

Abstract
Thirdhand cigarette smoke (THS) is a newly described toxin that lingers in the indoor environment long after cigarettes have been extinguished. Emerging results from both cellular and animal model studies suggest that THS is a potential human health hazard. DNA damage derived from THS exposure could have genotoxic consequences that would lead to development of diseases. However, THS exposure-induced interference with fundamental DNA transactions such as replication and transcription, and the role of DNA repair in ameliorating such effects, remain unexplored. Here we found that THS exposure increased the percentage of cells in S-phase, suggesting impaired S-phase progression. Key DNA damage response proteins including RPA, ATR, ATM, CHK1 and BRCA1 were activated in lung cells exposed to THS, consistent with replication stress. In addition, THS exposure caused increased 53BP1 foci, indicating DNA double-strand break (DSB) induction. Consistent with these results, we observed increased micronuclei formation, a marker of genomic instability, in THS-exposed cells. Exposure to THS also caused a significant increase in phosphorylated RNA Polymerase II engaged in transcription elongation, suggesting an increase in transcription-blocking lesions. In agreement with this conclusion, ongoing RNA synthesis was very significantly reduced by THS exposure. Loss of nucleotide excision repair (NER) exacerbated the reduction in RNA synthesis, suggesting that bulky DNA adducts formed by THS are blocks to transcription. The adverse impact on both replication and transcription supports genotoxic stress as a result of THS exposure, with important implications for both cancer and other diseases.
Keywords: Tobacco smoking, DNA double-strand breaks, micronuclei frequency, DNA damage response, DNA repair, stalled RNA polymerase
INTRODUCTION
Cigarette smoking is a global health problem and the leading cause of lung cancer, claiming nearly half a million lives each year, including ~50,000 from exposure to secondhand smoke (SHS). Most recently, potential health concerns have been raised about thirdhand smoke (THS), a much less understood type of smoke exposure defined as residual tobacco smoke adsorbed onto indoor surfaces after active smoking has ceased. The residual chemicals that build up over time onto indoor surfaces cannot be easily eliminated by airing out rooms, opening windows or by air conditioning. They remain, react and are re-emitted back into the environment as gases. Moreover, residual chemicals can undergo chemical transformation to generate secondary toxicants by interacting with specific air pollutants [Jacob III et al., 2017].
Chemical analysis revealed that THS contains toxic chemicals including volatile (VOCs) and semi-volatile organic compounds (SVOCs) that pose exposure risk to non-smokers [Jacob III et al., 2017]. Some of them are mutagenic and/or carcinogenic and are derived from mainstream smoke or SHS. However, specific compounds may be unique to THS [Jacob III et al., 2017]. For example, the surface-bound nicotine, the major THS constituent, interacts with the environmental pollutant nitrous acid (HONO) to generate tobacco-specific nitrosamines (TSNAs) [Sleiman et al., 2010]. HONO is a pollutant produced by indoor combustion appliances. Significant levels of several TSNAs, including NNA (4-(methylnitrosamino)-4-(3-pyridyl) butanal), NNK (4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone) and NNN (N-nitrosonornicotine), were detected inside households where active smoking takes place and in vehicles of smokers [Jacob III et al., 2017; Sleiman et al., 2010]. NNK and NNN are known human carcinogens [Lao et al., 2007] which can also cause deleterious effects to the developing lungs [Rehan et al., 2011]. NNK is known to form bulky DNA adducts associated with increased cancer risk [Ma et al., 2019; Lao et al., 2007]. NNA is rarely detected in mainstream smoke or freshly emitted SHS but is found in THS [Jacob III et al., 2017; Sleiman et al., 2010]. However, the genotoxicity of NNA has not been well investigated. We recently have shown that NNA can induce DNA strand breaks [Hang et al., 2013] and generates bulky DNA adducts in vitro [Hang et al., 2014]. These findings suggest that TSNAs in THS are likely play important roles in cigarette smoke-induced pathogenesis.
The adverse biological and health effects of active smoking and SHS have been extensively analyzed, but the potential health hazards due to exposure to THS remain largely unknown. Humans can be exposed to THS through ingestion, inhalation, or dermal contact, and exposures can be protracted compared to the typically acute exposure to SHS. Mice exposed to THS have been reported to have damage to multiple organs, impaired wound healing, and behavioral changes [Martins-Green et al., 2014; Dhall et al., 2016]. We previously reported that THS exposure induced DNA damage [Hang et al., 2013] and caused body weight change and impaired immune function [Hang et al., 2017]. Most recently, we reported an increased incidence of lung cancer following short-term early life exposure of A/J mice to THS [Hang et al., 2018].
THS exposure generates a variety of DNA lesions, including mutagenic base modifications (e.g., 8oxoG), strand-breaks and bulky DNA adducts, all of which are potentially deleterious [Hang et al., 2013; Dhall et al., 2016; Hang et al., 2018]. If unrepaired, bulky adducts may stall replication fork progression and thus cause replication stress [Zeman et al., 2014]. This is mainly due to the uncoupling of the replication machinery during DNA replication, thus resulting in the formation of stretches of single-stranded DNA (ssDNA) [Pacek et al., 2004]. Bulky DNA adducts and strand breaks are also known impediments for elongating RNA polymerase II (RNAPIIo), causing transcription stalling [Gregersen et al., 2018]. Both replication stress and stalled transcription are linked with elevated risk of diseases including cancer and premature aging [Edenberg et al., 2014; Lans et al., 2019].
To better understand the critical mechanism for THS-induced genotoxic effects and its potential relevance to biological effects and disease, we investigated the consequences for replication and transcription from exposure of human lung cells to laboratory-generated THS. We chose human primary lung fibroblasts (hPFs) and lung epithelial BEAS-2B cells for these studies since the lung is one of the major target organs for THS-induced health effects [Martins-Green et al., 2014].
MATERIALS AND METHODS
Human cell lines and antibodies used in this study.
hPFs were from Dr. Prudence Talbot (UC Riverside) and transformed nontumorigenic human lung epithelial cells (BEAS-2B) were from ATCC (Manassas, Virginia). Normal human skin fibroblasts (HCA2) were from J. Smith (University of Texas, USA) and were immortalized by infection with an hTERT expressing retrovirus [Rubio et al. 2002]. XPA deficient patient skin fibroblasts XP12BE were from Coriell (Camden, NJ).
Antibodies used were 53BP1 (A300–272A, Bethyl), phospho-RPA32 (S4/S8: A300–245A, Bethyl), RPA32 (A300–244A, Bethyl), phospho-H2AX S139-clone JBW301 (EMD Millipore), CHK1 (2345, Cell Signaling), pCHK1-Ser317 (2344S, Cell Signaling), pATM (S1981, Ab81292, Abcam), ATM (Cell Signaling), PCNA (Santa Cruz), pBRCA1 (S1423, A300–008A, Bethyl), pATR (Thr1989, GTX128145, GeneTex), Tubulin (ab4074, Abcam) and GAPDH (MAB374, EMD Millipore) and rabbit anti-XPA (sc853, Santa Cruz).
Generation of THS Samples and Extract Preparation
THS-exposed terry cloth was generated at the University of California, San Francisco (UCSF) using a previously described method and stored at −20°C before extraction into a cell culture medium [Hang et al., 2013; Schick et al. 2014]. Briefly, THS-exposed terry cloth (0.125 g of fabric/ml of medium) was soaked in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen) in 15 ml Falcon centrifuge tubes and rotated at 4°C overnight followed by centrifugation at 1200 × g for 5 minutes. The supernatant containing THS substances was recovered and filtered through 0.22 μM sterile filters and aliquoted into 1.5 ml vials for storage at −80°C. For chemical analysis, 0.8 ml of the media were taken and analyzed by LC-MS/MS as described [Hang et al., 2013]. Representative key constituents in the extract made from THS-exposed terry cloth as shown by LC-MS/MS included the following: nicotine 950 ng/ml; NNK 0.56 ng/ml (LOQ = 0.2 ng/ml); and nicotelline 1.52 ng/ml (LOQ = 1 ng/ml). None of these compounds were detected in the blank control sample.
Cell culture and cell cycle analysis
BEAS-2B cells were cultured in DMEM containing 10% FBS, 1% sodium pyruvate (Gibco), 2 mM L-Glutamine (Gibco) and 1% penicillin–streptomycin (Gibco). hPFs were maintained as described earlier [Bahl et al., 2016] with low serum medium. Cells were cultured at 37°C in 5% CO2 and 95% relative humidity. Medium was replaced after two to three days, and cells were used in experiments when confluency reached about 70–80%. For cell cycle analysis, cells were mock exposed or exposed to THS with doses and time as indicated in figure legend, trypsinized, collected by centrifugation, washed with PBS, fixed in ethanol, and treated with RNase (Trego et al. 2018). Cells were then stained with propidium iodide (PI) and analyzed by fluorescence activated cell sorting (FACSCalibur, Becton-Dickinson). Cell cycle analysis was performed using FlowJo (Tree Star) and the Watson (pragmatic) algorithm.
Cell Growth Rate Analysis and siRNA Knock-Down
Growth rate of BEAS-2B cells was determined by plating 4×104 cells in triplicate in 35 mm or 60 mm dishes. After 24 h cells were washed with PBS and exposed to THS-containing DMEM at the doses indicated, or were mock exposed (DMEM only). At each time point, samples were collected by trypsinization, counted by Coulter counter, and results were plotted as fold increase vs. time. Knock-down (KD) of XPA (xeroderma pigmentosum group A) gene expression was performed by transfection of specific siRNAs following the protocol as described [Trego et al. 2016]. siRNA sequence used was 5’-GCTACTGGAGGCATGGCTA-3’ for XPA.
Cell Extract Preparation and Western Blotting
Cells were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin and cultured at 37°C with 5% CO2. For induction of damage, cells were typically grown to 75% confluency, washed twice, and incubated with a range of THS extract concentrations diluted in serum-free DMEM. For western analysis of specific proteins, cells were trypsinized, washed with PBS and suspended in a buffer containing 100 mM HEPES-KOH (PH 7.5), 250 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.5% Igepal (v/v), 10% glycerol and a protease inhibitor cocktail (Roche). The DNA and RNA in the suspensions were digested with 5 units/ml benzonase (Novagene) for 30 min on ice and appropriate amount of SDS-loading dye added. The samples were heated at 94°C for 5 min and separated by either 4–8% or 4–12% SDS-acrylamide gels (Invitrogen). Proteins were transferred to a nitrocellulose membrane and probed with the indicated antibodies. Immunoblots were quantified by densitometry using ImageQuant or Versadoc 4000MP and Quantity One software (BioRad).
Indirect Immunofluorescence
Immunofluorescence analysis was carried out as described [Trego et al., 2016]. For 53BP1 immunostaining, cells grown in 4-well chamber slides (Nunc Lab-Tek) were exposed to THS or SHS and then washed three times with PBS. Cells were fixed with 4% para-formaldehyde (PFA) and 0.3% Triton-X-100 in PBS prior to permeabilization with PBS containing 0.5% Triton X-100. Cells were blocked with 2% BSA and incubated overnight at 4°C with rabbit anti-53BP1 (Bethyl, A300–272A) antibody. Following washes, samples were incubated with secondary antibodies conjugated to Alexa Fluor 488 (Molecular Probes) and with 4′,6-diamidino-2-phenylindole (DAPI) to stain nuclear DNA. Slides were mounted in Vectashield and images were captured using a Zeiss Axiovert epifluorescence microscope with a Zeiss plan-apochromat 406 dry lens and a 12-bit charged coupled device camera (ORCA AG Hamamatsu). Images within the same data set were captured with the same exposure time, so that the intensities were within the linear range and could be compared between samples. All modifications were applied to the whole image using Photoshop CS2 (Adobe).
DNA Synthesis Assay
DNA synthesis as measured by incorporation of 5-ethynyl-2’-deoxyuridine (EdU) was essentially the same as previously described [Nakazawa et al., 2010]. The Click-iT EdU cell proliferation assay (Invitrogen) for detection of cells undergoing DNA synthesis is based on incorporation of EdU into newly synthesized DNA and its recognition by azide dyes via a copper mediated “click” reaction. Cells were plated (25,000 cells/well) in a 4-well chamber slide and grown overnight followed by exposure to THS for 24 h at the indicated doses. DNA synthesis levels were determined following incubation with 10 μM EdU for 4 h [Nakazawa et al., 2010] by direct addition to the culture medium. Serum-free medium was used, as serum often contains thymidine which competes with EdU for incorporation into DNA. EdU incorporation was visualized using Click-iT conjugation of Alexa Fluor 488 (Invitrogen) according to the manufacturer’s protocol (Invitrogen, Cat #c10337). Briefly, cells were fixed with 3.7% paraformaldehyde for 15 min followed by permeabilization with 0.5% triton X-100 for another 20 min at room temp. After extensive washing with PBS, cells were blocked with 2% BSA (Gibco) for 1 h at room temperature followed by incubation for 30 min protected from light with reaction cocktail containing Alexa Fluor 488-azide following the Click-iT EdU imaging protocol (Invitrogen). Slides were washed and samples were counterstained with DAPI for nuclear staining to a final concentration of 1 μg/ml. Images were captured using a Zeiss LSM 710 confocal microscope (Carl Zeiss Inc.) and analyzed with ImageJ (NIH) software. In each experiment >100 cells per condition were analyzed to determine percentage of cells with the fluorescent signal.
RNA Synthesis Assay
Cells were grown in a 4-well chamber slide and exposed to THS for 24 h. RNA synthesis levels were determined by directly adding 5-ethynyl uridine (EU; 1 mM final concentration) to the culture medium and incubation for 2 h, with incorporation of EU visualized by Click IT conjugation of Alexa Fluor 488 according to the manufacturer’s protocol described earlier (Invitrogen Cat# c10329 and Nakazawa et al., 2010). Images were obtained using Zeiss LSM 710 confocal microscope as described above. Fluorescence-signal intensities were quantified using the ImageJ software (NIH). For each experiment, >100 cells were used for analysis.
Micronucleus Assay
The cytokinesis-blocked micronucleus assay [Fenech et al., 2003] was used to investigate formation of micronuclei (MN) in bi-nucleated cells [Rydberg et al., 2007]. BEAS-2B cells were exposed to THS or mock exposed (only DMEM) for 48 h and Cytochalasin B was added to both the mock-treated and THS-exposed cells at a concentration of 3 μg/ml immediately after exposure, followed by further incubation for 48 h. Cells were trypsinized, centrifuged and re-suspended in 7 ml of 0.075 M potassium chloride for 10 min followed by fixation with 3 ml of 100% methanol for 1 h at room temperature. The cells were centrifuged and again fixed twice with acetic acid/methanol (1:3). Fixed cells were dropped onto wet slides and stained with Diff Quick following the manufacturer’s protocol. For each sample, micronucleus formation in 100 bi-nucleated cells was scored and plotted. Sample identity was blinded prior to scoring.
Statistical Analysis
Mean value and SD or SEM error bars are shown. Unpaired, two-tailed t-test was used to determine statistical significance between groups. Significance was determined as a p value less than or equal 0.05. All analyses were performed using GraphPad PRIZM (GraphPad, CA) software.
RESULTS
THS Exposure Impairs Cell Growth and Cell Proliferation
To determine the effect of THS on cell growth, we seeded BEAS-2B cells, either exposed them to THS (5% THS; 47.5 ng/ml nicotine) or mock-treated (only DMEM) them, and monitored cell growth at 24, 48, 72, 96 and 144 h. Figure 1A shows the growth rate of BEAS-2B cells following exposure to 5% THS or mock exposure. We determined fold increase of the initial cell number vs time (24, 48, 72, 96 and 144 h) and observed a marked decrease in cell growth in the treated cultures after 96 h (12× vs 8×) and 144 h (30× vs 13×). Cell growth up to 72 h did not change significantly (Fig. 1A). These results prompted us to test the impact of THS exposure on cell cycle progression. Using FACS analysis with propidium iodide (PI) DNA staining on BEAS-2B cells exposed to 2.5% THS for 24 h, we found a decline in the fraction of cells in G1-phase (from 60 % to 47%) and a marked increase in S-phase cells (17% to 31%) (Fig.1B). The fraction of cells in the G2-phase was largely unaffected. These data suggest that a significant fraction of replicating BEAS-2B cells exposed to THS were not able to exit from S-phase.
Fig. 1.
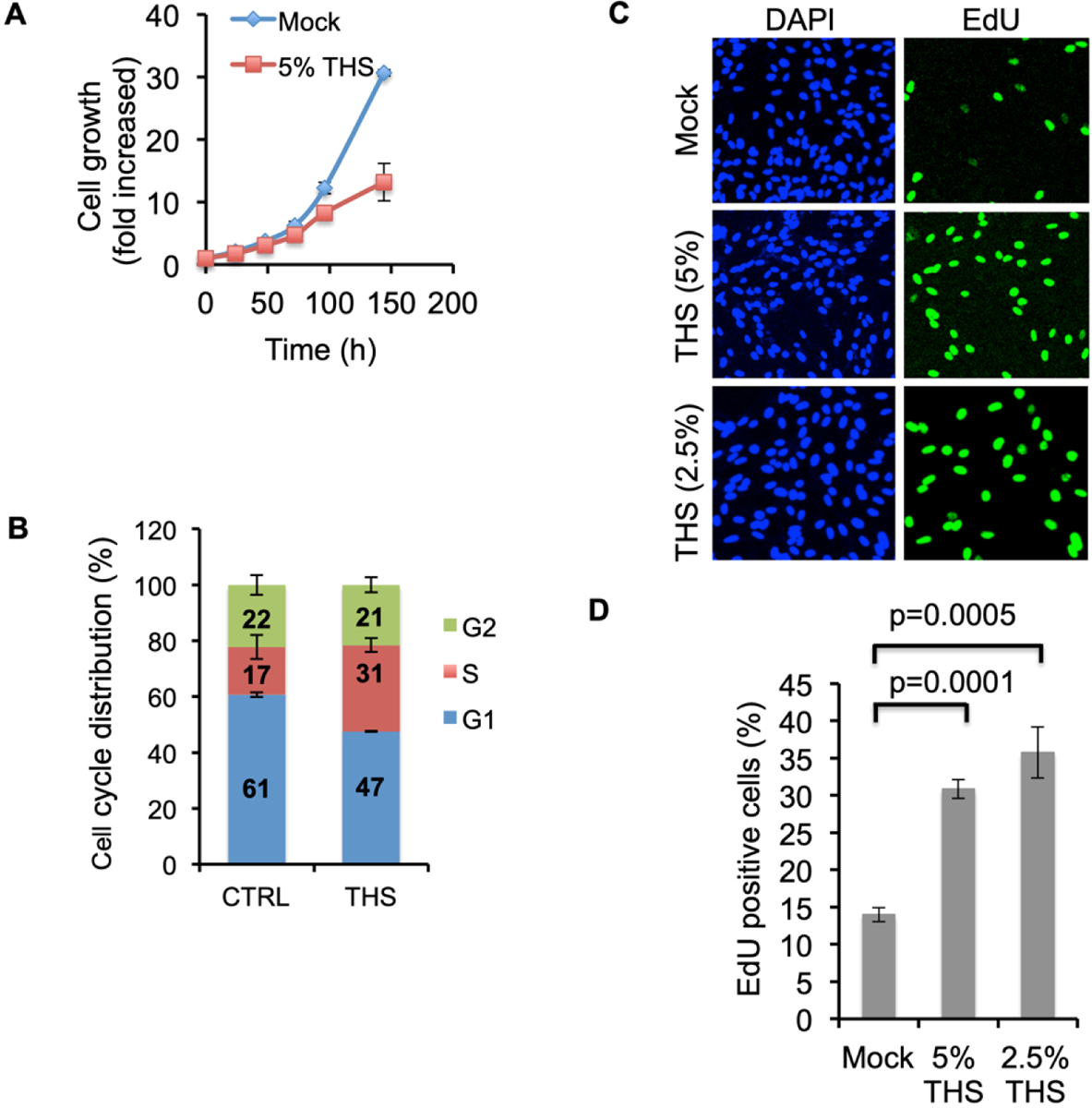
Growth rate, cell cycle distribution and proliferation analysis following exposure of BEAS-2B cells to THS. (A) Growth rate was analyzed following exposure to 5% THS. Cells were counted after 24, 48, 72, 96 and 144 h of exposure and plotted as fold increase vs time. (B) Cell cycle distributions were analyzed by PI staining protocol and plotted. Quantification shows accumulation of S-phase cells following exposure to 2.5% THS. CTRL represents mock exposure. Bars represent the mean from 3 experiments. (C) Cells were exposed to 5% or 2.5% THS for 24 h followed by EdU incorporation (4 h), fixed, permeabilized, conjugated with Alexa Fluor 488 by the Click-iT reaction, and imaged. Mock (only DMEM) exposure done in parallel served as control. (D) Quantification shown as percent EdU positive cells. Data are the mean ± SD from N=3.
To confirm and extend our results from cell cycle analysis, we then examined if the fraction of cells undergoing DNA synthesis was affected as a consequence of THS exposure. DNA replication was detected by incorporation of the thymidine analog EdU as measured by immunofluorescence (IF) following Click chemistry [Nakazawa et al., 2010]. BEAS-2B cells were exposed to 5% or 2.5% THS for 24 h followed by a 4 h pulse with 10 μM EdU. In mock control cells we observed only 13% EdU positive cells. In contrast, in 5% and 2.5% THS-exposed samples we observed 31% (2.3-fold) and 36% (2.6-fold) EdU positive cells, respectively (Fig. 1C & D). Since BEAS-2B cells are an immortalized cell line, we also investigated whether THS exposure exerts a similar effect on primary lung fibroblasts. We exposed hPFs to 5% THS for 24 h followed by EdU-labeling for 4 h as above and observed a 2.6-fold increase in EdU positive cells in THS-exposed hPFs compared to the mock exposed control (supplemental Fig. S1A). To test if such an effect is also present in other cell types, we exposed hTERT-immortalized skin fibroblast cells (hTERT-HCA2) to 5% THS for 24 h followed by EdU incorporation and detection. Similar to our results in BEAS-2B and hPFs cells, we observed a 2-fold increase in EdU positive cells following THS exposure (supplemental Fig. S1B).
Nucleotide Excision Repair Affects Consequences of THS Exposure
The consistently observed 2- to 3-fold increase in cells that are positive for DNA synthesis following exposure to THS could reflect a higher proportion of cells that are replicating their DNA, as implied by the increased fraction of cells in S-phase (Fig. 1B). Alternatively, however, it could reveal a large fraction of cells that are undergoing DNA repair synthesis during bulky lesion repair by the nucleotide excision repair (NER) pathway [Scharer et al., 2013]. TSNAs and PAHs, the constituent compounds found in THS, are known to produce bulky DNA lesions that are considered to play a major role in smoke-induced mutagenesis and carcinogenesis [Hang et al., 2010]. To examine the possibility that the increase we are detecting is actually repair synthesis, we tested whether loss of XPA protein, which is essential for NER, affects the incorporation of EdU after THS exposure. XPA was knocked down in BEAS-2B cells followed by THS exposure and EdU incorporation as described above. Loss of XPA protein was confirmed by Western analysis (Fig. 2C). As also observed in the experiments of Fig. 1C, D, control cells treated with a non-specific siRNA (siCTRL) had a highly significantly increased percentage of EdU positive cells as a result of THS exposure (Fig. 2 A, B). Strikingly, however, the opposite was observed when XPA was knocked down. In that case, DNA synthesis after THS exposure as marked by EdU incorporation was actually reduced relative to the mock-treated siXPA cells (Fig. 2A,B). Although this decrease in DNA synthesis between treated and mock-treated XPA knockdown cells did not reach statistical significance, the dramatic difference between the responses to THS exposure of XPA WT cells compared to the cells with XPA knocked down is striking. Importantly, there was no significant change in DNA synthesis in untreated control cells as a result of XPA loss (Fig. 2B). Furthermore, comparison of relative fluorescence intensity in EdU-positive cells reveals that loss of XPA resulted in significantly less incorporation per cell in response to THS exposure (Fig. S2), consistent with inability to carry out NER. These results strongly suggest that the increased DNA synthesis observed after THS exposure in control NER-competent cells represents EdU incorporation due to DNA repair synthesis during NER, rather than replicative synthesis. They further imply that NER capacity is likely to be an important factor in determining the consequences of THS exposure.
Fig. 2.
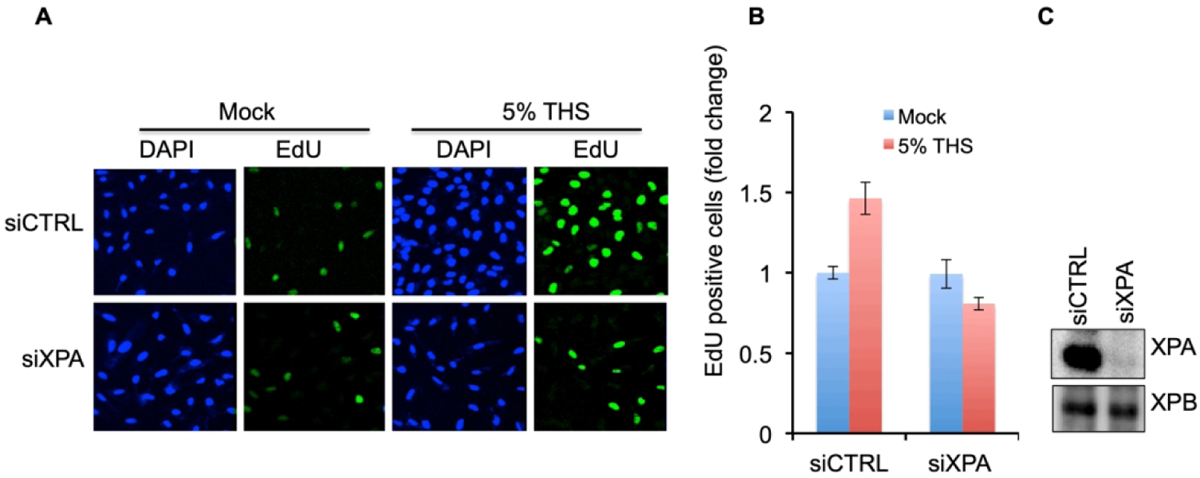
Loss of NER reduces the stimulation of DNA synthesis by THS exposure. (A) DNA synthesis following 24 h exposure to 5% THS was measured by EdU incorporation in BEAS-2B cells treated with a non-specific control siRNA (siCTRL) or with siRNA specific for XPA. (B) EdU fluorescence intensity of 100 cells each from siCTRL and siXPA cultures was monitored and plotted. Data represent the mean of ±SD for N=3. (C) Knock-down (KD) of XPA in BEAS-2B cells by treatment with siXPA vs siCTRL, analyzed by western. XPB was used as a loading control
THS Exposure Causes Replication Stress in Human Lung Cells
Slow growth rate and increased accumulation of cells in S-phase following exposure to THS led us to examine the possibility of increased replication stress. Specifically, we asked whether THS exposure causes increased RPA32 phosphorylation. RPA is a single-stranded DNA binding protein involved in chromosomal DNA replication and repair [Chen R and Wold MS, 2014]. Its RPA32 subunit is phosphorylated in response to replication stress at sites of ssDNA generated by resection to allow homologous recombination repair (HRR) of DSBs [Rothstein et al., 2013], and RPA32 phospho-S4/S8 is required for replication checkpoint arrest [Oakley et al., 2012]. Strikingly, both BEAS-2B and hPF cells exposed for 24 h to various doses of THS exhibited markedly increased pRPA32 by Western analysis (Fig. 3A,C). After normalization of the data with GAPDH, we found 3.3×, 5× and 2.6× increased RPA phosphorylation following 10%, 5% and 2.5% THS exposure, respectively, compared to mock exposure in BEAS-2B cells (Fig. 3A). The observed bell-shaped dose response in our study has been noted previously with THS compounds NNA and NNK in cellular strand-break studies (Hang et al., 2013). Increased RPA phosphorylation was also observed in hPFs with 10%, 5%, 2.5% and 1.25% THS exposure (1.4×, 7.5×, 5.8× and 4.2×, respectively) compared to mock exposure control (Fig. 3C). Exposure of BEAS-2B cells to 5% THS for various lengths of time showed that RPA32 phosphorylation started between 0.5 h to 3 h of exposure and increased further by 6–12 h, when the signal was increased by 5.9–6.3 fold, respectively, compared to mock-treated samples (Fig. 3B). Importantly, the levels of total RPA32 did not change with the increase in pRPA32 across the different time points after exposure (Fig. 3A–C). For comparison, treatment of hPFs with 20 nM camptothecin (CPT), a topoisomerase I inhibitor that is known to induce replication stress, caused a 3.9-fold increase in RPA phosphorylation (Supplemental Fig. S3), similar to 1.25% THS exposure (4.2×) dose in hPFs (Fig. 3C). These results are consistent with a high degree of replication stress in response even to relatively low concentrations of THS.
Fig. 3.
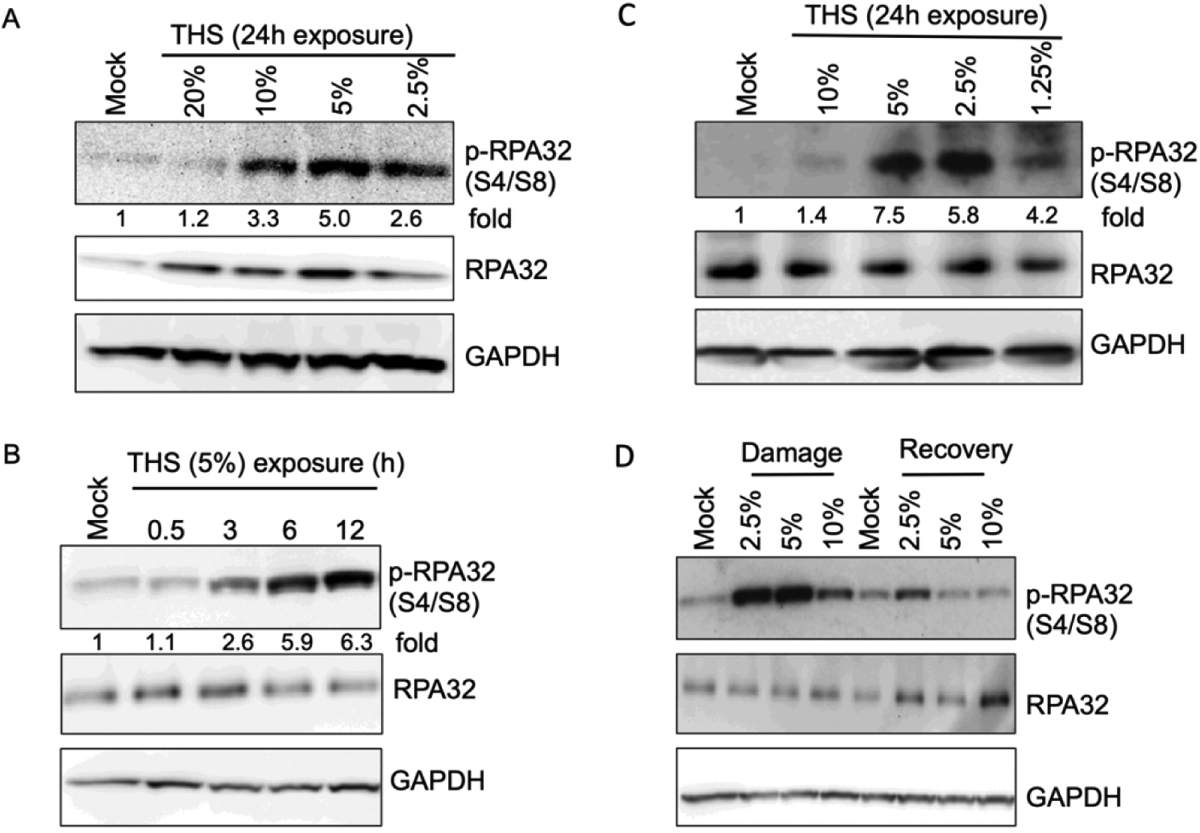
Replication stress following exposure of lung cells to THS as evidenced by RPA phosphorylation. (A) BEAS-2B cells were exposed to various doses of THS as indicated and RPA phosphorylation was monitored by western analysis with phospho-RPA32 (S4/S8) antibody. GAPDH was used as a loading control for normalization of pRPA32 signal at each time point in order to calculate fold increase for each THS dose compared to mock treated control. The same samples were also analyzed with anti-RPA32 antibody to assess the amount of total RPA32. (B). Time course of RPA phosphorylation in BEAS-2B cells exposed to 5% THS for various lengths of time. Cells were collected at the indicated treatment times followed by western analysis with phospho-RPA32 (S4/S8) antibody. Fold increase in RPA phosphorylation was calculated after normalization with GAPDH. (C) hPFs were exposed to the indicated doses of THS and RPA phosphorylation was monitored as in panel A. (D) Recovery of hPFs cells from replication stress following exposure to various doses of THS. Cells were treated with THS for 12h or mock treated, then allowed to recover for 12h by changing to complete medium. Induction of pRPA32 and recovery as indicated by its loss were followed by western analysis using phospho-RPA32 (S4/S8) antibody.
To address DNA damage repair kinetics for THS-induced lesions that cause replication stress, we measured recovery as indicated by decreased pRPA32. hPFs were exposed to various doses of THS for 12 h as indicated in Figure 3D. After exposure, THS was washed out and cells were allowed to recover for 12 h in complete DMEM medium containing 10% FBS, followed by Western analysis using anti-phospho-RPA32S4/S8 antibody. THS exposure induced a 7.2 and 7.3-fold increase in RPA32 phosphorylation after 2.5% and 5% THS exposure (Fig. 3D). As we observed previously, high dose THS exposure (10%) induced less RPA32 phosphorylation compared to lower doses of THS exposure (Fig. 3D). Nonetheless, RPA phosphorylation returned almost to baseline levels after a 12 h repair period independent of the THS dose (Fig. 3D). These results suggest that the effects on replicative stress of DNA damage produced by THS are reversed within 12 h after the exposure. Whether this is due primarily to DNA repair, or perhaps also reflects loss of the most heavily affected cells, requires further investigation.
THS Exposure Activates Replicative Stress and DNA Damage Response Machinery
RPA-coated ssDNA is the key structure for recruitment and activation of ataxia telangiectasia-mutated and Rad3-related (ATR) kinase [Kemp et al., 2017]. Phosphorylation of ATR at Thr-1989 is required for ATR activation in response to damage and has been observed within 1 h of exposure to either UV or ionizing radiation [Nam et al., 2011]. Using an antibody against phosphorylated ATR Thr-1989, we detected ATR activation in hPFs exposed for 12 h to either 5% or 2.5% THS, with some indication of an inverse dose response (Fig. 4A). ATR activation could be detected after only 3 h of exposure to 5% THS and reached a peak at 6 h of exposure (supplemental Fig. S4A). A commonly used marker of ATR activation is the phosphorylation of CHK1, and ATR-CHK1 signaling is required to prevent stalled replication forks from collapse [Ragland et al., 2013]. We observed CHK1 Ser-317 phosphorylation after 3 h of exposure, which increased until 6 h and then remained constant, thus paralleling ATR activation (Supplemental Fig. S4A). Total CHK1 did not significantly change.
Fig. 4.
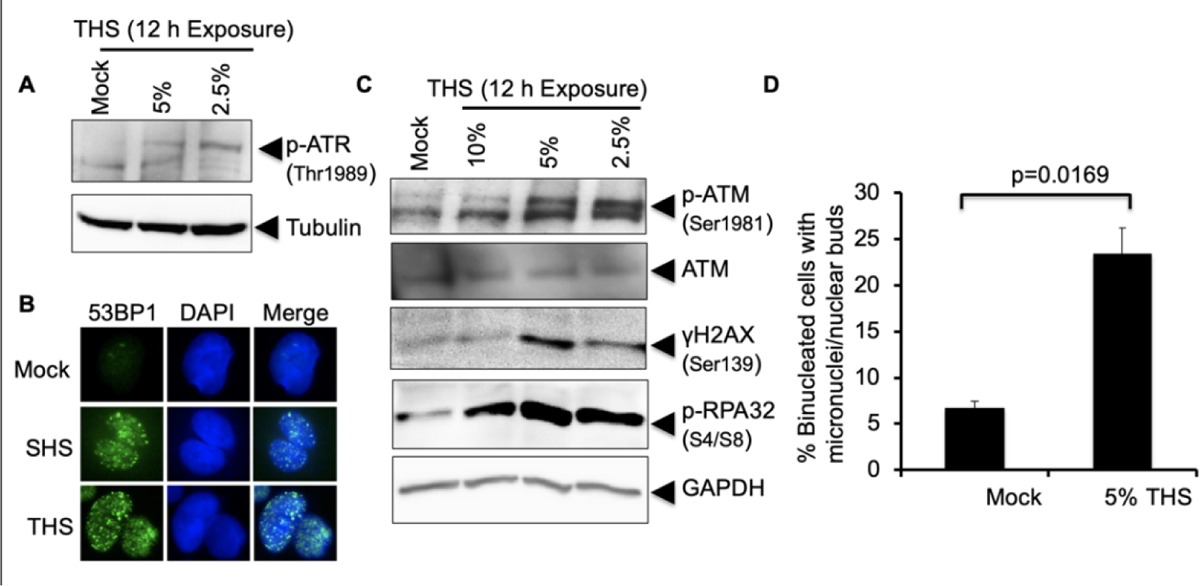
THS exposure causes replication stress and activates DSB response machineries. (A) Replication stress response was monitored following exposure of hPFs to the indicated doses of THS for 12 h followed by western analysis with anti-phosphoATR Thr1989 antibody. Tubulin was used as loading control. (B) Induction of DSBs by exposure of hPFs to 2.5% THS for 24 h was analyzed by IF detection of 53BP1 protein, which accumulates in foci at DSBs. SHS (0.4 PE) treatment was used as a positive control. (C) Activation of ATM following exposure of hPFs to 10%, 5% and 2.5% THS for 12 h was analyzed by western with anti-pATM (Ser1981). The same membrane was probed for phosphorylation of histone H2AX using anti-γH2AX antibody (Ser139) as an indicator of DSB formation and for pRPA32 (as in Fig. 3) to monitor replication stress. (D) Micronuclei formation in BEAS-2B cells following exposure to 5% THS for 48 h. Data represent the mean of ±SD for N=2.
We also investigated the generation of DNA double-strand breaks (DSBs) in response to THS using the formation of 53BP1 foci as a marker [Rogakou et al., 1999]. We have previously reported induction of DNA strand-breaks in human cells exposed to THS [Hang et al. 2013], but the assay employed did not distinguish between single-strand breaks (SSBs) and the much more deleterious DSBs. Dramatic formation of 53BP1 foci was observed in hPFs following exposure to 2.5% THS for 24 h, comparable to SHS (0.4 PE) exposure (Fig. 4B). The observed induction of DSBs suggested the activation of ATM, a master regulator of DNA damage responses. Upon DNA damage, ATM autophosphorylates its S1981 residue and provokes its dissociation from an inactive dimer to the active ATM monomer, which then phosphorylates downstream proteins involved in DNA repair, apoptosis and cell cycle checkpoints [Lavin et al., 2007]. To examine ATM activation by THS, hPFs were exposed to increasing doses of THS and ATM phosphorylation was monitored by western analysis using specific antibodies. Activation of ATM was detected at all three THS doses tested, again with an inverse dose response (Fig. 4C), as was also observed for RPA phosphorylation in hPFs (here and Fig. 3C). Western analysis with an antibody recognizing γH2AX S139 showed that H2AX was also phosphorylated in response to both 5% and 2.5% THS exposure for 12 h (Fig. 4C), consistent with DSB formation and ATM activation. To determine the time course of ATM activation in normal lung cells, we exposed hPFs to 5% THS for varying amounts of time. Strikingly, ATM S1981 phosphorylation was observed within 0.5 h of exposure (supplemental Fig. S4B).
Next we examined THS effects on BRCA1, a tumor suppressor protein with multiple roles in maintaining genomic integrity, including damage-induced cell cycle checkpoint activation and DSB repair by the homologous recombination repair (HRR) pathway [Chen C-C et al., 2018]. It is activated in response to DSB formation via phosphorylation by the ATM kinase. Western analysis using phospho-BRCA1 (BRCA1 S1423) antibody revealed that phosphorylation of BRCA1 started between 0.5 and 3 h of exposure, thus lagging slightly behind ATM activation (Supplemental Fig. S4B). Re-probing the same membrane with an antibody recognizing total BRCA1 revealed the induction of a second, slower-migrating band, clearly demonstrating the phosphorylation of BRCA1. Western analysis of the same blot with an antibody recognizing γH2AX showed that H2AX phosphorylation began within the first 0.5 h of THS exposure, congruent with ATM activation (Supplemental Fig. S4B). Together, these results demonstrate that exposure of lung cells to THS rapidly activates certain key DNA damage response proteins, consistent with induction of replication stress.
Induction of Micronuclei Following Exposure of Lung Cells to THS
Induction of DSBs and replication stress following exposure of lung cells to THS suggests that THS exposure may induce formation of micronuclei (MN), which are an indicator of genomic instability and cancer risk [Luzhna L., 2013]. To test for induction of MN, BEAS-2B cells were exposed to 5% THS for 48 h followed by addition of Cytochalasin-B to block cytokinesis, then further incubation for another 48 h in the presence of THS before collection and scoring for MN in bi-nucleated cells [Rydberg et al., 2007]. Significantly increased MN formation was observed following exposure of BEAS-2B cells to 5% THS compared to a mock exposed control (Fig. 4D). This striking effect strongly suggests that THS exposure induces genomic instability, as consistent with our report of increased incidence of lung cancer in THS-exposed mice [Hang et al., 2018].
THS Exposure Impairs Transcription and Stalls RNA Polymerase II
Bulky lesions in DNA, such as those produced by UV irradiation, pose a strong block to transcription by RNA polymerase II (RNAP II) and require repair by Transcription-Coupled NER (TC-NER) for efficient recovery of RNA synthesis and cell survival [Lans et al., 2019; Gregersen LH and Svejstrup JQ, 2018]. Transcription-blocking lesions result in accumulation of the elongating form of RNAP II, which is phosphorylated on Ser2 of the polymerase C-terminal domain [Donnio et al. 2019]. Because we have observed a strong DDR in response to THS exposure, including Ser-15 phosphorylation of p53 [Hang et al., 2018] and activation of both ATM and ATR (Fig. 4), we asked whether DNA damage induced by THS causes blocked transcription. That is indeed the case as was evident from western analysis of RNAP II in extracts prepared from BEAS-2B cells 12 h after exposure to various doses of THS. Our results revealed an increase in the hyper-phosphorylated form of RNAP II (RNAPIIo) with concomitant decrease of the hypo-phosphorylated form involved in transcription initiation (RNAP IIa) following exposure to THS, as shown by the calculated ratio between the two, expressed as % RNAPIIo (Fig. 5A). Interestingly, the dose-response for this endpoint was decidedly nonlinear, with a substantial but equivalent increase in % RNAPIIo for the two lowest doses compared to non-exposed control and a decrease for the highest dose tested. This inverse dose-response is similar to that observed for effects on DNA synthesis, phosphorylation of RPA32, and activation of ATM and ATR (Figs. 1, 3 and 4). To further examine the effect of THS exposure on transcription, we performed a time course experiment with cell extracts prepared at various times after exposure. Using an antibody selectively recognizing the elongating form of RNA polymerase (RNAPIIo-ser2), we detected its accumulation as early as 3 h after exposure to 5% THS (Fig. 5B). We then directly monitored the effect of THS on RNA synthesis by measuring the incorporation of EU into cells. Significantly reduced RNA synthesis as measured by fluorescence intensity was found after 24 h of exposure to either 2.5% or 5% THS (Fig. 5C, D). To verify that we were indeed observing damage-induced effects on transcription, we showed that this loss is comparable to that produced by a high dose (20 J/m2) of UV radiation when analyzed after 1 h of post-UV incubation (Fig. 5 C, D). These results suggest that bulky DNA lesions generated by exposure of lung cells to THS strongly block elongating RNAPIIo and thus reduce ongoing transcription of active genes.
Fig. 5.
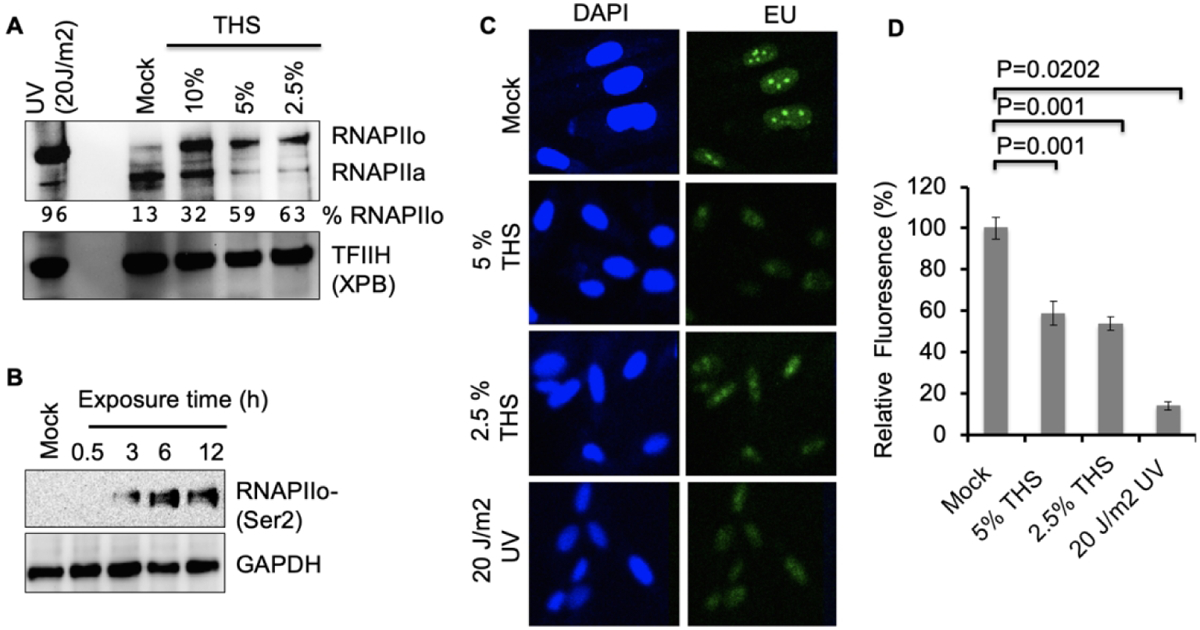
Basal transcription is impaired following exposure to THS. (A) BEAS-2B cells were exposed to THS for 24 h with the indicated doses or mock treated, and RNA polymerase II was analyzed by western using anti-RNAP II antibody (8wg16), which recognizes both hypo- and hyper-phosphorylated forms of the polymerase. 20 J/m2 UV- irradiation followed by 1h incubation was used as a positive control. TFIIH subunit XPB served as a loading control. Band intensities for RNAPIIo and IIa were quantified and the fraction of RNAP as the IIo form was calculated for each condition. (B) Time course of RNAPII phosphorylation in BEAS-2B cells treated for various times with 5% THS was analyzed by western with anti-RNAPII CTD phosphoSer2 antibody (Covance; H5), which recognizes the phosphorylated form of RNAP II that is engaged in transcription elongation. GAPDH was the loading control. (C) Ongoing RNA synthesis was measured by EU incorporation after exposing BEAS-2B cells to 5% or 2.5% THS for 24 h. As a positive control for transcription inhibition, cells were irradiated with 20 J/m2 UV and EU was added 1 h after exposure. Representative images obtained at 20x magnifications are shown. (D) Quantification by ImageJ of the EU fluorescence intensity of 100 cells from mock, THS exposed, or UV irradiated samples. Data represent the mean of ± SD for N=3
Having shown that NER operates in response to THS exposure (Fig. 2), we asked whether it is required for removal of THS-induced lesions during transcription elongation. We exposed NER-defective XPA patient (XP12BE) dermal fibroblasts to 2.5% and 5% THS for 24h and monitored RNA synthesis. These cells have no XPA protein detectable by western analysis (Fig. 6C). Compared to mock treated control cells, RNA synthesis was significantly reduced in the XP-A patient cells after both THS doses tested (Fig. 6A and 6B). Quantification of the fluorescence intensity (a measure of RNA synthesis) showed that 5% THS exposure reduced RNA synthesis to a much greater extent in the XPA-deficient cells (21%; Fig. 6B) as compared to the same dose in normal BEAS-2B cells (59%; Fig. 5D), strongly suggesting that intact NER is required for removal of THS induced lesions to allow unperturbed transcription.
Fig. 6.

THS exposure severely impairs basal transcription in NER defective cells. (A) Dermal fibroblasts from an XPA patient (XP12BE) were exposed to 5% or 2.5% THS for 24 h followed by EU incorporation and imaging as in Fig. 5. 40x magnification images are shown. (B) Quantification using ImageJ of the average fluorescence intensity of 10 cells from mock treated or THS-exposed XP12BE samples. Data represent the mean of ± SD for N=3. (C) Absence of XPA protein detectable with anti-XPA antibody in the XP-A patient cell line XP12BE is shown. The human fibroblast strain HCA2 was a normal control. XPB protein was used as a loading control.
DISCUSSION
In this study, we report for the first time that THS exposure impairs both DNA replication and transcription in cultured human lung cells. It is reasonable to suppose that such effects on cellular DNA transactions may underlie the adverse consequences of THS exposure that we reported in vivo [Hang et al., 2018].
Currently, nicotine concentration is used as a measure of THS contamination since it is the major THS constituent. The estimated surface and dust concentration of nicotine determined in a typical smoker’s home was 73 μg/m2 and 64 μg/m2 as reported [Focus, 2011; Matt et al., 2004]. In our experiments, we exposed cells to 24 (2.5% THS) to 96 (10% THS) ng/ml of THS extract over a 24 h time period. Although a direct comparison is not possible, the concentrations used in our study are considered to be comparable or even at the lower end of the estimated range of exposure from indoor contamination [Hang et al., 2013].
Our finding of an increased percentage of human lung cells in S-phase following exposure to the lowest dose of THS (2.5%, equivalent to 24 ng of nicotine level in THS sample) is unexpected. This observation is consistent with a previously observed exposure-induced increase in p21 and p53 levels [Hang et al., 2018]. The most likely explanation is that blocked replication as a result of THS-induced damage causes inability to exit from S-phase and thus S-phase accumulation. RPA32 binds to the ssDNA generated at stalled forks to prevent fork collapse and recruits ATR kinase through its binding partner ATRIP, followed by phosphorylation of RPA32 [Vassin et al., 2009]. Consistent with this notion, we found that THS exposure caused extensive hyper-phosphorylation of RPA32, as well as phosphorylation of ATR at Thr1989, a marker of replication stress (Fig. 3; Fig. 4A). THS exposure is known to induce DNA damage, including both base damage and strand breaks [Hang et al., 2013; Dhall et al., 2016; Hang et al., 2018]. Activation of ATM, phosphorylation of histone H2AX to γH2AX and increased 53BP1 foci formation demonstrated here are all consistent with induction of DSBs by exposure to THS (Fig. 4B, C). Correlating with the induction of DSBs and replication stress, a significant THS-induced increase in micronucleus formation was also observed (Fig. 4D). All of these cellular consequences are consistent with the induction of genomic instability, which is a hallmark of cancer and which may provide a mechanistic explanation for the increased incidence of lung cancer in THS-exposed A/J mice [Hang et al., 2018].
For many of the measured cellular consequences of THS exposure, most notably RPA32 phosphorylation but also including post-exposure DNA synthesis, activation of both ATR and ATM, and stalling of RNAPII during transcription, we observed a bell-shaped dose-response pattern with a decreased response at higher doses. This unexpected effect is similar to that in our previously reported DNA strand-break studies by NNA and NNK (Hang et al. 2013). While lower THS doses may not induce sufficient DNA damage for a measurable response (i.e., a threshold effect), it is more difficult to explain the decrease at higher doses. There are multiple possibilities, including attenuation by interaction of THS components or even inhibition of key protein activities, but further investigation is required to understand the effect.
NNK, a constituent of THS, has been shown to form bulky DNA lesions [Lao et al., 2007; Ma et al., 2019], which are known to block transcribing RNAP II and to cause transcriptional stress in addition to posing blocks to replication. The results reported here demonstrate both replication stress and transcription blockage in lung cells exposed to THS. Bulky DNA lesions generated as a consequence of UV or chemical exposure are repaired by the NER pathway, with lesions in transcribed strands being preferentially repaired by TC-NER at a much faster rate than the global genome-wide pathway [Lans H. 2019; Hanawalt PC and Spivak G. 2008; Gregersen LH and Svejstrup JQ. 2018; Scharer et al., 2013]. THS exposure arrested cells in S-phase, but paradoxically caused increased DNA synthesis as measured by incorporation of the DNA precursor EdU (Fig. 1). Since EdU incorporation after THS exposure was significantly reduced in XPA knockdown THS-exposure induced DNA damage, particularly the bulky adducts. Since THS cells that are unable to perform either global or transcription-coupled NER (Fig. 2), the observed increased DNA synthesis in NER-competent cells is likely due to DNA repair synthesis. This interpretation implies that the NER pathway is important for removing exposure reduced ongoing RNA synthesis and caused accumulation of the elongating form of RNA Pol II, it is likely that TC-NER in particular is required for effective cellular responses to DNA damage caused by THS.
CONCLUSIONS
In summary, we have demonstrated for the first time that THS exposure at a dose relevant to real-life exposure perturbs the progression of DNA replication and transcription, activates the DNA damage response, and causes both replication and transcriptional stress. The observed induction of DSBs, together with greatly increased MN formation, suggests that genomic instability is a consequence. These observations suggest a mechanism for the increased incidence of lung cancer that we have reported in THS-exposed A/J mice and point to the likelihood of a deleterious impact on human health. Moreover, these findings directly imply that THS exposure may accelerate the resulting pathologies in individuals with reduced DNA repair capacity. Since there is evidence for significant variability in the efficacy of DNA repair responses among individuals in human populations [Paz-Elizur T., 2005], the findings reported here suggest one basis for potential individual variation in risks associated with THS exposure.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Tapas Hazra (UTMB, TX) for intellectual advice during the course of the study. We thank Dr. Prudence Talbot (UC Riverside) for human lung fibroblasts. We thank David Acevedo for analysis of some IF data. We thank Polly Cheung at UCSF for carrying out the chemical analysis of THS exracts. This work was supported by Grant 26IR-0017 to AHS and Grant 28PT-0077 to PJ from the University of California Tobacco Related Disease Research Program (TRDRP) and by the NIH/NCI grant P01 CA092584 to PKC. Laboratory resources at UCSF for analytical chemistry were supported by NIH/NIDA grant P30 DA012393 to RT Jones.
REFERENCES
- Bahl V, Johnson K, Phandthong R, Zahedi A, Schick SF, Talbot P. 2016. From the Cover: Thirdhand Cigarette Smoke Causes Stress-Induced Mitochondrial Hyperfusion and Alters the Transcriptional Profile of Stem Cells. Toxicol Sci, 9; 153(1): 55–69. [DOI] [PubMed] [Google Scholar]
- Brueckner F and Cramer P. 2007. DNA photodamage recognition by RNA polymerase II. FEBS letters, 581:2757–2760. [DOI] [PubMed] [Google Scholar]
- Burton A, 2011. Does the smoke ever really clear. EHP, 119, 2, A71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Feng W, Lim PX, Kass EM and Jasin M. 2018. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol. March, 2: 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R and Wold MS. 2014. Replication Protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow Replication Protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays, December ; 36(12);1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhall S, Alamat R, Castro A, Sarker AH, Mao JH, Chan A, Hang B, Martins-Green M. 2016. Tobacco toxins deposited on surfaces (third hand smoke) impair wound healing. Clin Sci (Lond). July 1;130 (14):1269–84. doi: 10.1042/CS20160236. [DOI] [PubMed] [Google Scholar]
- Donnio LM, Lagarou A, Sueur G, Mari P-O, Giglia-Mari G. 2019. CSB-Dependent Cyclin-Dependent Kinase 9 Degradation and RNA Polymerase II Phosphorylation during Transcription-Coupled Repair. Mol. Cell Biol, V39 (6); e00225–18. doi: 10.1128/MCB.00225-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg ER, Downey M and Toczyski D. 2014. Polymerase stalling during replication, transcription and translation. Current Biol V24, 10, pp R445–R452. [DOI] [PubMed] [Google Scholar]
- Fenech M, Chang WP, Kirsch-Volders M, Holland N, Bonassi S, Zeiger E. 2003. HUMN project; detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte culture. Mutat Res, January 10; 534(1–2): 65–75. [DOI] [PubMed] [Google Scholar]
- Derheimer Frederick A., O’Hagan Heather M., Krueger Heather M., Hanasoge Sheela, Paulsen Michelle T., and Ljungman Mats. 2007. RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad Sci. USA, July 31, 104 (31); 12778–12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH and Svejstrup JQ. 2018. The cellular response to transcription-blocking DNA damage. Trends in Biochemical Sciences, May; 43(5); 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD and Paull TT. 2010. ATM activation by oxidative-stress. Science, October 22; 330(6003): 517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC and Spivak G. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol, December;9(12):958–70. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hang B, Sarker AH, Havel C, Saha S, Hazra TK, Schick S, Jacob P 3rd, Rehan VK, Chenna A, Sharan D, Sleiman M, Destaillats H, and Gundel LA. 2013. Thirdhand smoke causes DNA damage in human cells. Mutagenesis, 28, 381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang B, Snijders AM, Huang Y, Schick SF, Wang P, Xia Y, Havel C, Jacob P 3rd, Benowitz N, Destaillats H, Gundel LA, Mao JH. 2017. Early exposure to thirdhand cigarette smoke affects body mass and the development of immunity in mice. Sci. Rep 2017 February 3;7:41915. doi: 10.1038/srep41915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang B 2010. Formation and repair of tobacco carcinogen-derived bulky DNA adducts. J. Nuclei Acids. December 20; 2010:709521. doi: 10.4061/2010/709521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang B, Iavarone A, Havel C, et al. 2014. NNA, a thirdhand smoke constituent, induces DNA damage in vitro and in human cells. 247th National Meeting of the American Chemical Society (ACS) with press release; March 16–20 Dallas, TX. [Google Scholar]
- Hang B, Wang Y, Huang Y, Wang P, Langley SA, Bi L, Sarker AH, Schick SF, Havel C, Jacob P 3rd, Benowitz N, Destaillats H, Tang X, Xia Y, Jen KY, Gundel LA, Mao JH, Snijders AM. 2018. Short-term early exposure to thirdhand cigarette smoke increases lung cancer incidence in mice. Clin Sci (Lond)., February 28; 132(4):475488. doi: 10.1042/CS20171521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob III P, Benowitz NL, Destaillants H, Gundel L, Hang B, Martin-Green M, Matt GE, Quintana PJ, Samet JM, Schick SF, Talbot P, Aquilina NJ, Hovell MF, Mao JH and Whitehead TP. 2017. Thirdhand Smoke: New evidence, challenges and future directions. Chem Res Toxicol 17; 30 (1) 270–294. doi; 101021/acs.Chemrestox.6b00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp MG. 2017. DNA damage-induced ATM- and Rad-3-related (ATR) kinase activation in non-replicating cells is regulated by the XPB subunit of transcription factor IIH (TFIIH). J. Biol. Chem. June 7, doi; 10.1074/jbc.M117.788406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lans H, Hoeijmakers JH, Vermeulen W and Marteijin JA. 2019. The DNA damage response to transcription stress. Nat Rev Mol Cell Biol, December; 20(12);766–784. doi: 0.1093/nar/gkz977 [DOI] [PubMed] [Google Scholar]
- Lavin MF and Kozlov S. 2007. ATM activation and DNA damage. Cell cycle, April 15; 6(8): 931–42. [DOI] [PubMed] [Google Scholar]
- Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. 2007. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res. Toxicol, February; 20(2); 235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzhna L, Kathiria P and Kovalchuk O. 2013. Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Front Genet. 4:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Stepanov I, Hecht SS. 2019. Recent studies on DNA adducts resulting from human exposure to tobacco smoke. Toxics, March 19;7(1). pii: E16. doi: 10.3390/toxics7010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Green M, Adhami N, Frankos M, Valdez M, Goodwin B, Lyubovitsky J, Dhall S, arcia M, Egiebor I, Martinez B, Green HW, Havel C, Yu L, Liles S, Matt G, Destaillats H, Sleiman M, Gundel LA, Benowitz N, JacobIII P, Hovell M, Winickoff JP, Curras-Collazo M. 2014. Cigarette smoke toxins deposited on surfaces: Implications for human health. PLoS One, 2014, 9, e86391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt GE, et al. 2004. Households contaminated by environmental tobacco smoke: sources of infant exposure. Tob Control, 13(1): 29–37; doi; 10.1136/tc.2003.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Yamashita S, Lehmann AR and Ogi T. 2010. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair, 9 (2010) 506–516. [DOI] [PubMed] [Google Scholar]
- Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB and Cortez D. 2011. Thr-1989 Phosphorylation Is a Marker of Active Ataxia Telangiectasia-mutated and Rad3-related (ATR) Kinase. J. Biol. Chem June 24, doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley GG and Patrick SM. 2012. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci, 15: 883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek M and Walter JC. 2004. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. Sep 15;23 (18): 3667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Elizur T, Krupsky M, Elinger D, Schechtman E, Livneh Z. 2005. Repair of the oxidative DNA damage 8-oxoguanine as a biomarker for lung cancer risk. Cancer Biomark, 1 (2–3):201–5. [DOI] [PubMed] [Google Scholar]
- Ragland RL, Patel S, Rivard RS, Smith K, Peter AA, Bielinsky AK and Brown EJ. 2013. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev, October 15; 27(20); 2259–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan VK, Sakurai R, Torday JS. 2011. Thirdhand smoke: a new simension to the effects of cigarette smoke on the developing lung. Am J Physiol Lung Cell Mol Physiol. July; 301(1):L1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg B, Chun E, Groesser T. 2007. Relative Biological effectiveness of high-energy iron ions for micronucleus formation at low doses. Radiation Research, 168(6); 675–682. [DOI] [PubMed] [Google Scholar]
- Rothstein R and Jasin M. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harbor, 5;a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockx DA, Van-Hoffen A, Barton MC, Citterio E, Bregman DB, Van Zeeland AA,Vrieling H and Mullenders LH. 2000. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc. Natl. Acad Sci. USA, September 12; 97(19): 10503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. September 6;146 (5):905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MA, Kim SH, Campisi J. 2002. Reversible manipulation of telomerase expression and telomere length. Implications for the ionizing radiation response and replicative senescence of human cells. J Biol Chem, 277, 28609–28617. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. 2005. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB and TFIIH: Insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Sleiman M, Gundel LA, Pankow JF, Jacob P 3rd, Singer BC, Destaillats H. 2010. Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards. Proc Natl Acad Sci USA. April 13;107(15):6576–81.doi: 10.1073/pnas.0912820107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick SF, Farraro KF, Perrino C, Sleiman M, van de Vossenberg G, Trinh MP, Hammond SK, Jenkins BM, Balmes J. 2014. Thirdhand cigarette smoke in an experimental chamber: evidence of surface deposition of nicotine, nitrosamines and polycyclic aromatic hydrocarbons and de novo formation of NNK. Tobacco Control, March; 23(2):152–9. [DOI] [PubMed] [Google Scholar]
- Scharer OD. 2013. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb Perspect Biol., 2013;5:a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenberger JS and Dixon PS. 1999. Oxygen Induces S-Phase Growth Arrest and Increases p53 and p21 Expression in Human Bronchial Smooth-Muscle Cells. Am. J. Respir. Cell Mol. Biol, 21, pp. 395–402. [DOI] [PubMed] [Google Scholar]
- Tornaletti S, Patrick SM, Turchi JJ and Hanawalt PC. 2003. Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA. J. Biol. Chem, September 12; 278(37):35791–7. [DOI] [PubMed] [Google Scholar]
- Trego KS, Groesser T, Davalos AR, Parplys AC, Zhao W, Nelson MR, Hlaing A, Shih B, Rydberg B, Pluth JM, Tsai MS, Hoeijmakers JH, Sung P, Wiese C, Campisi J, Cooper PK. 2016. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol Cell, February 18;61(4):535–546. doi: 10.1016/j.molcel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassin VM, Anantha RW, Sokolova E, Kanner S and Brorwiec JA. 2009. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Science 122, 4070–80, doi; 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK and Cimprich KA. 2014. Cause and consequences of replication stress. Nat. Cell Biol, January;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J 2013. The role of BRCA1 in homologous recombination repair in response to replication stress: significance in tumorigenesis and cancer therapy. Cell Biosci, 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.