

Abstract
Selenoprotein I (SELENOI) is an ethanolamine phosphotransferase that catalyzes the third reaction of the Kennedy pathway for the synthesis of phosphatidylethanolamine. Since the role of SELENOI in murine embryogenesis has not been investigated, SELENOI−/+ mating pairs were used to generate global KO offspring. Of 323 weanling pups, no homozygous KO genotypes were found. E6.5-E18.5 embryos (165 total) were genotyped, and only two E18.5 KO embryos were detected with no discernable anatomical defects. To screen embryos prior to uterine implantation that occurs ~ E6, blastocyst embryos (E3.5-E4.4) were flushed from uteruses of pregnant females and analyzed for morphology and genotype. KO embryos were detected in 5 of 6 pregnant females, and 7 of the 32 genotyped embryos were found to be SELENOI KO that exhibited no overt pathological features. Overall, these results demonstrate that, except for rare cases (2/490 = 0.4%), global SELENOI deletion leads to early embryonic lethality.
Keywords: embryonic lethal, phosphatidylethanolamine, phospholipid transferase, selenium
Introduction
Phosphatidylethanolamine (PE) comprises 15–25% of cellular phospholipids, combining with other phospholipids, glycolipids, and cholesterol to constitute cellular membranes [1]. In addition to its role in membranes, PE serves as a precursor for the synthesis of other factors involved in cellular signaling and metabolism. For example, PE serves as a precursor for the synthesis of glycophosphatidylinositol (GPI) anchors that tether ~ 150 proteins to the surface of cells and play a wide variety of functions [2]. PE may also be converted into ether linked PEs, plasmenyl PE (a.k.a. plasmalogens), which function in synaptic vesicles and secretory granules as well as other less defined cellular processes [3]. In mammals, PE is synthesized via the Kennedy pathway in the endoplasmic reticulum, the phosphatidylserine (PS) decarboxylation pathway in the mitochondria [4], and perhaps yet to be identified pathways. While the PS decarboxylase pathway mainly synthesizes PE for the mitochondria, the Kennedy pathway in the ER supplies plasma and organelle membranes as well as other synthesis pathways with PE [5].
The Kennedy pathway is a three-step process in which ethanolamine is first phosphorylated by ethanolamine kinase 1 (ETNK1) in the cytosol to generate phosphoethanolamine. The second reaction is the rate-limiting step catalyzed by CTP-phosphoethanolamine cytidyltransferase (ECT) also located in the cytosol, which converts phosphoethanolamine to CDP-ethanolamine. The third step involves the reaction of the CDP-ethanolamine with 1,2-diacylglycerol catalyzed by selenoprotein I (SELENOI; ethanolamine phosphotransferase 1) to generate PE. SELENOI spans the ER membrane multiple times with its catalytic domain embedded in this pocket of the ER membrane [6]. This enzyme is a member of the selenoprotein family, in which all 25 members in humans (24 in mice) contain the signature selenocysteine amino acid. Recombinant SELENOI exhibits CDP ethanolamine-specific phosphatidyltransferase activity, and its expression has been detected in a wide variety of tissues [7].
The selenoprotein family of genes, including the SELENOI gene, was initially identified in 2003 as part of an in silico analyses of the human genome [8]. To date, 4 of the 24 mouse selenoproteins have been shown to be essential for life based on homozygous knockout (KO) models [9]. These include glutathione peroxidase-4, thioredoxin reductase-1 and −2, and selenoprotein T. While an essential role for SELENOI in mice has not yet been described, a rare case study described one patient with a mutation leading to nonfunctional SELENOI who exhibited severe neurodevelopmental defects, and fibroblasts derived from this patient had impaired ethanolamine phosphotransferase activity and reduced levels of PE species [10]. The patient was born to first cousin parents, presenting with severe complications 2 h post-partum and onward that required early and repeated intervention with medication (e.g. phenylbarbital) and surgery. These results support a critical role for SELENOI in early stages of human development, although it has been noted that the tolerance to loss of expression of individual selenoproteins may differ between human and mouse [11]. The latter study used bioinformatic approaches to estimate the impact of individual selenoproteins on human health and this group found that loss of functional SELENOI was strongly selected against, suggesting it may represent one of the most important selenoproteins for human health.
PE derived from the Kennedy pathway as well as the PS decarboxylase pathway have been shown to be essential in developing mice. In particular, the global knockout of ECT is embryonic lethal [12] as is the knockout of PS decarboxylase [13]. The ECT KO embryos die after uterine implantation (~E6), prior to embryonic day 8.5, which raises the question whether global deletion of SELENOI that catalyzes step 3 of the Kennedy pathway is also embryonic lethal in mice. To investigate this possibility, SELENO+/− mice were used to generate a global deletion of SELENOI in offspring. Results showed that homozygous deletion of SELENOI led to embryonic lethality prior to uterine implantation (~E6), with a rare detection of KO embryos at the E18.5 stage.
Materials and Methods
Cre/loxp-based generation of SELENOI KO mice.
Mice were housed in specific germ free conditions and fed a standard mouse chow containing 0.25 parts per million (ppm) selenium. Animal protocols were approved by the University of Hawaii Institutional Animal Care and Use Committee. A conditional SELENOI knockout mouse model was generated on a C57BL/6J background strain (Jackson Laboratories, Bar Harbor, ME, USA) using CRISPR/Cas9 technology to insert loxp sequences into intronic regions surrounding exons 2 and 3 of SELENOI (Applied Stem Cell, Inc., Milpitas, CA, USA). Founder mice with loxp flanked, or floxed (fl), SELENOI alleles were used to generate a SELENOIfl/fl a colony. The cmv-Cre and lck-Cre (distal promoter) mice were purchased from Jackson Laboratories and bred with SELENOIfl/fl mice. Timed pregnancies were carried out by inspecting for vaginal plugs and staging of embryos was confirmed using established criteria [14]. To genotype and detect the floxed SELENOI alleles in DNA extracted from mouse tails or embryos, PCR was carried out for 3’ loxp site using: fwd 5’- GTC TGT GTG AGG TTG TTG GAT CTC C −3’ and rev 5’- GCA TAT AGG TGT AGA GAA AAT AGG TAT GCA AAC C −3’. For the 5’ loxp site, the following primers were used: fwd 5’- GCA CTA GAG AGC CTA TAA ACC AAG ACT GC −3’ and rev 5’- CCA GAG GAT GTG AGC TTG GCG −3’. PCR products exhibit a 34 nucleotide difference with and without loxp sites. For the detection of nonexcised and excised alleles, respectively, the following PCR primers were used: fwd 5’- TTC CAG GGG TGC TTA GGT CT-3’ and rev 5’- AGA TCT GCC TGC CTA TGT GC-3’ (544 bp product); fwd 5’- TGT GAG TGT GCT GGG TTA GG-3’ and rev 5’- GGG TGG CAG ATG GGT ACA TAA-3’ (450 bp product). The PCR conditions were as follows: 94°C for 2 min; 10 cycles: 94°C for 20 s, 65°C for 15 sec, 68°C for 10 sec; 28 cycles: 94°C for 15 s, 60°C for 15 sec, 72°C for 20 s; 72°C for 2 min. For genotyping offspring, we analyzed all embryos and pups, including unhealthy or partially developed embryos and neonatal pups that died before reaching 3 wks of age.
Genotyping blastocyst embryos
Embryos were flushed from excised oviducts/uteruses and separated into PCR tubes. Individual embryos were for isolated as previously described [15] for cDNA synthesis using a SuperScript III Cells Direct cDNA synthesis kit (Invitrogen). PCR was conducted to detect the presence or absence of the floxed region (exons 2 and 3) using the following primers: fwd1 5’-CTT TGG ATA CCA ACC CAC TCT C-3’ and rev1 5’- CCA GTC AGG CAC ATG CTT AT −3’; fwd2 5’- CAT ACT TCG ACC CTG ACT TCT ATG-3’ and rev2 5’-CAC AAC CGC AGT CAC TAT GTA-3’. Primers for the cDNA region outside of the floxed regions: fwd3 5’- CTA GAT GGA GTG GAT GGA AAG C-3’ and rev3 5’- GAG CAA ACA CGG AGA GAA GAA-3’. PCR conditions were the same as described above.
Histology
Day 18.5 embryos were fixed with 10% buffered formalin and transferred to 70% ethanol, followed by stepwise increases in higher % ethanol to dehydrate the tissue. The embryos were embedded in paraffin, and 5 μm sections mounted onto slides and stained using standard haematoxylin and eosin (H&E) techniques. Dorsal, ventral, and axial sagital sectioning was carried out for each fixed embryo for H&E staining. Images were captured for serial sections using a using a Zeiss Axiovert 200M attached to a Zeiss LSM 5 Pascal imaging system.
Results
No SELENOI−/− weanling pups were detected
To study the effects SELENOI gene deletion on development, a cre-Lox recombination system was used. First, SELENOIfl/fl mice were generated with loxp sequences inserted into intronic regions surrounding exons 2 and 3, which upon excision by Cre recombinase would lead to a truncated mRNA with a frameshift downstream of this site to ensure that functional protein would be deleted (Figure 1A). Whole-body SELENOI deletion utilized cmv-driven expression of Cre, which excises the target region throughout tissues including germ cells [16]. Male and female SELENOI+/− mice were generated that were healthy and fertile, which allowed us to cross these mice for the generation of SELENOI−/− mice with an expected Mendelian frequency of 25%. However, with 323 pups produced and genotyped at 3 wks of age, no SELENOI−/− weanling mice were detected.
Figure 1.
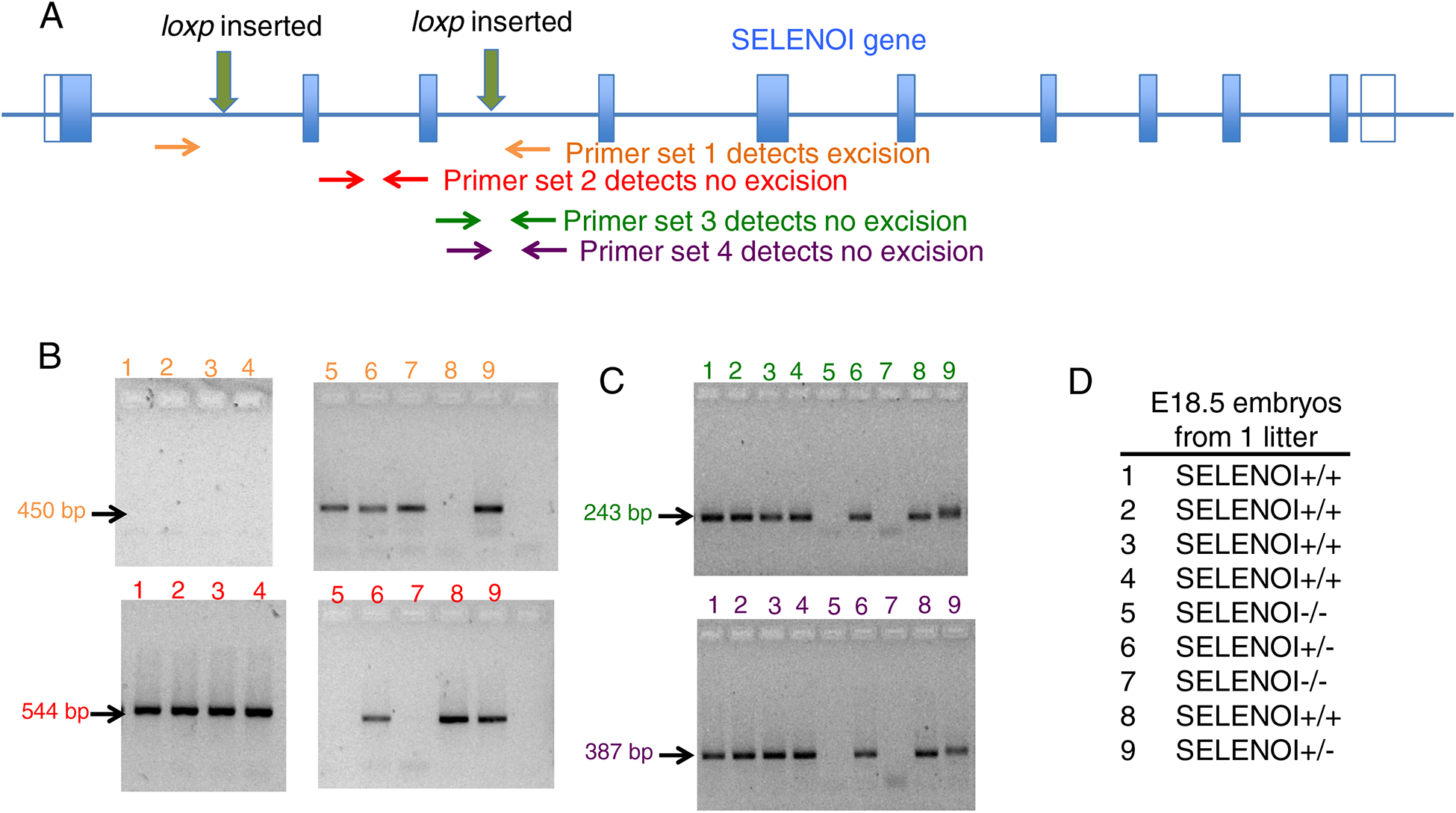
Strategy for generating and screening mice with SELENOI KO in all tissues. (A) A floxed mouse cell line was created on a C57BL/6 background using CRISPR/Cas9 to insert loxp sites flanking exons 2 and 3 in the SELENOI gene. PCR primer pairs were designed for genotyping the DNA of mouse tissues. (B) Results from one litter of E18.5 embryos show PCR results for primer pairs designed to detect the excised (top panel) and intact (bottom panel) SELENOI gene allele. (C) PCR results from the same litter show confirmation of intact SELENOI gene allele with two different primer pairs. (D) The combined PCR results allowed genotypes to be identified of all embryos from the E18.5 litter.
Analyses of E6.5-E18.5 embryos for SELENOI genotype
To examine pre-term embryos for SELENOI−/− genotypes, pregnant females were sacrificed and embryos collected from E6.5-E18.5 stages. Results revealed the presence of only SELENOI+/+ and SELENOI+/− genotypes, with the exception of one E18.5 litter that exhibited all three genotypes, including two SELENOI−/− embryos (Figure 1B–D). The pups from each genotype in this particular litter were analyzed by sagittal sectioning and histology for pathological features or malformed organ systems, and no discernable differences were found between genotypes (Figure 2). These data suggest that homozygous KO of SELENOI terminates development at a stage prior to E6.5, but there may be a rare escape of this termination with embryonic development proceeding to the perinatal stage (Table 1).
Figure 2.
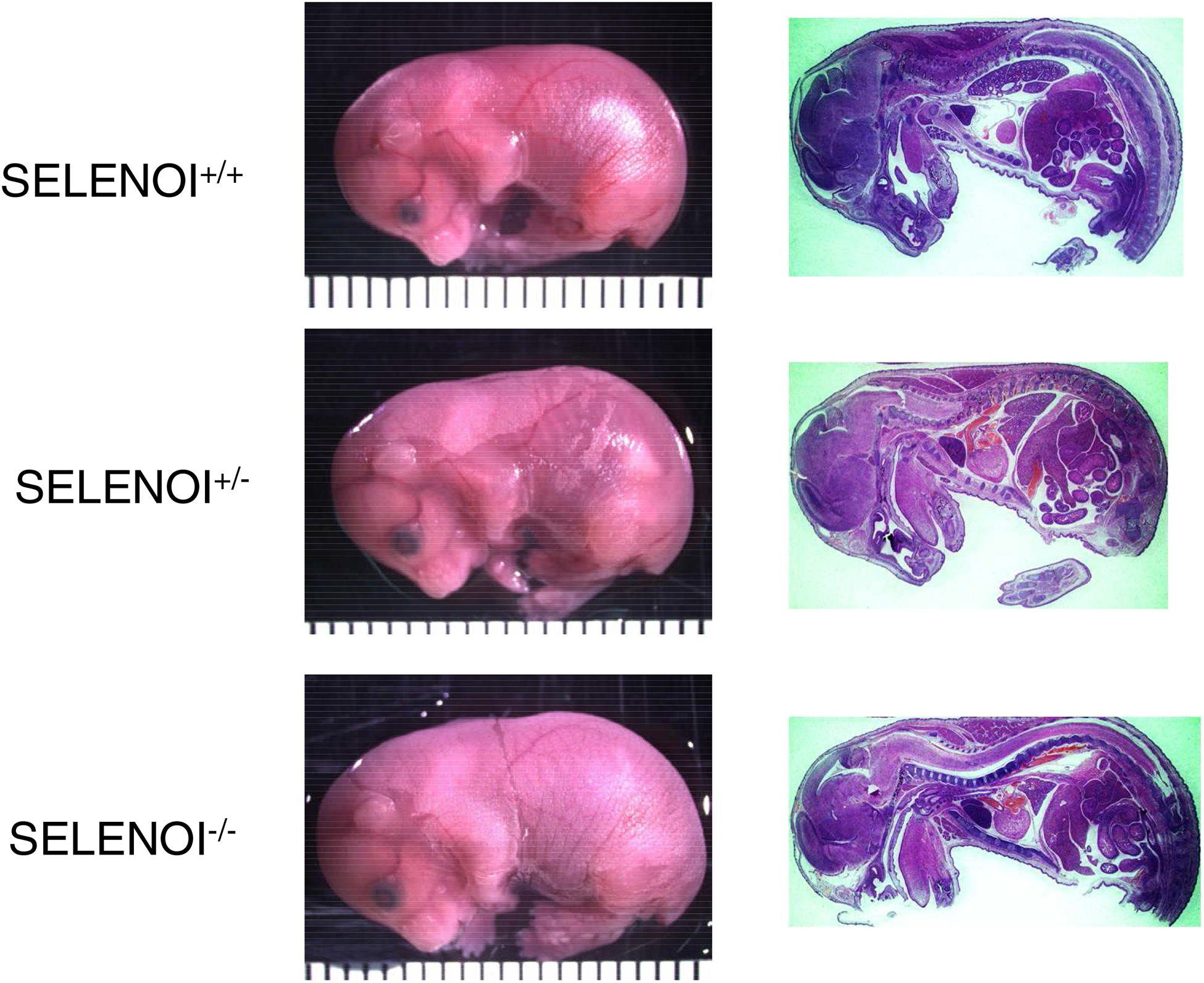
The rare E18.5 SELENOI KO embryo does not exhibit any pathological features. Representative sagittal sections from all three genotypes of E18.5 from the same litter are shown. No distinct features were found in SELENOI−/− embryos compared to SELENOI−/− and SELENOI−/− littermates.
Table 1.
Summary of Genotyping Results
| E6.5 | 10 | 19 | 0 |
| E7.5 | 5 | 3 | 0 |
| E8.5 | 0 | 1 | 0 |
| E9.5 | 0 | 0 | 0 |
| E10.5 | 4 | 12 | 0 |
| E11.5 | 0 | 0 | 0 |
| E12.5 | 10 | 26 | 0 |
| E13.5 | 4 | 15 | 0 |
| E14.5 | 0 | 0 | 0 |
| E15.5 | 0 | 0 | 0 |
| E16.5 | 0 | 0 | 0 |
| E17.5 | 5 | 8 | 0 |
| E18.5 | 14 | 29 | 2 |
| 3 wks old | 117 | 206 | 0 |
Genotyping of E3.5-E4.5 blastocysts reveals the presence of SELENOI−/− embryos
To analyze development prior to uterine implantation that occurs ~E6, blastocyst embryos were flushed from uteruses of pregnant females between E3.5 and E4.5 stages and genotyped (Figure 3A–C). Results from genotyping these early stage embryos showed the detection of SELENOI−/− in all but one pregnant female, with a total of 7 KO and 25 non-KO blastocyst embryos identified (Figure 3D). Altogether, these data reveal that whole animal SELENOI KO in mice leads to early-stage embryonic lethality with no detectable KO weanling pups with the exception of a very rare occurrence of KO embryos (1.2% of embryos; 0.4% of all genotyped offspring) surviving to a perinatal stage of development.
Figure 3.
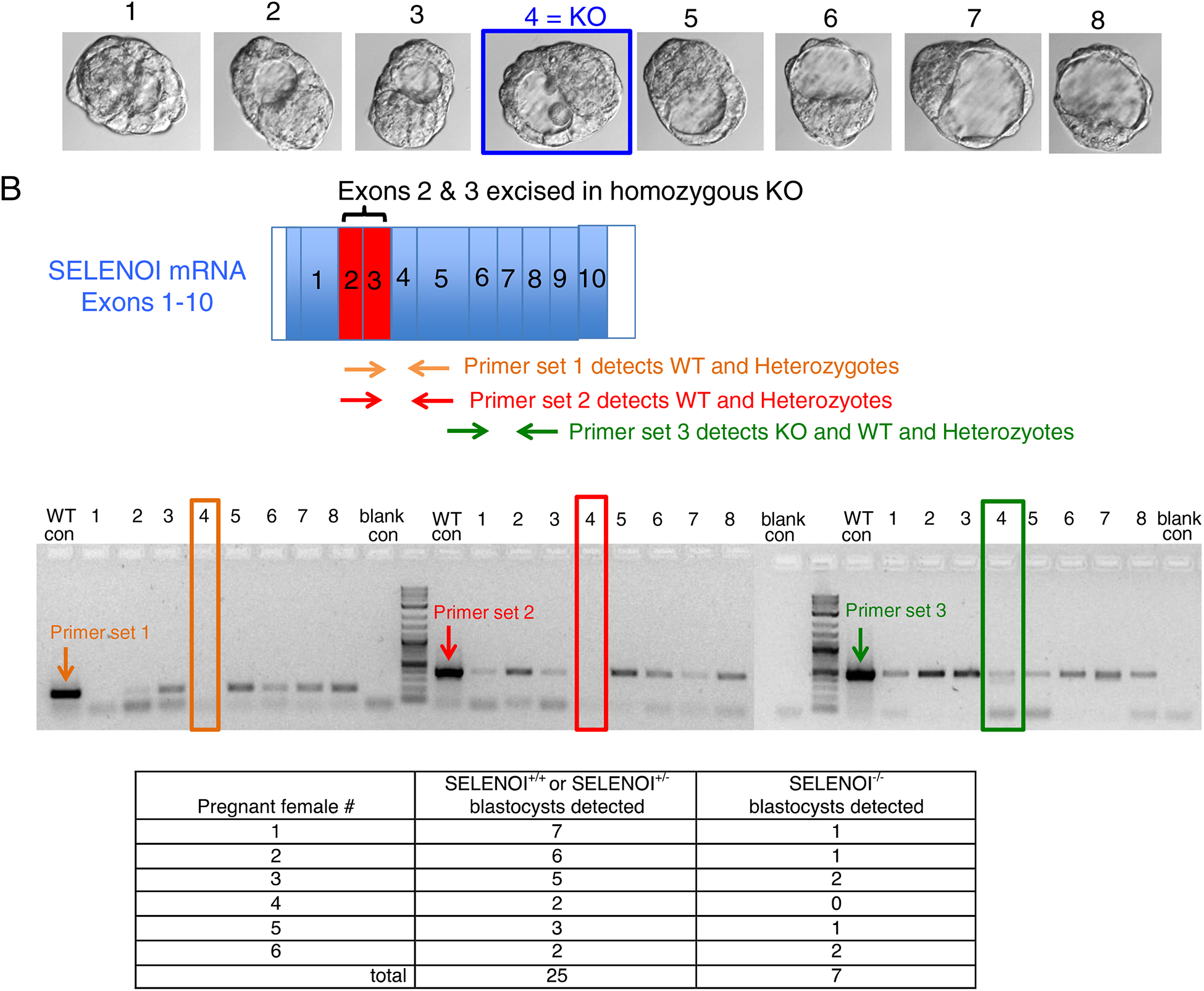
Genotyping blastocyst embryos prior to uterine implantation reveals the presence of SELENOI−/− embryos. (A) Pregnant female mice were euthanized and ovaries/ovaducts/uteruses removed. Blastocyste embryos were flushed from ovaducts and visualized for stage of development (E3.5–4.5). The morphology of the blastocyst embryos at this ~100 cell stage for SELENOI KO did not show any pathological features. (B) Primers were designed for distinguishing between KO (SELENOI−/−) and non-KO (SELENOI−/+ or +/+) using PCR analyses of cDNA. (C) PCR results showed the presence of 1 KO embryo out of 8 embryos from one pregnant female. (D) Cumulative results from 6 pregnant females shows the presence of 7 KO blastocysts.
Discussion
The 2018 case report of a young patient with a homozygous SELENOI mutation leading to nonfunctional enzyme demonstrated that this phosphoethanolamine transferase catalyzing the final step of the Kennedy pathway of PE synthesis was crucial for human health [7]. A lack of functional SELENOI in this patient led to multiple pathologies including reduced myelination and neurodevelopment, and cellular impairments in the maintenance of normal homeostasis of ether-linked phospholipids. Given that this isolated report of one child born to first cousin parents described his major complications 2 h post-birth and the patient required long-term medical intervention, there remain questions regarding the effects of SELENOI homozygous deletion on fetal development and post-birth survival. The data presented herein show that SELENOI homozygous deletion in mice leads to early embryonic lethality in mice. The data show that a lack of SELENOI expression terminates development prior to uterine implantation of embryos on E6. Since PE in cellular membranes is important for balanced membrane fluidity/rigidity and curvature [1, 17], SELENOI-dependent generation of adequate levels of PE may be required to facilitate membrane interactions between the embryo and uterus during implantation. Alternatively, cellular division may reach a point that SELENOI deficiency impedes cell cycle progression past the blastocyst stage. Our data also found rare perinatal homozygous SELENOI KO of E18.5 embryos (1.2% frequency) and this suggests that, in some cases, murine embryonic development in the absence of SELENOI expression can proceed to the perinatal stage development.
While homozygous embryos did not develop, our results show that SELENOI heterozygotes were not phenotypically different from WT mice. This differs with heterozygosity for the rate-limiting enzyme in the Kennedy pathway, ECT, which in mice resulted in hallmark symptoms of metabolic syndrome such as insulin resistance, obesity, dyslipidaemia and liver steatosis [18]. There also have been some tissue-specific KO studies of ECT in mice, including the skeletal muscle-specific knockout of ECT, which led to a build up of the substrate 1,2-diacylglycerol [19]. As other tissue-specific SELENOI KO mouse models are developed, it will be of interest to compare their phenotypes with the results from corresponding models of tissue-specific ECT KO. Interestingly, it has been noted that the tolerance to loss of expression of individual selenoproteins differs between human and mouse [11]. This same study used bioinformatic approaches to estimate the impact of individual selenoproteins on human health and this group found that loss of functional SELENOI was strongly selected against, suggesting it may represent one of the most important selenoproteins for human health. These findings along with our data suggest that SELENOI homozygous deletion in humans likely causes embryonic lethality with rare cases in which perinatal lethality may be observed that require severe therapeutic intervention for survival.
Selenium is an essential micronutrient, and selenoprotein expression is essential for life as demonstrated by the generation of mice lacking Sec-tRNA Sec required for translation of all selenoproteins, which selenoprotein family for which heterozygous deletion in mice leads to embryonic lethality [20]. In this previous report, the homozygous mutants died shortly after implantation, with embryos being resorbed before 6.5 days post coitum. The timing of the homozygous KO of SELENOI on embryonic lethality was similar (<E6.5), but one difference was the rare escape of KO to later stages of embryonic development. It is interesting to note that selenium supplementation for pregnant mice has been shown to improve the health of selenoprotein P KO offspring due to its role in distributing selenium throughout the body [21]. It may be possible that increased dietary selenium may mitigate the detrimental effects of SELENOI KO, but given that our chow had levels of selenium (0.25 ppm) far above the minimum required (0.1 ppm) [22] and the enzymatic role of SELENOI in cells, it is unlikely that this would lead to viable KO offspring. Our study is the first to publish results on homozygous KO of SELENOI in mice and the list of embryonic lethal selenoproteins now stands at 5 out of 25 family members.
Acknowledgements
We thank Ann Hashimoto for her mouse husbandry assistance.
Funding
This research was supported by NIH grants R01AI147496, P30GM131944.
Abbreviations
- ECT
ETP-phosphoethanolamine cytidyltransferase
- ETNK1
ethanolamine kinase 1
- fl
floxed
- GPI
glycophosphatidylinositol
- H&E
haematoxylin and eosin
- KO
knockout
- ppm
parts per million
- PE
phosphatidylethanolamine
- PS
phosphatidylserine
- SELENOI
selenoprotein I
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vance JE, Tasseva G, Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells, Biochim Biophys Acta 1831(3) (2013) 543–54. [DOI] [PubMed] [Google Scholar]
- [2].Menon AK, Eppinger M, Mayor S, Schwarz RT, Phosphatidylethanolamine is the donor of the terminal phosphoethanolamine group in trypanosome glycosylphosphatidylinositols, EMBO J 12(5) (1993) 1907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Braverman NE, Moser AB, Functions of plasmalogen lipids in health and disease, Biochim Biophys Acta 1822(9) (2012) 1442–52. [DOI] [PubMed] [Google Scholar]
- [4].Vance JE, Phospholipid synthesis and transport in mammalian cells, Traffic 16(1) (2015) 1–18. [DOI] [PubMed] [Google Scholar]
- [5].Bleijerveld OB, Brouwers JF, Vaandrager AB, Helms JB, Houweling M, The CDP-ethanolamine pathway and phosphatidylserine decarboxylation generate different phosphatidylethanolamine molecular species, J Biol Chem 282(39) (2007) 28362–72. [DOI] [PubMed] [Google Scholar]
- [6].Liu J, Rozovsky S, Membrane-bound selenoproteins, Antioxid Redox Signal 23(10) (2015) 795–813. [DOI] [PubMed] [Google Scholar]
- [7].Horibata Y, Hirabayashi Y, Identification and characterization of human ethanolaminephosphotransferase1, J Lipid Res 48(3) (2007) 503–8. [DOI] [PubMed] [Google Scholar]
- [8].Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN, Characterization of mammalian selenoproteomes, Science 300(5624) (2003) 1439–43. [DOI] [PubMed] [Google Scholar]
- [9].Schweizer U, Fradejas-Villar N, Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism, FASEB J 30(11) (2016) 3669–3681. [DOI] [PubMed] [Google Scholar]
- [10].Horibata Y, Elpeleg O, Eran A, Hirabayashi Y, Savitzki D, Tal G, Mandel H, Sugimoto H, EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans, J Lipid Res 59(6) (2018) 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Santesmasses D, Mariotti M, Gladyshev VN, Tolerance to selenoprotein loss differs between human and mouse, Mol Biol Evol 37(2) (2019) 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fullerton MD, Hakimuddin F, Bakovic M, Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2), Mol Cell Biol 27(9) (2007) 3327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE, Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects, J Biol Chem 280(48) (2005) 40032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fogelgren B, Polgar N, Lui VH, Lee AJ, Tamashiro KK, Napoli JA, Walton CB, Zuo X, Lipschutz JH, Urothelial Defects from Targeted Inactivation of Exocyst Sec10 in Mice Cause Ureteropelvic Junction Obstructions, PLoS One 10(6) (2015) e0129346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim SM, Yokoyama T, Ng D, Ulu F, Yamazaki Y, Retinoic acid-stimulated ERK1/2 pathway regulates meiotic initiation in cultured fetal germ cells, PLoS One 14(11) (2019) e0224628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schwenk F, Baron U, Rajewsky K, A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells, Nucleic Acids Res 23(24) (1995) 5080–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dawaliby R, Trubbia C, Delporte C, Noyon C, Ruysschaert JM, Van Antwerpen P, Govaerts C, Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells, J Biol Chem 291(7) (2016) 3658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pavlovic Z, Bakovic M, Regulation of Phosphatidylethanolamine Homeostasis - The Critical Role of CTP:Phosphoethanolamine Cytidylyltransferase (Pcyt2), Int J Mol Sci 14(2) (2013) 2529–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Selathurai A, Kowalski GM, Burch ML, Sepulveda P, Risis S, Lee-Young RS, Lamon S, Meikle PJ, Genders AJ, McGee SL, Watt MJ, Russell AP, Frank M, Jackowski S, Febbraio MA, Bruce CR, The CDP Ethanolamine Pathway Regulates Skeletal Muscle Diacylglycerol Content and Mitochondrial Biogenesis without Altering Insulin Sensitivity, Cell Metab 21(5) (2015) 718–30. [DOI] [PubMed] [Google Scholar]
- [20].Bosl MR, Takaku K, Oshima M, Nishimura S, Taketo MM, Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp), Proc Natl Acad Sci U S A 94(11) (1997) 5531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schweizer U, Michaelis M, Kohrle J, Schomburg L, Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency, Biochem J 378(Pt 1) (2004) 21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kasaikina MV, Hatfield DL, Gladyshev VN, Understanding selenoprotein function and regulation through the use of rodent models, Biochim Biophys Acta 1823(9) (2012) 1633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]