

Abstract
Cerebral vasospasm (VSP) is a common phenomenon after aneurysmal subarachnoid hemorrhage (aSAH) and contributes to neurocognitive decline. The natural history of the pro-inflammatory immune response after aSAH has not been prospectively studied in human cerebrospinal fluid (CSF). In this pilot study, we aimed to identify specific immune mediators of VSP after aSAH. Peripheral blood (PB) and CSF samples from patients with aSAH were prospectively collected at different time-points after hemorrhage: days 0–1 (acute); days 2–4 (pre-VSP); days 5–9 (VSP) and days 10 + (post-VSP peak). Presence and severity of VSP was assessed with computed tomography angiography/perfusion imaging and clinical examination. Cytokine and immune mediators’ levels were quantified using ELISA. Innate and adaptive immune cells were characterized by flow cytometry, and cell counts at different time-points were compared with ANOVA. Confocal immunostaining was used to determine the presence of specific immune cell populations detected in flow cytometry. Thirteen patients/aneurysms were included. Five (38.5%) patients developed VSP after a mean of 6.8 days from hemorrhage. Flow cytometry demonstrated decreased numbers of CD45+ cells during the acute phase in PB of aSAH patients compared with healthy controls. In CSF of VSP patients, NK cells (CD3-CD161 +) were increased during the acute phase and progressively declined, whereas CD8+CD161+ lymphocytes significantly increased at days 5–9. Microglia cells (CD45dimCD11b +) increased over time after SAH. This increase was particularly significant in patients with VSP. Levels of VEGF and MMP-9 were consistently higher in VSP patients, with the highest difference occurring at the acute phase. Confocal immunostaining demonstrated the presence of CD8+CD161+ lymphocytes in the arterial wall of two unruptured intracranial aneurysms. In this preliminary study, human CSF showed active presence of innate and adaptive immune cells after aSAH. CD8+CD161+ lymphocytes may have an important role in the inflammatory response after aneurysmal rupture and were identified in the aneurysmal wall of unruptured brain aneurysms. Microglia activation occurs 6 + days after aSAH.
Subject terms: Adaptive immunity, Chemokines, Cytokines, Inflammation, Innate immune cells, Innate immunity, Immunology, Neurology, Neurological disorders
Introduction
Brain injury after aneurysmal subarachnoid hemorrhage (aSAH) is a multimodal process that includes early brain injury and delayed cerebral ischemia (DCI). The mechanisms that lead to DCI are not fully understood1. The pathogenesis of DCI is hypothesized to include cerebral vasospasm (VSP), cortical spreading ischemia, microthrombosis and constriction of the microcirculation2.
About two-thirds of patients with aSAH develop VSP 3–14 days after initial rupture3. Heterogeneous evidence has suggested several mechanisms as potential contributors of VSP including thickness, density, location and persistence of subarachnoid blood4,5. Red blood cells in the subarachnoid space are hemolyzed, releasing hemoglobin, bilirubin and oxygen free radicals. These toxic molecules induce increased expression of endothelin-1 and reduced levels of nitrous oxide, leading to endothelial cell dysfunction and VSP6–8. Although angiographic VSP eventually resolves, arterial fibrosis, endothelial thickening and reduced arterial compliance may persist over time and contribute to the poor clinical outcomes observed in patients with DCI9.
The inflammatory response following aSAH leads to increased permeability of the blood–brain barrier (BBB) and increased expression of cellular adhesion molecules. These molecules allow infiltration of peripheral leukocytes that phagocytose red blood cells and debris in the cerebrospinal fluid (CSF)10–12. Activated leukocytes secrete several cytokines that promote activation of central nervous system-resident macrophages (microglia) and trigger secondary injury of the brain parenchyma13.
Cells of the innate immune system such as neutrophils, monocytes and macrophages, have been found in high numbers and in highly activated states in post-hemorrhagic CSF14. Increased CSF concentrations of neutrophils, myeloperoxidase and NADPH oxidase have been documented in patients with VSP15. On the other hand, little is known about the role of the adaptive immunity in aSAH. Histological studies from clipped aneurysms’ specimens and autopsies have found lymphocyte infiltration in the aneurysmal wall16,17. However, T and B cells have only rarely been found in the CSF of aSAH patients18,19. In this study, we aimed to characterize the evolution of the innate and adaptive immune responses after aSAH, and determine if there is a correlation between immune cell activation and the development of VSP.
Methods
Patients and data collection
After approval by the University of Iowa Institutional Review Board, CSF and peripheral blood (PB) samples of patients with aSAH were prospectively collected. All experiments were performed in accordance with relevant guidelines and regulations. Inclusion criteria were as follows: (1) clinical presentation within 24 h after aSAH, (2) evidence of aneurysmal etiology of the hemorrhage documented in computed tomography angiography (CTA), magnetic resonance angiography or digital subtraction angiography, and (3) placement of an external ventricular drain (EVD) for acute management of post-hemorrhagic hydrocephalus. Subjects with history of congenital or acquired immunosuppression, central nervous system infection, presumed bacteremia, endocarditis, autoimmune diseases, systemic inflammatory diseases or current treatment with immunosuppressants or immunomodulators were excluded.
CSF and PB sampling
Informed consent was obtained before sample collection. CSF samples were collected at four different time-points from the index hemorrhage (Supplemental Fig. 1): days 0–1 (acute phase); days 2–4 (pre-VSP); days 5–9 (VSP) and days 10 + (post-VSP peak). Five mL of CSF were slowly drawn from the EVD under continuous intracranial pressure monitoring and following standard aseptic techniques. The first 1.5 mL of CSF were discarded (dead space). A PB sample was collected simultaneously from the same patient at the first time-point, and PB samples from 6 control subjects without intracranial aneurysms were collected for comparison. The CSF was sampled until the EVD was discontinued (days 15–20 post-SAH).
Leukocyte subsets and gating strategy
Automated CSF leukocyte counts were performed on ABX micros 60 (Horiba, Montpellier, France). Cells were washed twice with phosphate-buffered saline and immune-stained for 30 min at 4 °C with the following panel of antibodies: anti-CD45-BV786, anti-CD8-BV510, antiCD66b-BV421, anti-CD45-PerCp5.5 (BD Bioscience, San Jose, CA, USA); anti-CD3-FITC, anti-CD19-PECy7, anti-CD11b-PECy5, anti-CD161-PE, anti-CD14-Alexa700, anti-CD11c-APC (Tonbo Biosciences, San Diego, CA, USA). Optimal antibody concentrations were previously defined by titration20. The cells were washed and suspended in 1% paraformaldehyde. Flow cytometry data was collected on a BD FACS LSR flow cytometer equipped with four lasers. Analyses of leukocyte subsets on CSF and PB samples were conducted by 13 flow cytometry within 30 min from collection. BD FACS Diva software was used for data acquisition and Flow Jo v10 (Becton–Dickinson Bioscience, San Jose, CA, USA) was used for data analysis. The gating strategy is depicted in Supplemental Fig. 2.
Enzyme linked immunoassay
CSF levels of several mediators and cytokines were quantified using Enzyme Linked Immunoassay (ELISA) arrays. This was done to better characterize the interactions and functional profile of immune cells after aSAH. Although several cytokines have been associated with occurrence of VSP, we analyzed the most often cited immune mediators as follows: vascular endothelial growth factor (VEGF), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α) (Tonbo Biosciences, San Diego, CA, USA), and matrix metalloproteinase-9 (MMP-9) (R&D Systems Minneapolis, MS, USA).
Immunofluorescence imaging
Samples of 2 previously clipped unruptured intracranial aneurysms were processed. Aneurysm tissue was fixed in 10% Neutral-Buffered Formalin within one hour of surgical excision. Formalin-fixed, paraffin-embedded tissue specimens were prepared, stored at room temperature and sectioned in slices of 4–8 μm thickness. Slides were deparaffinized and subjected to citrate buffer antigen retrieval. Then, tissue sections were stained using primary and secondary antibodies anti-human CDs to determine the presence of immune cells identified in flow cytometry. Images were acquired using a Zeiss LSM 710 confocal microscope with Zen 2010 software (Zeiss Group, Oberkochen, Germany).
Clinical outcome
VSP was defined as the development of a new focal neurological deficit that correlated with angiographic evidence of arterial narrowing on CTA/computed tomography perfusion imaging. Confounders of neurological deterioration such as hydrocephalus, fever, seizures and infection were ruled out. Based on previous literature and current guidelines21,22, degree of VSP was classified in three different categories using the following clinical and radiographic criteria: (1) mild = ≤ 33% reduction in arterial diameter and self-limited neurological deficits; (2) moderate = 34% to 66% reduction in arterial diameter and clinical improvement with triple-H therapy; and (3) severe = ≥ 67% reduction in arterial diameter requiring endovascular intra-arterial therapy. As standard of care, all patients were monitored in the neuro-intensive care unit and received nimodipine 60 mg every 4 h.
Statistical analysis
Measures of central tendency and dispersion were calculated using descriptive statistics. Brown-Forsythe test was performed to assess differences in standard deviations and confirm normality of our data distribution. One-way analysis of variance (ANOVA) tests (with Tukey’s multiple comparisons) were run to find any significant differences in the mean numbers of immune cell subpopulations at each time-point collection. Immune cell counts in patients who suffered symptomatic VSP were compared to those whom did not. Cytokine concentrations from ELISA essays were analyzed in a similar fashion. Due to small sample size, we did not perform comprehensive uni- or multi-variable logistic regression analyses correlating immune cell changes with severity of VSP. P-values < 0.05 were considered statistically significant. All statistical analyses were performed with GraphPad Prism v8 (San Diego, CA, USA).
Results
Patient and aneurysm characteristics
Twenty patients were screened. Six patients with non-aneurysmal SAH and one patient with human immunodeficiency virus were excluded. Thirteen patients/aneurysms were included over a period of seven months (Table 1). Five (38.5%) patients developed VSP: 2 cases mild and 3 cases moderate; no severe VSP cases were encountered. Mean time from aSAH to VSP was 163.2 ± 72.8 h (6.8 days). One patient died secondary to sudden cardiac arrhythmia.
Table 1.
Patients’ characteristics.
| N | Age/sex | Size | Location | mFS | WFNS | Treatment | VSP | Day of VSP |
|---|---|---|---|---|---|---|---|---|
| 1 | 65/M | 3 | Pericallosal | 1 | 1 | Coiling | No | – |
| 2 | 66/F | 6 | BA tip | 4 | 5 | Coiling + WEB | No | – |
| 3† | 88/F | 5.4 | PCOM* | 4 | 5 | Coiling | No | – |
| 4 | 67/F | 5 | ACOM* | 4 | 5 | Coiling | Moderate | 4 |
| 5 | 33/F | 7.4 | PCOM* | 1 | 1 | Coiling | No | – |
| 6 | 58/F | 6.5 | BA tip* | 4 | 2 | SAC | No | – |
| 7 | 50/M | 3 | ACOM | 1 | 1 | Coiling | No | – |
| 8 | 50/M | 5 | MCA* | 4 | 4 | Coiling | Moderate | 9 |
| 9 | 53/F | 3.2 | ACOM | 4 | 4 | Coiling | Mild | 9 |
| 10 | 45/F | 5 | ACOM | 3 | 2 | Coiling | No | – |
| 11 | 39/M | 3.8 | PCOM | 1 | 1 | Coiling + FD | Moderate | 9 |
| 12 | 53/F | 3.5 | BA tip* | 4 | 4 | BAC | No‡ | – |
| 13 | 65/M | 6.1 | MCA | 4 | 5 | Coiling | Mild | 3 |
ACOM anterior communicating artery; BA basilar artery; BAC balloon-assisted coiling; FD flow diversion; M male; MCA middle cerebral artery; mFS modified Fisher Scale; PCOM posterior communicating artery; SAC stent-assisted coiling; VSP cerebral vasospasm; WFNS World Federation of Neurological Surgeons grading scale.
*Aneurysm with bleb/daughter sac.
†Deceased.
‡Patient experienced clinically silent radiographic VSP.
Immune cell kinetics in aneurysmal subarachnoid hemorrhage
Brown-Forsythe test demonstrated no significant differences in standard deviations of immune cell counts at each time-point among patients with and without VSP (P > 0.05). ANOVA tests showed that during the acute phase (days 0–1), aSAH patients had significantly decreased numbers of lymphohematopoietic CD45+ cells when compared to healthy controls (P = 0.0101; Table 2, Supplemental Fig. 3A); this “acute immunosuppression” was particularly significant in patients without VSP (P = 0.0373; Supplemental Fig. 3B). CSF analysis demonstrated elevated counts of natural killer (NK) cells (CD3-CD161 +) during the acute phase, followed by a gradual reduction (P = 0.0419; Figs. 1, 2A) of cell numbers. During VSP, the number of CSF T-cells (CD3 +) was significantly higher when compared to the acute phase (P = 0.0333; Figs. 1, 2B). The number of CSF microglia (CD45dimCD11b +) cells progressively increased over time after aSAH (P = 0.0326; Figs. 1, 2C).
Table 2.
Evolution of immune cells and cytokine mediators in human CSF after aSAH.
| Immune cell/cytokine | Acute (days 0–1) | Pre-VSP (days 2–4) | VSP (days 5–9) | Post-VSP peak (days 10 +) |
|---|---|---|---|---|
| CD45 + Hematopoietic cells | ↓↓↓ | ↑ | ↑↑ | ↑↑↑ |
| CD3-CD161 + NK cells | ↑↑ | ↓ | ↓↓ | ↓↓↓ |
| CD3 + T-cells | ↑ | ↑↑ | ↑↑↑ | ↓ |
| CD8 + CD161 + Tc17 cells | ↑↑↑↑ | ↓ | ↓↓ | ↓↓↓ |
| CD45dimCD11b + Microglia | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ |
| VEGF | ↑↑ | ↓ | ↓↓ | ↑ |
| MMP-9 | ↑↑ | ↓ | ↓↓ | ↑ |
Highlighted in bold are the immune cell subpopulation with most significant count changes.
VSP cerebral vasospasm; MMP-9 matrix metalloproteinase-9; NK natural killer; VEGF vascular endothelial growth factor.
Figure 1.

Immune cell kinetics in aneurysmal subarachnoid hemorrhage. For the right column, patients with vasospasm (VSP) are depicted in continuous lines, whereas non-VSP patients are shown in dashed lines. PB peripheral blood; CSF cerebrospinal fluid.
Figure 2.
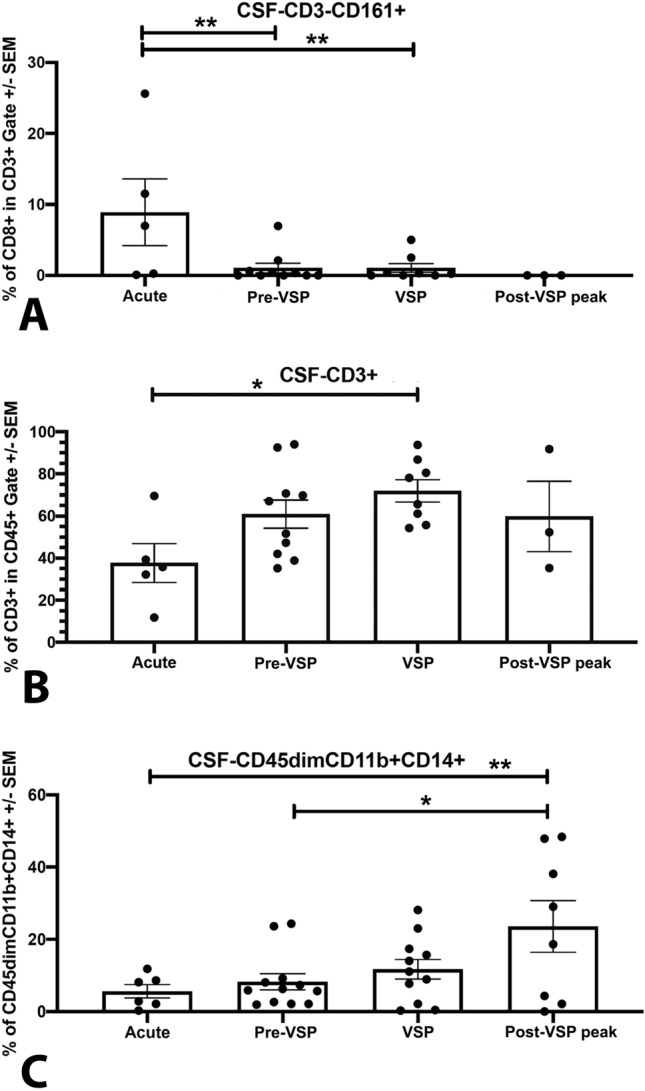
CSF cellularity for (A) CD3-CD161 + NK cells, (B) CD3 + T-cells, and (C) CD45dimCD11b + microglia.
CSF cellularity was compared among patients with and without VSP. Patients with VSP had lower numbers of CSF NK cells (CD3-CD161 +) in the acute phase compared to patients without VSP (P > 0.05; Figs. 1, 3A). The analysis of T cell subpopulations demonstrated significantly increased numbers of CD8+CD161+ cells during the acute phase in patients who developed VSP compared with those who did not (P < 0.0001; Figs. 1, 3B). In patients with VSP, the number of CSF microglial cells was low in the acute and pre-VSP phases, with a progressive increase during the post-VSP peak (P > 0.05; Figs. 1, 3C).
Figure 3.

Comparison of CSF cellularity among patients with and without VSP for (A) CD3-CD161 + NK cells, (B) CD8+ CD161+ Tc17 cells, and (C) CD45dimCD11b + microglia.
Inflammatory mediators and cytokine kinetics in aneurysmal subarachnoid hemorrhage
ELISA demonstrated higher levels of VEGF in the acute phase in aSAH patients compared with healthy controls (P > 0.05; Fig. 4 and Supplemental Fig. 4A). At all time-points, VEGF levels showed a higher trend in CSF from patients with VSP compared to those without VSP (P > 0.05; Supplemental Fig. 4B). Lower levels of MMP-9 were found in CSF from aSAH patients at pre-VSP and VSP time-points compared with the acute phase (P = 0.0248 and 0.0089, respectively; Fig. 4 and Supplemental Fig. 3C). There were no significant differences in the expression of IL-6 or TNF-α (Fig. 4 and Supplemental Fig. 5).
Figure 4.

Inflammatory mediators and cytokine kinetics in aneurysmal subarachnoid hemorrhage. For the right column, patients with vasospasm (VSP) are depicted in bold lines, whereas non-VSP patients are shown in dashed lines.
Confocal immunofluorescence imaging of unruptured intracranial aneurysms
Since the preliminary flow cytometry data demonstrated predominance of CD8+CD161+ cells in the CSF of patients with VSP, we performed immunostaining of histological samples previously collected from two unruptured aneurysms with the following primary antibodies: anti-human CD8 (mouse monoclonal, Cat # ab17147, Abcam, Cambridge, MA) and anti-human CD161 (rabbit polyclonal, Cat # ab197979, Abcam, Cambridge, MA). Tissue sections were washed twice in phosphate-buffered saline and incubated simultaneously with two secondary antibodies for immunofluorescence: goat anti-mouse Alexa Fluor 488 (Cat # 21042) and goat anti–rabbit Alexa Fluor 568 (Cat # 11011, ThermoFisher Scientific, Waltham, MA). Samples were washed three times with PBS and mounted (VECTASHIELD Antifade Mounting Medium with DAPI, Cat # H-1200, Vector Laboratories, Burlingame, CA). Confocal immunostaining indicated the presence of CD8+CD161+ cells in the arterial wall of both unruptured aneurysms (Fig. 5).
Figure 5.
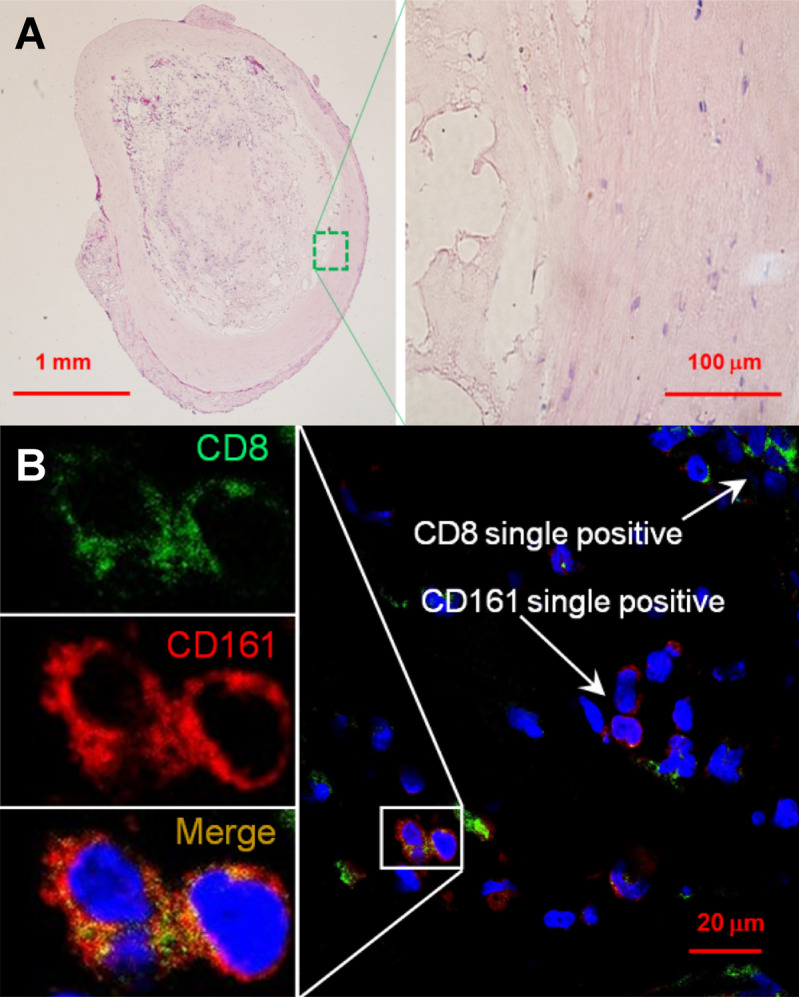
Confocal immunofluorescent analysis of unruptured intracranial aneurysms. (A) Hematoxylin and eosin staining of a specimen’s section. (B) Immunofluorescence imaging of anti-CD8 (green) and anti-CD161 (red) staining. Nuclei were stained by DAPI (blue). The area with the co-localization of CD8+ and CD161+ was zoomed-in to show CD8 and CD161 expression in separated and merged confocal images. Scales (in μm) are denoted by the bars.
Discussion
This preliminary study prospectively assessed the immunological response after aSAH in human CSF. Our results suggest that both innate and adaptive immune responses play pivotal roles after aSAH. The adaptive immune response seems to be primarily mediated by CD8+CD161+ cells, which significantly correlated with the occurrence of VSP. Confocal immunostaining suggested the presence of this subpopulation of cytotoxic T cells in the aneurysmal wall. Microglia proliferation seems to occur in a delayed fashion around 6 + days after aSAH (Fig. 6).
Figure 6.

The immune response following aSAH induces inflammation of the aneurysmal wall, apoptosis of endothelial cells and degradation of tight junctions. This increases the permeability of the BBB and allows active extravasation of immune cells into the subarachnoid space. Once in the CSF, cells of the innate immune system (mainly neutrophils and macrophages) phagocytose red blood cells/debris (dotted ellipse) and secrete multiple cytokines that stimulate CD4+ and CD8+ T-cells from the adaptive immunity. This might perpetuate the intrathecal inflammatory response by production of IL-17 from CD8+CD161+ cells. Finally, microglial cells in the brain parenchyma become activated and, instead of conferring protection from further damage, induce secondary neurotoxicity.
Acute immunosuppression after aneurysmal subarachnoid hemorrhage
The concentration of CD45+ cells in PB after aSAH was lower in aSAH patients compared with healthy controls. This finding is compatible with the acute systemic immunosuppressive response after aneurysmal rupture previously suggested by other studies. Sarrafzadeh et al. described T cell-lymphopenia and a decreased monocyte human leukocyte antigen-DR expression on PB of patients after aSAH (i.e. “aSAH-induced acute immunodepression state”)23. However, our results also suggest that the early decrease in CD45+ cells is particularly significant in patients without VSP, whereas patients with VSP show less immunosuppression during the acute phase. We hypothesize that the lack of systemic immunosuppression after aSAH may increase the risk of developing VSP due to a sustained pro-inflammatory response. This systemic effect may be magnified in the central nervous system after disruption of the BBB.
Innate immune system response after aneurysmal rupture
Innate immune cells enter the subarachnoid space after aSAH. This process occurs via increased expression of cellular adhesion molecules (E-selectin, VCAM-1, ICAM-1 and HMGB-1) and has been correlated with the occurrence of VSP24–27. Once in the subarachnoid space, activated cells secrete multiple cytokines such as IL-1β, IL-6, TNF-α, LFA-1, leukotrienes, arachidonic acid, von Willebrand factor, complement, MMP-9 and VEGF28–31. These chemo-attractants may trigger and/or worsen VSP32. We found a peak level of MMP-9 during the acute phase compared with the pre-VSP and VSP phases. MMP-9 is a key neurovascular protease that can induce BBB damage and cause edema, hemorrhage and neuronal death33. Egashira et al. found less white matter injury in MMP-9 knock-out mice compared with wild-type mice 8 days after induction of SAH34. Using quantitative reverse PCR in PB samples of 43 SAH patients and 23 healthy controls, Wang et al. detected increased levels of MMP-9 in SAH patients who developed VSP31.
Additionally, patients with VSP showed a trend to express higher levels of VEGF compared with those without VSP. VEGF is a key factor involved in revascularization, endothelial cell migration and proliferation. Several studies have suggested that increased VEGF levels reduce the integrity of the BBB by weakening tight junctions via the VEGFR-2 located on endothelial cells35. Liu et al. found that anti-VEGF antibodies decreased BBB permeability and ameliorated brain edema/injury after SAH induced in mice36,37. In conjunction with our pilot data, this evidence supports the hypothesis that active BBB injury mediated by MMP-9 and VEGF occurs early in aSAH, and may contribute to VSP and DCI by promoting neuroinflammation.
Adaptive immune system response after aneurysmal rupture
The role of the adaptive immunity after aSAH has not been thoroughly characterized. In 1993, Mathiesen et al. reported moderately increased levels of IL-2 receptor in patients after aSAH. No systemic changes in IL2 were documented, suggesting a selective intrathecal synthesis38. IL-2 receptor is expressed on the surface of lymphocytes, and its intracellular signal transduction has a crucial role in the development of memory T cells and terminal differentiation of effector T cells39. Although this study was the first to suggest activation of adaptive immunity in aSAH, a limitation was the inclusion of subjects with traumatic SAH and non-aneurysmal spontaneous SAH (n = 4/12). In 1996, the same group analyzed lymphocyte subpopulations from 10 CSF samples of aSAH patients and found an increase of CD3+ cells in 2 patients, CD4+ cells in 1 patient, CD8+ cells in 3 patients, and CD19+ cells in 3 patients18. The development of VSP was not assessed.
Recently, Moraes et al. analyzed CSF and PB samples from 12 aSAH patients and PB samples from 28 healthy controls. In aSAH patients, CD4+ and CD8+ T cells had decreased expression of CD3/CD28 and higher levels of CD69 in CSF compared with PB samples. This suggests a chronic activation of adaptive immune cells in the central nervous system19. The authors analyzed two CSF samples collected at different time-points after aSAH (1–3 and 4–6 days) in five patients: no differences in the number or activation status of immune cells were found.
Our study determined increased numbers of CSF T cells (CD3 +) during VSP compared with the acute phase. Specifically, CD8+CD161+ cells increased significantly in the CSF of patients who developed VSP compared to those who did not have clinical VSP. CD8 is a surface marker of cytotoxic T cells, whereas CD161 (a C-type lectin member of the human NKR-P1 family) is mainly expressed in NK cells and T cells. CD8+ T cells expressing high levels of CD161 synthesize a pattern of molecules similar to the type-17 phenotype40. Thus, these cells are also known as “Tc17” cells and have been shown to secrete IL-17 induced by activation of the transcription factor retinoic acid-related orphan receptor C (RORC) pathway41.
The main function of IL-17 is to increase the local production of chemokines and recruit monocytes to the site of inflammation42. IL-17 produced by CD8+CD161+ cells has been shown to contribute to the inflammatory response in experimental murine models of multiple sclerosis and autoimmune encephalitis43,44. However, the role of IL-17 in aSAH and VSP remains poorly understood. Our group has previously demonstrated increased concentrations of IL-17 in blood samples from the lumen of human cerebral aneurysms compared with samples from the femoral artery45. Chaudhry et al. also showed systemic upregulation of the IL-23/IL-17 inflammatory axis in human PB samples after aSAH46. Using an estrogen-deficient mice model, Hoh et al. recently demonstrated that IL-17A-blockade exerts a protective effect against aneurysm formation and rupture by increased E-cadherin expression58.
Previous evidence has shown active infiltration of the aneurysmal wall by cells of the innate immune system (e.g. macrophages, neutrophils)47. However, our flow cytometry analysis of post-hemorrhagic CSF from patients with VSP suggested activation of CD8+CD161+ “Tc17” lymphocytes. Thus, we aimed to identify the potential source of these cells by analyzing tissue of brain aneurysms. We demonstrated the presence of Tc17 cells in the wall of two unruptured aneurysms with confocal imaging. This finding suggests that: (1) the aneurysmal wall may be the source of Tc17 cells seen in the subarachnoid space after aneurysm rupture; and (2) Tc17 cells may be an important source of IL-17 and pro-inflammatory cytokines both in the vascular wall of unruptured aneurysms and CSF after aSAH. Expression of CD161 on the surface of CD8+ T cells stimulates high expression of transcription factors that induce differentiation of more Tc17 cells nearby48. Further research is needed to establish the role of CD8+CD161+ cells as a potential immunomodulatory target to prevent VSP after aSAH.
Microglia and secondary brain damage
VSP is not directly correlated with the development of DCI49. Several randomized controlled trials failed to demonstrate long-term improvement of clinical outcomes in patients with aSAH after administration of different anti-vasospasm medications21,50–54. DCI may be the result of a neuroinflammatory response mediated by microglial cells after aSAH. Microglia are the cellular mediators of inflammation in the central nervous system; they exist in a quiescent state and become activated following exposure to an insult or infection55. Hanafy et al. demonstrated in mice that neuronal apoptosis and VSP on day 7 after induction of SAH was mainly microglial-dependent, and became microglial-independent at latter stages56. Similarly, Schneider et al. showed a wave of Iba-1-positive cells spreading within mice brain tissue between days 4–28 after experimental SAH. The wave of immune cells was chronologically correlated with neuronal cell death13.
In our study, the number of CSF microglia was low immediately after aSAH. However, this cell population progressively increased during VSP and post-VSP peak periods. In chronic neurodegenerative processes such as Alzheimer’s/Parkinson’s disease and multiple sclerosis, it has been demonstrated that microglial cells remain activated for long periods of time and continue to release inflammatory mediators indefinitely57. These cells may have a role in the development of post-hemorrhagic VSP and DCI. The role of microglia in the secondary brain damage seen months to years after aSAH remains undetermined.
Limitations
The main limitation of this study is the small number of patients. However, we collected CSF samples at 4 different time-points and a PB sample at one point per study subject. The immunological profile changed over time and reached statistical significance despite the limited number of patients. The results of this pilot-exploratory study will need to be confirmed in a larger cohort.
Most patients had severe aSAH (61% = mFS 4), therefore the sample is biased towards sicker patients with larger hemorrhages. Additionally, one subject in the no-VSP group experienced clinically silent radiographic vasospasm (patient #12, Table 1). Whether these vascular changes are immune/inflammatorily mediated or not remains unknown2. Moreover, vasospasm is part of the spectrum of processes of DCI. Other events such as cortical spreading ischemia, microthrombosis and constriction of the microcirculation are difficult to quantify and were not assessed in this study.
Two patients underwent SAC and flow diversion for aneurysm treatment and required dual antiplatelet therapy. Aspirin may decrease the inflammatory response of these patients and may have altered the expansion of immune cell populations”58,59.
We did not quantify CSF IL-17 levels due to lack of enough sample volumes. Further investigation to better characterize the role of activated CD8+CD161+ cells in aSAH would require comparison of IL-17 production profiles using intracellular cytokine staining or gene expression patterns in RNA sequencing. The other limitation is the sampling of PB at only the first time-point of the study; future studies should sample PB longitudinally to compare immunological systemic and CSF responses.
Conclusion
Innate and adaptive immune cells play a pivotal role after aSAH. This preliminary study demonstrated that CD8+CD161+ lymphocytes increase over time in the CSF of patients after aSAH. The number of cells is higher during VSP. Moreover, these cells were identified in the wall of two unruptured intracranial aneurysms. Microglia activation occurs 6 + days after aSAH.
Supplementary information
Acknowledgments
Teresa Ruggle for graphic design and preparation. This work was supported by the 2019 Brain Aneurysm Research Grant from The Bee Foundation and by a Pilot Research Grant from the Society of Vascular and Interventional Neurology (SVIN), both granted to Edgar Samaniego.
Author contributions
Study conception and design: E.A.S., S.O.B. and J.A.R.; Collection of Data: D.S., J.A.R., S.O.B.; Collection and processing of biological samples: J.A.R., D.S. and S.O.B.; Drafting of the manuscript: E.A.S. and J.A.R.; Critical revision of the manuscript: M.Z., D.I., Y.L., N.J.K. and D.M.H.
Data availability
Data will be made available upon request from the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-68861-y.
References
- 1.Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (sah) Curr. Neuropharmacol. 2018;16:1385–1395. doi: 10.2174/1570159X16666180412110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014;10:44–58. doi: 10.1038/nrneurol.2013.246. [DOI] [PubMed] [Google Scholar]
- 3.Dorsch N. A clinical review of cerebral vasospasm and delayed ischaemia following aneurysm rupture. Acta Neurochir Suppl. 2011;110:5–6. doi: 10.1007/978-3-7091-0353-1_1. [DOI] [PubMed] [Google Scholar]
- 4.Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6:1–9. doi: 10.1227/00006123-198001000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Reilly C, Amidei C, Tolentino J, Jahromi BS, Macdonald RL. Clot volume and clearance rate as independent predictors of vasospasm after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2004;101:255–261. doi: 10.3171/jns.2004.101.2.0255. [DOI] [PubMed] [Google Scholar]
- 6.Geraghty JR, Davis JL, Testai FD. Neuroinflammation and microvascular dysfunction after experimental subarachnoid hemorrhage: Emerging components of early brain injury related to outcome. Neurocrit. Care. 2019;31:373–389. doi: 10.1007/s12028-019-00710-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyne-Geithman GJ, Morgan CJ, Wagner K, Dulaney EM, Carrozzella J, Kanter DS, et al. Bilirubin production and oxidation in csf of patients with cerebral vasospasm after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2005;25:1070–1077. doi: 10.1038/sj.jcbfm.9600101. [DOI] [PubMed] [Google Scholar]
- 8.Rapoport RM. Bilirubin oxidation products and cerebral vasoconstriction. Front. Pharmacol. 2018;9:303. doi: 10.3389/fphar.2018.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vorkapic P, Bevan JA, Bevan RD. Longitudinal in vivo and in vitro time-course study of chronic cerebrovasospasm in the rabbit basilar artery. Neurosurg Rev. 1991;14:215–219. doi: 10.1007/BF00310660. [DOI] [PubMed] [Google Scholar]
- 10.Provencio JJ, Altay T, Smithason S, Moore SK, Ransohoff RM. Depletion of ly6g/c(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J. Neuroimmunol. 2011;232:94–100. doi: 10.1016/j.jneuroim.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Testai FD, Valyi-Nagy T, Mani NP, Zhai F, Nanegrungsunk D, et al. Vap-1 blockade prevents subarachnoid hemorrhage-associated cerebrovascular dilating dysfunction via repression of a neutrophil recruitment-related mechanism. Brain Res. 2015;1603:141–149. doi: 10.1016/j.brainres.2015.01.047. [DOI] [PubMed] [Google Scholar]
- 12.Xu HL, Garcia M, Testai F, Vetri F, Barabanova A, Pelligrino DA, et al. Pharmacologic blockade of vascular adhesion protein-1 lessens neurologic dysfunction in rats subjected to subarachnoid hemorrhage. Brain Res. 2014;1586:83–89. doi: 10.1016/j.brainres.2014.08.036. [DOI] [PubMed] [Google Scholar]
- 13.Schneider UC, Davids AM, Brandenburg S, Muller A, Elke A, Magrini S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015;130:215–231. doi: 10.1007/s00401-015-1440-1. [DOI] [PubMed] [Google Scholar]
- 14.Gris T, Laplante P, Thebault P, Cayrol R, Najjar A, Joannette-Pilon B, et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J. Neuroinflamm. 2019;16:253. doi: 10.1186/s12974-019-1629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Provencio JJ, Fu X, Siu A, Rasmussen PA, Hazen SL, Ransohoff RM. Csf neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit. Care. 2010;12:244–251. doi: 10.1007/s12028-009-9308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery. 1999;45:1137–1146. doi: 10.1097/00006123-199911000-00024. [DOI] [PubMed] [Google Scholar]
- 17.Hughes JT, Schianchi PM. Cerebral artery spasm. A histological study at necropsy of the blood vessels in cases of subarachnoid hemorrhage. J. Neurosurg. 1978;48:515–525. doi: 10.3171/jns.1978.48.4.0515. [DOI] [PubMed] [Google Scholar]
- 18.Mathiesen T, Lefvert AK. Cerebrospinal fluid and blood lymphocyte subpopulations following subarachnoid haemorrhage. Br. J. Neurosurg. 1996;10:89–92. doi: 10.1080/02688699650040584. [DOI] [PubMed] [Google Scholar]
- 19.Moraes L, Grille S, Morelli P, Mila R, Trias N, Brugnini A, et al. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with aneurysmal subarachnoid hemorrhage. Springerplus. 2015;4:195. doi: 10.1186/s40064-015-0970-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ortega SB, Pandiyan P, Windsor J, Torres VO, Selvaraj UM, Lee A, et al. A pilot study identifying brain-targeting adaptive immunity in pediatric extracorporeal membrane oxygenation patients with acquired brain injury. Crit. Care Med. 2019;47:e206–e213. doi: 10.1097/CCM.0000000000003621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (conscious-1): Randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39:3015–3021. doi: 10.1161/STROKEAHA.108.519942. [DOI] [PubMed] [Google Scholar]
- 22.Connolly ES, Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- 23.Sarrafzadeh A, Schlenk F, Meisel A, Dreier J, Vajkoczy P, Meisel C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke. 2011;42:53–58. doi: 10.1161/STROKEAHA.110.594705. [DOI] [PubMed] [Google Scholar]
- 24.Kaynar MY, Tanriverdi T, Kafadar AM, Kacira T, Uzun H, Aydin S, et al. Detection of soluble intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in both cerebrospinal fluid and serum of patients after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2004;101:1030–1036. doi: 10.3171/jns.2004.101.6.1030. [DOI] [PubMed] [Google Scholar]
- 25.Mocco J, Mack WJ, Kim GH, Lozier AP, Laufer I, Kreiter KT, et al. Rise in serum soluble intercellular adhesion molecule-1 levels with vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2002;97:537–541. doi: 10.3171/jns.2002.97.3.0537. [DOI] [PubMed] [Google Scholar]
- 26.Polin RS, Bavbek M, Shaffrey ME, Billups K, Bogaev CA, Kassell NF, et al. Detection of soluble e-selectin, icam-1, vcam-1, and l-selectin in the cerebrospinal fluid of patients after subarachnoid hemorrhage. J Neurosurg. 1998;89:559–567. doi: 10.3171/jns.1998.89.4.0559. [DOI] [PubMed] [Google Scholar]
- 27.Rothoerl RD, Schebesch KM, Kubitza M, Woertgen C, Brawanski A, Pina AL. Icam-1 and vcam-1 expression following aneurysmal subarachnoid hemorrhage and their possible role in the pathophysiology of subsequent ischemic deficits. Cerebrovasc. Dis. (Basel, Switzerland). 2006;22:143–149. doi: 10.1159/000093243. [DOI] [PubMed] [Google Scholar]
- 28.Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J. Neurol. Neurosurg. Psychiatry. 2001;70:534–537. doi: 10.1136/jnnp.70.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou SH, Feske SK, Atherton J, Konigsberg RG, De Jager PL, Du R, et al. Early elevation of serum tumor necrosis factor-alpha is associated with poor outcome in subarachnoid hemorrhage. J. Investig. Med. 2012;60:1054–1058. doi: 10.231/JIM.0b013e3182686932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frijns CJ, Fijnheer R, Algra A, van Mourik JA, van Gijn J, Rinkel GJ. Early circulating levels of endothelial cell activation markers in aneurysmal subarachnoid haemorrhage: Associations with cerebral ischaemic events and outcome. J. Neurol. Neurosurg. Psychiatry. 2006;77:77–83. doi: 10.1136/jnnp.2005.064956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Gao Z. Expression of mmp-9 and il-6 in patients with subarachnoid hemorrhage and the clinical significance. Exp. Ther. Med. 2018;15:1510–1514. doi: 10.3892/etm.2017.5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochir. Suppl. 2013;115:233–238. doi: 10.1007/978-3-7091-1192-5_42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for mmps and timps in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- 34.Egashira Y, Zhao H, Hua Y, Keep RF, Xi G. White matter injury after subarachnoid hemorrhage: Role of blood-brain barrier disruption and matrix metalloproteinase-9. Stroke. 2015;46:2909–2915. doi: 10.1161/STROKEAHA.115.010351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu ZW, Zhao JJ, Pang HG, Song JN. Vascular endothelial growth factor a promotes platelet adhesion to collagen iv and causes early brain injury after subarachnoid hemorrhage. Neural Regen. Res. 2019;14:1726–1733. doi: 10.4103/1673-5374.257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Fujimoto M, Kawakita F, Ichikawa N, Suzuki H. Vascular endothelial growth factor in brain edema formation after subarachnoid hemorrhage. Acta Neurochir Suppl. 2016;121:173–177. doi: 10.1007/978-3-319-18497-5_31. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Fujimoto M, Kawakita F, Nakano F, Imanaka-Yoshida K, Yoshida T, et al. Anti-vascular endothelial growth factor treatment suppresses early brain injury after subarachnoid hemorrhage in mice. Mol. Neurobiol. 2016;53:4529–4538. doi: 10.1007/s12035-015-9386-9. [DOI] [PubMed] [Google Scholar]
- 38.Mathiesen T, Andersson B, Loftenius A, von Holst H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J Neurosurg. 1993;78:562–567. doi: 10.3171/jns.1993.78.4.0562. [DOI] [PubMed] [Google Scholar]
- 39.Waters RS, Perry JSA, Han S, Bielekova B, Gedeon T. The effects of interleukin-2 on immune response regulation. Math Med Biol. 2018;35:79–119. doi: 10.1093/imammb/dqw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, et al. Analysis of cd161 expression on human cd8+ t cells defines a distinct functional subset with tissue-homing properties. Proc. Natl. Acad. Sci. USA. 2010;107:3006–3011. doi: 10.1073/pnas.0914839107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. Cd161 is a marker of all human il-17-producing t-cell subsets and is induced by rorc. Eur. J. Immunol. 2010;40:2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 42.Borish LC, Steinke JW. Cytokines and chemokines. J Allergy Clin. Immunol. 2003;111:S460–475. doi: 10.1067/mai.2003.108. [DOI] [PubMed] [Google Scholar]
- 43.Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. Il-17a secretion by cd8+ t cells supports th17-mediated autoimmune encephalomyelitis. J. Clin. Investig. 2013;123:247–260. doi: 10.1172/JCI63681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annibali V, Ristori G, Angelini DF, Serafini B, Mechelli R, Cannoni S, et al. Cd161(high)cd8+t cells bear pathogenetic potential in multiple sclerosis. Brain. 2011;134:542–554. doi: 10.1093/brain/awq354. [DOI] [PubMed] [Google Scholar]
- 45.Chalouhi N, Points L, Pierce GL, Ballas Z, Jabbour P, Hasan D. Localized increase of chemokines in the lumen of human cerebral aneurysms. Stroke. 2013;44:2594–2597. doi: 10.1161/STROKEAHA.113.002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaudhry SR, Guresir E, Vatter H, Kinfe TM, Dietrich D, Lamprecht A, et al. Aneurysmal subarachnoid hemorrhage lead to systemic upregulation of il-23/il-17 inflammatory axis. Cytokine. 2017;97:96–103. doi: 10.1016/j.cyto.2017.05.025. [DOI] [PubMed] [Google Scholar]
- 47.Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Biology of intracranial aneurysms: role of inflammation. J. Cereb. Blood Flow Metab. 2012;32:1659–1676. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Y, Pan HF, Ye DQ. Tc17 cells in immunity and systemic autoimmunity. Int. Rev. Immunol. 2015;34:318–331. doi: 10.3109/08830185.2014.954698. [DOI] [PubMed] [Google Scholar]
- 49.Welling LC, Welling MS, Teixeira MJ, Figueiredo EG. Neuroinflammation after subarachnoid hemorrhage: a consolidated theory? World Neurosurg. 2016;85:8–9. doi: 10.1016/j.wneu.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Han D, Lee S, Lee S. Effect of nimodipine treatment on outcome in surgical cases of aneurysmal sah. J Neurosurg. 1993;78:346A. [Google Scholar]
- 51.Haley EC, Jr, Kassell NF, Torner JC. A randomized trial of nicardipine in subarachnoid hemorrhage: Angiographic and transcranial doppler ultrasound results. A report of the cooperative aneurysm study. J. Neurosurg. 1993;78:548–553. doi: 10.3171/jns.1993.78.4.0548. [DOI] [PubMed] [Google Scholar]
- 52.Reinert M, Wiest R, Barth L, Andres R, Ozdoba C, Seiler R. Transdermal nitroglycerin in patients with subarachnoid hemorrhage. Neurol. Res. 2004;26:435–439. doi: 10.1179/016164104225015976. [DOI] [PubMed] [Google Scholar]
- 53.Bradford CM, Finfer S, O'Connor A, Yarad E, Firth R, McCallister R, et al. A randomised controlled trial of induced hypermagnesaemia following aneurysmal subarachnoid haemorrhage. Crit Care Resusc. 2013;15:119–125. [PubMed] [Google Scholar]
- 54.Senbokuya N, Kinouchi H, Kanemaru K, Ohashi Y, Fukamachi A, Yagi S, et al. Effects of cilostazol on cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a multicenter prospective, randomized, open-label blinded end point trial. J Neurosurg. 2013;118:121–130. doi: 10.3171/2012.9.JNS12492. [DOI] [PubMed] [Google Scholar]
- 55.Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. 2019;25:112–123. doi: 10.1016/j.molmed.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Hanafy KA. The role of microglia and the tlr4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013;10:83. doi: 10.1186/1742-2094-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat. Neurosci. 2018;21:1359–1369. doi: 10.1038/s41593-018-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudson JS, Marincovich AJ, Roa JA, Zanaty M, Samaniego EA, Hasan DM. Aspirin and intracranial aneurysms. Stroke. 2019;50:2591–2596. doi: 10.1161/STROKEAHA.119.026094. [DOI] [PubMed] [Google Scholar]
- 59.Hasan DM, Chalouhi N, Jabbour P, Dumont AS, Kung DK, Magnotta VA, et al. Evidence that acetylsalicylic acid attenuates inflammation in the walls of human cerebral aneurysms: preliminary results. J. Am. Heart Assoc. 2013;2:e000019. doi: 10.1161/JAHA.112.000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available upon request from the corresponding author.