

Abstract
Purpose:
Over 60 % of melanoma patients respond to immune checkpoint inhibitor (ICI) therapy, but many subsequently progress on these therapies. Second-line targeted therapy is based on BRAF mutation status, but no available agents are available for NRAS, NF1, CDKN2A, PTEN, and TP53 mutations. Over 70% of melanoma tumors have activation of the MAPK pathway due to BRAF or NRAS mutations, while loss or mutation of CDKN2A occurs in ~40% of melanomas, resulting in unregulated MDM2-mediated ubiquitination and degradation of P53. Here we investigated the therapeutic efficacy of over-riding MDM2 mediated degradation of P53 in melanoma with an MDM2 inhibitor that interrupts MDM2 ubiquitination of P53, treating tumor bearing mice with the MDM2 inhibitor alone or combined with MAPK-targeted therapy.
Experimental Design:
To characterize the ability of the MDM2 antagonist, KRT-232, to inhibit tumor growth, we established patient-derived xenografts (PDX) from 15 melanoma patients. Mice were treated with KRT-232 or a combination with BRAF and/or MEK inhibitors. Tumor growth, gene mutation status, as well as protein and protein-phosphoprotein changes, were analyzed.
Results:
100% of the 15 PDX tumors exhibited significant growth inhibition either in response to KRT-232 alone or in combination with BRAF and/or MEK inhibitors. Only BRAFV600wt tumors responded to KRT-232 treatment alone while BRAFV600E/M PDXs exhibited a synergistic response to the combination of KRT-232 and BRAF/MEK inhibitors.
Conclusions:
KRT-232 is an effective therapy for the treatment of either BRAFwt or PANwt (BRAFwt, NRASwt) TP53WT melanomas. In combination with BRAF and/or MEK inhibitors, KRT-232 may an effective treatment strategy for BRAFV600 mutant tumors.
Keywords: Melanoma, MDM2, TP53, AMG 232, KRT-232
Translational Relevance:
While nearly 60% of melanoma patients respond to treatment with an immune checkpoint inhibitor (ICI) or targeted therapy, identification of therapeutic regimes that successfully treat the 40% of melanoma patients who fail to respond or progress after therapy, remains elusive. Here we evaluated the response of patient-derived xenograft tumors established from melanoma patients to a new therapeutic regimen: treatment with either MDM2 antagonist alone, or for tumors with a BRAFV600 mutation, treatment with KRT-232 in combination with BRAF/MEK inhibitors. Data show that non-BRAFV600E/M mutation tumors exhibited significant growth inhibition in response to MDM2 inhibitors, while those with BRAFV600E/M mutations responded to treatment with combined MDM2, BRAF, and MEK inhibition. These data provide key insight into the potential for using MDM2 inhibitors alone or combined with BRAF and/or MEK inhibitors for the treatment of melanoma.
Introduction:
The American Cancer Society estimates that there will be over 96,000 new cases of melanoma in the United States in 2019, with an estimated 7,230 deaths. The World Health Organization estimates nearly 300,000 melanoma skin cancers occur globally each year with the highest rates in Australia and New Zealand (1). The incidence of melanoma has risen rapidly over the past 30 years, especially in those over 50 years of age, which places melanoma as the 5th most common form of new cancer diagnoses. For the period between 2006–2015, the incidence rate increased by 3% per year (2). While new cancer diagnosis rates continue to rise, the five-year survival rate continues to improve (1). This improvement in survival reflects the recent emphasis in early detection combined with improvements in therapies, such as targeted and immune therapy.
Advances in metastatic melanoma treatment followed the identification and classification of melanoma driver mutations, particularly mutations in BRAF described in 2002 (3). Characterization of driver mutations paved the way for new treatment strategies focused on small molecule inhibitors targeting specific proteins involved in the melanoma pathogenesis (4–6). The most common oncogenic driver mutations are in the mitogen-activated protein-kinase (MAPK) pathway where BRAF mutations occur in approximately 50% of melanomas and NRAS mutations in 15–20% of melanomas, with neither BRAF nor NRAS mutations occurring concomitantly. Activating mutations in NRAS occur at either codon 12 or codon 61, or less frequently codon 13. Mutations at codon Q61, resulting in the Q61R/K/L substitutions, disrupts the GTPase activity of RAS, resulting in a constitutively active conformation; whereas, mutations at codon G12 or G13, affect the Walker A-motif of the protein, thus decreasing its sensitivity to GTPase-accelerating proteins (7). BRAF activating mutations resulting in V600E/K/M account for nearly 90% of all the BRAF mutations in melanomas. Inhibitors of BRAFV600E/K/M were developed including dabrafenib, vemurafenib, and encorafenib, and were successful in Phase III clinical trials (8–10). MEK kinases (MEK1 and MEK2), which function immediately downstream of BRAF, also have been studied as potential therapeutic targets for inhibition, especially in combination with BRAF inhibition. Three BRAF-MEK inhibitor combinations (dabrafenib-trametinib, vemurafenib-cobimetinib, and encorafenib-binimetinib) were successful in Phase III clinical trials (11–17). Treatment of patients with wild-type BRAF has proved more difficult, especially for those with NRAS mutations. There are currently no therapies that directly target mutant NRAS; however, MEK inhibitors (trametinib, binimetinib, and pimasertib) have been shown to have some effect in wild type BRAF and mutant NRAS melanoma (18,19).
The suboptimal response rates for targeted therapies, particularly within tumors not expressing the BRAFV600 mutation, have led to increased interest in other therapeutic options, including combination therapies targeting different pathways. The P53 tumor suppressor protein is one such option. In contrast to many other cancers, over 80% of human melanomas express TP53wt, but P53 degradation can be enhanced through overexpression of the murine double minute (MDM) proteins, MDM2 or MDMX (20–23). Loss or mutation of CDKN2A in ~40% of melanoma tumors results in loss of function of the MDM2 inhibitor, p14/p19, thus increasing MDM2 mediated degradation of P53. (24). In the past several years, several molecules have been developed to interrupt the interaction between P53 and MDM2. These include the cis-imidazolines “Nutlins” (RG7112 and RG7388), the spirooxindoles, MI-77301 and, MK-8242, as well as other recently developed compounds such as CGM097, HDM201, and the piperidinone, AMG 232/KRT-232 (25,26). Interestingly, KRT-232 was shown to have antitumor activity in cell and animal models of melanoma with BRAF and KRAS mutations. These studies showed that KRT-232 treatment led to increased P53, CDKN1A, MDM2, and PUMA proteins in TP53 wild-type cells (SJSA-1, HCT116), but not in TP53-mutant HT-29 cells. KRT-232 also has been reported to be an effective MDM2 inhibitor in glioblastoma cell lines, patient-derived stem cells (27), and a wide variety of TP53wt but not mutant TP53 tumor cell lines (28,29).
In the present study, we tested the anti-tumor efficacy of combining KRT-232 with BRAF and MEK inhibitors using melanoma patient-derived xenograft models (PDX). We utilized a well-characterized panel of melanoma PDX tumors, which represented an array of BRAF and NRAS genotypes. Others have demonstrated that melanoma PDX tumors accurately model the disease and respond to targeted therapies (30–32). When KRT-232 was tested as a single agent, only tumors that expressed wild type BRAFV600 (6/15 or 40%) had slower growth rates in response to KRT-232 compared to vehicle treated mice. The remaining nine PDX tumors were synergistically responsive to KRT-232 in combination with BRAF and MEK inhibition. These studies demonstrate that KRT-232 is an effective potential agent for the treatment of melanoma tumors that are either BRAFWT or PanWT (BRAFwtT, NRASWT, NF1WT). Furthermore, KRT-232 is an effective agent in combination with dabrafenib and trametinib for the treatment of BRAFV600MUT tumors.
Materials and Methods:
KRT-232 (AMG 232), navitoclax, dabrafenib and trametinib were provided by the NCI.
Mouse studies.
Animal studies were approved by the Vanderbilt IACUC. NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were purchased from Jackson Labs and Athymic Nude mice (Hsd: Athymic Nude-Foxn1nu) were purchased from Envigo. PDX tumors were established and propagated as described previously (33). The mice were all 3–6 month-old females weighing 20–26 grams. KRT-232 and navitoclax were prepared in 5% DMSO and 95% corn oil and administered five days a week by oral gavage in a total volume of 100–200uL based upon the weight of the mouse. Corn oil with 5% DMSO was used as the vehicle control. Dabrafenib and trametinib were prepared in 0.5% hydroxypropyl methylcellulose. At least 5 mice per treatment group were used, with each mouse bearing 2 tumors. Mouse body weight was measured daily for gavage dosing and recorded twice a week and tumor measurements were taken twice a week with micro-calipers. Tumor volume was estimated as 0.5 × length × width × width. Treatment began when tumors reached 50–100mm3 volume on average and continued until tumors reached 15mm in diameter or became perforated. At the end of the experiment, the final weight of the tumors was recorded, and tumors were either flash-frozen for RPPA analysis or fixed in 10% buffered formalin for paraffin embedding. PDX models 1577, 1668, 1767, 1595, 2316 and 2252 were provided to the Patient-Derived Models Repository at NCI-Frederick. All models were established according to the recommended Minimal Information Standard (34).
Targeted DNA sequencing.
DNA from patient tumor or early passage (P2) PDX tumor (PDX1577, 1668, 2316, and 2552) and from leukocytes isolated from peripheral blood from the PDX donor were submitted for QC and DNAseq using the Human Comprehensive Cancer Panel (QIAGEN Sciences, Frederick, MD). Identification of tumor-associated somatic mutations and copy number alterations was performed using the QIAGEN NGS Data Analysis Web Portal. A color-coded map of genetic alterations in PDXs was constructed using OncoPrinter available through cBioPortal (35,36)
STR Profiling.
DNA extraction from tumor tissue was achieved using standard phenol: chloroform extraction methods and ethanol precipitation or using the DNeasy Blood and Tissue Kit. For a subset of tumors with high melanin content, the DNA was further purified using the Qiagen DNeasy PowerClean Pro Cleanup K Kit (QIAGEN Sciences, Frederick, MD). DNA from matching blood samples, when available, was extracted using the QIAamp DNA Mini and Blood Mini Kit. DNA from matched patient blood, when available, P0 (patient’s original tumor), and passage two of patient-derived xenografts tumors (P2A and P2B) were subject to STR Profiling leveraging the PowerPlex 16 HS System (Promega). A total of 16 STR loci (D351358, TH01, D18551, Penta E, D55828, D135317, D75820, D165539, CSF1P0, Dmelogenin, vWA, D851179, TPOX, FGA) were co-amplified in each sample. PDX specimens were considered a match to the patient specimen when one allele from each 16 STR locus was present in the P0 and P2 specimen. The amelogenin alleles for all the specimens match the patient sex that was reported.
Pathology/histology assessment.
Immunohistochemical (IHC) analyses were performed using the Leica Bond Max IHC stainer. All steps besides dehydration, clearing and cover-slipping were performed on the Bond Max. Slides were deparaffinized and heat-induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 30 minutes. The sections were incubated with Ready-to-Use antibody as indicated below. The Bond Refine Polymer detection system was used for visualization. Slides were then dehydrated, cleared and coverslipped. IHC slides were scanned at the Digital Pathology Shared resource. The automated quantification of the percentages of the KI67-positive cells was performed by Leica Biosystems’ Digital Image Hub, using the software available with the Leica SCN400 Slide Scanner. The antibodies used were as follows: anti-p53 (PA0057, Leica Biosystems, Newcastle, United Kingdom) for 30 minutes; anti-MelanA (PA0233, Leica, Buffalo Grove, IL) for 15 minutes; anti-Ki67 (RM47–26, StatLab, McKinney, TX) for 30 minutes; anti-SOX-10 (PA0813, Cell Marque, Rocklin, CA) for one hour.
Proteomics.
Flash-frozen tumor tissue was processed in RIPA buffer using a Precellys Homogenizer. Lysates were analyzed with a 382 antibody panel. For each condition, three independent tumors were analyzed from three PDX tumors, resulting in nine samples analyzed for each treatment. The only exception to this was for the KRT-232 + dabrafenib +trametinb treatment of PDX1351 where only two independent tumors were analyzed. RPPA data was analyzed after a log2 transformation. All comparisons were performed based on a mixed-effect model to account for the correlation structure with the measured data from the sample PDX. Using model-based (least-square) means, the average adjusted difference (log2FC: fold change in a log2 scale) between treatments (or groups) was estimated and compared using the Wald test. Bonferroni correction was used to adjust for multiple testing. Changes in protein expression were considered to be significant (shown in red) based upon two criteria, the significance of the change (false discovery rate (FDR) < 0.05) and the degree of change (when log2(FC)=+/− 0.4). A heat map was used for the visualization of proteins with significantly different expression across treatments. Hierarchical clustering analysis using Ward’s distance was used to measure the dissimilarity between two clusters of observations.
Statistical Analysis.
For a comparison of more than two groups, one-way analysis of variance (ANOVA) with pairwise t test comparison was used. For in vivo experiments, tumor volume or tumor weight were analyzed on the natural log scale to better meet the normality assumption. The mixed-effects model with posthoc tests was used to compare the difference in tumor growth or tumor weight between treatments. P-value was adjusted using the Bonferroni method. With pairwise comparison, the p-value was adjusted using the Bonferroni method to correct for inflated Type I error (the higher the change for a false positive). For each PDX, initial statistical and data analysis was based on all treatment groups, which included three experimental agents tested individually and in combination compared to vehicle controls, but data analyses are presented based upon the statistical analysis for the subset of experimental agents. For synergy analysis, a mixed-effect model with individual effect and interactive effect was used to assess treatment differences in tumor volume over days. Each observation was classified according to the PDX. Tukey’s adjusted p-value was used for multiple comparisons. All tests of statistical significance were two-sided. Unless otherwise noted in the manuscript, we use the word “significant” to represent statistical significance (p<0.05). *** for p<0.001, ** for p<0.01, * for p<0.05. Analyses were performed using R version 3.4.3 (37,38)
Results:
Patient Demographics:
Melanoma patient-derived xenografts (PDX) were established from melanoma tumors obtained from patients who were referred for surgical resection or biopsy. From a larger set of PDX tumors, 15 PDXs (Table 1A) were chosen for this study to approximately represent the range of mutations found in melanoma tumors. The patient population was 47% male and 53% female, represented ages from 29 to 77 and were largely American Joint Committee on Cancer (AJCC) (39) Stage IV (although two patients with locally advanced stage III melanoma; PDX2316 and 2552). The prior systemic therapies are listed in Table 1A, in the order in which they were received. Four patients were not treated systemically before resection (PDX2195, 2252, 1668, and 2316). The remaining 11 patients had tumor progression after targeted therapy or immunotherapy; both patients with BRAFV600 mutations treated with dabrafenib and trametinib (D+T) had progression on therapy.
Table 1A, PDX Tumors: Patient Demographics and Molecular Characterization.
A panel of 15 PDX melanoma tumors is arranged by distinct genetic phenotypes based on the mutation status. For each PDX, the patient demographics are shown including Clark’s Level tumor stage and prior treatments. Prior treatments are list in the order they were received. The gene mutation status was determined by NGS of the primary tumor sample or PDX P2 passage (PDX1577, 1668, 2316 and 2552, indicated by a *) using a Human Comprehensive Cancer Panel (QIAGEN Sciences, Frederick, MD), and the results for 10 melanoma driver mutations are shown. The metastatic tumor was used to generate the PDX. Abbreviations for therapies patients received are as follows: D,dabrafenib (Tafinlar), V600B-Raf mutant Inhibitor; G, GM-CSF (Oncovax); I,interferon; Ipi,ipilimumab (Yervoy), monoclonal Ab CTLA-4; N,nivolumab (Opdivo), monoclonal Ab PD-1; Pem,pembrolizumab (Keytruda), IgG4 isotype antibody PD-1; S,sunitinib (Sutent), multi-targeted receptor tyrosine kinase (RTK) inhibitor; S+T, sorafenib (Nexavar) + tivantinib, tyrosine-protein kinase inhibitor, MET inhibitor; T, trametinib (Mekinist), MEK1/2 inhibitor; V, vemurafenib (Zelboraf), B-Raf inhibitor. Table 1B, TP53 Mutation Status.The specific TP53 mutations detected by tumor NGS are shown for each PDX. NON_SYN, indicates a non-synonomous mutation, STOP, indicates a nonsense mutation resulting in a stop codon and SPL_SITE indicates a mutation (deletion) involving a splice site. The identification of the mutations, the small nucleotide polymorphism (SNP) effect and impact, and the designation of germline or somatic mutation was determined by using QIAGEN NGS Data Web Analysis Web Portal based on sequence analysis of the tumor and patient blood when available. SnpEff is an open-source tool that annotates variants and predicts their effects on genes by using an interval forest approach (48). The variant frequency is also listed for each non-synonymous mutation (NSM). The determination of somatic versus germline was based upon analysis of the patient’s blood, when available. The patient tumor (P0) was used to determine TP53 mutation status, except for PDX1577, 1668, 2316, and 2552. For these four PDX, the second PDX passage (P2) was sequenced.
| Patient Information | Gene Mutation Status | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDX | Sex | Age | Clark Stage | Prior Treatment | BRAF | NRAS | TP53 | NF1 | CDKN2A | PTEN | MTOR | MAP2K1 | KIT | CDK4 |
| 1351 | F | 61 | V | Pem, V | V600E Stop Lost | N | N | A1226T | N | Stop Lost | H1366Y | E203K | Stop Lost | N |
| 2552* | M | 40 | IIIB | Pem | V600E | N | N | N | Stop Lost | T398S | G580A N382S | N | G265S | I51V G45A G44A |
| 1577* | F | 38 | IV | D+T | V600E | N | N | N | N | T398S | T1837S G580A N382S | N | N | I51V G45A G44A |
| 2195 | M | 45 | V | None | V600E | N | N | V530I | N | N | N | N | N | N |
| 1839 | F | 38 | IV | I | V600E | N | N | N | N | Stop Lost | N | N | N | N |
| 1946 | F | 32 | IV | D+ T,Ipi | V600E | N | N | N | N | G129E | N | N | N | N |
| 9164 | M | n/a | n/a | Pem | V600E | N | Stop Lost | N | N | N | N | N | N | N |
| 2252 | M | 34 | IV | None | V600M V600E | N | Stop Lost | N | Stop Lost | Stop Lost | N | N | N | N |
| 0807 | F | 29 | IV | D, D+T | P192Q | N | A138ADG T140S | N | N | N | LR282L | D67N P124S | N | N |
| 1668* | M | 61 | IV | None | S147N N140T | N | N | Stop Lost | Stop Lost | T398S | T1837S G580A N382S | N | M541L | I51V G45A G44A |
| 1129 | F | 69 | V | Ipi | D549N K483T | N | N | N | N | N | N | N | N | N |
| 1767 | F | 49 | n/a: spindle | Ipi | N | N | N | N | N | N | N | N | N | N |
| 2316* | F | 71 | IIIB | None | N | N | N | Stop Lost | Stop Lost | Y | T1837S G580A | N | M541L K642E | N |
| 1595 | M | 33 | IV | S, G | P334S | Q61R | N | N | N | N | T1837S G580A N382S | N | N | I51V G45A G44A |
| 1179 | M | 77 | likely V | S+T, Pem | N | Q61H | N | N | N | N | K1981E | N | N | N |
| PDX | Gene Name | Germline/Somatic | Ref | Variant | Codon Change | AA Change | Variant Frequency | snpEff Effect | snpEff Impact |
|---|---|---|---|---|---|---|---|---|---|
| 0807 | TP53 | G | CCGATCT | p.A138ADG | 0.265 | NON_SYN | MOD | ||
| 0807 | TP53 | G | C | c.419C>G | p.T140S | 0.269 | NON_SYN | MOD | |
| 0807 | TP53 | G | C | c.215C>G | p.P72R | 0.458 | NON_SYN | MOD | |
| 1129 | TP53 | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD | |
| 1179 | TP53 | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD | |
| 1351 | TP53 | germline | G | C | c.215C>G | p.P72R | 0.58 | NON_SYN | MOD |
| 1577 | TP53 | germline | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD |
| 1595 | TP53 | G | C | c.215C>G | p.P72R | 0.486 | NON_SYN | MOD | |
| 1668 | TP53 | somatic | C | <DEL> | SPL_SITE | HIGH | |||
| 1668 | TP53 | germline | G | C | c.215C>G | p.P72R | 0.986 | NON_SYN | MOD |
| 1767 | TP53 | G | C | c.215C>G | p.P72R | 0.556 | NON_SYN | MOD | |
| 1839 | TP53 | somatic | G | A | c.916C>T | p.R306* | 0.484 | STOP | HIGH |
| 1839 | TP53 | somatic | G | A | c.483C>T | p.A161 | 0.468 | NON_SYN | LOW |
| 1839 | TP53 | somatic | A | G | c.706T>C | p.Y236H | 0.235 | NON_SYN | MOD |
| 1839 | TP53 | germline | G | C | c.215C>G | p.P72R | 0.359 | NON_SYN | MOD |
| 1946 | TP53 | somatic | C | <DEL> | SPL_SITE | HIGH | |||
| 1946 | TP53 | germline | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD |
| 2195 | TP53 | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD | |
| 2252 | TP53 | somatic | C | <DEL> | SPL_SITE | HIGH | |||
| 2252 | TP53 | germline | GG | AA | p.IR0I* | 0.607 | STOP | HIGH | |
| 2252 | TP53 | somatic | G | A | c.586C>T | p.R196* | 0.607 | STOP | HIGH |
| 2252 | TP53 | germline | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD |
| 2316 | TP53 | germline | G | C | c.215C>G | p.P72R | 1 | NON_SYN | MOD |
| 2552 | TP53 | germline | G | C | c.215C>G | p.P72R | 0.605 | NON_SYN | MOD |
| 9164 | TP53 | G | A | c.574C>T | p.Q192* | 1 | STOP | HIGH | |
| 9164 | TP53 | C | T | c.108G>A | p.P36 | 0.939 | NON_SYN | LOW | |
| 9164 | TP53 | A | G | 1 | INTRON | MODIFIER |
Genetic Analysis:
Except for patient 2316, all tumors were screened by either SNaPshot analysis or in the case of patient 2552 by Onkosight as standard of care in the clinic (40). SNaPshot analyses indicated that three tumors (1129, 1668 and 1767) were BRAFwt, NRASwt, and NF1wt for specific mutations in the nucleotides queried. To further understand the genetic make-up of the PDX tumors utilized in this study, either the original patient tumor or in the case of PDX1577, 1668, 2316 and 2552, second passage PDX aliquots were analyzed using a comprehensive cancer next generation sequencing (NGS) panel that targets exonic regions of genes with known associations to cancer. In all cases, the mutations noted in the patients’ tumors by SNaPshot and Onkosight sequencing obtained as standard of care in the clinic were confirmed by NGS in the laboratory.
NGS provided additional genetic information relevant to this study. The UV carcinogenic etiology of melanoma results in melanoma having among the highest prevalence of somatic mutation across all cancer types (41). The number of mutations between samples sequenced was variable. PDX1767 had the fewest non-synonymous mutations (NSMs), with 295 high confidence variants at a mean read depth in the target region of 83, while PDX1668 had the most NSMs with 2,352 high confidence variants at a mean read depth of 199 (Supplementary Table S1A&B). Neither of these tumors had hot spot mutations in BRAF and NRAS. For two of the tumors where no mutations were identified by SNaPshot (1129 and 1668), NGS identified additional mutations in driver genes. For PDX1668, NGS identified a loss of the stop codon in NF1, and two mutations in BRAF, S147N, and N140T, with unknown significance. Further analysis of PDX1129 by NGS identified two mutations in BRAF (D549N and K483T). While the BRAF p.D549N is of unknown significance, it has been previously identified in the NZM41 cell line and in other melanomas, colorectal and lung carcinomas, in addition to hairy cell leukemia (42) (43–45). PDX2316 also was retrospectively analyzed by NGS and was found to lack mutations in BRAF and NRAS, although mutations in NF1, CDKN2A, MTOR, and KIT were detected. The tumors used in this study closely mimic the expected distribution of mutations within the driver mutation genes BRAF, NRAS, and NF1, with 53% exhibiting mutated BRAFV600 and 13% exhibiting mutated NRASQ61 mutations. Furthermore, PDX2316 was BRAFwt, NRASwt and PDX1767 was BRAFwt, NRASwt, and NF1wt.
Given the high number of tumor variants, we queried the cBioPortal for Cancer Genomics (35,36) to identify the 10 most frequently mutated genes in melanoma. The data set included 1414 patient/case sets for melanoma, including acral, desmoplastic and lentigo maligna but not uveal melanoma. The distribution of occurrence of mutations in these top 10 genes is shown in Table 1A and Supplementary Table S1B and the hotspot mutations identified in the PDX panel are listed. A hotspot in the cBioPortal is defined as, “this mutated amino acid was identified as a recurrent hotspot (statistically significant) and a 3D clustered hotspot in a population-scale cohort of tumor samples of various cancer types using a previously described methodology(46,47)”. Close attention was paid to mutations and copy number variations (CNV) within the TP53 locus since MDM2 is an important negative regulator of P53 (Table 1B and Supplementary Table S1B). By far the most prevalent TP53 variant was the P72R polymorphism, which was present in all the PDXs, except PDX9164. Four PDX tumors, PDX9164, 1839, 2252, and 0807, had nonsynomonous mutations (NSM) in the TP53 locus. PDX9164, PDX1839 and PDX2252 had mutations resulting in early translation termination and PDX2252 also had decreased TP53 copy number (CNV could not be determined for PDX9164). PDX0807 had two TP53 mutations, an insertion at codon 138 (A—ADG) and an NSM at codon T140 (T—S). The mutations in PDX0807 occurred at a low frequency (26–27%). In addition to these NSMs, PDXs 1668, 1839, and 1946 had mutations within TP53 splice sites. While the exact nature and frequency of these mutations is not reported in the NGS data, these three PDX tumors also had a loss of heterozygosity in the TP53 locus. All mutations and CNVs within the 10 focus genes and MDM2, regardless of SnpEff scores (48), for each PDX are listed in Supplementary Table S1B. Interestingly, copy number alterations, but not mutations, were noted for MDM2.
Verification of the PDX Panel:
Eight PDX tumors were characterized by Short Tandem Repeat (STR) analysis to confirm that the resulting PDX tumors were derived from the patient sample (Supplementary Figure S1A). The STR profiles demonstrate that each analyzed PDX was derived from the primary specimen. As previously reported (49), mouse PDX tumors do undergo genetic evolution, and in accordance with this, there were changes noted between the STR profiles of the human tumor sample and the PDX, with occasional loss of an allele, consistent with clonal selection.
For each PDX tumor, pathology and melanoma IHC markers were confirmed relative to the patient diagnosis of melanoma by a clinical pathologist. Representative results are shown for PDXs 2552, 1767, 1668 and 1595 (Supplementary Figure S1B). The markers used for melanocytic differentiation were Melan-A/Mart 1 and SOX10. Also, patient tumors (P0) and the second passage PDX (P2) were stained by H&E and for Ki-67, to determine the mitotic rate. All melanoma tumors and resulting PDXs were positive for SOX10 (50) while a subset was positive for Melan-A, consistent with prior results (51). In addition to the correlation between SOX10 and Melan-A expression between the tumor sample and PDX P2 passage, we also saw a correlation with melanin expression, as shown for PDX1767. PDX tumors derived from tumors with high expression of melanin also had high expression of melanin in the PDX. Likewise, if a tumor did not express detectable melanin, the PDX did not either, as shown for PDX2552 and PDX1668. PDX1595 had a low but detectable level of melanin as did the patient tumor. All tumors and patient-derived PDXs stained positive for Ki-67.
Effects of KRT-232 on Growth of Melanoma PDX tumors:
To evaluate the effects of KRT-232 on the melanoma PDX panel, 50 mg/kg KRT-232, which is comparable to a human dose of 250mg, was administered by oral gavage to the mice once the PDX tumor reached a volume of 50–100mm3. Mice were treated 5-days/week and treatment was continued until one or more tumors reached the endpoint size limit defined by the IACUC protocol (1.5 cm diameter). We compared the KRT-232 response to an FDA approved therapy, either dabrafenib (30 mg/kg) and trametinib (1 mg/kg) (D+T) or trametinib (T) alone (for NRAS mutant tumors) administered by oral gavage 5-days/week, once daily(QD). For mice harboring mutated BRAFV600 tumors, dabrafenib and trametinib (D+T) were administered in vivo either with or without 50 mg/kg KRT-232 QD. The response to all drugs was compared to the appropriate vehicle control group. Toxicity was not detected with any treatment group based on AST and ALT levels, weight loss or morbidity. Representative data from these studies for PDX1839, PDX1946, PDX2316, PDX1668, and PDX1595 are shown in Figure 2. (Data from treatment effects on tumor growth for all the PDX tumors are shown in Supplemental Figure S2). For each drug study, the tumor growth rate was statistically determined, the final tumor weight was measured, and FFPE sections were stained and quantified to measure proliferation based on Ki67 expression. To facilitate comparisons between PDX tumors and the responses to therapy, a summary of t-ratios for treatment group comparisons across 15 PDX tumors is shown in Figures 2 and 3A. The t-ratio is a ratio of the difference in the slope of tumor growth between control and a treatment group relative to its standard error, calculated by estimated marginal means based on the mixed effect model. For each PDX, response to a drug was based on a positive t-ratio greater than 2.6. The lowest positive t-ratio considered was 2.598 for the KRT-232 treatment of PDX0807, which correlated with an adjusted p-value of 0.063. Any treatment which resulted in a decreased tumor growth compared to vehicle control is indicated by a positive t-ratio. Likewise, any treatment which resulted in increased tumor growth compared to vehicle control is indicated by a negative t-ratio.
Figure 2A, Statistical Summary Table of T-ratio.
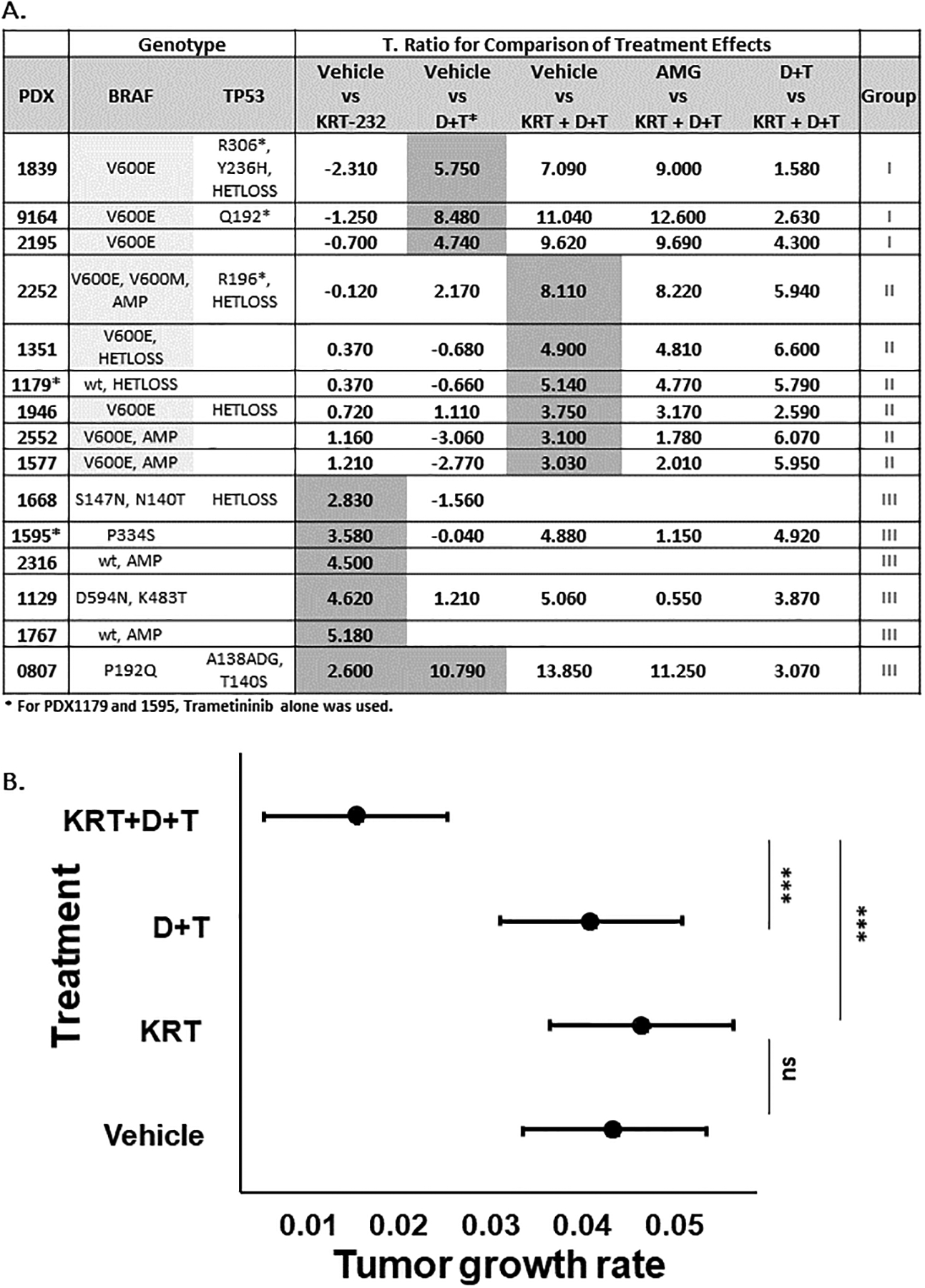
The t-ratio, obtained from the statistical analysis of treatment difference comparisons of the tumor growth rate based on the tumor volume, for each PDX treatment comparison, is listed. The BRAF and TP53 mutational status of each PDX is also listed. The group assignment, shown in the final column, is based on the response to KRT-232 and T+/−D treatment. Figure 2B, Analysis of Synergy. The mean estimated tumor growth rate of 4 independent PDX Group II lines is graphed with a 95% confidence interval by treatment. Mice implanted with PDX1351, 1577, 1946, and 2552 were treated in vivo with Vehicle, KRT-232, D+T, or KRT-232+D+T as described in Figure 1.
Figure 3A, Alterations in Cell Morphology Associated with Synergism.
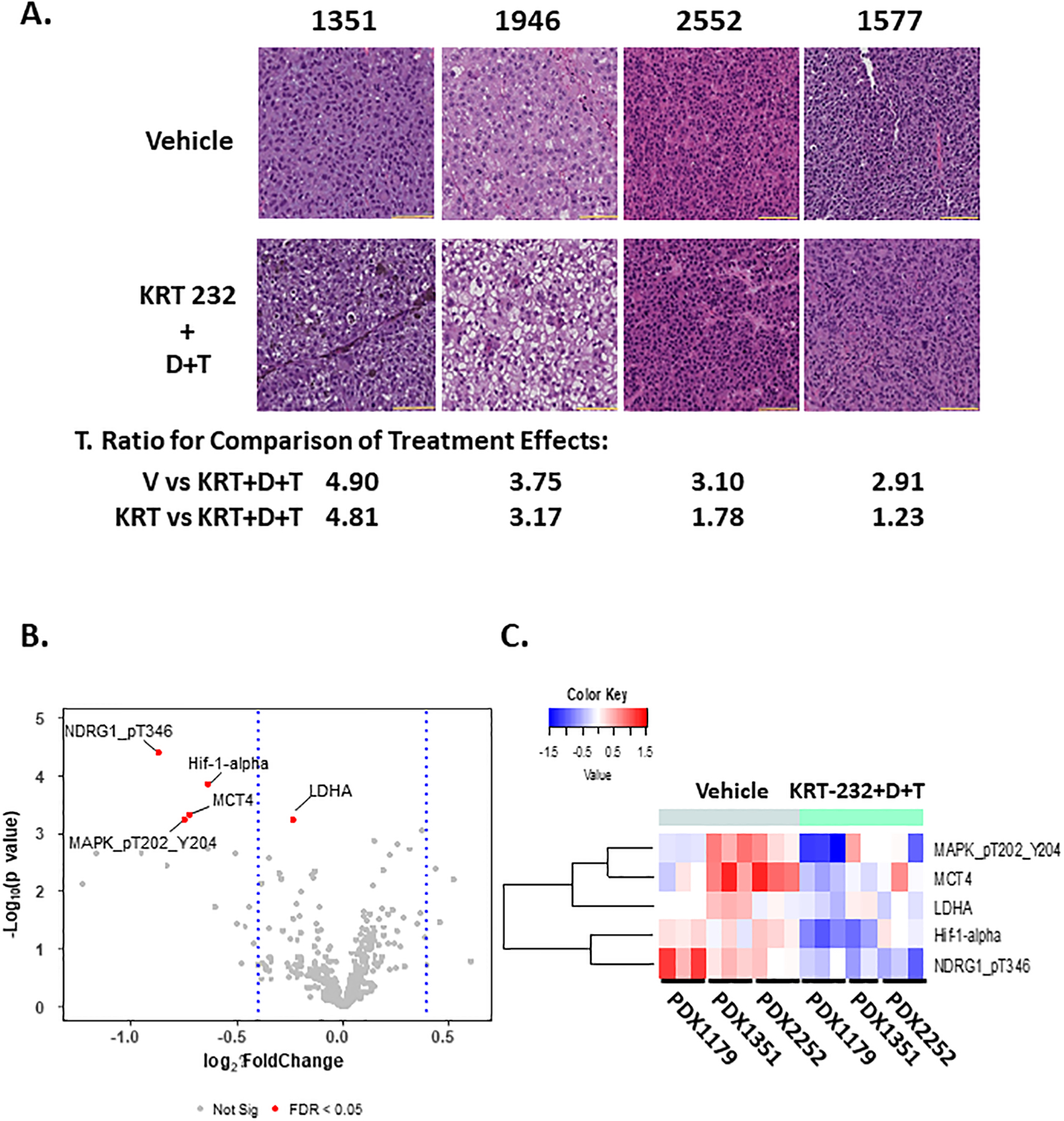
H&E is shown for Vehicle and KRT-232 + D+T treated mice implanted with PDX1351, PDX1946, PDX2552 and PDX1577 (Group III). The t-ratio for the statistical difference between the vehicle or KRT-232 treatment group compared to the KRT-232+D+T treatment group is shown. 20X images are shown and the scale marker is 100μm. Figure 3 B & C, Alterations in Protein Expression Associated with Synergism. A volcano plot (B) and a heat map (C) of RPPA data obtained from vehicle-treated and KRT-232 + D+T treated Group III PDX tumors samples for PDX1179, PDX1351, and PDX2252 is shown. Three tumors were analyzed for each PDX treatment. Only those proteins with an FDR <0.05 and log2(FC)=+/− 0.4 were included in the heat map.
The response to drug treatment could be classified into three distinct categories. Figure 1A, with PDX1839 as an example, represents those PDX tumors that responded to the standard therapy (D+T), but not to KRT-232. This group of PDX tumors is referred to as Group I. In fact, in the case of PDX1839 in vivo administration of KRT-232 caused a slight, albeit insignificant, increase in growth (Bonferroni adjusted (adj.) p=0.138) (Figure 1A). In contrast, D+T, an FDA approved therapy for BRAFV600 mutated melanoma, did decrease the growth rate of the Group I PDX tumors (t-ratio=5.4741, adj. p <0.001). The combination of KRT-232 with D+T resulted in a further reduction in the growth rate compared to KRT-232 alone treatment (t-ratio=9.00, adj. p<0.001), but not compared to D+T standard therapy (t-ratio=1.58, adj. p<0.7). A decrease in tumor size was also observed when comparing the response to KRT-232 alone versus KRT-232 + D+T (adj. p = 0.014). Also shown in Figure 1, Ki67 levels were not affected by in vivo administration of either KRT-232 or D+T (adj. p=0.714 and 0.184 respectively); however, a significant decrease in Ki67 staining compared to the vehicle control was seen with the combination therapy of KRT-232 and D+T (adj. p<0.001). A significant decrease was also observed when comparing the combination therapy (KRT-232 + D+T) to KRT-232 alone treatment (adj. p< 0.001), but not when compared to D+T therapy (adj. p=0.999). In summary, in Group I PDX tumors, D+T has a predominant effect on decreasing melanoma PDX tumor growth while KRT-232 therapy does not inhibit tumor growth in Group I PDX tumors.
Figure 1. Response to KRT-232.
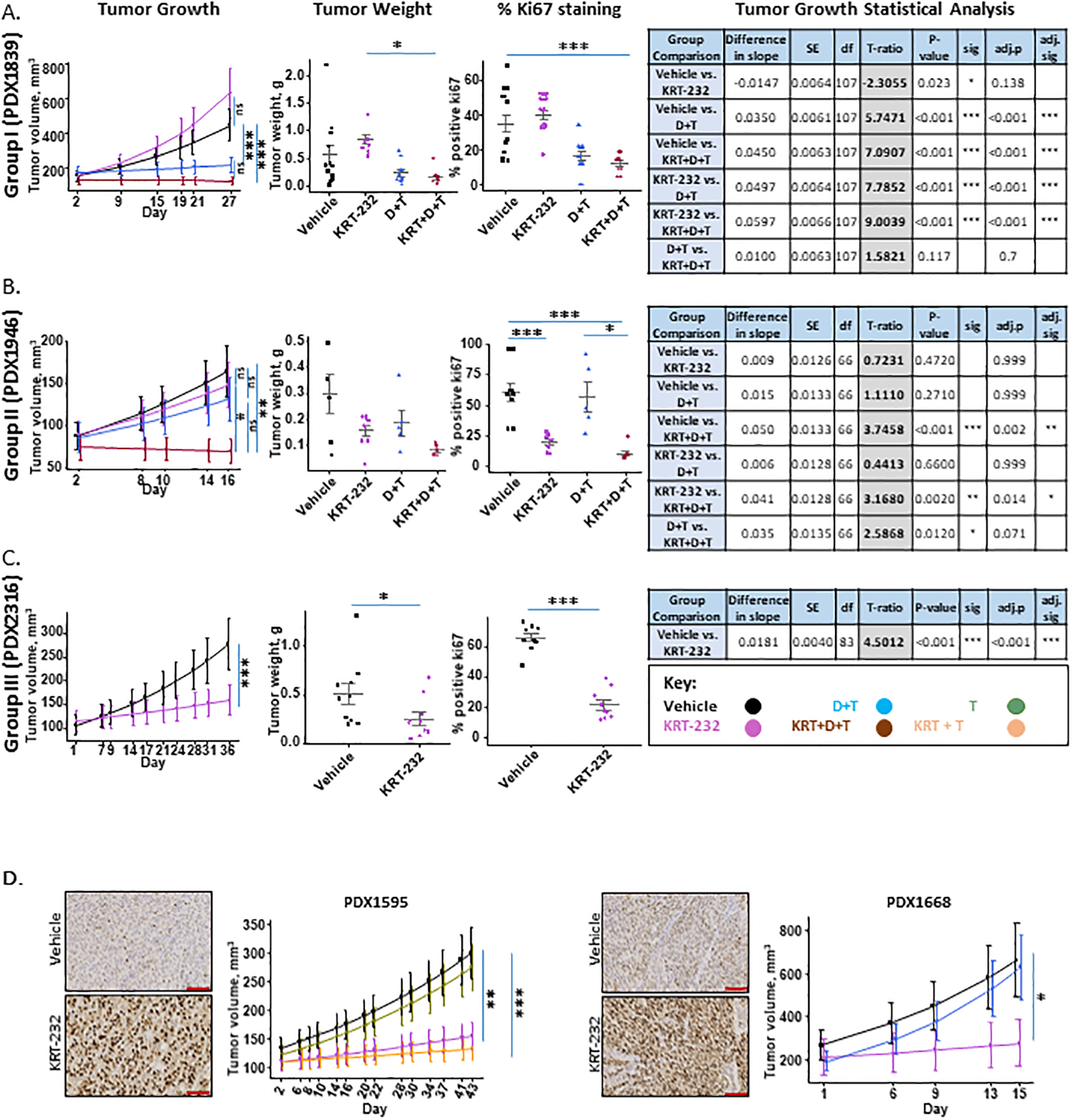
The response to drug treatment was divided into three separate categories. Figure 1A, exemplified by PDX1839 represents those PDX tumors that responded to dabrafenib and trametinib (D+T) but not KRT-232 (Group I). Figure 1B, exemplified by PDX1946, represents those PDX tumors that did not respond to D+T or KRT-232 but did respond synergistically to both (Group II). Figure 1C, exemplified by PDX2316, and Figure 1D, with PDX1668 and PDX1595, represent those PDX tumors that responded to KRT-232 but not D+T (Group III). Each panel in A-C includes data showing the effect of drug treatments on Tumor Growth, Final Tumor Weight, %Ki67 Staining and the Tumor Growth Statistical Analysis. Tumor volume was analyzed on the natural log scale to better meet normality assumptions and the predicted mean and standard error of tumor volume over time for each treatment group is shown. Dot plot of tumor weight (g) by treatment and % positive Ki67 by treatment (gray line: mean with standard error) are shown. A t-ratio table for pairwise comparison in tumor growth rate between treatments based on the mixed-effect model with post hoc tests is shown. Figure 1D, KRT-232 treatment increases the nuclear localization of P53. IHC staining of huP53 in melanoma PDX tumors from mice treated with the vehicle or KRT-232. 20X images are shown and the scale marker is 100μm. Tumor growth is also shown. The standard therapy for PDX1595 was trametinib and for PDX1668 the standard therapy was dabrafenib + trametinib.
The second group of tumors (Group II) shown in Figure 1B includes those PDX tumors that did not respond to either KRT-232 or the standard therapy alone but responded synergistically to the combination of KRT-232 and the standard therapy. Group II was comprised of 6 PDX tumors; five of the six PDX tumor lines exhibited BRAFV600 mutations. The sixth line, PDX 1179, had an NRASQ61 mutation and a BRAF copy number of 1.36 (see Supplementary Table S1B). Therefore, PDX1179 was treated with trametinib but not dabrafenib. As shown in Figure 1B, as exemplified with PDX1946 neither KRT-232 nor the standard therapy (D+T or T) resulted in a significant decrease in either growth rate (adj. p=0.999) or final tumor size for Group II PDX tumors. However, in vivo administration of KRT-232 in combination with the standard therapy, resulted in a significant decrease in the tumor growth rate compared to the vehicle control (t-ratio=3.75, adj. p=0.002) and a significant decrease in Ki67 positive cells (Figure 1B and Supplementary Figure S2).
The third group of PDX tumors (Group III), tumors 0807, 1129, 1595, 1668, 1767, and 2316, did respond to KRT-232 alone with growth inhibition (Figure 1C and D). Two of the lines, PDX2316 and PDX1767, were BRAFwt and NRASwt so mice were treated only with KRT-232. One line, PDX1595, exhibited an NRASQ61R mutation, so the mice implanted with this PDX were treated with either KRT-232, trametinib, or the combination. The final three PDX tumor lines, 0807, 1129 and 1668, had BRAF mutations outside the V600 position (Table 1 and Figure 2), so mice carrying these three PDX tumors were treated with KRT-232, D+T alone, and the combination of KRT-232 plus D+T, except for 1668, which was only treated with KRT-232 alone or D+T alone. PDX2316, PDX1668, and PDX1595 are shown as examples of the three different genetic subgroups within the group that responded to KRT-232 [Figure 1C (PDX2316) and D (PDX1595 and PDX1668)]. For PDX2316, PDX1668, and PDX1595, KRT-232 treatment resulted in a significant decrease in tumor growth rate (adj. p<0.001 for PDX2316 and 1595 and adj. p=0.018 for PDX1668), and as shown for PDX2316, KRT-232 alone also decreased the final tumor weight (adj. p=0.042) and the level of Ki-67 staining (adj. p<0.001). When KRT-232 was used in combination with the standard therapy, as shown in Figure 1D for PDX1595, the standard therapy alone did not affect the growth rate (adj. p=0.999), except for PDX0807 and there was no further decrease in growth rate comparing the rate with KRT-232 to the combination therapy (adj. p=0.999). In summary, in Group III PDX tumors, KRT-232 has a predominant effect on tumor growth inhibition that is not enhanced by D+T therapy.
Effect of KRT-232 on P53 Expression and Localization:
KRT-232 previously has been shown to increase P53 protein levels in cell lines and mouse xenografts (28,29). To determine whether KRT-232 was affecting P53 expression and localization in the melanoma PDX models, the P53 protein level was monitored by IHC in two Group III PDX tumors that had been treated in vivo with KRT-232 (Figure 1D). For both PDX1595 and PDX1668, the P53 protein level was very low in tumors from the vehicle-treated mice, but KRT-232 treatment resulted in a substantial increase in P53 protein and nuclear localization. In contrast, neither dabrafenib or trametinib altered P53 levels or localization.
Synergy in PDX tumors with BRAFv600 Mutation:
As exemplified in Figure 1 and summarized in Figure 2, the 15 PDXs studied here were classified into three subgroups: Group I PDXs did not respond to KRT-232, responded to D+T, and treatment with the combination of D+T and KRT-232 resulted in a small augmentation of tumor growth inhibition. Group II PDXs did not respond to either KRT-232 alone or D+T alone but responded to the combination of KRT-232 and D+T (KRT+D+T). Group III PDXs responded to KRT-232 monotherapy, as indicated by the gray shading. A larger t-ratio indicates that there was a larger difference between the treatment group and the comparison group. Interestingly, PDX 1767 was the only PDX with MDM2 amplification, and this PDX had the largest t-ratio when comparing the effect of KRT-232 to vehicle treatment (t-ratio=5.182). This finding is consistent with prior findings in glioblastoma cell lines, where lines with MDM2 amplification were the most sensitive to KRT-232 (27).
In the Group II PDX tumors, the range of t-ratios for the combined treatment with KRT-232 and D+T was 3.028 to 8.106, which is suggestive of a synergistic relationship between KRT-232 and D+T treatments. The synergistic effect was defined as an interaction between KRT-232 and D+T treatments that causes the total effect of the drugs to be greater than the sum of the individual effects of each drug. This analysis did not include data from PDX1179, since this NRASQ61H tumor was treated only with trametinib, in contrast to the other Group II PDX tumors that were treated with dabrafenib and trametinib. The analysis showed a significant interaction effect of KRT-232 and D+T for four independent PDX tumors over time (p=0.004) using the mixed-effect model. With model-based means, tumor growth rate with 95% confidence intervals for each treatment was estimated (Figure 2B). The multiple comparison graph showed that the tumor growth rate in the combination group (KRT+D+T) was significantly reduced compared to either the KRT-232 group (Tukey’s adjusted p<0.001) or the D+T group (Tukey’s adjusted p<0.001). There was no difference in the tumor growth rate between KRT-232 alone and D+T groups (Tukey’s adjusted p=0.696). In other words, the Group II PDX tumors did not respond to KRT-232 or D+T alone, but when administered together there was synergistic inhibition of tumor growth. The power of this analysis is based on analyzing treatment effectiveness for multiple independent PDX tumors (Figure 2).
Morphological Changes Associated with Synergism:
Interestingly, analysis of H&E staining of those PDX tumors that had the highest t-ratios when comparing the effect of KRT-232 treatment to the effect of KRT-232 + D+T revealed morphologic differences after KRT-232 and D+T therapy (Figure 3). H&E staining of tumors isolated from vehicle-treated and D+T treated cells revealed that the tumor cells were tightly packed with minimal stroma, were of uniform size and appearance, and had large nuclei with a high mitotic rate and a high Ki67 expression. In contrast, PDX tumors exhibiting a synergistic growth-inhibitory response to KRT-232 and D+T were characterized by mostly karyorrhectic nuclei with vacuolar changes around the karyorrhectic debris. This change in histology was only present in PDX tumors where the t-ratio comparing the effect of KRT-232 treatment to the effect of KRT-232 combined with D+T was greater than 2.6 (PDX1351 and 1946).
Biomarkers of Treatment Response: Identifying Proteins with Altered Expression in Tumors Responding Synergistically to KRT-232 and D+T
To identify changes in protein expression associated with the synergistic response to KRT-232 and D+T in Group II tumors, RPPA analysis of protein and phosphoprotein expression was conducted (Figure 3B). Unexpectedly, protein markers for different modalities of cell death, including bcl-2-like protein 4 (BAX), B-cell lymphoma 2 (BCL2), p53 upregulated modulator of apoptosis (PUMA), poly (ADP-ribose) polymerase (PARP), beclin, janus kinase (JAK), induced myeloid leukemia cell differentiation protein (MCL1), and the caspase, signal transducer and activator of transcription (STAT), and heat shock proteins (HSP) families of proteins, while present in the antibody panel, were notably absent from the list of positive hits when comparing vehicle-treated PDX tumors to KRT-232 + D+T treated tumors. In contrast, a decreased expression hypoxia-inducible factor 1 subunit alpha (HIF-1α), monocarboxylate transporter 4 (MCT4), phosphorylated N-myc downstream-regulated gene-1 (pNDRG1), and phosphorylated mitogen-activated protein kinase (pMAPK) were noted in the KRT-232 + D +T treatment group as compared to vehicle-treated tumors. Lactate dehydrogenase A (LDHA) was also decreased, although the fold change was small and below the cutoff of log2(FC)=+/− 0.4. The decreased levels of phosphorylated MAPK kinase were expected since these tumors were treated with dabrafenib and trametinib, both of which regulate the phosphorylation of MAPK. In contrast, in response to the combination of KRT-232 and D+T, the observed decreased expression of HIF-1-α, MCT-4, LDHA, and decreased phosphorylation of NDRG was unexpected and indicate treatment-mediated decreased rates of glycolysis (52–54). The change in expression of HIF-1-α, pNDRG and LDHA was only seen in PDX tumors treated with the combination of KRT-232 and D+T (Group II: PDX1179, 1351, and 2252) where synergy was exhibited. Changes in MCT-4 expression were also observed when KRT-232 was used as a single agent (Figure 4D and E). Furthermore, Group I PDX tumors 1839, 2195, and 2252 responded to D+T as a single agent, and exhibited decreased phosphorylation of MAPK, but did not exhibit altered expression of HIF-1-α, MCT-4, LDHA, nor was there a decrease in phosphorylation of NDRG in response to D+T without KRT-232 (Supplementary Figure S3). These data suggest that the synergistic action of KRT-232 and D+T results in metabolic reprogramming of the tumor cells.
Figure 4A, Biomarkers as Predictors of KRT-232 Response. Oncoprint Cluster Analysis.
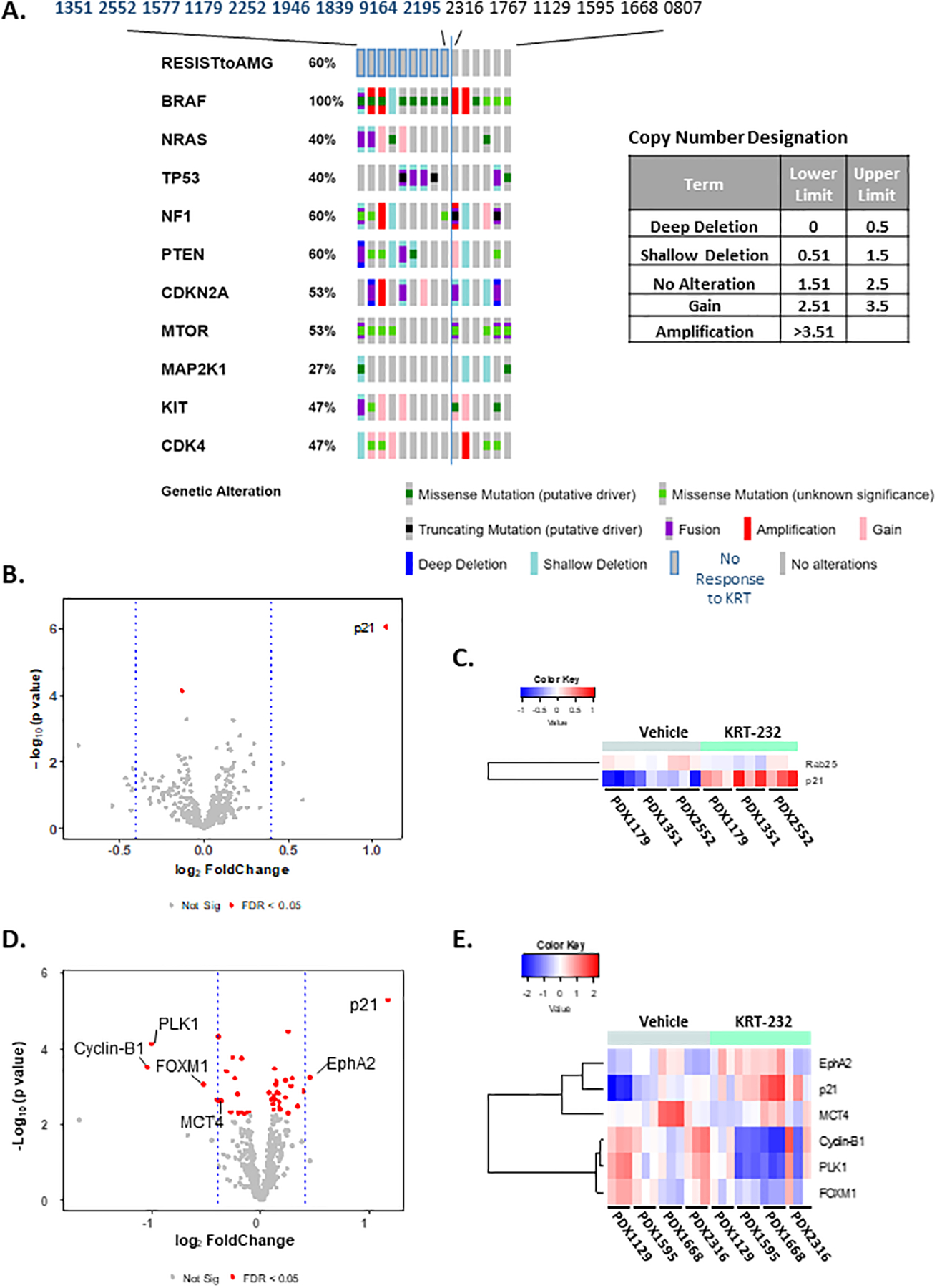
DNA sequence analysis was performed using NextGen sequencing. Paired targeted analysis for all 15 PDX tumors was performed using Oncoprint on the cBioPortal (http://www.cbioportal.org/) hosted by Sloan Kettering Institute. These results show the analysis of the 10 genes listed in Table 1. The inset table lists the brackets and terms used to describe the copy number variations. Figure 4 B-E, Comparison of RPPA Analysis Between Group II and III PDX tumors Treated with KRT-232. Volcano plots (B&D) and heat maps (C&E) of RPPA data obtained from vehicle-treated and KRT-232 treated Group II (B&C) and Group III PDX (D&E) tumors samples. Three tumors were analyzed for each PDX treatment. Group II PDX tumors were PDX1179, 1351 and 2552 and Group III PDX tumors were PDX1129, 1595, 1668 and 2316. Only those proteins with an FDR <0.05 and log2(FC)=+/− 0.4 were included in the heat map.
Biomarkers of Treatment Response: Identifying Tumors That Respond to KRT-232 Monotherapy.
To identify potential biomarkers for KRT-232 response we examined both the genetic mutation and copy number variation data obtained by NGS and RPPA analysis (Figure 4). Analysis and comparison of basal (vehicle-treated) protein and phosphoprotein expression in PDX tumors that were either resistant or responsive to KRT-232 did not identify any significant proteins or phosphoproteins (FDR >0.05) that could be used as predictors of KRT-232 responsiveness, as shown in the Volcano Plot comparing vehicle-treated samples from PDX tumors in Groups I and II compared to those in Group III (Supplementary Figure S4). While protein expression patterns were not a predictor of KRT-232 responsiveness, the BRAF mutation status was a strong predictor of KRT-232 responsiveness.
The Oncoprint Cluster Analysis shown in Figure 4 represents the analysis with the 10 most relevant genetic alterations, as discussed in Table 1. No further insights were gained by including all 300 genes analyzed by NGS (data not shown). This analysis indicates that the mutational status of BRAF is highly correlated with responsiveness to KRT-232. Of the 9 PDX tumors that were resistant to KRT-232 (Groups I and II), all but one had a V600 mutation in BRAF (Table 1).
In contrast to the high prevalence of V600 BRAF mutations in the PDX tumors that were resistant to KRT-232, all of the PDX tumors that responded to KRT-232 lacked the BRAFV600 mutation, although other mutations were noted in the BRAF gene. PDX1129 had two coding changes, D594N and K483T and PDX1668 and 1595 had missense mutations, of unknown significance. For PDX1668 the two non-synonymous BRAF mutations were S147N and N140T and for PDX1595 the BRAF mutation was P334S. While there are reports of the D594N mutation in melanoma, colorectal and lung carcinomas and hairy cell leukemia, none of the other mutations have been reported and there are no data on the biological significance of these mutations (43–45). Amplification of the BRAF gene also occurred in the PDX tumors, but occurred in equal percentages in both the PDX tumors that were resistant to KRT-232 (3/9 or 33.3%) and those that were sensitive (2/6 or 33.3%). Statistical analysis of the correlation between the BRAFV600 mutation and the in vivo effectiveness of KRT-232 in decreasing tumor growth, as measured by the t-ratio, was analyzed using the Wilcoxon rank sums test and Pearson’s product-moment correlation. The Point-Biserial correlation coefficient (a special case of Person’s correlation coefficient) was −0.7938, with a Bonferroni adjusted p-value of <0.001. A dot plot showing the correlation is shown in Supplementary Figure S5.
RPPA analysis of Group III PDX tumors (1129, 1595, 1668 and 2316) revealed altered levels of six proteins in KRT-232 monotherapy responsive tumors, based on the limits of an FDR < 0.5 and log2(FC)>+/− 0.4. The P53-inducible P21 protein (CDKN1A) exhibited the most significant increase in expression in response to KRT-232 treatment. The other notable protein induced by KRT-232 was the receptor tyrosine kinase ephrin type-A receptor 2, also known as (erythropoietin-producing hepatocellular receptor A2 (EPHA2), which has been reported to play a critical role in oncogenic signaling in many types of solid tumors (55). This increase in EPHA2 was offset by KRT-232-induced decreases in the cell cycle regulatory protein Cyclin B1 (CCNB1), polo-like kinase 1 (PLK1) which belongs to the CDC5/Polo subfamily, and forkhead box protein M1 (FoxM1) (56,57). These three proteins are involved in the regulation of M-phase of the cell cycle (58,59). (Figure 4 D&E). Finally, the monocarboxylate transporter 4 (MCT4), which releases lactate from glycolytic tumors (60) was decreased by KRT-232 treatment. Altogether, these changes in protein expression in response to KRT-232 should result in a reduction in tumor growth.
Interestingly, RPPA analysis of PDX tumors that did not respond to KRT-232 treatment had a similar increase in p21 expression (Figure 4 B&C) but the increase in P21 was not accompanied by significant changes in the downstream pathways regulated by MDM2 including cell cycle regulatory proteins or proteins involved in the regulation of apoptosis (61). Since all these PDX tumors have activating mutations in the MEK/ERK pathway, these data suggest that activation of BRAF or NRAS interferes with the pathways associated with the growth inhibitory effects of KRT-232.
Role of TP53 in Responsiveness to KRT-232.
Detailed analysis of the TP53 status in the panel of 15 melanoma PDX tumors revealed 6 PDXs had either a non-synonymous mutation in the TP53 gene and/or a loss of heterozygosity (Table 1A, B and Supplementary Table S1B). As shown in Figure 2A, the PDX tumors with alterations in TP53 status still responded to KRT-232 either as a single agent (PDX0807 and PDX1668) or in combination with D+T (PDX2252 and PDX1946), but the response was considerably reduced compared to the TP53wt tumors and was at the lower limits of significance. For example, PDX0807 and PDX1668, both of which responded to KRT-232 as a single agent had t-ratios of 2.598 and 2.829, respectively, while the TP53wt PDX tumors in Group III, PDX 1595, 2316, 1129 and 1767, had t-ratios greater than 3.5 (Figure 2), indicating stronger inhibition of tumor growth in TP53wt tumors.
Effects of Navitoclax on Growth of Melanoma PDX tumors:
BRAFV600 mutant melanoma tumors often develop resistance to MAPK inhibitors like dabrafenib and trametinib. We postulated that D+T resistant tumors might respond to a Bcl-2 inhibitor to block tumor growth and proliferation through an alternate pathway. Indeed, it has been demonstrated that combining the MDM2 inhibitor, idasanutlin RG7388, with the BCL-2 inhibitor, ABT-199, provides better therapeutic efficacy than either drug alone in acute myeloid leukemia (AML) (62). To determine if similar effects occur in melanoma, two BRAFmut and two BRAFwt tumors were treated with navitoclax, a BCL2 inhibitor, either as a single agent or in combination with KRT-232. The summary data for navitoclax treatment are shown in Figure 5C. Navitoclax slightly enhanced tumor growth for two PDX tumors (PDX1351 and 1577), as indicated by the negative t-ratio. While navitoclax was not effective as a single agent in any PDX tumor, navitoclax in combination with KRT-232 in BRAFV600E (PDX1351 and 1577) tumors was more effective than either drug alone and resulted in tumor regression in three PDX models, as shown by example for PDX1577 (Figure 5). Interestingly, the growth of only mutant BRAFV600E tumors was inhibited by the combination of navitoclax and KRT-232. In BRAFV600wt tumors (PDX1129 and PDX1668), the effects of KRT-232 and navitoclax were not more effective than KRT-232 alone as indicated by a t-ratio of less than 2.6 when comparing KRT-232 + navitoclax treatment groups to KRT-232 alone. Similar effects were noted in the weights of the final tumors.
Figure 5A, Response to Navitoclax. Tumor Growth of PDX1577.
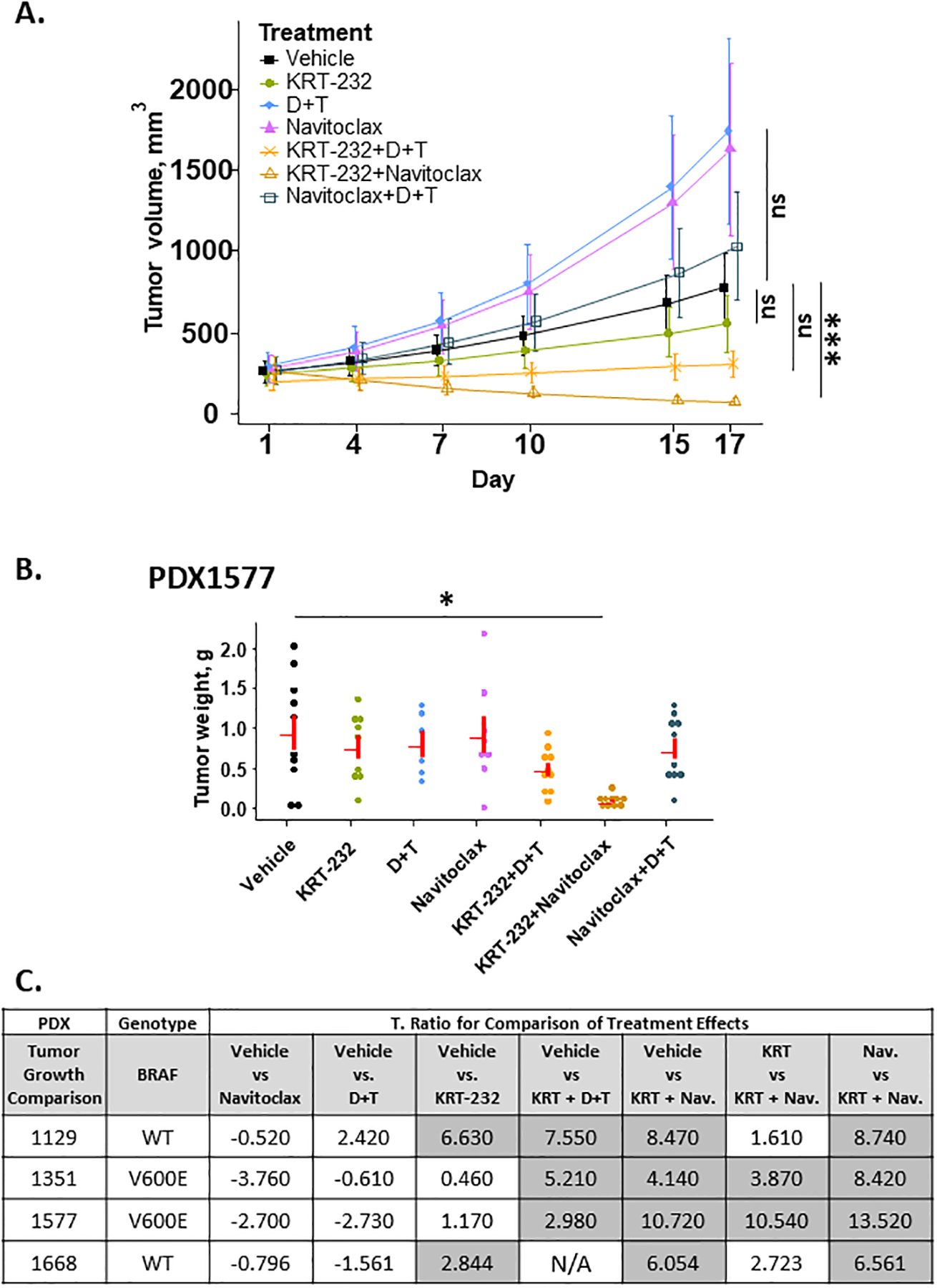
Tumor volume was analyzed on the natural log scale to better meet normality assumptions and the predicted mean and standard error of tumor volume over time for each treatment group is shown. Figure 5B, Final Tumor Weight of PDX 1577. Dot plot of tumor weight (g) by treatment (gray line: mean with standard error) Figure 5C, T-ratio Table for PDX tumors Treated with Navitoclax. A t-ratio table for pairwise comparison in tumor growth rate between treatments based on the mixed-effect model with post hoc tests is shown.
Discussion:
While recent therapeutic advances for metastatic melanoma have significantly increased the overall survival of melanoma patients, a subset of patients does not respond to immunotherapy or targeted therapies. In this study, utilizing a panel of melanoma PDX tumors we observed that therapy with KRT-232, based upon mutational status of the tumors, either singularly or in combination with dabrafenib and trametinib treatment effectively decreased tumor growth in 100% of the tumors tested. Similar results were obtained in a small Phase I/2 clinical trial testing KRT-232 with trametinib and/or dabrafenib (NCT02110355), where patients with TP53wt metastatic cutaneous melanoma were treated with KRT-232 at 120, 180 or 240 mg QD (7 days of each 3 week cycle) in combination with T (BRAFnonV600-mutant) or D+T (BRAFV600-mutant). KRT-232 + T was found to reduce tumor size in 73% of the patients with metastatic cutaneous melanoma expressing BRAFnonV600-mutant, with 13% (2/15) showing a greater than 30% reduction in size according to the RECIST criteria (44,45). Furthermore, 100% (6/6) of patients with BRAFV600-mutant melanoma treated with KRT-232 in combination with trametinib or dabrafenib had a reduction in tumor size, with 67% (4/6) patients having a decrease in tumor size of greater than 30%. Based on preliminary results, the KRT-232 maximally tolerated dose was 180 mg, based on observed adverse effects at 240 mg in the KRT-232 + T treatment arm. The incidence of adverse effects was notable, albeit acceptable, with 1 patient having a grade 3 adverse event and all remaining patients have either a G1 or G2 adverse event (64). Similarly, based on the calculations described by Nair and Jacob (65), the human equivalent dose for 50 mg/kg KRT-232 used in mice is ~250 mg KRT-232 for an average 62 kg individual.
In our study, tumors responding to KRT-232 plus dabrafenib and/or trametinib could be classified into three separate groups. Group I, consisted of BRAFV600E mutant PDX tumors, which responded to dabrafenib and trametinib but not to KRT-232 as a single agent. In combination, these agents further inhibited tumor growth. Group II, consisted of BRAFV600E/M and NRASQ61H mutant tumors that did not respond to either KRT-232 or dabrafenib and/or trametinib alone, but when used in combination, the inhibition of tumor growth was demonstrated to be statistically significant and synergistic. Finally, in Group III, the BRAFV600wt PDX tumors responded to KRT-232 alone with significant inhibition of tumor growth. The mutational status of BRAF was an effective predictor of KRT-232 monotherapy response. In our PDX panel, only those tumors that were BRAFV600wt were responsive to KRT-232 alone, while, except for PDX1179, tumors with BRAFV600 mutations were resistant to KRT-232, but responded synergistically to KRT-232 combined with dabrafenib and trametinib.
In another tumor type, acute myeloid leukemia (AML), Pan et al. reported that some AML cell lines are sensitive to the MDM2 inhibitor, RG7388 (idasanutlin) which is a more potent second-generation nutlin, while other AML cell lines were resistant (62). As seen with KRT-232, in both the RG7388 sensitive and resistant cell lines, the protein level of P21 is abundantly induced. Through a series of elegant experiments using RG7388 in combination with a Bcl-2 inhibitor, ABT-199 (venetoclax), they demonstrated that P53 plays a pivotal role in regulating the Ras/Raf/MEK/ERK signaling cascade (62). While Pan et al. did not explore the mechanistic role of P53 in regulating the MAPK pathway, others have identified P53-regulated transcription of four phosphatases, wild-type p53-induced phosphatase 1 (Wip1), mitogen-activated protein kinase phosphatase 1 (MKP1), phosphatase of activated cells 1, also known as dual specificity phosphatase 2 (PAC1/DUSP2), and DUSP5, that negatively regulate MAPK signaling (66). While further experiments are needed, we propose that in tumors with BRAFV600 mutations, activation of P53 through MDM2 inhibition will not inhibit the MAPK pathway; therefore, those tumors with BRAFV600 mutations are resistant to the effects of KRT-232 and other MDM2 inhibitors. However, in the presence of dabrafenib and trametinib, two inhibitors of the MAPK pathway, inhibition of the constitutively activated MAPK pathway reverses the resistance to MDM2 inhibition, resulting in a decrease in MAPK T202/Y204 phosphorylation. The MAPK pathway is also a key regulator of the transcription factor Myc through both phosphorylation and transcription control (67–70). While our RPPA panel did not include p-Myc or N-Myc, NDRG1, an N-Myc downstream-regulated gene, was decreased by KRT-232 plus D+T treatment. Additionally, Myc has been shown to stabilize HIF-1α (71). Consistent with the concept that KRT-232 plus D+T treatment down regulates Myc-mediated transcription, we observed a decrease in HIF-1α in KRT-232 plus D+T treated BRAFV600 tumors. Therefore, our studies suggest that the treatment of melanoma PDX tumors expressing the activating BRAFV600 mutation with the combination of dabrafenib, trametinib and KRT-232, modulates MYC and HIF-1. These proteins play a key role in the regulation of the Warburg phenomenon (72–75) and are key regulators of the glycolytic switch in tumors (76).
It has also been reported that like dabrafenib and trametinib, Bcl-2 inhibitors can overcome the resistance to MDM2 inhibitors (62). Preclinical studies have shown that mice implanted with OCL-AML3 leukemic cells that are resistant to both the MDM2 inhibitor, RG7388, and the Bcl-2 inhibitor, ABT-199, exhibited enhanced overall survival with the two agents combined. Similarly, we observed that treatment of BRAFV600 mutant PDX tumors PDX1351 and 1577 that are resistant to KRT-232, with KRT-232 combined with navitoclax significantly inhibited tumor growth when neither agent was effective alone. These data suggest that, as in AML, co-treatment with Bcl2 inhibitors can overcome the resistance to MDM2 inhibitors in tumors in which the MAPK pathway is constitutively activated. It is interesting to note that in the PDX tumors that were treated with KRT-232 and navitoclax or KRT-232 and D+T, the combination of KRT-232 with navitoclax was more effective and not only inhibited tumor growth but caused tumor regression.
While the combination of KRT-232 plus dabrafenib and trametinib in BRAFmutV600 tumors resulted predominantly in a decreased expression of key proteins involved in the regulation of glycolysis, KRT-232 monotherapy treatment of BRAFwtV600 tumors resulted in a decrease in expression of a triad of proteins regulating the G2/M checkpoint in the cell cycle. The G2/M DNA damage checkpoint serves to prevent the cell from entering mitosis (M-phase) with genomic DNA damage and the key regulators of this process are FoxM1, PLK1, and Cyclin B1, all of which were decreased by KRT-232 treatment. FoxM1 expression is increased in a variety of solid tumors, including melanoma, and inhibition of FoxM1 leads to a decrease in cell proliferation and migration, metastasis and angiogenesis. Furthermore, analysis of the TGCA database illustrated that high levels of FoxM1 are related to poor prognosis in most solid tumors (58). P53, both directly and indirectly through p21, has been reported to decrease FoxM1 expression (77,78). FoxM1 induces the expression of the Cdk1 activators, cyclin B and Cdc25 (79), so a decrease in FoxM1 expression should coincide with a decrease in cyclin B and Cdc25, as we observed in the RPPA data. Interestingly, cyclin B and polo-like kinases (PLK) are proposed as critical transcriptional targets of FoxM1 at the G2/M transition and PLK1 phosphorylates FoxM1 to further activate it as a transcription factor (59,80). Therefore, KRT-232, through the P53 dependent inhibition of FoxM1 expression, has a direct effect in regulating the G2/M checkpoint.
It is important to note that the tumors used in this study were heterogeneous and all PDX tumors included some mouse and potentially human stromal tissue. The lack of homozygosity for driver mutations likely reflected the heterogeneous nature of tumors instead of the presence of non-tumor tissue within our samples since the NGS panel was specific for human genes, though we cannot fully rule out a contribution of stroma to wild type allele detection (Supplementary Figure S1B). Similar heterogeneity was seen with TP53. In this study, we included four PDX tumors with mutations in TP53. Only one of the lines, PDX9164 had a heterozygous TP53 mutation, with a frequency of 1, at codon 192 (Q192*) and this mutation has a FATHMM pathogenic score of 0.93 suggesting it is highly pathogenic. The other mutations occurred at lower frequencies, 0.265 for PDX0807 (A138ADG and T140S); 0.607 for PDX 2252 (R196*); and 0.484 and 0.235 for PDX1839 (R306* and Y236H). This heterogeneity complicates the interpretation of the effectiveness of KRT-232 in TP53 mutant versus wild type tumors. It is interesting to note, however, that while KRT-232 was somewhat effective in melanoma PDX tumors with mutant TP53, the effect was at the lower limit of significance. Furthermore, while TP53mut and BRAFV600 mutant, PDX9164 did not respond to KRT-232 monotherapy, it did respond synergistically to KRT-232 plus dabrafenib and trametinib. Similar results were noted in a Phase 1 trial of RG7112, a member of the Nutlin family of MDM2 inhibitors, in leukemia. In this study, 3 of 19 patients with TP53 mutations showed evidence of response to RG7112 as demonstrated by a decrease peripheral blast count and one patient was reported to have stable disease for more than 2 years (81). While neither study was designed to specifically address the effectiveness of KRT-232 in TP53 mutant tumors, these data suggest that KRT-232 may have limited effectiveness in TP53 mutant tumors.
We demonstrate here that 60% of melanoma PDX tumors have an inherent resistance to KRT-232 and this correlates with the incidence of BRAFV600 mutations. In this study, of the eight BRAFV600 mutant PDX tumors treated with dabrafenib and trametinib, five (62.5%) were resistant to dabrafenib and trametinib. Two of the five resistant PDX tumors came from patients previously treated with dabrafenib and trametinib who had progressed on treatment. In these two MAPKi-resistant PDX tumors, the resistance is undoubtedly acquired. The study of MAPKi-resistant melanoma has been extensively studied and the mechanism of resistance is complex and multifactorial. The causative factors that contribute to MAPKi-resistance can be broadly classified into three categories: mutational events and non-mutational events, which are tumor inherent and lead to either MAPK pathway reactivation or activation of a parallel signaling pathway, and changes in the surrounding microenvironment (82–84). Based on the pleiotropic nature of drug resistance it is interesting, although not surprising, the two PDX tumors previously treated with D+T remained resistant. It is also interesting that none of the BRAFV600E mutant tumors that responded to D+T were derived from patients who had prior exposure to D+T.
Overall, our results demonstrate the efficacy of the MDM2 antagonist as a single agent or as a part of a therapeutic combination with either MAPK pathway or Bcl2–targeting agents in preclinical melanoma models. We have previously shown in preclinical studies that melanoma tumors exhibit a limited response to CDK4/6 inhibitors but the addition of an MDM2 antagonist results in significantly increased inhibition of tumor growth (85). Notably, tumors that acquired resistance to the current standard of care approaches such as immunotherapy and BRAF/MEK targeted therapy, are responsive to the MDM2 antagonist given alone or as a part of combination treatment. These findings support the clinical development of MDM2 antagonists as a second-line treatment for metastatic melanoma.
Supplementary Material
Acknowledgments:
Animal husbandry and animal care was performed by the Division of Animal Care, Vanderbilt University Medical Center (https://www.vumc.org/acup/system/files/DAC%20Brochure.pdf)
Histology and immunostaining were performed in the Translational Pathology Shared Resource at Vanderbilt University Medical Center, supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485-14 and the Vanderbilt Mouse Metabolic Phenotyping Center Grant 5U24DK059637-13 (https://ww2.mc.vanderbilt.edu/TPSR/30962). The authors wish to acknowledge helpful discussions with Drs. Kelli Boyd and Lauren E. Himmel.
Whole slide imaging and quantification of immunostaining were performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center (www.mc.vanderbilt.edu/dhsr).
Next-Generation sequencing was provided by Vanderbilt Technologies for Advanced Genomics (VANTAGE: http://vantage.vanderbilt.edu/).
RPPA analysis was performed at the Reverse Phase Protein Array (Core Facility at MD Anderson Cancer Center. This facility is funded by NCI# CA16672.
The DNA extraction was performed in the Vanderbilt Innovative Translational Research Shared Resource supported by the Vanderbilt-Ingram Cancer Center (5P30CA068485-21,) the TJ Martell Foundation, and the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation.
Short tandem repeat (STR) profiling was performed by the Interdisciplinary Center for Biotechnology Research at the University of Florida. The authors wish to acknowledge the technical expertise of the Gene Expression and Genotyping Core of the Interdisciplinary Center for Biotechnology at the University of Florida, Gainesville for performing the STR Assay
Human Melanoma samples were obtained through the Melanoma BioRepository supported by James C. Bradford Jr. Melanoma Fund.
The authors wish to acknowledge Dr. Chi Yan for his helpful discussions and invaluable help with the figures.
Support: 5P30 CA068485-S3, CA116021(AR), VA SRCS Award (AR), NCI-K23CA204726(DJ), Harry J Lloyd Charitable Trust (AV), BCRF IIDRP-16-001 (AV), NCI-R37CA233770 (AV).
Footnotes
In experiments involving animals, the experiments were executed in compliance with institutional guidelines and regulations and after approval from the appropriate institutional review board (IACUC protocol number M1600236). The studies involving human tissue were conducted in accordance with the ethical principles and guidelines for research outlined in The Belmont Report (Vanderbilt IRB# M030220), and informed written consent was obtained from each subject or each subject’s guardian.
Conflicts of Interest: Doug Johnson is on Advisory Boards for Array Biopharma, BMS, Jansen Merck, Novartis and received research funding from BMS and Incyte. Ann Richmond is on the Advisory Board for Polyphor. Kim Dahlman received research funding from Kadmon, Inc.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA JA. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Glob Cancer Stat 2018 [Internet]. 2018. Available from: http://gco.iarc.fr [DOI] [PubMed] [Google Scholar]
- 2.Facts C. Cancer Facts & Figures. Am Cancer Soc [Internet]. 2019; Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf [Google Scholar]
- 3.Davies Helen, Bignell Graham R., Cox Charles S P, Edkins Sarah, Clegg Sheila, Teague Jon W H, Garnett Mathew J., Bottomley William, Davis Neil D E, Ewing Rebecca, Floyd Yvonne, Gray Kristian H S, Hawes Rachel, Hughes Jaime, Kosmidou Vivian M A, Mould Catherine, Parker Adrian, Stevens Claire W S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 4.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy BY, Miller DM, Tsao H. Somatic driver mutations in melanoma. Cancer. 2017;123:2104–17. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: Targeted strategies for melanoma. Nat Rev Cancer. Nature Publishing Group; 2012;12:349–61. [DOI] [PubMed] [Google Scholar]
- 7.Muñoz-Couselo E, Adelantado EZ, Ortiz C, García JS, Perez-Garcia J. NRAS-mutant melanoma: Current challenges and future prospect. Onco Targets Ther. 2017;10:3941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet [Internet]. Elsevier Ltd; 2012;380:358–65. Available from: 10.1016/S0140-6736(12)60868-X [DOI] [PubMed] [Google Scholar]
- 9.Chapman P, Hauschild A, Robert C, Haanen J, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JT, Li L, Brafford PA, Van Den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23:820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreno B, Larkin JMG, Wu FS, Hassel JC, Robert C, Becker JC, et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N Engl J Med. 2012;367:107–14. [DOI] [PubMed] [Google Scholar]
- 12.Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined Vemurafenib and Cobimetinib in BRAF -Mutated Melanoma. N Engl J Med. 2014;371:1867–76. [DOI] [PubMed] [Google Scholar]
- 13.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600Emelanoma. Sci Transl Med. 2015;7:279ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mai R, Zhou S, Zhong W, Rong S, Cong Z, Li Y, et al. Therapeutic efficacy of combined BRAF and MEK inhibition in metastatic melanoma: a comprehensive network meta-analysis of randomized controlled trials. Oncotarget. 2015;6:28502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol Off J Eur Soc Med Oncol. 2017;28:1631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hauschild A, Dummer R, Schadendorf D, Santinami M, Atkinson V, Mandala M, et al. Longer Follow-Up Confirms Relapse-Free Survival Benefit With Adjuvant Dabrafenib Plus Trametinib in Patients With Resected BRAF V600–Mutant Stage III Melanoma. J Clin Oncol. 2018;36:3441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. Elsevier Ltd; 2018;19:1315–27. [DOI] [PubMed] [Google Scholar]
- 18.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Echevarría-Vargas IM, Villanueva J. Combating NRAS mutant melanoma: from bench to bedside. Melanoma Manag. 2017;4:183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polsky D, Bastian BC, Hazan C, Melzer K, Pack J, Houghton A Busam K, Cordon-cardo C, Osman I HDM2 Protein Overexpression, but not Gene Amplificaion is Related to Tumorgenesis of Cutaneous Melanoma. Cancer Res. 2001;61:7642–6. [PubMed] [Google Scholar]
- 22.Bastian BC. The Molecular Pathology of Melanoma: An Integrated Taxonomy of Melanocytic Neoplasia. Annu Rev Pathol Mech Dis. 2014;9:239–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jochemsen AG. Reactivation of p53 as therapeutic intervention for malignant melanoma. Curr Opin Oncol. 2014;26:114–9. [DOI] [PubMed] [Google Scholar]
- 24.Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–9. [DOI] [PubMed] [Google Scholar]
- 25.Merkel O, Taylor N, Prutsch N, Staber PB, Moriggl R, Turner SD, et al. When the guardian sleeps: Reactivation of the p53 pathway in cancer. Mutat Res - Rev Mutat Res. Elsevier B.V; 2017;773:1–13. [DOI] [PubMed] [Google Scholar]
- 26.Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. Journal of Hematology & Oncology; 2017;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Her NG, Oh JW, Oh YJ, Han S, Cho HJ, Lee Y, et al. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. Springer US; 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol Cancer Ther. 2015;14:649–58. [DOI] [PubMed] [Google Scholar]
- 29.Werner L, Huang S, Francis DM, Armstrong EA, Ma F, Li C, et al. Small Molecule Inhibition of MDM2-p53 Interaction Augments Radiation Response in Human Tumors. Mol Cancer Ther. 2015;14:1994–2003. [DOI] [PubMed] [Google Scholar]
- 30.Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep. ElsevierCompany; 2017;21:1953–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garman B, Anastopoulos IN, Krepler C, Brafford P, Sproesser K, Jiang Y, et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017;21:1936–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truvé K, Nilsson LM, Lopez MD, Nilsson JA, Ny L, Nilsson O, et al. Abstract B38: Melanoma patient-derived xenografts accurately models the disease and develop fast enough to guide treatment decisions. Cancer Res. 2015;75:B38–B38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, et al. Mdm2 and aurora kinase A inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res. 2015;75:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meehan TF, Conte N, Goldstein T, Inghirami G, Murakami MA, Brabetz S, et al. PDX-MI: Minimal information for patient-derived tumor xenograft models. Cancer Res. 2017;77:e62–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Aksoy B arman, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6:pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenth R. Estimated Marginal Means, aka Least-Squares Means. R package version [Internet] 2019. Available from: https://cran.r-project.org/package=emmeans
- 38.Cameron AC, Trivedi PK. Microeconometrics: Methods and Applications. New York: Cambridge University Press; 2005. [Google Scholar]
- 39.Hartman RI, Lin JY. Cutaneous Melanoma—A Review in Detection, Staging, and Management. Hematol Oncol Clin North Am. Elsevier Inc; 2019;33:25–38. [DOI] [PubMed] [Google Scholar]
- 40.Lovly CM, Dahlman KB, Fohn LE, Su Z, Dias-Santagata D, Hicks DJ, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7:e35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stones CJ, Kim JE, Joseph WR, Leung E, Marshall ES, Finlay GJ, et al. Comparison of responses of human melanoma cell lines to MEK and BRAF inhibitors. 2013;4:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyd EM, Bench AJ, van ‘t Veer MB, Wright P, Bloxham DM, Follows GA, et al. High resolution melting analysis for detection of BRAF exon 15 mutations in hairy cell leukaemia and other lymphoid malignancies. Br J Haematol. 2011;155:609–12. [DOI] [PubMed] [Google Scholar]
- 44.Boursault L, Haddad V, Vergier B, Cappellen D, Verdon S, Bellocq JP, et al. Tumor Homogeneity between Primary and Metastatic Sites for BRAF Status in Metastatic Melanoma Determined by Immunohistochemical and Molecular Testing. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mufti M, Ching S, Farjami S, Shahangian S, Sobnosky S. A Case Series of Two Patients Presenting With Pericardial Effusion as First Manifestation of Non-Small Cell Lung Cancer With BRAF Mutation and Expression Of PD-L1. World J Oncol. 2018;9:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46:1258–63. [DOI] [PubMed] [Google Scholar]
- 47.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. Nature Publishing Group; 2016;34:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin). 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. Nature Publishing Group; 2017;49:1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willis BC, Johnson G, Wang J, Cohen C. SOX10: A useful marker for identifying metastatic melanoma in sentinel lymph nodes. Appl Immunohistochem Mol Morphol. 2015;23:109–12. [DOI] [PubMed] [Google Scholar]
- 51.Berset M, Cerottini J-P, Guggisberg D, Romero P, Burri FO, Rimoldi D, et al. Expression of Melan-a / Mart-1 Antigen As a Prognostic Factor in. Int J Cancer. 2001;77:73–7. [DOI] [PubMed] [Google Scholar]
- 52.Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (review). Oncol Lett. 2012;4:1151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warburg O. On the Origin of Cancer Cells. Science (80-). 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 54.Tran Q, Lee H, Park J, Kim S, Park J. Targeting Cancer Metabolism - Revisiting the Warburg Effects. Toxicol Res. 2016;32:177–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y, Sakurai H. Emerging and diverse functions of the EphA2 noncanonical pathway in cancer progression. Biol Pharm Bull. 2017;40:1616–24. [DOI] [PubMed] [Google Scholar]
- 56.Stark GR, Taylor WR. Analyzing the G2 / M Checkpoint. Method Mol Biol. 2004;280:51–82. [DOI] [PubMed] [Google Scholar]
- 57.Down CF, Millour J, Lam EWF, Watson RJ. Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. Biochim Biophys Acta - Gene Regul Mech. Elsevier B.V; 2012;1819:855–62. [DOI] [PubMed] [Google Scholar]
- 58.Liao G Bin, Li XZ, Zeng S, Liu C, Yang SM, Yang L, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. Cell Communication and Signaling; 2018;16:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murakami H, Aiba H, Nakanishi M, Murakami-Tonami Y. Regulation of yeast forkhead transcription factors and FoxM1 by cyclin-dependent and polo-like kinases. Cell Cycle. 2010;9:3233–42. [DOI] [PubMed] [Google Scholar]
- 60.Draoui N, Feron O. Lactate shuttles at a glance: From physiological paradigms to anti-cancer treatments. DMM Dis Model Mech. 2011;4:727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.El-Deiry WS. p21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016;76:5189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell. Elsevier Inc; 2017;32:748–760.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eisenhauer E a, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. Elsevier Ltd; 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 64.Moschos SJ, Sandhu SK, Lewis KD, Sullivan RJ, Johnson DB, Zhang Y, et al. Phase 1 study of the p53-MDM2 inhibitor AMG 232 combined with trametinib plus dabrafenib or trametinib in patients (Pts) with TP53 wild type (TP53WT) metastatic cutaneous melanoma (MCM). J Clin Oncol. 2017;35:2575–2575. [Google Scholar]
- 65.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharmacol. 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gen SW. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3:156–61. [DOI] [PubMed] [Google Scholar]
- 67.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–79. [DOI] [PubMed] [Google Scholar]
- 68.Wei Z, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. [DOI] [PubMed] [Google Scholar]
- 69.Zhao Q, Assimopoulou AN, Klauck SM, Damianakos H, Chinou I, Kretschmer N, et al. Inhibition of c-MYC with involvement of ERK/JNK/MAPK and AKT pathways as a novel mechanism for shikonin and its derivatives in killing leukemia cells. Oncotarget. 2015;6:38934–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105:6584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012;72:949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - The seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. Elsevier Inc; 2018;49:178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Melotte V, Qu X, Ongenaert M, Van Criekinge W, De Bruïne AP, Baldwin HS, et al. The N-myc downstream regulated gene (NDRG) family: Diverse functions, multiple applications. FASEB J. 2010;24:4153–66. [DOI] [PubMed] [Google Scholar]
- 75.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell. 2007;12:108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: How many players are involved? J Cancer. 2017;8:3430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW, et al. P53 Regulates a Mitotic Transcription Program and Determines Ploidy in Normal Mouse Liver. Hepatology. 2013;57:2004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barsotti AM, Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. Nature Publishing Group; 2009;5:4295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laoukili J, Kooistra MRH, Brás A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36. [DOI] [PubMed] [Google Scholar]
- 80.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10:1076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the phase i trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22:868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahmed F, Haass NK. Microenvironment-driven dynamic heterogeneity and phenotypic plasticity as a mechanism of melanoma therapy resistance. Front Oncol. 2018;8:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manzano JL, Layos L, Bugés C, de los Llanos Gil M, Vila L, Martínez-Balibrea E, et al. Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl Med. 2016;4:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kozar I, Margue C, Rothengatter S, Haan C, Kreis S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim Biophys Acta - Rev Cancer. 2019;1871:313–22. [DOI] [PubMed] [Google Scholar]
- 85.Vilgelm AE, Saleh N, Shattuck-Brandt R, Riemenschneider K, Slesur L, Chen SC, et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci Transl Med. 2019;11:eaav7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.