

Abstract
Background/Objectives
Hypothalamic neurons play a major role in the control of body mass. Obese subjects present radiologic signs of gliosis in the hypothalamus, which may reflect the damage or loss of neurons involved in whole-body energy homeostasis. It is currently unknown if hypothalamic gliosis (1) differs between obese nondiabetic (ND) and obese diabetic subjects (T2D) or (2) is modified by extensive body mass reduction via Roux-n-Y gastric bypass (RYGB).
Subjects/Methods
Fifty-five subjects (all female) including lean controls (CT; n = 13), ND (n = 28), and T2D (n = 14) completed at least one study visit. Subjects underwent anthropometrics and a multi-echo MRI sequence to measure mean bilateral T2 relaxation time in the mediobasal hypothalamus (MBH) and two reference regions (amygdala and putamen). The obese groups underwent RYGB and were re-evaluated 9 months later. Analyses were by linear mixed models.
Results
Analyses of T2 relaxation time at baseline showed a group by region interaction only in the MBH (P < 0.0001). T2D had longer T2 relaxation times compared to either CT or ND groups. To examine the effects of RYGB on hypothalamic gliosis a three-way (group by region by time) mixed effects model adjusted for age was executed. Group by region (P < 0.0001) and region by time (P = 0.0005) interactions were significant. There was a reduction in MBH relaxation time by RYGB, and, although the T2D group still had higher T2 relaxation time overall compared to the ND group, the T2D group had significantly lower T2 relaxation time after surgery and the ND group showed a trend. The degree of reduction in MBH T2 relaxation time by RYGB was unrelated to clinical outcomes.
Conclusion
T2 relaxation times, a marker of hypothalamic gliosis, are higher in obese women with T2D and are reduced by RYGB-induced weight loss.
Introduction
Hypothalamic neurons play a major role in the control of body mass in both experimental models and humans [1, 2]. Obesity is usually characterized by hypothalamic resistance to the adipostatic hormones leptin and insulin, which leads to an imbalance between food intake and energy expenditure [1, 3]. In animal models, this dysfunction may result from hypothalamic inflammation and injury, which involves neuron apoptosis and reactive gliosis—the activation of astrocyte and microglial cell populations [4–7]. Although plausible, there is no direct evidence to date implying such a mechanism in the pathogenesis of human obesity. A few functional magnetic resonance image (fMRI) studies suggested that hypothalamic activity is dysfunctional in subjects with obesity or type 2 diabetes, compared to lean controls [8–11]. Furthermore in humans, Thaler et al. reported an increased signal intensity in the mediobasal hypothalamus (MBH) of obese subjects in T2-weighted magnetic resonance images, indicative of local gliosis [6], a finding that was subsequently replicated [12].
These findings raise the possibility that obesity affects the human hypothalamus similarly to animal models of this disease, but whether these structural alterations are reversible is unknown. The dysfunctional brain activity observed in severely obese subjects is partially restored after bariatric surgery induced weight loss, as assessed by fMRI [10]. Also, it was recently demonstrated that in rodents, MBH gliosis associated with high-fat feeding is largely reversible after return to standard chow diet and reversal of the obesity phenotype [13].
Another area of uncertainty is whether hypothalamic gliosis is associated with obesity itself or obesity-related disorders, such as insulin resistance (IR). Metabolic phenotype varies considerably among obese individuals. While most of them develop IR and present a range of comorbidities, including diabetes, dyslipidemia, and hypertension, some remain metabolically healthy despite similar fat mass [14]. In rodents, hypothalamic injury is related to high fat feeding-induced inflammation, not adiposity [4–6, 15]. Also in humans, a link between MBH gliosis and IR was demonstrated, which is independent of body mass index (BMI) [16]. Along with abnormal insulin secretion, IR is a hallmark of type 2 diabetes. It is currently unknown if hypothalamic gliosis differs between obese nondiabetic (ND) and obese diabetic subjects or is modified by body mass reduction.
We, therefore, employed a quantitative MRI approach that has been validated in mice [17] and humans [16] to detect and quantify MBH gliosis in obese subjects with and without type 2 diabetes, before and after massive weight loss induced by a Roux-en-Y gastric bypass (RYGB), as compared to lean healthy subjects.
Results
Subjects baseline features
Groups differed slightly in age with T2D being significantly older than CT (Supplemental Table 1). Participants with obesity (ND and T2D) weighed more, had a higher BMI and higher plasma concentrations of fasting insulin, CRP, and AST compared with CT. Furthermore, compared to ND, T2D had higher weight, BMI, fasting glucose and insulin, HbA1c, and HOMA-IR scores (Supplemental Table 1).
Association of age, BMI, and T2D with hypothalamic gliosis at baseline
Among all participants, there were normal [18] age-related negative correlations with bilateral T2 relaxation time in the reference regions (amygdala: β = −0.19 ms, P = 0.001; putamen: β = −0.19 ms, P = 0.003), but within the bilateral MBH T2 relaxation times tended to be longer with increasing age (β = 0.36 ms, P = 0.06), therefore all models are adjusted for age. At baseline, there was a significant group by region interaction (χ2(4) = 48.3, PInt < 0.0001 adjusted), and group differences in T2 relaxation time were only present in the MBH and not in the reference regions. T2D had longer T2 relaxation times in the MBH compared to either the CT or ND group (Fig. 1a, b). In age-adjusted models across all participants, longer T2 relaxation time in the MBH was associated with higher weight and BMI (β = 0.12 ms, P = 0.04; β = 0.34 ms, P = 0.02, respectively), log fasting insulin and HOMA-IR scores (Fig. 1c, d), and tended to be related to fasting glucose (β = 0.16 ms, P = 0.05). However, HbA1c (β = 0.25, P = 0.91), triglycerides (β = 0.04, P = 0.16), CRP (β = 1.03, P = 0.42), and markers of liver function (AST (β = 0.22, P = 0.18), ALT (β = −0.05, P = 0.58)) were unrelated to MBH T2 relaxation time. Adjusting models for T2 relaxation times in control regions did not substantively change results (data not shown), although the relationship with weight was altered from significant to a trend (β = 0.11 ms, P = 0.08).
Fig. 1.
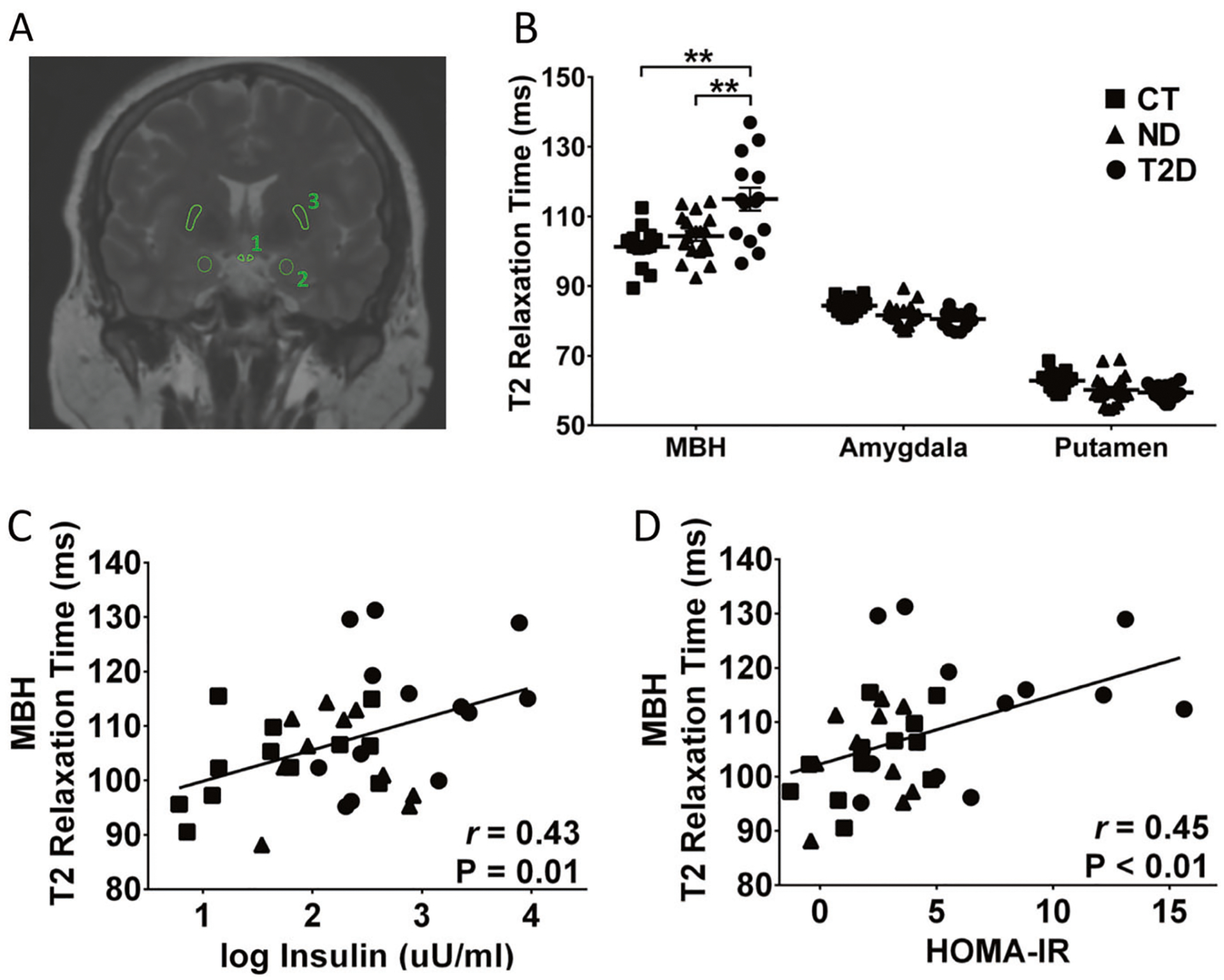
T2 relaxation time and correlation analysis. Schematic representation of the regions of interest; 1, mediobasal hypothalamus (MBH); 2, amygdala; 3, putamen (a). T2 relaxation time in lean controls (CT), obese non-diabetic (ND) and patients with type 2 diabetes (T2D) obtained for MBH, amygdala and putamen (b). Correlations between T2 relaxation time in MBH and blood insulin (c) and HOMA-IR (d). HOMA-IR, homeostatic model assessment for insulin resistance. **P < 0.01
Effect of RYGB on hypothalamic gliosis
Twenty-six ND and fourteen T2D underwent RYGB and were re-evaluated 276.4(32.2) days post treatment. A group by time interaction was present for weight, BMI, fasting glucose, fasting insulin, HbA1c, HOMA-IR (Fig. 2), and CRP (χ2(1) = 4.2, P = 0.04), but no interactions were present for triglycerides (χ2(1) = 0.2, P = 0.64), markers of liver function (AST: χ2(1) = 0.3, P = 0.60; GPT/ALT: χ2 (1) = 0.8, P = 0.37), or M-value. In both groups, weight, BMI, fasting insulin, HbA1c, and HOMA-IR were reduced by bariatric surgery, whereas fasting glucose was significantly reduced in T2D and CRP was significantly reduced in ND (z = −3.37, P = 0.001; Fig. 2). Postsurgery values of fasting insulin and HOMA-IR scores were not significantly different between groups but weight, BMI, and M-value tended to remain higher in T2D compared to ND after RYGB. Fasting glucose and HbA1c values remained significantly higher in T2D compared to ND after RYGB (Fig. 2d, e). Triglyceride values were equally reduced by surgery in both groups (main effect of time χ2(1) = 19.9, P < 0.0001), in contrast to markers of liver function which were unchanged by surgery (no main effect of time or group for GOT or GPT, data not shown). Using a three-way (group by region by time) mixed effects model adjusted for age, we examined the effect of bariatric surgery on hypothalamic gliosis. Significant interactions for group by region (χ2(2) = 25.4, P < 0.0001) and region by time (χ2(2) = 15.1, P = 0.0005) were present. The latter indicates that changes in T2 relaxation time by RYGB significantly differed by region, and the former that group differences were also limited to a distinct region. There were no significant changes in T2 relaxation time by RYGB in either group in reference regions nor were there group differences (Fig. 2a; amygdala and putamen). Within the MBH, T2 relaxation time was significantly reduced by RYGB among T2D; and among ND there was a trend toward reduced MBH T2 relaxation time (Fig. 2a). Post-RYGB, MBH T2 relaxation times in T2D patients remained significantly higher than the post-RYGB ND group.
Fig. 2.
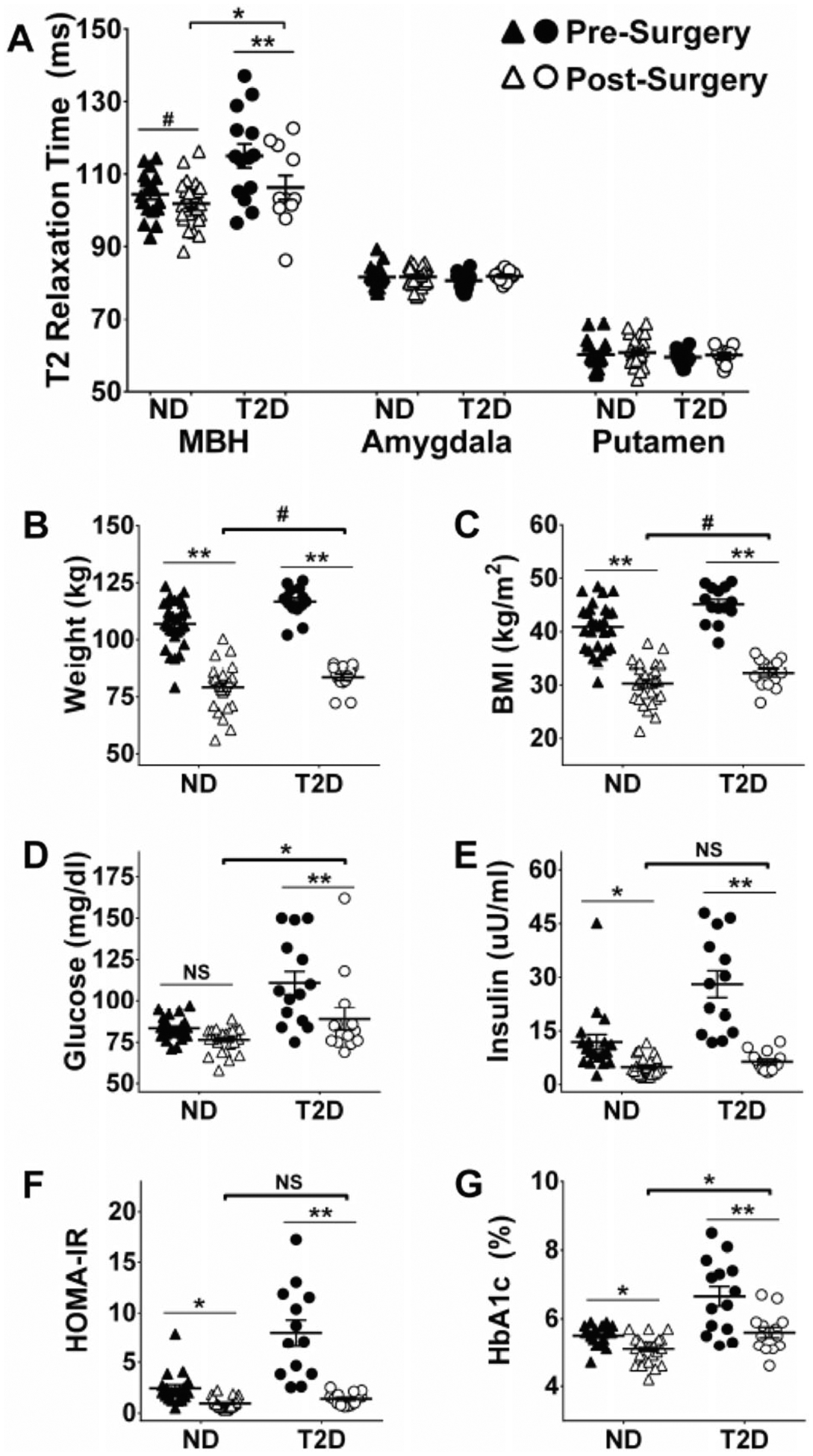
Effect of body mass reduction following Roux-in-Y gastric bypass on T2 relaxation time, body mass and markers of insulin sensitivity. T2 relaxation time before and after surgery in obese non-diabetic (ND) and patients with type 2 diabetes (T2D) obtained for mediobasal hypothalamus (MBH), amygdala and putamen (a). Body mass (b), body mass index (BMI) (c), blood glucose (d), blood insulin (e), homeostatic model assessment for insulin resistance (HOMA-IR) (f) and blood hemoglobin A1c (g) before and after surgery in obese nondiabetic (ND) and patients with type 2 diabetes (T2D). *P < 0.05 pre-surgery versus post-surgery within group (line) or ND versus T2D post-surgery (bracket); **P < 0.0001 pre-surgery versus post-surgery within group (line); #P < 0.01 pre-surgery versus post-surgery within group (line) or ND versus T2D post-surgery (bracket)
Association of change in T2 relaxation time with clinical outcomes
For participants who underwent RYGB and completed pre- and post-surgery MRI scans (n = 17 NS, n = 11 T2D), the change in MBH T2 relaxation time and clinical outcomes were calculated. In age-adjusted models, the degree of reduction in MBH T2 relaxation time by RYGB was unrelated to reductions in weight (β = 0.01, P = 0.91), BMI (β = −0.01, P = 0.85), glucose (β = 0.22, P = 0.64), insulin (β = 0.27, P = 0.19), HbA1c (β = 0.002, P = 0.88) or HOMA-IR (β = 0.10, P = 0.11), or improvements in insulin sensitivity (by M-value (β = −0.23, P = 0.33)). Adjusting models for changes in T2 relaxation times in control regions did not alter results (data not shown).
Evaluation of gray and white matter density using voxel-based morphometry (VBM) [19]. Comparing pre- and post-surgery MRIs for the T2D group, we found a reduction of gray matter density in the temporal pole and increases in cerebellar areas. In the opposite direction, we found reduced white matter density in adjacent cerebellar areas. For the ND group, we found increased gray matter density in the right precuneus and bilateral cerebellum. For the white matter we found reduced density in the right precuneus, inferior temporal gyrus, and right cerebellum and increased white matter density on the left cerebellum. All findings considered significant respected a voxel level statistical threshold of p < 0.001 with subsequent data-driven FDR-cluster-level correction with p < 0.05. There were no changes in gray or white matter densities in the hypothalamic area (Supplementary Data).
We also performed VBM analyses comparing each group of patients versus controls, and again, without changes in the hypothalamic area. We found increased white matter density in the preoperative MRI of ND group compared to controls as shown in the Supplemental Data, but we found no differences in the T2D group versus controls (Supplementary Data).
We also performed a separate VBM ROI analysis including only the hypothalamic area. There were no differences for gray matter density in the longitudinal analysis for both groups of patients. However, in the transversal baseline ROI analyses, when compared to CT and T2D groups, the ND group had areas of decrease gray matter in the dorsal-superior hypothalamic areas and increased gray matter in the ventral hypothalamus. We found no differences between CT and the T2D groups in the hypothalamic ROI transversal analyses (the statistical threshold applied was voxel level p < 0.001, uncorrected, given the lower number of voxels [730] tested and to the smoothing factor applied) See Supplementary Data.
Discussion
In the present study, we asked if hypothalamic gliosis, as determined by quantitative T2 MRI, differed between obese participants with and without T2D, and if hypothalamic gliosis could be modified by body mass reduction via RYGB. At baseline, obese participants with T2D demonstrated significantly greater T2 relaxation times (a radiologic sign of gliosis) as compared to ND and nonobese participants. Control brain regions outside the hypothalamus were unaffected. After RYGB there was a significant reduction in the MBH T2 relaxation times in T2D, but the values still remained longer than both ND and CT groups. Our study provides the second report of the effect of massive body mass reduction on the hypothalamic T2 relaxation time in ND and the first report in T2D.
One of the major challenges in studying the structure and function of the hypothalamus in human obesity is the impossibility of obtaining tissue specimens from living subjects for microscopic, cellular, and molecular tests. Therefore, most of the current concepts that support the existence of a hypothalamic abnormal function in obesity were obtained in experimental studies, particularly in rodents. However, this obstacle has been partially overcome by the rapid advance in the development of methods employed in neuroimaging [20–22]. A wide variety of neuroimaging approaches have been utilized to query hypothalamic function and structure in obesity (Supplemental Table 2). In all studies, the presence of obesity was associated with the modification of at least one parameter evaluated in the hypothalamus, the most common being the attenuation of the fMRI signal in obese subjects. Most of the studies used fMRI to determine oscillations in blood oxygen level-dependent signals, a parameter expected to reflect regional neuronal activity [23]. Using fMRI, obese subjects present attenuated hypothalamic signals in response to glucose [8, 10], cold [11], overfeeding [24], and food image cues [25]. Available data also suggest that such obesity-associated abnormalities in hypothalamic responses can be ameliorated following body mass reduction obtained as a result of a RYGB [10, 26].
In addition to the fMRI studies that aim at determining regional brain function, recent studies have employed quantitative structural MRI approaches to indirectly determine the magnitude of regional gliosis, which could reflect structural damage [27, 28]. Using this strategy, several studies support the presence of gliosis in the MBH region of the hypothalamus by demonstrating that hypothalamic signal intensity positively correlated with BMI [6] and with the magnitude of IR [16]. In addition, it was shown that abnormal hypothalamic diffusivity was associated with greater systemic inflammation [29]. To date, only one MRI study performed a longitudinal analysis [12]; Kreutzer et al. initially evaluated 57 obese/overweight subjects and found significant increase in the left, but not the right hypothalamus T2-weighted MRI signal [12]. A subset of ten obese subjects were reimaged after body mass reduction following a RYGB, showing no significant change in hypothalamic T2-weighted MRI signal [12]. It is important to note that there was a reduction in T2-weighted MRI signal that was on the order of magnitude of the difference between obese and normal weight groups at baseline. The difference in number of subjects at baseline and after RYGB could explain the negative finding.
Studies in which patients with T2D were evaluated are more limited [9, 30]. Vidarsdottir et al. used fMRI to determine the hypothalamic responsiveness to glucose in ten lean controls and seven patients with T2D, and found attenuation of the glucose-induced hypothalamic signal in T2D. Lips et al. determined the hypothalamic connectivity with other brain regions in response to a liquid meal; as a whole, obese subjects, including patients with and without T2D, presented an abrogation of the change in connectivity between the hypothalamus and frontal regions; however, there were no differences in connectivity when all three groups were evaluated simultaneously.
Our sample differs from that of Kreutzer et al. [12] in that we detected no baseline abnormalities in the hypothalamus of obese ND participants pre-RYGB as compared to nonobese controls. As an attempt to identify the reasons that could explain the reported differences between these two study results, we compared the metabolic parameters of the patients of the two studies; our ND patients were leaner (BMI 40 versus 43 kg/m2), had lower glucose (83 versus 101 mg/dL) and insulin (8.7 versus 24 uU/mL) levels and were more sensitive to insulin (HOMA-IR 2.1 versus 5.8). Thus, we suspect that the metabolically healthier status of our patients may have attenuated the impact of obesity on MBH gliosis. This interpretation is supported by the robust elevations in hypothalamic T2 relaxation time we observed in T2D patients that were partially corrected after metabolically impactful surgery. Moreover, blood insulin concentrations and HOMA-IR, an index of IR, directly correlated with T2 relaxation time in the hypothalamus at baseline, further supporting this hypothesis and adding to existing findings [16] relating IR to more extensive evidence of MBH gliosis.
Regarding the baseline hypothalamic T2 relaxation time in T2D, despite the fact that ours is the first study to report this fact, we can compare it with the findings of two other studies that evaluated the hypothalamus of T2D patients using fMRI [9, 30]. In both cases, there were changes in the responsiveness to the stimuli (glucose and liquid meal) as compared to lean subjects. Thus, it seems that the hypothalamus of obese subjects with T2D is affected both functionally and structurally, providing additional indirect evidence (now in humans) to support the hypothesis that the hypothalamus belongs to an octet that play a role in the pathogenesis of T2D as proposed by Defronzo [31]. In addition, it is important to state that changes in T2 signal could be due to regional brain atrophy; however, in the present study, we do not believe this could be the case because: (1) in VBM analysis we found no changes in hypothalamic volume; (2) T2 relaxation times were generally higher in T2D; (3) we have observed a significant decrease in T2 relaxation time after surgery in those patients (Fig. 2a) and the timeframe of 9 months is short for chronic atrophic changes, particularly as the individuals were becoming metabolically healthier as opposed to progressing.
In conclusion, this is the first report showing the partial reversibility of hypothalamic gliosis, as determined by MRI, in T2D patients submitted to RYGB. Despite the apparent evidence for a role of whole body metabolic status, particularly insulin sensitivity, in the magnitude of the hypothalamic abnormality, we cannot propose causality to explain this finding. Further studies with larger cohorts and different approaches to correct metabolic status and IR may provide additional evidence to support this hypothesis.
Materials and methods
Subjects
Forty-two female obese subjects, 14 with type 2-diabetes (T2D) and 28 without diabetes (ND) were recruited from the Obesity Clinic at the University of Campinas Clinics Hospital. All patients were selected for bariatric surgery according to the National Institutes of Health Consensus Statement [32]. The surgical technique used was always the RYGB, performed as previously described [33]. In addition, 13 lean healthy control subjects (CT) were selected among students of the University. Female subjects aged 18–60 were considered eligible for the study. Diabetes mellitus was diagnosed according to the American Diabetes Association criteria [34]. Patients were not taking insulin and had an HbA1c ≤ 8.0%. Exclusion criteria were pregnancy or breastfeeding, inflammatory or infectious diseases, neurologic or psychiatric illnesses, alcohol consumption of more than 30/15 g per day for men and women, respectively, smoking, use of psychotropic or anti-inflammatory drugs and MRI contraindications including weight >120 kg. The study was approved by the University of Campinas Ethics Committee, and written informed consent was obtained from all patients (293/2011).
Procedures and final sample size
Once included, subjects underwent anthropometric evaluation, fasting blood collection, and MRI; a subset underwent hyperinsulinemic clamp. Obese patients were evaluated before and ~9 months after surgery and lean subjects were evaluated once. Fifty-five subjects completed at least one study visit, including n = 13 CT, n = 28 ND, and n = 14 T2D. CT (n = 13) were evaluated once and all underwent an MRI, however n = 3 did not undergo clamp procedures. Prior to RYGB, n = 19 ND and n = 14 T2D underwent MRI procedures and were included in baseline analyses. After RYGB, n = 26 ND underwent an MRI (n = 9 subjects did not undergo or had unusable baseline MRI data, n = 17 returned for the postsurgery visit), and n = 16 underwent clamp procedures; n = 14 T2D returned for follow-up, and n = 11 had usable MRI data. Analyses considering the effects of treatment included subjects with obesity (ND and T2D) who had usable MRI data at either time point (baseline n = 19 ND, n = 14 T2D; postsurgery n = 26 ND, n = 11 T2D; statistical models were applied account for missing data). Only subjects with obesity who underwent RYGB and had MRI data at both time points were included in analyses related to the change in T2 relaxation time by surgery (n = 17 ND, n = 11 T2D). Some participants had missing data for certain plasma measures, specific Ns are reported in tables and graphs.
Blood biochemistry, hormonal measurements, and hyperinsulinemic-euglycemic clamp
Blood biochemistry measurements were performed at the Clinical Laboratory of University of Campinas Clinics Hospital using automated methods. Plasma glucose levels were measured in fasting state using a glucose analyzer (YSI 2700; YSI Life Sciences, Yellow Spring, OH, USA), reference values 60–99 mg/dL. Plasma insulin and leptin were determined by ELISA (Phoenix Pharmaceuticals, Burlingame, CA, USA). Homeostatic model assessment (HOMA-IR and HOMA-ß) were calculated as previously described [14]. Hyperinsulinemic-euglycemic clamp has performed as previously described [15]. In short, insulin was continuously infused (40 mU m−2 min−1) into a peripheral vein. Blood glucose was held constant (variation <5%) by a glucose infusion according to the results of blood glucose determination every 5 min. Insulin sensitivity was calculated as the glucose infusion rate in the last 60 min, corrected for the glucose distribution space and adjusted to fat-free mass, resulting in the M-value.
Image acquisition
Scans were acquired on a 3-Tesla Philips Achieva MR scanner (version 3.2, Philips Medical Systems, Best, The Netherlands) using a 32-channel head radiofrequency coil. Sequences included a T1-weighted scan and a quantitative multi-slice/multi-echo T2-weighted sequence with 16 echoes (interecho spacing of 10 ms; TR/TE/NSA: 2000/20–170/1). Slices were acquired from the optic chiasm through the mammillary bodies (9–12 slices/subject, slice thickness 2.5 mm, and interslice gap 0.0–0.2 mm). In-plane pixel resolution was 0.70–0.80 × 0.75–0.86 mm resulting in an acquired voxel size of 1.313–1.720 mm3. T2 relaxation time was calculated from the signal decay curve of the 16 echoes on a pixel-by-pixel basis and displayed as a quantitative T2 relaxation time parametric map to evaluate regional tissue T2 values.
MRI analysis
The coronal slice immediately posterior to the optic chiasm encompassing the rostral arcuate nucleus was identified for each subject. The ROI location was selected and the ROI shape was designed according to published references for arcuate nucleus size, shape, and position [28, 35]. Within the same slice, reference regions of interest (ROIs) in the putamen and amygdala were identified. ROIs were placed on high-resolution coronal images and then were transferred to the T2 parametric map. Mean ± SD T2 relaxation time per ROI was recorded (OsiriX Imaging Software, version 8.0.2). In addition, it was performed longitudinal VBM analyses (SPM12/CAT12—longitudinal pairwise affine regularization followed by nonlinear spatial registration to DARTEL MNI152 template, tissues segmentations, and modulation) based on anatomical images (3D T1-weighted 1 × 1 × 1mm), as previously described (Statistical Parametric Mapping. 2007, Friston et al. Editors, Academic Press. https://doi.org/10.1016/B9788-0-12-372560-8.X5000-1).
Statistical analysis
Data are reported as mean ± SEM, unless otherwise noted. χ2 tests of proportions were used for categorical variables. Linear mixed models capable of accounting for repeated measures and missing data were used to evaluate regional, laterality and group differences in mean T2 relaxation time. Group differences were tested by using multiple linear regression to adjust for covariates. Least square means were calculated for adjusted models. Simple and multiple linear regression models were applied to evaluate associations and Pearson’s correlation coefficients were calculated for descriptive purposes. Analyses were performed with STATA v.13.1 (College Station, TX, USA).
Supplementary Material
Acknowledgements
We thank ER, GF, and LS for technical assistance. This work was supported by funding provided by the Fundação de Amparo a Pesquisa do Estado de São Paulo, Conselho Nacional de Desenvolvimento Cientifico e Tecnologico, grants from the Trust in Science Initiative from Glaxo-Smithkline, UK, and the National Institutes of Health (DK089036, DK098466; EAS). The Laboratories of Cell Signaling and Experimental Endocrinology belong to the Obesity and Comorbidities Research Center and the National Institute of Science and Technology—Diabetes and Obesity.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41366-019-0399-8) contains supplementary material, which is available to authorized users.
References
- 1.Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nat Rev Neurosci. 2014;15:367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velloso LA, Schwartz MW. Altered hypothalamic function in diet-induced obesity. Int J Obes. 2011;35:1455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van de Sande-Lee S, Velloso LA. Hypothalamic dysfunction in obesity. Arq Bras Endocrinol Metabol. 2012;56:341–50. [DOI] [PubMed] [Google Scholar]
- 4.Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE. 2009;4:e5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morari J, Anhe GF, Nascimento LF, de Moura RF, Razolli D, Solon C, et al. Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes. 2014;63:3770–84. [DOI] [PubMed] [Google Scholar]
- 6.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9:2124–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuda M, Liu Y, Mahankali S, Pu Y, Mahankali A, Wang J, et al. Altered hypothalamic function in response to glucose ingestion in obese humans. Diabetes. 1999;48:1801–6. [DOI] [PubMed] [Google Scholar]
- 9.Vidarsdottir S, Smeets PA, Eichelsheim DL, van Osch MJ, Viergever MA, Romijn JA, et al. Glucose ingestion fails to inhibit hypothalamic neuronal activity in patients with type 2 diabetes. Diabetes. 2007;56:2547–50. [DOI] [PubMed] [Google Scholar]
- 10.van de Sande-Lee S, Pereira FR, Cintra DE, Fernandes PT, Cardoso AR, Garlipp CR, et al. Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes. 2011;60:1699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rachid B, van de Sande-Lee S, Rodovalho S, Folli F, Beltramini GC, Morari J, et al. Distinct regulation of hypothalamic and brown/beige adipose tissue activities in human obesity. Int J Obes. 2015;39:1515–22. [DOI] [PubMed] [Google Scholar]
- 12.Kreutzer C, Peters S, Schulte DM, Fangmann D, Turk K, Wolff S, et al. Hypothalamic inflammation in human obesity is mediated by environmental and genetic factors. Diabetes. 2017;66:2407–15. [DOI] [PubMed] [Google Scholar]
- 13.Berkseth KE, Guyenet SJ, Melhorn SJ, Lee D, Thaler JP, Schur EA, et al. Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: a combined immunohistochemical and magnetic resonance imaging study. Endocrinology. 2014;155:2858–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samocha-Bonet D, Dixit VD, Kahn CR, Leibel RL, Lin X, Nieuwdorp M, et al. Metabolically healthy and unhealthy obese— the 2013 Stock Conference report. Obes Rev. 2014;15:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Souza GF, Solon C, Nascimento LF, De-Lima-Junior JC, Nogueira G, Moura R, et al. Defective regulation of POMC precedes hypothalamic inflammation in diet-induced obesity. Sci Rep. 2016;6:29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schur EA, Melhorn SJ, Oh SK, Lacy JM, Berkseth KE, Guyenet SJ, et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity. 2015;23:2142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee D, Thaler JP, Berkseth KE, Melhorn SJ, Schwartz MW, Schur EA. Longer T(2) relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am J Physiol Endocrinol Metab. 2013;304:E1245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasan KM, Walimuni IS, Abid H, Frye RE, Ewing-Cobbs L, Wolinsky JS, et al. Multimodal quantitative magnetic resonance imaging of thalamic development and aging across the human lifespan: implications to neurodegeneration in multiple sclerosis. J Neurosci. 2011;31:16826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuulari JJ, Karlsson HK, Antikainen O, Hirvonen J, Pham T, Salminen P, et al. Bariatric surgery induces white and grey matter density recovery in the morbidly obese: a voxel-based morphometric study. Hum Brain Mapp. 2016;37:3745–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polimeni JR, Wald LL. Magnetic resonance Imaging technology-bridging the gap between noninvasive human imaging and optical microscopy. Curr Opin Neurobiol. 2018;50:250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akassoglou K, Merlini M, Rafalski VA, Real R, Liang L, Jin Y, et al. In vivo imaging of CNS injury and disease. J Neurosci. 2017;37:10808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patriarca L, Magerowski G, Alonso-Alonso M. Functional neuroimaging in obesity. Curr Opin Endocrinol Diabetes Obes. 2017;24:260–5. [DOI] [PubMed] [Google Scholar]
- 23.Ogawa S, Lee TM, Nayak AS, Glynn P. Oxygenation-sensitive contrast in magnetic resonance image of rodent brain at high magnetic fields. Magn Reson Med. 1990;14:68–78. [DOI] [PubMed] [Google Scholar]
- 24.Cornier MA, Salzberg AK, Endly DC, Bessesen DH, Rojas DC, Tregellas JR. The effects of overfeeding on the neuronal response to visual food cues in thin and reduced-obese individuals. PLoS ONE. 2009;4:e6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher PC, Napolitano A, Skeggs A, Miller SR, Delafont B, Cambridge VC, et al. Distinct modulatory effects of satiety and sibutramine on brain responses to food images in humans: a double dissociation across hypothalamus, amygdala, and ventral striatum. J Neurosci. 2010;30:14346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoon HFA, de Bruijn SEM, Jager G, Smeets PAM, de Graaf C, Janssen IMC, et al. Altered neural inhibition responses to food cues after Roux-en-Y Gastric bypass. Biol Psychol. 2018;137:34–41. [DOI] [PubMed] [Google Scholar]
- 27.Dallaire-Theroux C, Callahan BL, Potvin O, Saikali S, Duchesne S. Radiological-pathological correlation in Alzheimer’s disease: systematic review of antemortem magnetic resonance imaging findings. J Alzheimers Dis. 2017;57:575–601. [DOI] [PubMed] [Google Scholar]
- 28.Berkseth KE, Rubinow KB, Melhorn SJ, Webb MF, Rosalynn BDLM, Marck BT, et al. Hypothalamic gliosis by MRI and visceral fat mass negatively correlate with plasma testosterone concentrations in healthy Men. Obesity. 2018;26:1898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puig J, Blasco G, Daunis IEJ, Molina X, Xifra G, Ricart W, et al. Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. J Clin Endocrinol Metab. 2015;100:E276–81. [DOI] [PubMed] [Google Scholar]
- 30.Lips MA, Wijngaarden MA, van der Grond J, van Buchem MA, de Groot GH, Rombouts SA, et al. Resting-state functional connectivity of brain regions involved in cognitive control, motivation, and reward is enhanced in obese females. Am J Clin Nutr. 2014;100:524–31. [DOI] [PubMed] [Google Scholar]
- 31.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gastrointestinal surgery for severe obesity: National Institutes of Health Consensus Development Conference Statement. Am J Clin Nutr. 1992;55(Suppl 2):615S–9S. 10.1093/ajcn/55.2.615s. (Abstract). [DOI] [PubMed] [Google Scholar]
- 33.de Carvalho CP, Marin DM, de Souza AL, Pareja JC, Chaim EA, de Barros Mazon S, et al. GLP-1 and adiponectin: effect of weight loss after dietary restriction and gastric bypass in morbidly obese patients with normal and abnormal glucose metabolism. Obes Surg. 2009;19:313–20. [DOI] [PubMed] [Google Scholar]
- 34.Diabetes A 2. Classification and diagnosis of diabetes. Diabetes Care. 2017;40(Suppl 1):S11–24. American [DOI] [PubMed] [Google Scholar]
- 35.Abel TW, Rance NE. Stereologic study of the hypothalamic infundibular nucleus in young and older women. J Comp Neurol. 2000;424:679–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.