy P, Obier N & Bonifer C The RUNX1-PU.1 axis in the control of hematopoiesis. International journal of hematology 101, 319–329 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Soares E & Zhou H Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 75, 1179–1190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watt AJ, Garrison WD & Duncan SA HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 37, 1249–1253 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Lefterova MI, Haakonsson AK, Lazar MA & Mandrup S PPARγ and the global map of adipogenesis and beyond. Trends in Endocrinology & Metabolism 25, 293–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge Z, Quek BL, Beemon KL & Hogg JR Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. eLife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature Cell Biology 16, 191–198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goh K et al. The human disease network. Proc. Natl. Acad. Sci. U. S. A 104, 8685–8690 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 348 (6235): 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nature genetics 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerstberger S, Hafner M & Tuschl T A census of human RNA-binding proteins. Nat. Rev. Genet 15, 829–845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaiteri C, Ding Y, French B, Tseng GC & Sibille E Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 13, 13–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crow M, Lim N, Ballouz S, Pavlidis P & Gillis J Predictability of human differential gene expression. Proc. Natl. Acad. Sci. U. S. A 116, 6491–6500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergstra J, Komer B, Eliasmith C, Yamins D & Cox DD Hyperopt: a Python library for model selection and hyperparameter optimization. Computational Science & Discovery 8, 014008 (2015). [Google Scholar]
- 34.Korotkevich G, Sukhov V & Sergushichev A Fast gene set enrichment analysis. bioRxiv, 060012 (2019).
- 35.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merico D, Isserlin R, Stueker O, Emili A & Bader GD Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PloS one 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dawes R, Lek M & Cooper ST Gene discovery informatics toolkit defines candidate genes for unexplained infertility and prenatal or infantile mortality. NPJ genomic medicine 4, 8–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CL, Blake JA, Kadin JA, Richardson JE & Bult CJ Mouse Genome Database (MGD)-2018: knowledgebase for the laboratory mouse. Nucleic Acids Res. 46, D836–D842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koscielny G et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 42, 802 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 44, 90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clarke DJB et al. eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res 46, W171–W179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robin X et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sources of GTEx data, TF and RBP binding peaks, miRNA binding locations, disease-related genes, protein-protein interaction data, pathways, gene ontology, and the DEprior rank that were used for model training and interpretation are available in Supplementary Table 8. Processed data are available through our Code Ocean capsule (https://doi.org/10.24433/CO.0084803.v1).