

Brief Summary Statement
The first basket clinical trial testing the BRAF inhibitor vemurafenib resulted in evidence of activity in 13 unique cancer types with BRAFV600 mutations, but the response rates were variable. Therefore, different cancer histologies with the same driver oncogene display different degrees of oncogenic pathway addiction.
It seems logical to want to inhibit an activating mutation in a cancer-driving oncogene (e.g., BRAFV600 mutations) to treat a patient whose cancer has that genetic event, regardless of the cancer’s tissue of origin. However, it took over 10 years and the development of a new type of clinical trial, the “basket trial,” to facilitate this goal. It turns out that the degree to which a cancer is addicted to an oncogene depends on the tissue of origin. In addition to this biologic complexity, the regulatory path for testing therapies targeting a specific genetic alteration across different cancer histologies was not cleared until years after initial clinical development of specific BRAF inhibitors.
In this issue of Cancer Discovery, Subbiah et al. report on the full cohort of patients enrolled in the first ever basket trial designed to test a single drug, the BRAFV600 mutant inhibitor vemurafenib, in multiple indications that share the same genetic marker (1). The path to the clinical testing of BRAF inhibitors in BRAFV600MUT-driven cancers, which should have been a slam dunk, detoured in order to develop a new paradigm for drug development and understand the nuanced biology underlying different clinical outcomes across early indications tested.
Before the clinical development of BRAF inhibitors, there had been already other recognized driver oncogenes expressed by cancers of different histologies. A notable example is the Her2/neu amplification present in approximately one fourth of breast cancers and also some cases of ovarian and gastric cancers (2). The development of Her2/neu antibody trastuzumab focused on each separate cancer histology against a competing drug in the same indication, instead of focusing on the same genetic event across multiple cancer histologies. Evidence of the exquisite sensitivity of BRAFV600MUT metastatic melanoma to vemurafenib (3) prompted a logical desire to test this drug in patients with any cancer that harbors BRAFV600 mutations. But there was no path for such clinical development in the early 2010s.
This scenario led to the initial concept of a “basket trial,” where patients are selected based on a genetic driver event (BRAF, NTRK, Ret) present across cancers of different histologies and tissues of origin, sometimes at very low frequencies, and then treated with the same targeted drug (4). During the planning of the basket trial for vemurafenib, there was no regulatory path for drug approval based on BRAFV600 mutations. There was no precedent for any drug that had received a license based on studies conducted across multiple cancer histologies, even if the drug is a highly targeted agent that would only work when a specific genetic event is present. The precedents came in 2017, with the approval of the anti-PD-1 antibody pembrolizumab in patients with cancers that harbor very high mutational load due to microsatellite instability (MSI-high) (5), and then in 2018, with the approval of larotrectinib in patients with cancers that harbor NTRK gene fusions (6).
The initial development of vemurafenib in BRAFV600MU cancers met hurdles which were somewhat mitigated by the time pembrolizumab and larotrectinib were being developed for MSI-high and NTRK cancers, respectively. For instance, because MSI-high was already recognized to have a distinct cancer biology even within the same cancer histology, MSI status was already a standard of care test in many centers before anti-PD-1 antibodies were found to have high activity against cancer types with high mutational burden. Moreover, prospective testing of multiple genetic alterations in cancers had been implemented in several referral cancer centers using multigene sequencing panels by the time NTRK inhibitors entered clinical testing, facilitating the identification of very rare patients whose cancers harbor NTRK fusions. In 2012, finding the rare patients across non-melanoma cancer histologies to be enrolled in the newly open vemurafenib basket trial was very slow. Compounding this problem was the lack of response to vemurafenib in the initial cohort of patients with colon cancer harboring BRAFV600 mutations. The combination of slow accrual and evidence of biologic complexity behind cancer addiction to BRAFV600 mutations resulted in a long delay in acquiring the antitumor data of vemurafenib in BRAFV600 mutated cancers across wide-ranging cancer histologies.
The report by Subbiah et al. is one of several articles published from the vemurafenib basket trial. The first report differentiated a higher response rate in patients with BRAFV600MUT non–small-cell lung cancer and Langerhans’-cell histiocytosis (both with response rates in the range of 45%), from anecdotal and lowe rates of responses among patients with several other BRAFV600MUT cancers (including colorectal cancer, pleomorphic xanthoastrocytoma, anaplastic thyroid cancer, cholangiocarcinoma, salivary-duct cancer, ovarian cancer, and clear-cell sarcoma) (7,8). All the non-melanoma response rates were lower than that for melanoma (3), but the highest response rate exceeded 95% in patients with BRAFV600MUT-driven hairy cell leukemia, which was based on a different clinical trial that followed the report that all hairy cell leukemias harbor the BRAFV600E oncogene (9).
At the end of the day, the significance of the Subbiah et al. report lies in the finding that BRAFV600 mutations are clinically actionable in 13 unique cancer types and not in the semantic portrayal of overall response rates across diverse cancer types as an indication of lineage-independent oncogene or mitogen activated protein kinase (MAPK) pathway addiction. The ultimate arbiter of lineage-dependence or -independence of BRAF-MAPK addiction will rest with which major pathway the underlying resistance mechanisms point to. Oncogene (or pathway) addiction refers to the exquisite (clinical) dependence of cancer cell growth and survival on a singular or select few oncogenic alteration(s), in the context of a much larger number (one to two orders of magnitude) of somatic alterations. This concept justifies a limited gene panel approach for sequencing to guide the clinical choice of pathway-directed agents for cancer treatment. Thus, if the simple majority of adaptive or acquired resistance mechanisms reactivate the MAPK pathway among the 13 cancer types where responses were observed (or innate resistance mechanisms among the cancers that did not respond), then a strong case for lineage-independent BRAFV600MUT or MAPK pathway addiction could be made. Unfortunately, tumor samples for the analysis of innate, adaptive and acquired resistance mechanisms, through interrogation of the proteome, transcriptome, epigenome and genome, are unavailable.
Since the initial discovery of a genetic mechanism (i.e., acquisition of RAS co-mutations) of MAPK reactivation driving acquired vemurafenib resistance in BRAFV600MUT melanoma (10), landscape studies of clinical tumor samples support MAPK reactivation as a convergent mechanism in a simple majority of patient (11,12). Even with the combination of a type I RAF inhibitor (vemurafenib, dabrafenib, encorafenib) and an allosteric MEK inhibitor (cobimetinib, trametinib and binimetinib), clinical acquired resistance occurs frequently and commonly through reactivation of the MAPK pathway (13). The specific alterations that mediate reactivation vary but converge to upregulate two key protein/protein interactions: RAF/RAF and RAF/MEK (Figure 1). Thus, alternative (combination) strategies that can better combat these upregulated protein interactions are needed.
Figure 1. Resistance to BRAFV600 mutation inhibition driven by MAPK reactivation in melanoma.
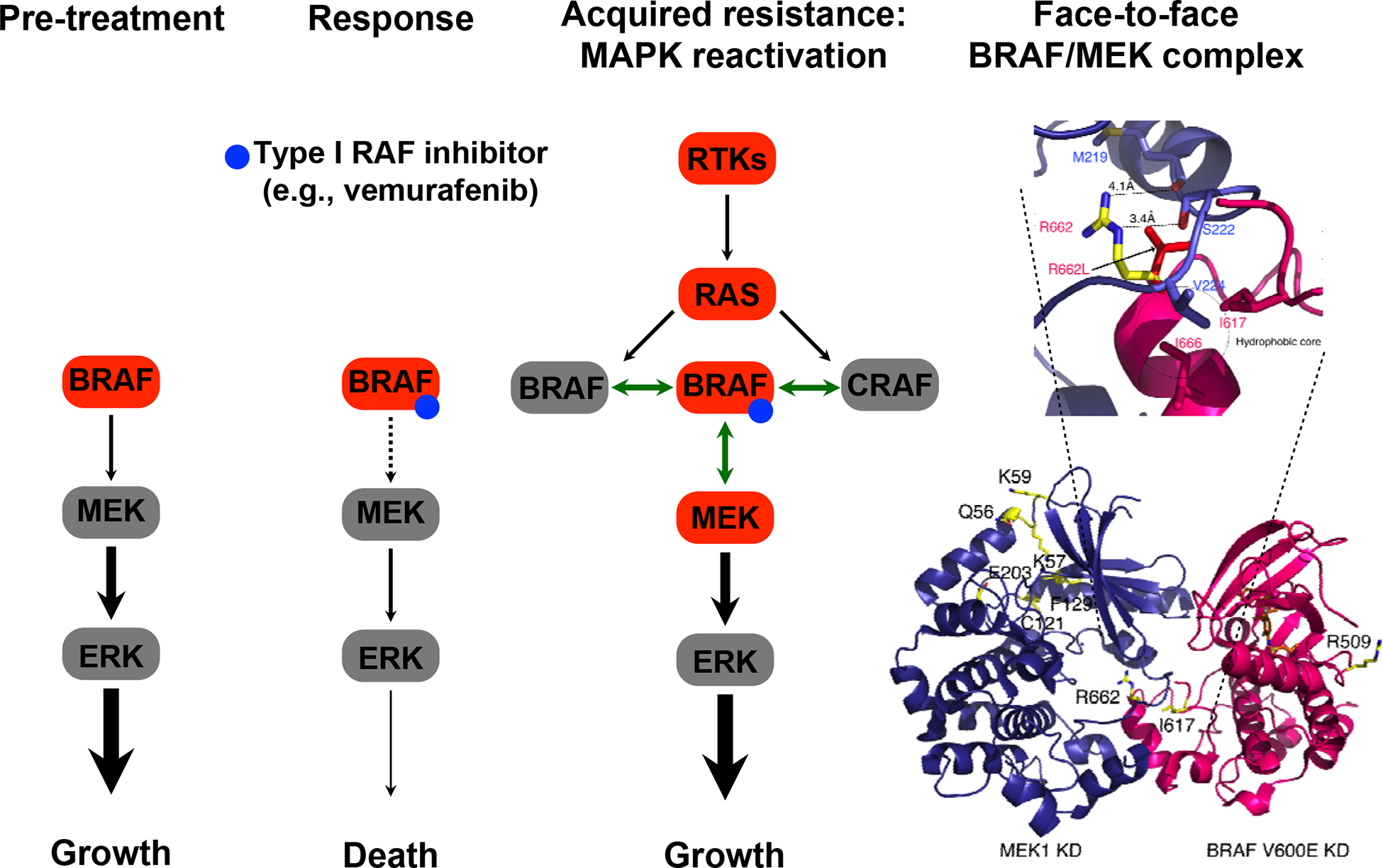
Alterations in genes (red symbols) thought to contribute to acquired resistance by turning the MAPK pathway back-on after prolonged selection with BRAF inhibitors. These alterations include transcriptional upregulation of multiple receptor tyrosine kinases, transcriptional/mutational upregulation of KRAS or NRAS, BRAFV600 mutant amplification or alternative splicing, and MEK1/2 mutations that result in so-called RAF-regulated MEK mutants. These genomic or non-genomic alterations converge on and augment two protein/protein interactions (shown as double-headed green arrows): “back-to-back” BRAF/BRAF (BRAF/CRAF) or “face-to-face” BRAF/MEK (CRAF/MEK) interactions. Adaptive resistance occurs early during the course of treatment and can result from relief of negative feedback, resulting in rebound levels of signals, including RTK-mediated RAS/RAF activation. The RTK implicated in adaptive vemurafenib resistance in colon cancer is thought to be EGFR, although multiple receptor tyrosine kinases can be upregulated simultaneously and redundantly activate the MAPK and/or PI3K-AKT pathways.
Varying tumor immune microenvironments across a wide range of tumor types can contribute potentially to disparate response rates or durability, regardless of the debate over lineage-dependent or -independent oncogene addiction. In BRAFV600MUT melanoma, acquired resistance to MAPK-targeted therapy selects strongly for loss of intratumoral CD8 T cells (14), and a pretreatment inflammation signature is associated with complete responses (15). Future developments of MAPK-targeted regimens should consider cancer histology-specific tumor immune microenvironments and the dynamic impact of MAPK-targeted agents on the evolving profile of systemic and local immunity. Therefore, a BRAF slam dunk may ultimately require an assist from next-generation MAPK-targeted or an immunotherapy teammate.
Funding:
NIH grants R35 CA197633 (A.R.), P01 CA244118 (A.R. and R.S.L) and P30 CA016042 (A.R.), R01CA176111 (R.S.L), R21CA215910-01 (R.S.L), The Parker Institute for Cancer Immunotherapy (A.R.), American Skin Association (R.S.L.), and The Ressler Family Fund (R.S.L and A.R.).
Footnotes
Conflict of Interest Disclosure: R.S.L. receives research funding from Merck, Array BioPharma, and OncoSec Medical and consulted in the last three years with Amgen, Shire, Novartis, Array BioPharma and Merck. A.R. has received honoraria from consulting with Amgen, Bristol-Myers Squibb, Chugai, Genentech, Merck, Novartis, Roche and Sanofi, is or has been a member of the scientific advisory board and holds stock in Advaxis, Apricity, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix and Tango Therapeutics, has received research funding from Agilent and from Bristol-Myers Squibb through Stand Up to Cancer (SU2C).
References
- 1.Subbiah vea. Pan-cancer efficacy of vemurafenib in BRAFV600-mutant non-melanoma cancers. Cancer Discovery 2020;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989;244(4905):707–12. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363(9):809–19 doi 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willyard C ‘Basket studies’ will hold intricate data for cancer drug approvals. Nat Med 2013;19(6):655 doi 10.1038/nm0613-655. [DOI] [PubMed] [Google Scholar]
- 5.Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res 2019;25(13):3753–8 doi 10.1158/1078-0432.CCR-18-4070. [DOI] [PubMed] [Google Scholar]
- 6.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018;378(8):731–9 doi 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med 2015;373(8):726–36 doi 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF Inhibition in BRAF(V600)-Mutant Gliomas: Results From the VE-BASKET Study. J Clin Oncol 2018:JCO2018789990 doi 10.1200/JCO.2018.78.9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiacci E, Park JH, De Carolis L, Chung SS, Broccoli A, Scott S, et al. Targeting Mutant BRAF in Relapsed or Refractory Hairy-Cell Leukemia. N Engl J Med 2015;373(18):1733–47 doi 10.1056/NEJMoa1506583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468(7326):973–7 doi nature09626 [pii] 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4(1):80–93 doi 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4(1):94–109 doi 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015;27(2):240–56 doi 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015;162(6):1271–85 doi 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Y, Wongchenko MJ, Robert C, Larkin J, Ascierto PA, Dreno B, et al. Genomic Features of Exceptional Response in Vemurafenib +/− Cobimetinib-treated Patients with BRAF (V600)-mutated Metastatic Melanoma. Clin Cancer Res 2019;25(11):3239–46 doi 10.1158/1078-0432.CCR-18-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]