

Abstract
Maples (Acer) are among the most diverse and ecologically important tree genera of the north-temperate forests. They include species highly valued as ornamentals and as a source of timber and sugar products. Previous phylogenetic studies employing plastid markers have not provided sufficient resolution, particularly at deeper nodes, leaving the backbone of the maple plastid tree essentially unresolved. We provide the plastid genome sequences of 16 species of maples spanning the sectional diversity of the genus and explore the utility of these sequences as a source of information for genetic and phylogenetic studies in this group. We analyzed the distribution of different types of repeated sequences and the pattern of codon usage, and identified variable regions across the plastome. Maximum likelihood and Bayesian analyses using two partitioning strategies were performed with these and previously published sequences. The plastomes ranged in size from 155,212 to 157,023 bp and had structure and gene content except for Acer palmatum (sect. Palmata), which had longer inverted repeats and an additional copy of the rps19 gene. Two genes, rps2 and rpl22, were found to be truncated at different positions and might be non-functional in several species. Most dispersed repeats, SSRs, and overall variation were detected in the non-coding sequences of the LSC and SSC regions. Fifteen loci, most of which have not been used before in the genus, were identified as the most variable and potentially useful as molecular markers for barcoding and genetic studies. Both ML and Bayesian analyses produced similar results irrespective of the partitioning strategy used. The plastome-based tree largely supported the topology inferred in previous studies using cp markers while providing resolution to the backbone relationships but was highly incongruous with a recently published nuclear tree presenting an opportunity for further research to investigate the causes of discordance, and particularly the role of hybridization in the diversification of the genus. Plastome sequences are valuable tools to resolve deep-level relationships within Acer. The variable loci and SSRs identified in this study will facilitate the development of markers for ecological and evolutionary studies in the genus. This study underscores the potential of plastid genome sequences to improve our understanding of the evolution of maples.
Keywords: Acereae, Plastomes, Phylogenomics, Repeated sequences, SSRs
Introduction
Acer L., the maple genus, is the third most species-rich genus of trees in the northern hemisphere after Quercus L. and Salix L., and the fourth largest genus (in terms of species number) in the Sapindaceae (Acevedo-Rodríguez et al., 2011). It includes over 150 species distributed across all northern continents (De Jong, 2004). The greatest diversity is found in eastern Asia, particularly in China, where ca. 100 species have been reported (Xu et al., 2008). A few species extend into Central America (Guatemala and Honduras) and northern Africa, and only one extends beyond the Equator into Java and Sulawesi in Indonesia (De Jong, 1976).
Maples are popular and widely planted in temperate areas as decorative trees for their characteristic leaf shapes and showy fall foliage (Harris, 1975). Some species provide good-quality timber and hardwood for flooring, furniture, and many other applications (e.g., musical instruments, barrels, boxes, and woodenware) (Betts, 1959). The sugary sap of members of the Acer saccharum complex is used to produce the highly valued maple syrup (Ball, 2007).
Maples are among the most important components in the north-temperate deciduous forest biome. They can be found in a diverse range of habitats, from sea-level flatlands to higher than 3,000 m in the Himalayan forests. Several species, e.g., Acer saccharum Marshall, can be very abundant and are recognized as keystone species within their communities (Bishop et al., 2015; Minorsky, 2003). Many others are considered rare or threatened, mainly due to habitat loss and overexploitation. Fifty–four taxa (or about one–third of the genus) are included in the IUCN red list of maples with some category of threat (Gibbs & Chen, 2009).
The taxonomy of maples has always been considered as complicated due to the presence of extensive morphological variation in vegetative characters and the propensity of species to hybridize (De Jong, 1976; Grimm, Denk & Hemleben, 2007; Liao et al., 2010). This is reflected in the wide range of estimates of species numbers (and infraspecific taxa) reported in the literature, e.g., from 110 to over 155 spp. (Acevedo-Rodríguez et al., 2011; De Jong, 1976; De Jong, 2004; Xu et al., 2008). About a dozen classification systems have attempted to organize the species into subgenera, sections and series, mainly based on characters of leaves and the structure of inflorescences and flowers including the sex expression (De Jong, 1976; De Jong, 1994; De Jong, 2004; Momotani, 1962; Murray, 1970; Ogata, 1967; Pax, 1885; Pax, 1902; Pojarkova, 1933; Xu, 1996). The most recent system classifies the 156 species into 19 sections, six of which were subdivided into series (De Jong, 2004). However, the monophyly of some of these infrageneric groups is not or weakly supported by molecular data, and thus the classification of the genus remains only partially resolved (Harris et al., 2017; Li, 2011; Li, Yue & Shoup, 2006).
Phylogenetic studies in maples conducted over the past two decades (Ackerly & Donoghue, 1998; Grimm, Denk & Hemleben, 2007; Grimm et al., 2006; Harris, Frawley & Wen, 2017; Li, 2011; Li, Yue & Shoup, 2006; Renner et al., 2007; Renner et al., 2008; Suh, Heo & Park, 2000; Tian, Guo & Li, 2002; Zhang, Li & Li, 2010) have been based on a limited number of loci and/or have centered on specific taxonomic sections. Only recently, the first comprehensive phylogenomic study of Acer was published (Li et al., 2019). It was based on sequences of over 500 nuclear loci generated with hybrid enrichment for 65 species of Acer. Most of the 16 sections represented in their study were recovered as monophyletic with relatively high support, whereas three sections: Acer, Lithocarpa, and Trifoliata, were non-monophyletic.
Several Acer plastomes (e.g., Jia et al., 2016; Kim et al., 2019; Li et al., 2015; Wang, Chen & Zhang, 2017; Zhang et al., 2016) have been recently published. However, to our knowledge, no study has explored the structure and variation of the chloroplast genome across the genus. The plastid genome, which is maternally inherited in Acer (Corriveau & Coleman, 1988), has many advantages for phylogenetic inference and genetic studies over the nuclear and mitochondrial genomes (Daniell et al., 2016; Gitzendanner et al., 2018). A comparison between plastid and nuclear trees is relevant to our understanding of relationships in maples and could also provide evidence for hybridization and other processes in their evolutionary history. Here, we explore the diversity of plastomes in Acer as a first step towards generating a plastome-based tree for the genus. We assess the variation and compare structural features among 16 newly sequenced plastomes, each belonging to a different section in the genus. We investigate the effect of partitioning on tree inference using these and previously published plastid sequences and compare our results with the recently published nuclear tree. Finally, we discuss the utility of this genome as a source of information for genetic studies and phylogenetic inference in this valuable group of trees.
Materials & Methods
Taxon sampling
Leaf samples of 16 species of maples were obtained from cultivated trees in private and state gardens of Europe and China (Table S1). The identity of all samples was verified with experts from these institutions. Herbarium vouchers were deposited in the BGT herbarium, Guangxi University, China.
Each of the 16 species selected belongs to a different section in the genus (sensu De Jong, 2004), and 13 are the type of their respective section. Section Hyptiocarpa was recently merged with section Rubra (Harris et al., 2017) and was not considered in this study. Acer oblongum, placed in section Pentaphylla series Trifida by De Jong (2004), was included as a representative of section Oblonga, which was recognized by Xu et al. (2008) in Flora of China. Only sections Spicata, Wardiana, and Macrophylla could not be sampled.
For the phylogenomic reconstruction, plastome sequences from six additional species of maples were obtained from GenBank (see accessions in Table S1). We also retrieved sequences of the two species of Dipteronia (the sister genus of Acer), Litchi chinensis J. F. Gmel (Sapindaceae) and Spondias mombin L. (Anacardiaceae), which were incorporated as outgroups.
DNA sequencing and plastome assembly and annotation
DNA extraction, library construction, and sequencing were performed by Annoroad Gene Technology (Beijing, PR China) Co., Ltd. Genomic DNA was isolated from frozen leaves using the DNAquick Plant system (TianGen Biotech, Beijing) following the manufacturer’s protocol. DNA degradation was assessed on 1% agarose gels. Purity and concentration were determined with a NanoPhotometer (Implen, USA) and a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Massachusetts). Total DNA was fragmented to approximately 350 bp on an Ultrasonic Processor. Libraries were constructed using the NEBNext Ultra II DNA Library Prep Kit (Ipswich, Massachusetts) according to the manufacturer’s protocol, and subsequently diluted to 1 ng/µL. The final concentration and fragment sizes were verified on an Agilent 2100 Bioanalyzer (Agilent Technologies, California). Sequencing was performed on an Illumina HiSeq X Ten System (San Diego, California). Approximately one GB of 150 bp paired-end reads was generated for each sample.
Plastomes were assembled from cleaned reads using NOVOPlasty v. 2.7.2 (Dierckxsens, Mardulyn & Smits, 2017). When multiple contigs were obtained (rather than a single circularized assembly), we mapped them contigs against one of several Acer plastomes from GenBank (see accessions in Table S1) using Geneious v. 11.0.4 and merged the ones that overlapped. Because in several instances the assemblies resulted in short non-overlapping contigs, we mapped our reads against the contigs to extend their ends until the gap was closed. Mapping was performed with medium-low sensitivity for 100 iterations.
The annotation of the assemblies was performed with GeSeq (Tillich et al., 2017). We selected ARAGORN as third party tRNA annotator and the plastome sequence of Acer miyabei Maxim. subsp. miaotaiense (P.C. Tsoong) A.E. Murray (NC_030343) as the reference genome. The annotated sequences were aligned and verified with published Acer plastomes using MAFFT v. 7.450 (Katoh & Standley, 2013) in Geneious and submitted to GenBank (Table S1).
Plastome comparison
The boundaries between the four plastome regions were inspected with the online tool IRscope (Amiryousefi, Hyvonen & Poczai, 2018), which allows visualizing the position of genes in the vicinity of these sites across species. We compared sequences and identified regions of variability with mVISTA (Frazer et al., 2004) using the annotated plastome of Acer acuminatum Wall. ex D. Don as a reference. Additionally, we performed a sliding window analysis as implemented in DnaSP v. 6.12.03 (Rozas et al., 2017) to locate genomic regions with high levels of variation. The alignment of the 16 Acer plastomes obtained with MAFFT (with default settings) was used as input file. The window length and step size were set to 600 bp and 100 bp, respectively. Using window’s position, we identified regions with 60 or more variable (polymorphic) sites (S). These regions were extracted from the alignment and analyzed individually to estimate their number of variable sites and parsimony-informative sites.
A codon usage analysis for protein-coding genes was also performed in DnaSP. We extracted the CDSs using Geneious and calculated the codon frequency and the relative synonymous codon usage (RSCU) values as a measure of codon usage bias (Sharp, Tuohy & Mosurski, 1986).
REPuter (Kurtz et al., 2001) was used to detect various types of repeated sequences (forward, reverse, complement, and palindromic) with a minimum size of 25 bp and sequence identity greater than 90%. Simple sequence repeats (SSRs) were identified with MISA-web (Beier et al., 2017). We used the default values of 10, 6, 5, 5, 5, and 5 to set the minimum number of repetitions for mono, di-, tri-, tetra-, penta-, and hexanucleotide repeats, respectively.
Phylogenomic reconstruction
Phylogenomic reconstruction was performed on a dataset consisting of 22 Acer plastomes (six retrieved from GenBank), and four outgroup species (Table S1). We aligned the annotated sequences using MAFFT with default parameters and then removed one inverted repeat. Maximum likelihood and Bayesian analyses were performed on this data set using two partitioning strategies to explore the effect of partitioning on tree topology. In the first one, the alignment was fully partitioned into coding and non-coding regions with protein-coding genes further divided by codon position. We used Geneious to extract the protein-coding sequences to define the codon positions in the data blocks and then concatenated these with the non-protein-coding regions. In the second strategy, the whole plastome was treated as a single partition (i.e., unpartitioned).
We used PartitionFinder2 (Lanfear et al., 2016) to select the best partitioning scheme and best-fit substitution models for the partitioned dataset. Branch lengths were set as ‘linked’ and the AICc was used for model selection. The search was performed using ‘rcluster’, a fast algorithm recommended for large datasets with a high number of partitions (Lanfear et al., 2014). We defined 324 data blocks which were reduced to 93 subsets in the best-fit scheme. These subsets and their corresponding substitution models were specified in both ML and Bayesian partitioned analyses. The unpartitioned analysis was run using GTR+I+G as a substitution model, which was selected using both PartitionFinder2 and ModelTest-NG (Darriba et al., 2019). The maximum likelihood analysis was performed in RaxML-NG (Kozlov et al., 2019) with 1,000 bootstrap replicates. The Bayesian analysis was conducted in MrBayes v. 3.2.7 (Ronquist et al., 2012) on the CIPRES Science Gateway (Miller, Pfeiffer & Schwartz, 2010). We ran two runs of 100 million generations and four chains, sampling trees every 4,000 generations and discarding the 20% as burn-in. Tracer v. 1.6.0 (Rambaut et al., 2018) was used to verify that both runs reached stationarity and converged on the same distribution.
Results
Size and composition of Acer plastomes
The 16 plastomes generated in this study varied in length from 155,212 bp in Acer carpinifolium Siebold & Zucc. to 157,023 bp in Acer palmatum Thunb.var. palmatum (Table 1). All exhibited the typical tetrapartite organization, with the large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions ranging from 85,313 to 86,147, 17,724 to 18,232, and 26,020 to 26,757 bp, respectively. The largest genome size of A. palmatum is due to its expanded IRs relative to the other species.
Table 1. Main features of the chloroplast genomes generated in this study.
| Species | Section | Total length (bp) | LSC (bp) | SSC (bp) | IR (bp) | GC% | # of coding loci | # of tRNA loci | # of rRNA loci | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | LSC | SSC | IR | |||||||||
| Acer acuminatum | Arguta | 155,548 | 85,358 | 18,046 | 26,072 | 37.9 | 36.1 | 32.1 | 42.9 | 89 | 38 | 8 |
| Acer carpinifolium | Indivisa | 155,212 | 85,448 | 17,724 | 26,020 | 38.0 | 36.2 | 32.5 | 43.0 | 89 | 38 | 8 |
| Acer glabrum | Glabra | 156,373 | 86,034 | 18,211 | 26,064 | 37.9 | 36.0 | 32.2 | 42.9 | 89 | 38 | 8 |
| Acer maximowiczianum | Trifoliata | 156,082 | 85,865 | 18,147 | 26,035 | 37.9 | 36.1 | 32.3 | 42.9 | 89 | 38 | 8 |
| Acer micranthum | Macrantha | 156,399 | 86,147 | 18,128 | 26,062 | 37.9 | 36.0 | 32.1 | 42.9 | 89 | 38 | 8 |
| Acer negundo | Negundo | 155,938 | 85,678 | 18,092 | 26,084 | 37.9 | 36.1 | 32.3 | 42.9 | 89 | 38 | 8 |
| Acer nipponicum | Parviflora | 156,225 | 85,823 | 18,232 | 26,085 | 37.8 | 35.9 | 32.0 | 42.9 | 89 | 38 | 8 |
| Acer oblongum | Oblonga | 155,686 | 85,665 | 17,821 | 26,100 | 38.0 | 36.1 | 32.4 | 42.9 | 89 | 38 | 8 |
| Acer palmatum var. palmatum | Palmata | 157,023 | 85,342 | 18,167 | 26,757 | 37.9 | 36.0 | 32.2 | 42.8 | 90 | 38 | 8 |
| Acer pentaphyllum | Pentaphylla | 156,220 | 85,938 | 18,148 | 26,067 | 37.9 | 36.0 | 32.3 | 42.9 | 89 | 38 | 8 |
| Acer pilosum | Pubescentia | 155,586 | 85,313 | 18,139 | 26,067 | 38.0 | 36.2 | 32.1 | 42.9 | 89 | 38 | 8 |
| Acer platanoides | Platanoidea | 156,385 | 86,098 | 18,107 | 26,090 | 37.9 | 36.0 | 32.1 | 42.9 | 89 | 38 | 8 |
| Acer pseudoplatanus | Acer | 155,933 | 85,812 | 17,971 | 26,075 | 37.9 | 36.1 | 32.2 | 42.9 | 89 | 38 | 8 |
| Acer rubrum | Rubra | 155,683 | 85,383 | 18,086 | 26,107 | 37.9 | 36.1 | 32.2 | 42.9 | 89 | 38 | 8 |
| Acer sterculiaceum subsp. sterculiaceum | Lithocarpa | 156,258 | 86,014 | 18,048 | 26,098 | 38.0 | 36.1 | 32.3 | 42.9 | 89 | 38 | 8 |
| Acer tataricum subsp. ginnala | Ginnala | 155,667 | 85,404 | 18,061 | 26,101 | 38.0 | 36.2 | 32.4 | 42.9 | 89 | 38 | 8 |
Notes.
- IR
- inverted repeat
- LSC
- large single copy
- SSC
- small single copy
The GC content was similar for all species (37.8–38%). As is common in plastomes of seed plants, the IRs had the highest GC content (42.8–43%) due to the presence of the GC-rich rRNA genes, whereas the SSC region had the lowest values (35.9–36.2%, Table 1).
All plastomes contained 117 different genes, of which 19 are duplicated for a total of 136 genes (Table 2). Acer palmatum is the only species with 137 genes due to an entire additional copy of the ribosomal rps19. Eighty-one of the 117 genes are protein-coding genes, 31 are transfer RNA genes, and four are ribosomal RNA genes. One gene, infA, appears as a pseudogene and is likely to be non-functional in all species. Sixteen genes contain a single intron, and two have two introns (Table 2).
Table 2. List of genes annotated in the plastomes of Acer species generated in this study.
A total of 136 genes are present in all species except for A. palmatum which has two copies of the ribosomal protein gene rps19. Genes marked with one asterisk contain one intron; two asterisks indicate two introns.
| Gene class | # of genes | Gene name |
|---|---|---|
| Photosystem I | 5 | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | 15 | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Photosystem assembly factors | 2 | ycf3 **, ycf4 |
| Cytochrome b/f complex | 6 | petA, petB *, petD *, petG, petL, petN |
| ATP synthase complex | 6 | atpA, atpB, atpE, atpF *, atpH, atpI |
| NADH dehydrogenase complex | 12 | ndhA *, ndhB * (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Large subunit of RuBisCO | 1 | rbcL |
| Maturase | 1 | matK |
| RNA polymerase subunits | 4 | rpoA, rpoB, rpoC1 *, rpoC2 |
| Small subunit ribosomal proteins | 14 (15) | rps2, rps3, rps4, rps7 (×2), rps8, rps11, rps12 * (×2), rps14, rps15, rps16 *, rps18, rps19 (×2 in A. palmatum) |
| Large subunit ribosomal proteins | 11 | rpl2 * (×2), rpl14, rpl16 *, rpl20, rpl22, rpl23 (×2), rpl32, rpl33, rpl36 |
| Subunit of acetyl-CoA-carboxylase | 1 | accD |
| Subunit of Clp-protease | 1 | clpP ** |
| Translation initiation factor | 1 | infAΨ |
| Inner envelope membrane protein | 1 | cemA |
| Cytochrome c biogenesis protein | 1 | ccsA |
| Genes of unknown function | 8 | orf42 (×2), ycf1 , ycf1a, ycf2 (×2), ycf15 (×2) |
| Ribosomal RNAs | 8 | rrn4.5 (×2), rrn5 (×2), rrn16 (×2), rrn23 (×2) |
| Transfer RNAs | 38 | trnA-UGC * (×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU (×2), trnI-GAU * (×2), trnK-UUU *, trnL-CAA (×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU (×2), trnP-GGG, trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnV-UAC *, trnW-CCA, trnY-GUA |
| TOTAL | 136 (137) |
The size of homologous genes was very similar across species except for a few cases. For example, the rps2 gene was found to be truncated at different positions in 11 species due to mutations leading to premature stop codons. It was significantly shorter (72–264 bp) and perhaps non-functional in A. acuminatum, A. carpinifolium, A. micranthum, A. negundo, A. nipponicum, A. palmatum, A. pilosum, A. pseudoplatanus, A. rubrum, and A. tataricum. The other six species had lengths of 588 and 633 bp. The rpl22 gene was also notably shorter (183–225 bp) in two species (A. sterculiaceum and A. pentaphyllum) compared to the rest whose length ranged from 480 and 498 bp.
In all species, with the exception of A. maximowiczianum and A. palmatum, the boundary between the LSC and IRb regions is located in the 3′ end of the rps19 gene (Fig. 1). In A. maximowiczianum, the entirety of rps19 lies in the LSC region, whereas in Acer palmatum, it is found within the inverted repeats. This species is atypical in that the boundary LSC-IRb is situated in the rpl22 gene, which results in a duplicated fragment of this gene in the IRa adjacent to the junction with the LSC region (Fig. 1).
Figure 1. Schematic diagram of the boundaries between the four regions (LSC, large single copy; SSC, short single copy; IR, inverted repeat) in the 16 plastid genomes sequenced in this study.

See Table 1 for full species names.
A truncated but seemingly functional copy of the ycf1 gene is present in the IRb of all plastomes extending to various lengths into the SSC region. The ndhF gene also spans the boundary SSC-IRb in the opposite direction, partially overlapping with the 3′ end of ycf1. The only exception is Acer nipponicum, in which the entire ndhF sequence lies within the SSC region (Fig. 1).
Sequence variation
The plastomes of Acer are very similar and largely conserved as shown in the mVISTA alignment, with most of the variation found in the non-coding sequences of the LSC and SSC regions (Fig. 2). Fifteen most variable regions (S ≥ 60) were identified in the sliding window analysis. All but one, the ycf1 gene, were or contained intergenic spacers. Three compound regions, formed by one protein-coding gene (rps2, rpl32 and ccsA) plus one or both flanking spacers, were detected (Table 3, Fig. 3). When these regions were analyzed separately, the number of polymorphic sites varied from 36 (ccsA–ndhD) to 438 (ycf1). The percent of variability and parsimony-informative sites ranged from 5.3% (rps16–trnQ-UUG) to 16.7% (ccsA–ndhD), and from 1.0% (accD–psaI and rps16–trnQ-UUG) to 8.8% (ccsA–ndhD), respectively (Table 3). The five most variable regions, both in percent of variability and total number of parsimony-informative sites were the spacers ccsA–ndhD, psbZ–trnG-GCC, ndhC–trnV-UAC, trnE-UUC–trnT-GGU, and the ycf1 gene. This gene exhibited greater variation in the SSC portion of the genome (9.7%) compared to the IR portion (1.1%).
Figure 2. mVISTA alignment comparing the plastomes of Acer species against A. acuminatum.

The vertical scale to the right shows the percent of identity (50–100%) between the species compared. The black arrows indicate the boundaries of the inverted repeats. Coding regions are indicated in blue and non-coding regions in pink.
Table 3. List of most variable regions in Acer plastomes.
| Region | Length (bp) | Aligned length (bp) | No. of variable (polymorphic) sites | No. of parsimony- informative sites |
|---|---|---|---|---|
| ycf1 (SSC portion) | 4205–4282 | 4361 | 423 (9.7%) | 93 (2.1%) |
| ycf1 (whole) | 5484–5541 | 5673 | 438 (7.7%) | 99 (1.7%) |
| ndhF–rpl32+rpl32+ | 1529–2061 | 2453 | 146 (5.9%) | 42 (1.7%) |
| rpl32–trnL-UAG | ||||
| rpl32–trnL-UAG | 833–1159 | 1334 | 98 (7.3%) | 30 (2.2%) |
| trnT-GGU–psbD | 1454–1511 | 1565 | 119 (7.6%) | 21 (1.3%) |
| trnK-UUU–rps16 | 776–1022 | 1127 | 118 (10.5%) | 22 (1.6%) |
| atpH–atpI | 1117–1178 | 1274 | 103 (8.1%) | 14 (1.1%) |
| trnE-UUC–trnT-GGU | 761–818 | 866 | 95 (11%) | 18 (2.1%) |
| ccsA+ccsA–ndhD | 1150–1162 | 1173 | 93 (7.9%) | 24 (2.0%) |
| ccsA–ndhD | 193–205 | 216 | 36 (16.7%) | 19 (8.8%) |
| rpoB–trnC-GCA | 1161–1189 | 1265 | 92 (7.3%) | 15 (1.2) |
| ndhC–trnV-UAC | 884–927 | 1040 | 84 (8.1%) | 39 (3.75) |
| atpI–rps2+rps2 +rps2–rpoC2 | 790–1195 | 1243 | 76 (6.1%) | 18 (1.4%) |
| petN–psbM | 762–796 | 867 | 73 (8.4%) | 15 (1.7%) |
| psbZ–trnG-GCC | 516–604 | 685 | 71 (10.4%) | 27 (3.9%) |
| rps16–trnQ-UUG | 783–1120 | 1252 | 66 (5.3%) | 13 (1.0%) |
| accD–psaI | 607–707 | 759 | 59 (7.8%) | 8 (1.0%) |
| trnH-GUG–psbA | 272–435 | 529 | 52 (9.8%) | 7 (1.3%) |
Figure 3. Sliding window analysis of polymorphic sites (S) for plastomes of 16 species of Acer (window length: 600 bp, step size: 100 bp).

Regions with the highest number of S are indicated. 1: trnH-psbA, 2: atpH-atpI, 3: atpI-rps2+rps2+rps2-rpoC2, 4: petN-psbM, 5: trnT -psbD (see Table 3 for details). One inverted repeat not shown.
Codon usage
The total number of codons (including stop codons) of the protein-coding regions of the plastomes ranged from 26,678 in A. psudoplatanus to 26,865 in A. glabrum. Codon frequency and RSCU values were very similar across species (Table 4, Table S2). The most frequent codons were AUU-Ile (1084-1112), AAA-Lys (1,060–1,079), and GAA-Glu (1,011–1,024), the three accounting for about 11% of the total number of codons in all species. The three least frequent were UGC-Cys (81–86), AGC-Ser (124–131), and CGC-Arg (127–136). The most commonly specified amino acids were leucine and isoleucine, encoded by about 10% and 8% of codons, respectively (Table 4). Codons that have T or A in their third position had RSCU >1, whereas codons ending in C or G had RSCU <1 indicating a strong bias in favor of codons ending with T and A. The only exceptions to this pattern were UUG-Leu with RSCU values between 1.20 and 1.23, CUA-Leu with 0.83–0.85, and AUA-Ile with 0.92–0.94. The two codons with the highest RSCU values (both with 1.78–1.81) were UUA-Leu and AGA-Arg, and the two with the lowest were AGC-Ser (0.36–0.38) and UAC-Tyr (0.37–0.39) (Table 4, Table S2).
Table 4. Codon usage in Acer plastomes.
| Codon | Aa | No. | RSCU | Codon | Aa | No | RSCU | Codon | Aa | No | RSCU | Codon | Aa | No | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU | Phe | 999-1012 | 1.28-1.29 | UCU | Ser | 548-556 | 1.58-1.61 | UAU | Tyr | 771-788 | 1.61-1.63 | UGU | Cys | 211-224 | 1.42-1.47 |
| UUC | 556-565 | 0.71-0.72 | UCC | 348-358 | 1.00-1.03 | UAC | 177-187 | 0.37-0.39 | UGC | 81-86 | 0.53-0.58 | ||||
| UUA | Leu | 835-853 | 1.78-1.81 | UCA | 433-447 | 1.25-1.28 | UAA* | Stop | 48-52 | 1.62-1.75 | UGA* | Stop | 14-17 | 0.47-0.57 | |
| UUG | 563-576 | 1.20-1.23 | UCG | 196-206 | 0.57-0.60 | UAG* | 22-25 | 0.74-0.84 | UGG | Trp | 452-460 | 1.00 | |||
| CUU | 576-588 | 1.23-1.25 | CCU | Pro | 411-421 | 1.46-1.50 | CAU | His | 491-508 | 1.48-1.50 | CGU | Arg | 317-323 | 1.16-1.19 | |
| CUC | 213-223 | 0.45-0.47 | CCC | 227-238 | 0.81-0.85 | CAC | 164-173 | 0.50-0.52 | CGC | 127-136 | 0.47-0.50 | ||||
| CUA | 389-400 | 0.83-0.85 | CCA | 310-323 | 1.11-1.16 | CAA | Gln | 716-728 | 1.52-1.54 | CGA | 360-376 | 1.33-1.37 | |||
| CUG | 207-218 | 0.44-0.46 | CCG | 151-164 | 0.54-0.58 | CAG | 218-227 | 0.46-0.48 | CGG | 139-148 | 0.51-0.55 | ||||
| AUU | Ile | 1084-1112 | 1.45-1.47 | ACU | Thr | 513-523 | 1.52-1.56 | AAU | Asn | 958-977 | 1.50-1.52 | AGU | Ser | 398-412 | 1.15-1.19 |
| AUC | 445-460 | 0.59-0.61 | ACC | 258-269 | 0.77-0.81 | AAC | 307-318 | 0.48-0.50 | AGC | 124-131 | 0.36-0.38 | ||||
| AUA | 696-709 | 0.92-0.94 | ACA | 396-406 | 1.18-1.20 | AAA | Lys | 1060-1079 | 1.47-1.48 | AGA | Arg | 481-492 | 1.78-1.81 | ||
| AUG | Met | 611-623 | 1.00 | ACG | 155-168 | 0.46-0.50 | AAG | 374-384 | 0.52-0.53 | AGG | 180-191 | 0.67-0.70 | |||
| GUU | Val | 524-534 | 1.43-1.46 | GCU | Ala | 608-625 | 1.72-1.75 | GAU | Asp | 838-862 | 1.57-1.58 | GGU | Gly | 586-598 | 1.28-1.30 |
| GUC | 185-191 | 0.50-0.52 | GCC | 228-238 | 0.64-0.67 | GAC | 226-233 | 0.42-0.43 | GGC | 185-193 | 0.40-0.42 | ||||
| GUA | 539-549 | 1.47-1.50 | GCA | 378-388 | 1.07-1.09 | GAA | Glu | 1011-1024 | 1.47-1.48 | GGA | 720-733 | 1.57-1.59 | |||
| GUG | 199-206 | 0.55-0.56 | GCG | 187-196 | 0.52-0.55 | GAG | 356-370 | 0.52-0.53 | GGG | 326-340 | 0.71-0.74 |
Notes.
- Aa
- Amino acid
- No
- Number (minimum-maximum values)
- RSCU
- Relative synonymous codon usage (minimum-maximum values)
Repeat sequence analysis
The analysis with REPuter detected between four and 18 repeats with a length ≥25 bp in all plastomes (Fig. 4, Table S3). Acer platanoides L. had the highest number while all other species ranged between four and eight repeats. The most common type were forward sequences in the range of 40–49 bp followed by palindromes between 30–39 bp (Fig. 4). No complement repeats were found in any of the plastomes analyzed.
Figure 4. Number of repeats found in Acer. plastomes using REPuter.
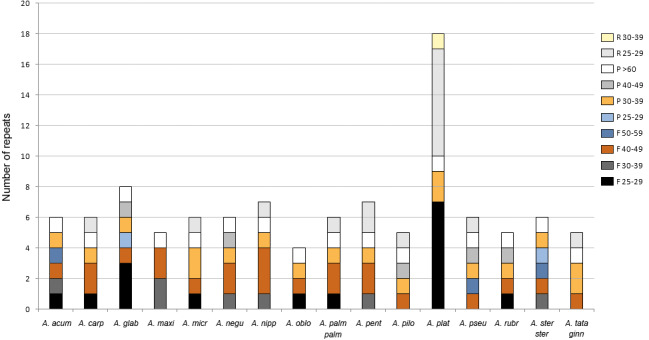
Only repeats ≥25 bp were considered. F, forward; P, palindrome; R, reverse. See Table 1 for full species names.
The majority (about 67%) of these repeated sequences were identified in the intergenic spacers of the LSC and SSC regions; nearly a third were detected in the most variable spacers (Table 3). Only two were located in intergenic spacers of the IRs in one species (Acer glabrum Torr.). The remaining repeats were detected in five protein-coding genes (psaA, psaB, rpoC1, rpl22, rpl32) and one transfer RNA gene (trnS-GGA). The repeats found in the rpoC1 gene were located in its intron sequence (Table S3).
The number of SSR loci ranged from 60 in the plastomes of A. acuminatum and Acer pilosum Maxim. to 92 in A. glabrum. Ninety-seven percent were mononucleotide repeats of up to 28 bp long. A few dinucleotide repeats were found in 13 of the 16 species, whereas tri- and tetranucleotide repeats were detected in only five and one species, respectively (Fig. 5). The most frequent motifs were (A/T)10 and (A/T)11, which collectively accounted for up to 78–79% of all SSRs in some species. C and G repeats were rare, with only one to three per species (Table S4).
Figure 5. Number of simple sequence repeats (SSRs) found in Acer plastomes using MISA-web.
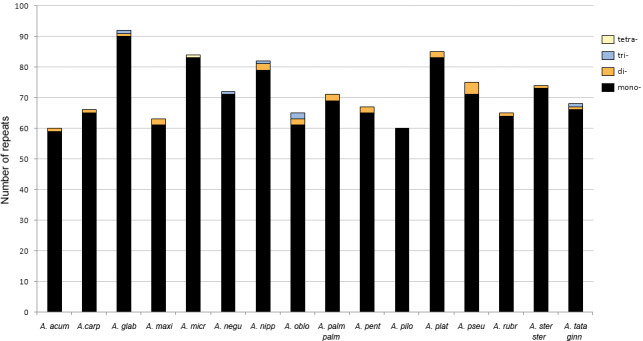
See Table 1 for full species names.
The percentage of the cp genome containing SSRs varied from 0.48 to 0.94% (average = 0.7%). Most loci (90%) were identified in the LSC and SSC regions (Fig. 6). About 58% were located in the intergenic spacers of the genome, whereas 42% were detected in 25 different genes. The ycf1 gene had the highest number of SSRs with six to ten per species (Table S5).
Figure 6. Number of simple sequence repeats (SSRs) found in Acer plastomes according to their location.
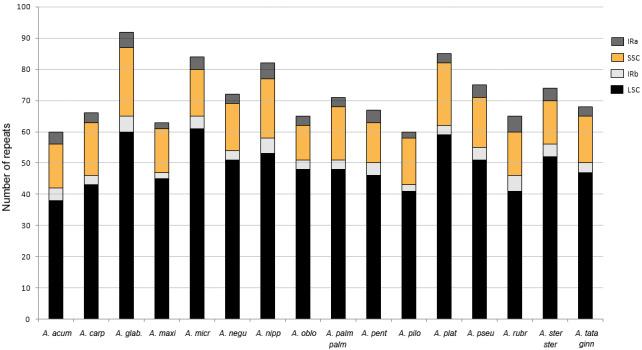
IR, inverted repeat; LSC, large single copy, SSC, short single copy. See Table 1 for full species names.
Phylogenomic reconstruction
The plastome data set (with one inverted repeat removed) consisted of 153,383 aligned sites, of which 20,478 (13.4%) were variable, and 5,774 were parsimony-informative. Pairwise percent identity for all species is provided in Table S6. Both ML and Bayesian analyses on the fully partitioned and the unpartitioned dataset produced trees with identical topologies, very short internal branches, and similar support values (Fig. 7).
Figure 7. Maximum likelihood tree for Acer. plastomes inferred with RaxML-NG.

The topology is identical to the Bayesian 50% majority rule consensus tree. Numbers above and below branches are bootstrap values and posterior probabilities, respectively. Names on the right are Acer sections.
All analyses provided maximum support for the monophyly of Acer and its sister relationship to Dipteronia. Relationships within the genus were fully resolved with strong support; only two branches were weakly to moderately supported in the ML analysis with BS values of 67 and 83%, respectively (Fig. 7). The earliest diverging lineages within the genus included species of sections Acer (Acer pseudoplatanus L.), Glabra (A. glabrum), Pubescentia (A. pilosum), Negundo (Acer negundo L.) and Rubra (Acer rubrum L.). The remaining species were grouped into three mutually exclusive clades, one consisting of A. nipponicum and A. carpinifolium (sections Parviflora and Indivisa, respectively), another formed by species of sections Palmata and Lithocarpa, and the other composed by members of the remaining sections. Within this clade, A. tataricum subsp. ginnala is sister to all other species. Section Platanoidea was recovered as sister to section Macrantha with A. acuminatum (section Arguta) as sister to this clade. The species of sections Trifoliata, Oblonga, and Pentaphylla formed another clade that is sister to the Platanoidea-Macrantha-Arguta clade. All sections represented by more than one species were recovered as monophyletic with high support (Fig. 7).
Discussion
Plastome composition and variable regions
The plastomes of Acer generated in this study are very homogeneous in size and structure as well as gene content. Of the 16 species sampled, A. palmatum (section Palmata) is the only one that differed from the rest by having much longer IRs and an extra copy of the rps19 gene (Fig. 1, Table 1). A survey of plastomes of other members of section Palmata, including Acer wilsonii Rehder (MG012225) and 16 additional species assembled by the authors (Data S2) shows that all share these IRs of about 700 bp longer. Expanded IRs containing whole copies of rps19 are also present in Dipteronia (NC_029338, NC_031899), Dodonaea viscosa Jacq. (NC_036099), Dimocarpus longan Lour. (NC_037447), Koelreuteria paniculata Laxm. (NC_037176), Litchi sinensis (NC_035238), Sapindus mukorossi Gaertn. (NC_025554), and Xanthoceras (NC_037448), but are absent in Aesculus wangii Hu (NC_035955). They have also been detected in other families of Sapindales, including Meliaceae, Rutaceae, Nitrariaceae, and Simaroubaceae (Lu et al., 2017; Mader et al., 2018; Saina et al., 2018). In the case of Acer, this feature appears to be exclusive to species of section Palmata.
In the 16 plastomes studied, the infA gene was found as a pseudogene, with no start codon and the open reading frame disrupted by internal stop codons. In all other Sapindaceae surveyed (listed above), infA also appears as a pseudogene suggesting that it became obsolete in the ancestor of this family or perhaps of Sapindales. It has been reported as missing or as a pseudogene, in several genera of Meliaceae (Mader et al., 2018), Simaroubaceae (Saina et al., 2018), Nitrariaceae (Lu et al., 2017), and Anacardiaceae (Wang et al., 2020). The loss of this gene from cpDNA has been documented in many other angiosperm lineages, and there is even indication of its transfer to the nuclear genome in a number of species (Daniell et al., 2016; Millen et al., 2001).
The ribosomal protein genes rps2 and rpl22 exhibited considerable variation in length and might be non-functional in several species. In A. acuminatum, A. carpinifolium, A. micranthum, A. negundo, A. nipponicum, A. palmatum, A. pilosum, A. pseudoplatanus, A. rubrum, and A. tataricum, rps2 was 60–80% shorter compared to the other Acer species and genera of Sapindales. In the case of rpl22, it was 50 and 60% shorter in A. sterculiaceum and A. pentaphyllum, respectively. The likely loss of function in these species is notable because both genes are considered essential for plant survival (Daniell et al., 2016; Tiller & Bock, 2014). A possible explanation is that they have been transferred to the nucleus, as documented for rpl22 in Fabaceae and Fagaceae (Jansen et al., 2011). Another possibility is their substitution by a nuclear gene that encodes chloroplast (and mitochondrion) targeted proteins as is the case of the rps16 gene in some plant lineages (Keller et al., 2017; Ueda et al., 2008). A search for these genes in the nuclear and mitochondrial genomes will reveal insights into their evolutionary fate in Acer.
In addition to providing insight into the structure and gene organization, genome comparisons are useful for identifying variable regions suitable for the development of molecular markers. In Acer, only a few cpDNA loci (atpB-rbcL, ndhF, psbA-trnH, psbM-trnD, rbcL, rpl16, trnD-trnT, trnL and trnL-trnF) have been employed in phylogenetic (Li, 2011; Li, Yue & Shoup, 2006; Pfosser et al., 2002; Renner et al., 2007; Renner et al., 2008; Tian, Guo & Li, 2002; Zhang, Li & Li, 2010) and phylogeographic studies (Guo et al., 2014; Saeki et al., 2011). The maximum number included in a single dataset is six, but the resulting trees failed to provide adequate resolution and support for many major clades (Renner et al., 2007; Renner et al., 2008). Two additional loci, matK and trnS-trnG, were explored along with rbcL as barcodes to distinguish among 85 Acer species, but they also showed low discrimination power even when used in combination (Han et al., 2016). Only one of these commonly used markers, psbA-trnH, was identified in this study among the most variable in the genus. We found that 14 other loci (Table 3) exhibited greater variation and are thus potentially more informative than the previously used sequences. This supports the notion that universal markers may not be variable enough for many groups and underscores the importance of identifying specific loci for phylogenetic studies at the genus and family level (Cvetkovic, Hinsinger & Strijk, 2019).
Codon usage and repeat sequence analysis
The pattern of codon usage in Acer plastomes is very similar to that of other members of Sapindales. The three most frequent codons (AUU, AAA, and GAA) and the least frequent (UGC, AGC, and CGC) are shared, for example, with Ailanthus (Simaroubaceae), Nitraria (Nitrariaceae) and Toxicodendron (Anacardiaceae) (Lu et al., 2017; Saina et al., 2018; Wang et al., 2020). The most commonly specified amino acids, leucine and isoleucine, are also the most commonly specified in these and many other plant genera (Li et al., 2018; Silva et al., 2018; Yang et al., 2018).
Codon usage was strongly biased toward codons ending in A or T, i.e., there are more codons ending in A or T (68–69% of all codons), and these are used more often (RSCU>1) than the ones ending in C or G (Table 4). This pattern has been observed in most chloroplast genomes of plants (Morton, 2000; Shimada & Sugiura, 1991), and has been related to the high AT composition bias of the protein-coding genes (Morton, 2000). However, selection for translation efficiency is likely the most important factor driving the codon usage of plastid genes (Suzuki & Morton, 2016).
Repetitive elements are a common feature of plastomes, although the amount and distribution can vary widely among plant groups (Sveinsson & Cronk, 2014); (Wicke et al., 2011). In the Acer plastomes analyzed, dispersed repeats of 25 bp and longer were few, ranging between four and eight in most species (Fig. 4). Except for the IRs, all had less than 60 bp in length and were more often located in the non-coding regions of the plastome conforming to the general pattern in angiosperms (Wicke et al., 2011). In the case of SSRs, T and A mononucleotide repeats were more frequent than C/G repeats and than more complex motifs, and were more abundant in non-coding DNA, also conforming to the trend observed in many plant groups (Ebert & Peakall, 2009).
SSRs are among the most variable components of the genome, and constitute an invaluable source of information for population genetic studies, DNA fingerprinting, and plant breeding programs (Nybom, Weising & Rotter, 2014; Wheeler et al., 2014). They have been used to characterize the genetic diversity in Acer species of economic and conservation interest (e.g., Acer campestre L., Acer capillipes Maxim., Acer mono Maxim. Acer opalus Mill., A. pseudoplatanus, A. saccharum and Acer yangbiense Y. S. Chen & Q. E. Yang). However, most studies have employed nuclear (nSSRs) microsatellite loci (Chybicki, Waldon-Rudzionek & Meyza, 2014; Graignic, Tremblay & Bergeron, 2013; Kikuchi & Shibata, 2008; Liu et al., 2014; Neophytou, Konnert & Fussi, 2019; Pandey et al., 2012; Segarra-Moragues, Gleiser & González-Candelas, 2008; Terui et al., 2006; Zhao, Sun & Yang, 2011), and very few have used cpSSRs (Petit et al., 2003; Saeki et al., 2011; Neophytou, Konnert & Fussi, 2019). Our SSR analysis revealed a large number of these repeats distributed across non-coding and protein-coding regions of the Acer plastomes (Table S5) providing an opportunity for the development of new cpSSRs markers for the genus. Because plastids are maternally inherited in Acer (Corriveau & Coleman, 1988), cpSSRs can be particularly useful for studies of gene flow through seed dispersal, and for tracing the maternal lineage in space and time (Nybom, Weising & Rotter, 2014; Petit et al., 2003; Provan, Powell & Hollingsworth, 2001).
The locus with the highest number of SSRs was ycf1. This gene was also identified among the most variable in Acer. It has been reported as among the most variable of the plastome in many plant groups (Jiang, Hinsinger & Strijk, 2016; Kumar et al., 2009; Neubig et al., 2009; Silva et al., 2018; Thomson, Vargas & Dick, 2018), and has even been recommended as a barcode marker (Dong et al., 2015). However, it has been rarely used for phylogenetic inference and barcoding compared to other loci. Future studies should investigate the potential of this gene as a phylogenetic marker in Acer and other plant groups.
Phylogenomic reconstruction
We conducted ML and Bayesian analyses with 22 species of Acer using two different partitioning strategies (i.e., a fully partitioned and an unpartitioned dataset) to explore the effect of partitioning on tree inference. We found that both methods produced identical trees with similar support values regardless of the partitioning strategy used.
It is widely accepted that partitioning is important to account for rate heterogeneity and patterns of substitution among sites and that the choice of partitioning scheme can affect the phylogenetic inference (Brown & Lemmon, 2007; Kainer & Lanfear, 2015; Lanfear et al., 2014). A number of studies that have investigated the effect of partitioning have reported improvement in tree topology, branch lengths, and branch support when the data is appropriately partitioned. In contrast, unpartitioned or poorly partitioned analyses may lead to well-supported but incorrect relationships (Brandley, Schmitz & Reeder, 2005; Kainer & Lanfear, 2015; Tao, Mayden & He, 2013; Ward et al., 2010). In our case, partitioning the cp dataset into 93 subsets (based on spacers, genes, introns, exons, and codon position) did not have an effect on the phylogenetic reconstruction. A similar outcome was reported by Fu et al. (2017) using plastid genomes of Cornales and by Dong et al. (2018) with Saxifragales. The results of these studies, including our own, suggest that the effect of partitioning might be less important in large datasets, presumably due to the increase in phylogenetic signal. Kainer & Lanfear (2015) examined the impact of partitioning scheme choice by analyzing 34 datasets, the largest of which had over 25,000 sites. They found that “the longer the alignment, the less the results depend on the partitioning scheme”. With typically more than 130 kb in length, plastid genome data sets appear to be large enough (and to contain enough phylogenetic signal) to converge on the correct tree irrespective of the partitioning scheme used. It would be interesting, nonetheless, to explore further the effect of partitioning on tree inference using a diverse set of complete plastid genome data.
Our phylogenetic analyses confirmed many of the relationships inferred in previous studies using cp markers (Renner et al., 2007; Renner et al., 2008). These include the earliest-diverging position of the clade formed by A. glabrum and A. pseudoplatanus, the close relationship between A. nipponicum and A. carpinifolium (sections Parviflora and Indivisa respectively), the sister relationship of section Pentaphylla to the Trifoliata-Oblonga clade, and the sister relationship of sections Macrantha and Platanoidea (Fig. 7). Previously unresolved or unsupported relationships in the cp tree were also clarified in our analysis. For example, section Lithocarpa, represented here by Acer sterculiaceum Wall. was resolved as sister to the clade comprising members of section Palmata with maximum support. Section Rubra, A. negundo, and A. pilosum, whose positions were also unresolved, were placed among the early-diverging lineages of the genus (Fig. 7). The placement of A. tataricum, the only species of sect. Ginnala, differed from previous studies based on cp data. This species was recovered by Renner et al. (2007) and Renner et al. (2008) as sister to the clade comprising members of section Platanoidea, Macrantha, Arguta, and part of Negundo, although with no support. In our study, A. tataricum was placed as sister to a major clade formed by species of sections Platanoidea, Macrantha, Arguta, Trifoliata, Oblonga, and Pentaphylla with maximum support (Fig. 7). Overall, these results represent a significant improvement over previous studies based on cp markers, which have been characterized by poor resolution and statistical support (Harris et al., 2017; Li, Yue & Shoup, 2006; Renner et al., 2007; Renner et al., 2008).
A comparison with the phylogenomic analysis by Li et al. (2019) based on nuclear sequences reveals very different topologies between the plastid and nuclear trees. In the nuclear tree, Acer species were grouped into two main lineages, one comprising members of sections Spicata, Palmata, Negundo and Arguta, and the other including the remaining sections. Relationships among sections within each of these two clades also differed markedly between the two trees. For example, sect. Platanoidea and Macrantha were not sister groups in the nuclear tree as was inferred in our cp tree (Fig. 7). Section Macrantha was recovered as sister to a clade comprising species of sect. Ginnala, Indivisa, Lithocarpa, Platanoidea, Rubra, Acer, Trifoliata, and Pentaphylla, whereas monotypic sect. Glabra was sister to sect. Parviflora (Li et al., 2019).
Unlike nuclear trees, which typically tend to be consistent with taxonomy (as is the case in Acer), cp-based trees often correlate with geographic patterns, which can be explained by the occurrence of hybridization and introgression (Albaladejo et al., 2005; McKinnon et al., 1999; Rautenberg et al., 2010). Natural hybridization has been documented in Acer (e.g., Liao et al., 2010). However, few studies have employed molecular tools to study its contribution to the evolution of the genus. For example, Grimm, Denk & Hemleben (2007), using nuclear markers, found evidence of ancient hybridization and introgression in Acer section Acer, a group exhibiting high morphological variability. Also, polyploidization has been suggested to have played a role in the diversification of section Rubra, which includes diploid, tetraploid, hexaploid, and octoploid species (Harris et al., 2017), although no study has been conducted to test this hypothesis. Unfortunately, the limited sampling of cp genomes in this study does not allow detecting any apparent geographic pattern, except perhaps for the position of the three North American species (A. glabrum, A. negundo, and A. rubrum) which were placed among the earliest-diverging lineages (Fig. 7).
There are three polyploid species in our study, and their discordant positions in the plastid and nuclear trees (Li et al., 2019) might indicate past hybridization and introgression. Acer rubrum is hexaploid and octoploid, whereas Acer carpinifolium and A. pseudoplatanus are both tetraploids (Contreras & Shearer, 2018). Acer pseudoplatanus (section Acer) is an autotetraploid (Grimm, Denk & Hemleben, 2007; Pandey et al., 2012) i.e., its polyploidy did not result from hybrid speciation. Yet, its different placement in the plastid and nuclear trees suggests a hybrid origin for this species. In the cp tree, this species was placed as sister to A. glabrum (section Glabra) in the earliest diverging lineage of Acer with maximum support (Fig. 7). It was also recovered as sister to A. glabrum in the analyses of Renner et al. (2007) and Renner et al. (2008) and outside the clade comprising most species of section Acer. However, in the nuclear tree of Li et al. (2019), A. pseudoplatanus was placed in a monophyletic Acer section, also with strong support. This incongruence might be explained by chloroplast capture via hybridization between these two lineages (but also by the retention of ancestral polymorphism, i.e., incomplete lineage sorting). Further analyses with an increased sampling within sections (and populations) will likely reveal important clues on the causes of discordance between plastid and nuclear trees, and on the role of hybridization in the diversification of the genus.
The very short internal branches in our trees suggest a rapid differentiation of the main lineages of Acer within a short period of time. This is consistent with earlier studies that have analyzed the distribution of the rich fossil record (Boulter et al., 1996; Manchester, 1999; Wolfe & Tanai, 1987), which have suggested a burst of diversification during the second half of the Eocene. Based on nuclear data, Li et al. (2019) estimated that most sections in Acer had originated by the late Eocene (33–38 Mya). However, their biogeographic analysis was limited by a narrow sampling (30 taxa) and only two calibration points. Dating analyses incorporating a denser sampling of both plastid and nuclear genomic data are needed to improve our understanding of the diversification and biogeographic history of this diverse genus.
Conclusions
In this study we assembled, annotated, and compared 16 plastid genomes of maples, each belonging to a different section of the 18 that are currently recognized for the genus. We found that Acer plastomes are very similar in structure and gene content. Expanded IRs with two whole copies of the ribosomal rps16 gene distinguished Acer palmatum from the rest of species. Variation in length, possibly accompanied by a loss of function in several species, was detected in the rps2 and rpl22 genes. We confirmed that the greater interspecific variation is located in non-coding sequences of the LSC and SSC regions, and identified variable and potentially informative loci that will facilitate the development of markers for species identification, population genetics, and evolutionary studies in the genus.
Our phylogenetic analysis showed that plastome sequences are valuable tools to resolve deep-level relationships that were unclear or poorly supported in earlier studies using cp markers. Future studies with increased taxon sampling are needed to generate a robust plastid tree that, in combination with nuclear data, will contribute to improve our understanding of the evolution of this diverse and economically important group.
Supplemental Information
Plastomes generated in this study have voucher information and accession numbers in boldface.
Aa: Amino acid, No: Number of codons, RSCU= Relative synonym codon usage.
Only sequences ≥25 bp were considered. F: forward, P: palindrome, R: reverse; IGS: intergenic spacer, IR: inverted repeat, LSC: large single copy, SSC: small single copy.
See Table S1 for full species names.
C: compound repeat, p1: monomeric repeat, p2: dimeric repeat, p3: trimeric repeat, p4: tetrameric repeat; IGS: intergenic spacer, IR: inverted repeat, LSC: large single copy, SSC: small single copy.
See Table S1 for full species names.
Acknowledgments
We are grateful to Iturraran Botanical Garden (Pagoeta Natural Park, Basque country, Spain), Arboretum de Passadoux (France), and Hangzhou Botanical Garden (China), for their help in data sampling.
Funding Statement
This work was supported by funding through the Bagui Scholarship team funding under Grant No. C33600992001 and the government of Guangxi Province (Provincial 100 Talent Program—recruitment of overseas talents for colleges and universities in Guangxi) to Joeri S. Strijk, and China Postdoctoral Science Foundation Grants (No. 2015M582481 and 2016T90822) to Damien D. Hinsinger. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests
Author Contributions
Fabiola Areces-Berazain performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, and approved the final draft.
Yixi Wang analyzed the data, prepared figures and/or tables, and approved the final draft.
Damien D. Hinsinger and Joeri S. Strijk conceived and designed the experiments, authored or reviewed drafts of the paper, and approved the final draft.
Data Availability
References
- Acevedo-Rodríguez et al. (2011).Acevedo-Rodríguez P, Van Welzen PC, Adema F, Van der Ham RWJM. Sapindaceae. In: Kubitzki K, editor. The families and genera of vascular plants. Vol. 10. Springer; Berlin: 2011. pp. 357–407. [Google Scholar]
- Ackerly & Donoghue (1998).Ackerly DD, Donoghue MJ. Leaf size, sapling allometry, and Corner’s rules: phylogeny and correlated evolution in maples (Acer) The American Naturalist. 1998;152(6):767–791. doi: 10.2307/2463600. [DOI] [PubMed] [Google Scholar]
- Albaladejo et al. (2005).Albaladejo RG, Aguilar JF, Aparicio A, Feliner GN. Contrasting nuclear-plastidial phylogenetic patterns in the recently diverged Iberian Phlomis crinita and P. lychnitis lineages (Lamiaceae) Taxon. 2005;54(4):987–998. doi: 10.2307/25065483. [DOI] [Google Scholar]
- Amiryousefi, Hyvonen & Poczai (2018).Amiryousefi A, Hyvonen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018;34(17):3030–3031. doi: 10.1093/bioinformatics/bty220. [DOI] [PubMed] [Google Scholar]
- Ball (2007).Ball DW. The chemical composition of maple syrup. Journal of Chemical Education. 2007;84(10):1647–1650. doi: 10.1021/ed084p1647. [DOI] [Google Scholar]
- Beier et al. (2017).Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33(16):2583–2585. doi: 10.1093/bioinformatics/btx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts (1959).Betts H. Maple (Acer species). American wood series. US Department of Agriculture. US Government Printing Office; Washington DC: 1959. [Google Scholar]
- Bishop et al. (2015).Bishop DA, Beier CM, Pederson N, Lawrence GB, Stella JC, Sullivan TJ. Regional growth decline of sugar maple (Acer saccharum) and its potential causes. Ecosphere. 2015;6(10):a179. doi: 10.1890/es15-00260.1. [DOI] [Google Scholar]
- Boulter et al. (1996).Boulter MC, Benfield JN, Fisher HC, Gee DA, Lhotak M. The evolution and global migration of the Aceraceae. Philosophical Transactions of the Royal Society B: Biological Sciences. 1996;351(1340):589–603. doi: 10.1098/rstb.1996.0058. [DOI] [Google Scholar]
- Brandley, Schmitz & Reeder (2005).Brandley MC, Schmitz A, Reeder TW. Partitioned bayesian analyses, partition choice, and the phylogenetic relationships of Scincid lizards. Systematic Biology. 2005;54(3):373–390. doi: 10.1080/10635150590946808. [DOI] [PubMed] [Google Scholar]
- Brown & Lemmon (2007).Brown JM, Lemmon AR. The importance of data partitioning and the utility of bayes factors in bayesian phylogenetics. Systematic Biology. 2007;56(4):643–655. doi: 10.1080/10635150701546249. [DOI] [PubMed] [Google Scholar]
- Chybicki, Waldon-Rudzionek & Meyza (2014).Chybicki IJ, Waldon-Rudzionek B, Meyza K. Population at the edge: increased divergence but not inbreeding towards northern range limit in Acer campestre. Tree Genetics and Genomes. 2014;10(6):1739–1753. doi: 10.1007/s11295-014-0793-2. [DOI] [Google Scholar]
- Contreras & Shearer (2018).Contreras RN, Shearer K. Genome size, ploidy, and base composition of wild and cultivated Acer. Journal of the American Society for Horticultural Science. 2018;143(6):470–485. doi: 10.21273/JASHS04541-18. [DOI] [Google Scholar]
- Corriveau & Coleman (1988).Corriveau JL, Coleman AW. Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. American Journal of Botany. 1988;75(10):1443–1458. doi: 10.1002/j.1537-2197.1988.tb11219.x. [DOI] [Google Scholar]
- Cvetkovic, Hinsinger & Strijk (2019).Cvetkovic T, Hinsinger DD, Strijk JS. Exploring evolution and diversity of Chinese Dipterocarpaceae using next-generation sequencing. Scientific Reports. 2019;9:11639. doi: 10.1038/s41598-019-48240-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell et al. (2016).Daniell H, Lin CS, Yu M, Chang WJ. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biology. 2016;17(1):134. doi: 10.1186/s13059-016-1004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba et al. (2019).Darriba D, Posada D, Kozlov AM, Stamatakis A, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. BioRxiv. 2019;37(1):291–294. doi: 10.1101/612903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong (1976).De Jong P. Flowering and sex expression in Acer L.: a biosystematic study. Vol. 2. H. Veenman & Zonen B. V; Wageningen: 1976. [Google Scholar]
- De Jong (1994).De Jong P. Taxonomy and reproductive biology of maples. In: Van Gelderen D, De Jong P, Oterdoom H, editors. Maples of the World. Oregon: Timber Press; 1994. pp. 69–99. [Google Scholar]
- De Jong (2004).De Jong P. World maple diversity. In: Wiegrefe SJ, editor. Proceedings of the International Maple Symposium. Westonbirt Arboretum and Royal Agricultural College; Gloucestershire: 2004. pp. 69–99. [Google Scholar]
- Dierckxsens, Mardulyn & Smits (2017).Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research. 2017;45(4):e18. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong et al. (2015).Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S. ycf1, the most promising plastid DNA barcode of land plants. Scientific Reports. 2015;5:8348. doi: 10.1038/srep08348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong et al. (2018).Dong W, Xu C, Wu P, Cheng T, Yu J, Zhou S, Hong D. Resolving the systematic positions of enigmatic taxa: manipulating the chloroplast genome data of Saxifragales. Molecular Phylogenetics and Evolution. 2018;126:321–330. doi: 10.1016/j.ympev.2018.04.033. [DOI] [PubMed] [Google Scholar]
- Ebert & Peakall (2009).Ebert D, Peakall R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Molecular Ecology Resources. 2009;9:673–690. doi: 10.1111/j.1755-0998.2008.02319.x. [DOI] [PubMed] [Google Scholar]
- Frazer et al. (2004).Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleid Acids Research. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu et al. (2017).Fu C-N, Li H-T, Milne R, Zhang T, Ma P-F, Yang J, Li D. Comparative analyses of plastid genomes from fourteen Cornales species: inferences for phylogenetic relationships and genome evolution. BMC Genomics. 2017;18:956. doi: 10.1186/s12864-017-4319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs & Chen (2009).Gibbs D, Chen Y. The Red List of maples. Botanic Gardens Conservation International; Richmond: 2009. [Google Scholar]
- Gitzendanner et al. (2018).Gitzendanner MA, Soltis PS, Yi TS, Li DZ, Soltis DE. Plastome phylogenetics: 30 years of inferences into plant evolution. In: Chaw S-M, Jansen R, editors. Advances in Botanical Research. Vol. 85. Academic Press; Massachusetts: 2018. pp. 293–313. [DOI] [Google Scholar]
- Graignic, Tremblay & Bergeron (2013).Graignic N, Tremblay F, Bergeron Y. Development of polymorphic nuclear microsatellite markers in sugar maple (Acer saccharum Marsh.) using cross-species transfer and SSR-enriched shotgun pyrosequencing. Conservation Genetics Resources. 2013;5(3):845–848. doi: 10.1007/s12686-013-9923-7. [DOI] [Google Scholar]
- Grimm, Denk & Hemleben (2007).Grimm GW, Denk T, Hemleben V. Evolutionary history and systematics of Acer section Acer—a case study of low-level phylogenetics. Plant Systematics and Evolution. 2007;267(1–4):215–253. doi: 10.1007/s00606-007-0572-8. [DOI] [Google Scholar]
- Grimm et al. (2006).Grimm GW, Renner SS, Stamatakis A, Hemleben V. A nuclear ribosomal DNA phylogeny of Acer inferred with maximum likelihood, splits graphs, and motif analysis of 606 sequences. Evolutionary Bioinformatics. 2006;2:7–22. doi: 10.1177/117693430600200014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo et al. (2014).Guo X, Wang H, Bao L, Wang T, Bai W, Ye J, Ge J. Evolutionary history of a widespread tree species Acer mono in East Asia. Ecology and Evolution. 2014;4(22):4332–4345. doi: 10.1002/ece3.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han et al. (2016).Han Y, Duan D, Ma X, Jia Y, Liu Z, Zhao G, Li Z-H. Efficient identification of the forest tree species in Aceraceae using DNA barcodes. Frontiers in Plant Science. 2016;7:1707. doi: 10.3389/fpls.2016.01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris et al. (2017).Harris AJ, Chen Y, Olsen RT, Lutz S, Wen J. On merging Acer sections Rubra and Hyptiocarpa: molecular and morphological evidence. PhytoKeys. 2017;86:9–42. doi: 10.3897/phytokeys.86.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, Frawley & Wen (2017).Harris AJ, Frawley E, Wen J. The utility of single-copy nuclear genes for phylogenetic resolution of Acer and Dipteronia (Acereae, Sapindaceae) Annales Botanici Fennici. 2017;54(4–6):209–222. doi: 10.5735/085.054.0603. [DOI] [Google Scholar]
- Harris (1975).Harris J. Tree genera—3. Acer—of the maple. Arboricultural Journal: The International Journal of Urban Forestry. 1975;2:361–369. doi: 10.1080/03071375.1975.10590443. [DOI] [Google Scholar]
- Jansen et al. (2011).Jansen RK, Saski C, Lee S, Hansen AK, Daniell H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Molecular Biology and Evolution. 2011;28(1):835–847. doi: 10.1093/molbev/msq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia et al. (2016).Jia Y, Yang J, He Y-L, He Y, Niu C, Gong L-L, Li Z-H. Characterization of the whole chloroplast genome sequence of Acer davidii Franch (Aceraceae) Conservation Genetics Resources. 2016;8(2):141–143. doi: 10.1007/s12686-016-0530-2. [DOI] [Google Scholar]
- Jiang, Hinsinger & Strijk (2016).Jiang GF, Hinsinger DD, Strijk JS. Comparison of intraspecific, interspecific and intergeneric chloroplast diversity in Cycads. Scientific Reports. 2016;6:31473. doi: 10.1038/srep31473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainer & Lanfear (2015).Kainer D, Lanfear R. The effects of partitioning on phylogenetic inference. Molecular Biology and Evolution. 2015;32(6):1611–1627. doi: 10.1093/molbev/msv026. [DOI] [PubMed] [Google Scholar]
- Katoh & Standley (2013).Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller et al. (2017).Keller J, Rousseau-Gueutin M, Martin GE, Morice J, Boutte J, Coissac E, Ourari M, Aïnouche M, Salmon A, Cabello-Hurtado F, Aïnouche A. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Research. 2017;24(4):343–358. doi: 10.1093/dnares/dsx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi & Shibata (2008).Kikuchi S, Shibata M. Development of polymorphic microsatellite markers in Acer mono Maxim. Molecular Ecology Resources. 2008;8(2):339–341. doi: 10.1111/j.1471-8286.2007.01948.x. [DOI] [PubMed] [Google Scholar]
- Kim et al. (2019).Kim S, Shin S, Lee M, Lee J. Complete chloroplast genome of Acer tegmentosum and phylogenetic analysis. Mitochondrial DNA Part B. 2019;4(2):2555–2556. doi: 10.1080/23802359.2019.1640646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov et al. (2019).Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar et al. (2009).Kumar S, Hahn FM, McMahan CM, Cornish K, Whalen MC. Comparative analysis of the complete sequence of the plastid genome of Parthenium argentatum and identification of DNA barcodes to differentiate Parthenium species and lines. BMC Plant Biology. 2009;9:131. doi: 10.1186/1471-2229-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz et al. (2001).Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research. 2001;29(22):4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear et al. (2014).Lanfear R, Calcott B, Kainer D, Mayer C, Stamatakis A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evolutionary Biology. 2014;14:82. doi: 10.1186/1471-2148-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear et al. (2016).Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution. 2016;34(3):772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- Li (2011).Li J. Phylogenetic evaluation of series delimitations in Section Palmata (Acer, Aceroideae, Sapindaceae) based on sequences of nuclear and chloroplast genes. Aliso. 2011;29(1):43–49. doi: 10.5642/aliso.20112901.05. [DOI] [Google Scholar]
- Li et al. (2018).Li ZZ, Saina JK, Gichira AW, Kyalo CM, Wang QF, Chen JM. Comparative genomics of the Balsaminaceae sister genera Hydrocera triflora and Impatiens pinfanensis. International Journal of Molecular Sciences. 2018;19:319. doi: 10.3390/ijms19010319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al. (2019).Li J, Stukel M, Bussies P, Skinner K, Lemmon AR, Lemmon EM, Moriarty Lemmon E, Brown K, Bekmetjev A, Swenson NG. Maple phylogeny and biogeography inferred from phylogenomic data. Journal of Systematics and Evolution. 2019;57:594–606. doi: 10.1111/jse.12535. [DOI] [Google Scholar]
- Li, Yue & Shoup (2006).Li J, Yue J, Shoup S. Phylogenetics of Acer (Aceroideae, Sapindaceae) based on nucleotide sequences of two chloroplast non-coding regions. Harvard Papers in Botany. 2006;11(1):101–115. doi: 10.3100/1043-4534(2006)11[101:poaasb]2.0.co;2. [DOI] [Google Scholar]
- Li et al. (2015).Li ZH, Xie YS, Zhou T, Jia Y, He YL, Yang J. The complete chloroplast genome sequence of Acer morrisonense (Aceraceae) Mitochondrial DNA Part A. 2015;28(3):309–310. doi: 10.3109/19401736.2015.1118091. [DOI] [PubMed] [Google Scholar]
- Liao et al. (2010).Liao P-C, Shih H-C, Yen T-B, Lu S-Y, Cheng Y-P, Chiang Y-C. Molecular evaluation of interspecific hybrids between Acer albopurpurascens and A. buergerianum var. formosanum. Botanical Studies. 2010;51:413–420. [Google Scholar]
- Liu et al. (2014).Liu C, Tsuda Y, Shen H, Hu L, Saito Y, Ide Y. Genetic structure and hierarchical population divergence history of Acer mono var. mono in South and Northeast China. PLOS ONE. 2014;9(1):e87187. doi: 10.1371/journal.pone.0087187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu et al. (2017).Lu L, Li X, Hao Z, Yang L, Zhang J, Peng Y, Xu H, Lu Y, Zhang J, Shi J, Jinhui Chen Cheng T. Phylogenetic studies and comparative chloroplast genome analyses elucidate the basal position of halophyte Nitraria sibirica (Nitrariaceae) in the Sapindales. Mitochondrial DNA Part A. 2017;29(5):745–755. doi: 10.1080/24701394.2017.1350954. [DOI] [PubMed] [Google Scholar]
- Mader et al. (2018).Mader M, Pakull B, Blanc-Jolivet C, Paulini-Drewes M, Henri-Noel Bouda Z, Degen B, Small I, Kersten B. Complete chloroplast genome sequences of four Meliaceae species and comparative analyses. International Journal of Molecular Sciences. 2018;19:701. doi: 10.3390/ijms19030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchester (1999).Manchester SR. Biogeographical relationships of north American tertiary floras. Annals of the Missouri Botanical Garden. 1999;86(2):472–522. doi: 10.2307/2666183. [DOI] [Google Scholar]
- McKinnon et al. (1999).McKinnon GE, Steane DA, Potts BM, Villancourt RE. Incongruence between chloroplast and species phylogenies in Eucalyptus subgenus Monocalyptus (Myrtaceae) American Journal of Botany. 1999;86(7):1038–1046. doi: 10.2307/2656621. [DOI] [PubMed] [Google Scholar]
- Millen et al. (2001).Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L, Kavanagh TA, Hibberd JM, Gray JC, Morden CW, Calie PJ, Jermiin LS, Wolfe KH. Many parallel losses of infA from chloroplast DNA during Angiosperm evolution with multiple independent transfers to the nucleus. The Plant Cell. 2001;13:645–658. doi: 10.1105/tpc.13.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, Pfeiffer & Schwartz (2010).Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE); 2010. pp. 1–8. [DOI] [Google Scholar]
- Minorsky (2003).Minorsky PV. The decline of sugar maples (Acer saccharum) Plant Physiology. 2003;133:441–442. doi: 10.1104/pp.900091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momotani (1962).Momotani Y. Taxonomic study of the genus Acer, with special reference to the seed proteins III. System of Aceraceae. Memoirs of the College of Science Kyoto University, Ser. B. 1962;29:177–189. [Google Scholar]
- Morton (2000).Morton BR. Codon Bias and the context dependency of nucleotide substitutions in the evolution of plastid DNA. In: Hecht MK, MacIntyre RJ, Clegg MT, editors. Evolutionary Biology. Vol. 31. Springer; Boston, Massachusetts: 2000. pp. 55–103. [DOI] [Google Scholar]
- Murray (1970).Murray E. A monograph of the Aceraceae. D. Phil. Thesis, Pennsylvania State University,; USA: 1970. [Google Scholar]
- Neophytou, Konnert & Fussi (2019).Neophytou C, Konnert M, Fussi B. Western and eastern post-glacial migration pathways shape the genetic structure of sycamore maple (Acer pseudoplatanus L.) in Germany. Forest Ecology and Management. 2019;432:83–93. doi: 10.1016/j.foreco.2018.09.016. [DOI] [Google Scholar]
- Neubig et al. (2009).Neubig KM, Whitten WM, Carlsward BS, Blanco MA, Endara L, Williams NH, Moore M. Phylogenetic utility of ycf1 in orchids: A plastid gene more variable than matK. Plant Systematics and Evolution. 2009;277(1–2):75–84. doi: 10.1007/s00606-008-0105-0. [DOI] [Google Scholar]
- Nybom, Weising & Rotter (2014).Nybom H, Weising K, Rotter B. DNA fingerprinting in botany: past, present, future. Investigative Genetics. 2014;5:1. doi: 10.1186/2041-2223-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata (1967).Ogata K. A systematic study of the genus Acer. Bulletin of the Tokyo University Forests. 1967;63:89–206. [Google Scholar]
- Pandey et al. (2012).Pandey M, Gailing O, Hattemer HH, Finkeldey R. Fine-scale spatial genetic structure of sycamore maple (Acer pseudoplatanus L.) European Journal of Forest Research. 2012;131:739–746. doi: 10.1007/s10342-011-0546-9. [DOI] [Google Scholar]
- Pax (1885).Pax F. Monographie der Gattung Acer. Engler’s Botanische Jahrbücher. 1885;6:287–347. [Google Scholar]
- Pax (1902).Pax F. Aceraceae. In: Engler A, editor. Das Pflanzenreich, heft. Vol. 8. Wilhelm Engelmann; Leipzig: 1902. pp. 1–89. [Google Scholar]
- Petit et al. (2003).Petit RJ, Aguinagalde I, De Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Pedro Martn J, Rendell S, Vendramin GG. Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 2003;300(5625):1563–1565. doi: 10.1126/science.1083264. [DOI] [PubMed] [Google Scholar]
- Pfosser et al. (2002).Pfosser M, Wrobelska JG, Sun B-Y, Stuessy TF, Sugawara T, Fujii N. The origin of species of Acer (Sapindaceae) endemic to Ullung Island, Korea. Systematic Botany. 2002;27(2):351–367. [Google Scholar]
- Pojarkova (1933).Pojarkova AI. Botanico-geographical survey of the maples in USSR, in connection with the history of the whole genus. Acta Institute of Botany Academy of Sciences, USSR, Ser. 1. 1933;1:225–374. [Google Scholar]
- Provan, Powell & Hollingsworth (2001).Provan J, Powell W, Hollingsworth PM. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends in Ecology and Evolution. 2001;16(3):142–147. doi: 10.1016/S0169-5347(00)02097-8. [DOI] [PubMed] [Google Scholar]
- Rambaut et al. (2018).Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology. 2018;67(5):901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautenberg et al. (2010).Rautenberg A, Hathaway L, Oxelman B, Prentice HC. Geographic and phylogenetic patterns in Silene section Melandrium (Caryophyllaceae) as inferred from chloroplast and nuclear DNA sequences. Molecular Phylogenetics and Evolution. 2010;57(3):978–991. doi: 10.1016/j.ympev.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Renner et al. (2007).Renner SS, Beenken L, Grimm GW, Kocyan A, Ricklefs RE. The evolution of dioecy, heterodichogamy, and labile sex expression in Acer. Evolution. 2007;61(11):2701–2719. doi: 10.1111/j.1558-5646.2007.00221.x. [DOI] [PubMed] [Google Scholar]
- Renner et al. (2008).Renner SS, Grimm GW, Schneeweiss GM, Stuessy TF, Ricklefs RE. Rooting and dating maples (Acer) with an uncorrelated-rates molecular clock: implications for North American/Asian disjunctions. Systematic Biology. 2008;57(5):795–808. doi: 10.1080/10635150802422282. [DOI] [PubMed] [Google Scholar]
- Ronquist et al. (2012).Ronquist F, Teslenko M, Van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck J. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Systematic Biology. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas et al. (2017).Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution. 2017;34(12):3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Saeki et al. (2011).Saeki I, Dick CW, Barnes BV, Murakami N. Comparative phylogeography of red maple (Acer rubrum L.) and silver maple (Acer saccharinum L.): impacts of habitat specialization, hybridization and glacial history. Journal of Biogeography. 2011;38:992–1005. doi: 10.1111/j.1365-2699.2010.02462.x. [DOI] [Google Scholar]
- Saina et al. (2018).Saina JK, Li Z, Gichira AW, Liao Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima Mill.) (Sapindales: Simaroubaceae), an important pantropical tree. International Journal of Molecular Sciences. 2018;19:929. doi: 10.3390/ijms19040929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segarra-Moragues, Gleiser & González-Candelas (2008).Segarra-Moragues JG, Gleiser G, González-Candelas F. Isolation and characterization of microsatellite loci in Acer opalus (Aceraceae), a sexually-polymorphic tree, through an enriched genomic library. Conservation Genetics. 2008;9(4):1059–1062. doi: 10.1007/s10592-007-9451-7. [DOI] [Google Scholar]
- Sharp, Tuohy & Mosurski (1986).Sharp PM, Tuohy TMF, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Research. 1986;14(13):5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada & Sugiura (1991).Shimada H, Sugiura M. Fine structural features of the chloroplast genome: comparison of the sequenced chloroplast genomes. Nucleic Acids Research. 1991;19(5):983–995. doi: 10.1093/nar/19.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva et al. (2018).Silva SR, Michael TP, Meer EJ, Pinheiro DG, Varani M, Miranda VFO. Comparative genomic analysis of Genlisea (corkscrew plants—Lentibulariaceae) chloroplast genomes reveals an increasing loss of the ndh genes. PLOS ONE. 2018;13(1):e0190321. doi: 10.1371/journal.pone.0190321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh, Heo & Park (2000).Suh Y, Heo K, Park C-W. Phylogenetic relationships of maples (Acer L.; Aceraceae) implied by nuclear ribosomal ITS sequences. Journal of Plant Research. 2000;113(2):193–202. doi: 10.1007/pl00013914. [DOI] [Google Scholar]
- Suzuki & Morton (2016).Suzuki H, Morton BR. Codon adaptation of plastid genes. PLOS ONE. 2016;11(5):e0154306. doi: 10.1371/journal.pone.0154306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveinsson & Cronk (2014).Sveinsson S, Cronk Q. Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium) BMC Evolutionary Biology. 2014;14(1):228. doi: 10.1186/s12862-014-0228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Mayden & He (2013).Tao W, Mayden RL, He S. Remarkable phylogenetic resolution of the most complex clade of Cyprinidae (Teleostei: Cypriniformes): A proof of concept of homology assessment and partitioning sequence data integrated with mixed model Bayesian analyses. Molecular Phylogenetics and Evolution. 2013;66(3):603–616. doi: 10.1016/j.ympev.2012.09.024. [DOI] [PubMed] [Google Scholar]
- Terui et al. (2006).Terui H, Lian C, Saito Y, Ide Y. Development of microsatellite markers in Acer capillipes. Molecular Ecology Notes. 2006;6:77–79. doi: 10.1111/j.1471-8286.2005.01144.x. [DOI] [Google Scholar]
- Thomson, Vargas & Dick (2018).Thomson AM, Vargas OM, Dick CW. Complete plastome sequences from Bertholletia excelsa and 23 related species yield informative markers for Lecythidaceae. Applications in Plant Sciences. 2018;6(5):e1151. doi: 10.1002/aps3.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Guo & Li (2002).Tian X, Guo Z-H, Li D-Z. Phylogeny of Aceraceae based on ITS and trnL-F data sets. Acta Botanica Sinica. 2002;44(6):714–724. [Google Scholar]
- Tiller & Bock (2014).Tiller N, Bock R. The translational apparatus of plastids and its role in plant development. Molecular Plant. 2014;7(7):1105–1120. doi: 10.1093/mp/ssu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillich et al. (2017).Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Research. 2017;45:W6–W11. doi: 10.1093/nar/gkx391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda et al. (2008).Ueda M, Nishikawa T, Fujimoto M, Takanashi H, Arimura SI, Tsutsumi N, Kadowaki KI. Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Molecular Biology and Evolution. 2008;25(8):1566–1575. doi: 10.1093/molbev/msn102. [DOI] [PubMed] [Google Scholar]
- Wang et al. (2020).Wang L, He N, Li Y, Fang Y, Zhang F. Complete chloroplast genome sequence of Chinese lacquer tree (Toxicodendron vernicifluum, Anacardiaceae) and its phylogenetic significance. Biomed Research International. 2020;1:1–13. doi: 10.1155/2020/9014873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Chen & Zhang (2017).Wang WC, Chen SY, Zhang XZ. The complete chloroplast genome of the endangered Chinese paperbark maple, Acer griseum (Sapindaceae) Conservation Genetics Resources. 2017;9:527–529. doi: 10.1007/s12686-017-0715-3. [DOI] [Google Scholar]
- Ward et al. (2010).Ward PS, Brady SG, Fisher BL, Schultz TR. Phylogeny and biogeography of Dolichoderine ants: effects of data partitioning and relict taxa on historical inference. Systematic Biology. 2010;59(3):342–362. doi: 10.1093/sysbio/syq012. [DOI] [PubMed] [Google Scholar]
- Wheeler et al. (2014).Wheeler GL, Dorman HE, Buchanan A, Challagundla L, Wallace LE. A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Applications in Plant Sciences. 2014;2(12):1400059. doi: 10.3732/apps.1400059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke et al. (2011).Wicke S, Schneeweiss GM, DePamphilis CW, Mueller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe & Tanai (1987).Wolfe JA, Tanai T. Systematics, phylogeny, and distribution of Acer (maples) in the Cenozoic of Western North America. Journal of the Faculty of Science, Hokkaido University, Series 4, Geology and Mineralogy. 1987;22:1–246. [Google Scholar]
- Xu (1996).Xu T-Z. A new system of the genus Acer. Acta Botanica Yunnanica. 1996;18(3):277–292. [Google Scholar]
- Xu et al. (2008).Xu T-Z, Chen Y, De Jong P, Oterdoom H, Chang C-S. Aceraceae. In: Wu Z, Raven P, Hong D, editors. Flora of China. Vol. 11. Missouri Botanical Garden Press; St. Louis: 2008. pp. 515–553. [Google Scholar]
- Yang et al. (2018).Yang Y, Zhu J, Feng L, Zhou T, Bai G, Yang J, Zhao G. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Frontiers in Plant Science. 2018;9:82. doi: 10.3389/fpls.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al. (2016).Zhang Y, Li B, Chen H, Wang Y. Characterization of the complete chloroplast genome of Acer miaotaiense (Sapindales: Aceraceae), a rare and vulnerable tree species endemic to China. Conservation Genetics Resources. 2016;8(4):383–385. doi: 10.1007/s12686-016-0564-5. [DOI] [Google Scholar]
- Zhang, Li & Li (2010).Zhang Z, Li C, Li J. Conflicting phylogenies of Section Macrantha (Acer Aceroideae, Sapindaceae) based on chloroplast and nuclear DNA. Systematic Botany. 2010;35(4):801–810. doi: 10.1600/036364410x539871. [DOI] [Google Scholar]
- Zhao, Sun & Yang (2011).Zhao L, Sun W, Yang J. Development and characterization of microsatellite markers in the critically endangered species Acer yangbiense (Aceraceae) American Journal of Botany. 2011;98:e247–e249. doi: 10.3732/ajb.1100142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plastomes generated in this study have voucher information and accession numbers in boldface.
Aa: Amino acid, No: Number of codons, RSCU= Relative synonym codon usage.
Only sequences ≥25 bp were considered. F: forward, P: palindrome, R: reverse; IGS: intergenic spacer, IR: inverted repeat, LSC: large single copy, SSC: small single copy.
See Table S1 for full species names.
C: compound repeat, p1: monomeric repeat, p2: dimeric repeat, p3: trimeric repeat, p4: tetrameric repeat; IGS: intergenic spacer, IR: inverted repeat, LSC: large single copy, SSC: small single copy.
See Table S1 for full species names.
Data Availability Statement
The following information was supplied regarding data availability:
The annotated sequences of the 16 plastomes are available at GenBank: MN864496–MN864511 and in Data S1.