

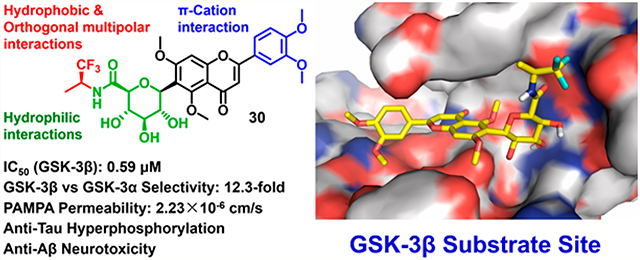
Abstract
Glycogen synthase kinase-3β (GSK-3β) is a key enzyme responsible for tau hyperphosphorylation and is a viable therapeutic target of Alzheimer’s disease (AD). We developed a new class of GSK-3β inhibitors based on the 6-C-glycosylflavone isoorientin (1). The new inhibitors are passive membrane permeable and constitutively attenuate GSK-3β mediated tau hyperphosphorylation and amyloid neurotoxicity in an AD cellular model. Enzymatic assays and kinetic studies demonstrated that compound 30 is a GSK-3β substrate-competitive inhibitor with distinct kinase selectivity, isoform-selectivity and over 310-fold increased potency as compared to 1. Structure–activity relationship analyses and in silico modeling suggest the mechanism of actions by which the hydrophobic, π–cation, and orthogonal multipolar interactions of 30 with the substrate site are critical for the GSK-3β inhibition and selectivity. The results provide new insights into GSK-3β drug discovery. The new inhibitors are valuable chemical probes and drug leads with therapeutic potential to tackle AD and other GSK-3β relevant diseases.
Keywords: C-Glycosylflavone, computer-aided drug design, glycogen synthase kinase-3, substrate-competitive inhibitor, kinase selectivity, tauopathy, Alzheimer’s disease
Graphical Abstract
■ INTRODUCTION
Alzheimer’s disease (AD) is an irreversible and progressive neurological disorder that causes memory loss, cognitive decline, and eventually death due to severe brain degeneration.1 This devastating and fatal disease, unfortunately, cannot be prevented, cured, or even slowed. Although the exact cause of AD remains unclear, increasing evidence suggests that β-amyloid (Aβ) is plausibly the pathogenic initiator of AD and tau aggregation and distribution in the brain cortices correlate closely with neuronal loss and cognitive decline in AD progression.1,2 The tau protein stabilizes the structural integrity of microtubules in neurons to maintain healthy axonal transport. In AD, aberrant phosphorylation of tau proteins disrupts their association, leading to destabilization of microtubules and mislocalization of the abnormal tau to the soma and dendrites.3 The subsequent aggregation of hyperphosphorylated tau results in formation and disposition of toxic neurofibrillary tangles (NFTs), triggering synaptic dysfunction, neuronal cell death and cognitive impairment. This aberrant signaling cascade gives rise to the tauopathy in AD.4 The continued failures of anti-Aβ approaches in AD clinical studies5,6 drive researchers to embrace an alternative approach to investigate anti-tau strategies for AD treatment.4,7
Glycogen synthase kinase-3β (GSK-3β) is a key protein kinase in the cascade leading to aberrant tau hyperphosphorylation. Elevated GSK-3β activity has been implicated in AD and other tauopathies.8 Overactive GSK-3β hyperphosphorylates over 70% of the potential phosphorylation sites on tau proteins in AD brains, which impairs the healthy interactions of tau proteins with microtubules.9 The development of selective GSK-3β inhibitors modulating aberrant tau phosphorylation is therefore a promising strategy for AD chemotherapy.10,11 To date, the United States Food and Drug Administration (FDA) has approved four AD drugs, but they only ameliorate temporary symptoms, and none target GSK-3β.10
Traditional GSK-3β inhibitors target the highly conserved ATP site. However, the limited selectivity of those inhibitors raises safety concerns owing to off-target effects and, therefore, remains a major challenge in GSK-3β based drug development.11 Despite substantial efforts in developing GSK-3β inhibitors in the past decades, to date only lithium carbonate and tideglusib (a TDZD compound) have been studied in clinical trials for AD.10 Lithium carbonate shows a weak inhibition (IC50, 2 mM),11 while tideglusib (IC50, 100 nM) is an irreversible and time-dependent inhibitor of GSK-3β.12,13 In recent years, strategies have been employed to search for selective and reversible GSK-3β inhibitors, particularly those that are not ATP-site directed. It is known that the substrate domain of GSK-3β is less conserved with a unique folding different from other kinases.11,14 Inhibitors targeting this site are thought to be more specific and selective than the ATP-competitive inhibitors.11,14 Yet few substrate-competitive inhibitors of GSK-3β have been reported.15–18 New, potent, selective and reversible inhibitors targeting the substrate site on GSK-3β are potential disease-modifying therapies for AD.
We have undertaken a different approach to discover potential substrate-competitive inhibitors of GSK-3β from natural sources. Natural products are valuable starting points for drug discovery as they have been naturally selected and optimized under evolutionary pressure and obtained privileged structures for protein binding.19 C-Glycosylflavones and their aglycones (Figure 1) omnipresent in plants are important phytochemicals noted for antioxidation, anti-inflammation, anticancer, anticardiovascular diseases, and cognitive enhancement. 20–22 We recently screened corn silks for GSK-3β inhibitors and isolated a 6-C-glycosylflavone, isoorientin (1).23 Compound 1 alleviates tau hyperphosphorylation and amyloid neurotoxicity through selective and reversible GSK-3β inhibition,23 by which the mechanism of action is substrate competition rather than the common ATP competition.23 In addition, a recent study showed that 1 and related natural flavones attenuate Aβ burden and neuroinflammation in an APPswe/PSEN1dE9 mouse model of AD.24 1 from maize crop23 is conceivably safe as supported by in vivo subchronic toxicity studies of corn silk-derived flavones in mice and rats.25 1 is a promising medicinal natural product with a novel mode of action for reducing AD burdens.23–25 However, the lack of druggable potency (IC50, 185 μM), commonly suffered by bioactive natural products, makes it challenging in pharmaceutical applications. Potency improvement, through structure–activity relationship (SAR)-based optimization, pharmacological and pharmacokinetic evaluations, as well as testing administration routes in vivo are therefore necessary to develop analogues of 1 with therapeutic potential.
Figure 1.

Natural C-glycosyl and aglycosyl flavones 1–4.
In our recent paper, preliminary examination of the predicted 3D structure of the GSK-3β–1 complex revealed that the substrate pocket of GSK-3β favors specific interactions with both the C-glycone and flavone moieties in 1.23 In particular, a cleft composed of Phe67, Val87, Leu88, and Phe93 on GSK-3β is critical for substrate recognition.26 This concave cleft could accommodate a hydrophobic moiety favoring ligand binding, which is in the vicinity of the primary hydroxyl group on Cglycone of 1. This raises our hypothesis that introduction of a hydrophobic functional group at this primary alcohol enhances affinity and selectivity of 1 to GSK-3β. In addition, methylation of the phenolic hydroxyls can improve bioavailability, metabolic stability, and potency of flavones.27
In the present study, a new series of 1 analogues containing a 6-C-glycosylflavone scaffold were semisynthesized to target the substrate site on GSK-3β. Potency, selectivity, passive membrane permeability, anti-tau hyperphosphorylation, and anti-Aβ neurotoxicity of the new inhibitors were evaluated with molecular and cellular studies, SAR analysis, and molecular modeling.
■ RESULTS
Chemistry
Design and Synthesis
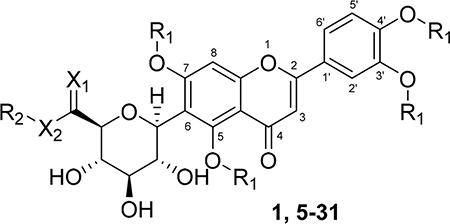
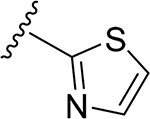
A series of new analogues of 1 were designed and synthesized (Table 1). The semisynthesis of 6-C-glycosylflavones from 1 was carried out according to the route outlined in Scheme 1. The four phenolic hydroxyls on the flavone core of 1 were first selectively methylated by TMSCHN2 in a methanolic toluene solution according to a previously described method.28 The resulting tetramethylated product (5) then underwent an oxoammonium salt-mediated oxidation. We optimized an oxidation protocol29 using a [TEMPO]+[BF4]− salt in a pyridine base solution at room temperature, which chemoselectively transformed the primary alcohol to a carboxylic acid without affecting the secondary hydroxyls on the C-glycone. This method successfully afforded the desired product (6) in over 95% yield which was confirmed by ESI-TOF-MS and NMR analyses. The carboxylic acid derivative 6 was then subjected to methyl esterification to afford 7. It is known that an ester bond is susceptible to metabolic hydrolysis. Our chemical design thus focused on amide analogues with better physiochemical stability. A small library of hydrophobic amides (8-31) was generated by coupling 6 with a series of organic amines via HCTU catalysis. Aliphatic, alicyclic, aromatic and fluorinated amines were selected for the solution-phase amidation (Table 1). The resulting hydrophobic amides were evaluated on their SAR, cytotoxicity, anti-AD activity, and passive membrane permeability. All final products were characterized by NMR and HRMS, and were over 95% purity determined by HPLC-UV at 210, 254, and 340 nm.
Table 1.
Chemical Structures of Natural and Semisynthetic C-Glycosylflavones
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| cmpd no. | Functional group |
cmpd no. | Functional group |
||||||
| R1 | X1 | X2 | R2 | R1 | X1 | X2 | R2 | ||
| 1 | H | H2 | O | H | 19 | CH3 | O | NH | 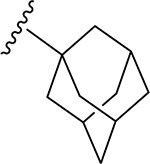 |
| 5 | CH3 | H2 | O | H | |||||
| 6 | CH3 | O | O | H | 20 | CH3 | O | NH | 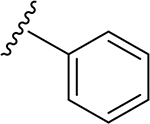 |
| 7 | CH3 | O | O | CH3 | |||||
| 8 | CH3 | O | NH | 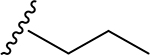 |
21 | CH3 | O | NH | 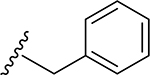 |
| 9 | CH3 | O | NH | 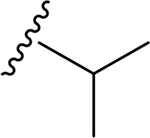 |
22 | CH3 | O | NH | 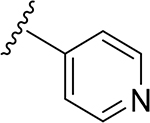 |
| 10 | CH3 | O | NH | 23 | CH3 | O | NH | ||
| 11 | CH3 | O | NH | 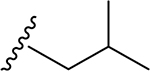 |
 |
||||
| 12 | CH3 | O | NH | 24 | CH3 | O | NH | 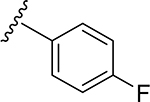 |
|
| 13 | CH3 | O | NH | ||||||
| 14 | CH3 | O | NH | 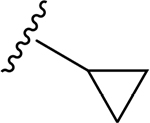 |
25 | CH3 | O | NH | 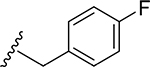 |
| 15 | CH3 | O | NH | 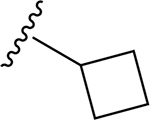 |
26 | CH3 | O | NH | 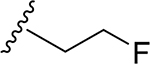 |
| 27 | CH3 | O | NH | 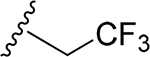 |
|||||
| 16 | CH3 | O | NH | 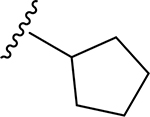 |
28 | CH3 | O | NH | |
| 17 | CH3 | O | NH | 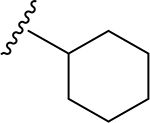 |
29 | CH3 | O | NH | 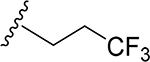 |
| 30 | CH3 | O | NH | 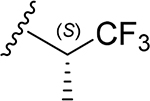 |
|||||
| 18 | CH3 | O | NH | 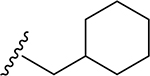 |
31 | CH3 | O | NH | 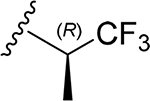 |
Scheme 1. Chemical Modifications Beginning with Isoorientina.

aReagents and conditions: (a) TMSCHN2, toluene, methanol, rt, 80%; (b) [TEMPO]+[BF4]−, DCM, pyridine, rt, 95%; (c) R-NH2, HCTU, DIPEA, DMF, DCM, rt, 80–90%.
Biological Activities and Relevance
Structure–Activity Relationship and Ligand-Lipophilic Efficiency of C-Glycosylflavones to GSK-3β
The semisynthetic 6-C-glycosylflavones (5-31) were assayed on GSK-3β inhibition in comparison to four natural flavones with structural similarities (two 6-C-glycosylflavones isoorientin 1 and isovitexin 2, one 8-C-glycosylflavone orientin 3, and a flavone aglycone luteolin 4). Among the four flavones (1–4, Figure 1), 4 showed the highest potency against GSK-3β (IC50, 3.1 μM in Table 2), but it is nonspecific and promiscuous as noted in the literature and our previous studies.23,30 1 and 2 with C-glycosides at 6-position showed a moderate potency against GSK-3β with an IC50 value of 185 and 194 μM, respectively (Figure 1 and Table 2). In contrast, 3 with an 8-C-glycoside was inactive (IC50, >5 mM). The results demonstrated that the presence and position of C-glycone on the flavone core are critical for GSK-3β inhibition, which agrees with our previous observation.23
Table 2.
Comparison of Natural and Semisynthetic C-Glycosylflavones on GSK-3β Inhibition and CLogP
| cmpd no. | GSK-3β inhibition, IC50 (μM)aj | CLogPb | cmpd no. | GSK-3β inhibition, IC50 (μM)a | CLogPb |
|---|---|---|---|---|---|
| 1 | 184.9 ± 1.4 | 0.21 | 17 | 9.0 ± 1.3 | 2.55 |
| 2 | 194.1 ± 1.0 | 0.80 | 18 | 26.0 ± 1.2 | 3.16 |
| 3 | 5153 ± 31 | 0.21 | 19 | 33.5 ± 0.8 | 3.17 |
| 4 | 3.1 ± 1.3 | 2.31 | 20 | 19.7 ± 1.1 | 2.21 |
| 5 | 239.2 ± 1.2 | 1.28 | 21 | 15.8 ± 1.2 | 2.34 |
| 6 | 237.3 ± 1.4 | 0.80 | 22 | 35.6 ± 1.3 | 1.54 |
| 7 | 135.0 ± 1.3 | 1.21 | 23 | 35.1 ± 1.0 | 1.45 |
| 8 | 29.2 ± 1.1 | 1.57 | 24 | 56.2 ± 1.2 | 2.61 |
| 9 | 5.4 ± 0.1 | 1.35 | 25 | 21.7 ± 1.2 | 2.48 |
| 10 | 33.3 ± 1.1 | 2.10 | 26 | 34.8 ± 1.0 | 0.77 |
| 11 | 31.1 ± 1.2 | 1.97 | 27 | 19.3 ± 0.8 | 1.31 |
| 12 | 28.0 ± 1.1 | 2.63 | 28 | 49.5 ± 1.2 | 1.00 |
| 13 | 9.4 ± 0.9 | 3.16 | 29 | 17.2 ± 0.9 | 1.29 |
| 14 | 25.8 ± 1.1 | 1.10 | 30 | 0.59 ± 0.04 | 1.62 |
| 15 | 61.9 ± 1.2 | 1.43 | 31 | 2.3 ± 0.5 | 1.62 |
| 16 | 13.1 ± 1.1 | 1.99 |
IC50 values were the mean of quadruplicate of each of two independent experiments.
CLogP values were calculated by a fragment-based method.34
cmpd, compound.
The tetramethylated alcohol (5) and tetramethylated carboxylic acid (6) slightly decreased the potency (IC50, 237 and 239 μM, respectively) in comparison with 1, indicating a trivial contribution of the phenolic hydroxyl groups to GSK-3β inhibition. However, a methyl ester (7) (IC50, 135 μM) increased the potency by 1.37-fold as compared to 1 (IC50, 185 μM), suggesting hydrophobic groups at the primary hydroxyl position are preferred for GSK-3β inhibition. Remarkably, transforming the primary alcohol to corresponding hydrophobic amides (8–31) (Table 2) significantly increased the potency against GSK-3β as most analogues displayed IC50 values less than 50 μM and 10 of them (9, 13, 16, 17, 20, 21, 27, 29, 30 and 31) were less than 20 μM. As shown in Figure 2A, the aliphatic (e.g., 9 and 13) and alicyclic amides (e.g., 16 and 17) exhibited a higher affinity to GSK-3β than the aromatic amides (e.g., 20–23). Small (14 and 15) or large (18 and 19) alicyclic rings showed a less affinity than the cyclopentyl (16) or cyclohexyl (17) analogues, plausibly due to the size of the hydrophobic concave cleft in the substrate site on GSK-3β. A branched isopropyl group (9, 30, and 31) showed a higher affinity than a linear propyl group (8, 28, and 29). Monofluorination of phenyl (24), benzyl (25), ethyl (26) or propyl (28) groups did not improve affinity as compared to nonfluorinated counterparts (e.g., 8, 20, and 21). It is interesting that a trifluoromethyl (CF3) group has a significant effect on potency. Compounds 27, 29, 30 and 31 containing a CF3 moiety consistently improved binding affinity to GSK-3β in comparison with no fluorine or monofluorinated counterparts (e.g., 8, 9, 26, and 28) (Figure 2A). In particular, 30 (IC50, 0.59 μM) with a (S)-CF3 group increased the potency against GSK-3β by 310-fold in comparison with 1, and is about 4-fold more potent than its epimer 31 with a (R)-CF3 group (IC50, 2.3 μM). All new analogues were not pan-assay interference compounds to GSK-3β as determined with a detergent-based assay.23,30,31
Figure 2.

Analyses of GSK-3β inhibitory activities for compounds 1–31. (A) Scatter plot of pIC50 (−log IC50) for GSK-3β inhibitors 1 and 8–31. The parent compound isoorientin 1 is shown in red, aliphatic amide analogues are shown in cyan, alicyclic amide analogues are shown in blue, aromatic amide analogues are shown in yellow, and fluorinated amide analogues are shown in purple. (B) Plot of CLogP versus pIC50 for GSK-3β inhibitors 1–31. Diagonal lines represent areas of the same LiPEs to estimate druglikeness. LiPE = pIC50 – CLogP. Solid circle, natural flavones; open circle, semisynthetic flavones; cmpd, compound.
Ligand-lipophilic efficiency (LiPE) is a parameter commonly used in drug design to assess the quality of compound candidates. Lipophilicity is the most important druglike physiochemical property that highly correlates to absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles and ultimately to the pharmacological response for oral drugs.32 High potency (large pIC50) is a desirable feature in drug candidates, as it reduces the risk of off-target and nonspecific pharmacology. Correlation between lipophilicity (CLogP) and potency (pIC50) provides a valuable parameter to estimate druglikeness.33 Many of the semisynthetic analogues clustered in an upper-right range of the LiPEs between 2 and 4, indicating that both lipophilicity (CLogP) and GSK-3β inhibitory potency (pIC50) have been increased relative to 1 (Figure 2B). Particularly, 30 has the highest LiPE value (>4) among the analogues, suggesting a unique contribution of the CF3 moiety of 30 to improving potency and lipophilicity.
Evaluations of 1, 9, 17, 21, and 30 on Anti-Tau Hyperphosphorylation Mediated by GSK-3β
Compounds 9, 17, 21, and 30 were selected for further evaluation on their effect against GSK-3β mediated tau hyperphosphorylation relative to 1, as they are the most potent GSK-3β inhibitors within each of the aliphatic, alicyclic, aromatic and fluorinated amide analogues, respectively. We recently established an in vitro GSK-3β assay using a whole-cell lysate of human SH-SY5Y neuroblastomas and demonstrated that the pS396 site on tau protein is GSK-3β specific.23 Direct GSK-3β inhibition by isoorientin 1 led to its consequent effect against GSK-3β mediated tau hyperphosphorylation on the pS396 site in an ex vivo protein matrix of the SH-SY5Y lysate.23 In analogy, an aliquot of lysate was fortified with GSK-3β (wt/wt 0.25%), and incubated with 9, 17, 21, or 30 at different concentrations (2, 10, and 50 μM) for 2 h followed by an enzyme linked immunosorbent assay (ELISA) with an anti-tau pS396 antibody. TDZD-8 (10 μM) and 1 (100 μM) were used as reference controls. Quantitative ELISA measurements substantiated that introducing exogenous GSK-3β significantly increased phosphorylation by approximately 2.5-fold (p < 0.0001) at the site pS396 on tau proteins as compared to their basal levels (lysate fortified with heat-inactivated GSK-3β) (Figure 3). In contrast, treatment of 9, 17, 21, or 30 effectively attenuated tau hyperphosphorylation in a dose-dependent manner (p < 0.0001). All four analogues showed vast improvements of anti-tau hyperphosphorylation in comparison to the natural product 1. Compound 30 at 10 μM exerted an anti-tau effect comparable to TDZD-8 (a known potent GSK-3β inhibitor, IC50 = 2 μM) at the same concentration (p < 0.05). These results demonstrated that the new 6-C-glycosylflavone analogues indeed alleviate tau hyperphosphorylation, of which 30 is the most promising candidate suitable for further in vivo pharmacological assessments.
Figure 3.

Compounds 9, 17, 21, and 30 attenuate GSK-3β-mediated tau phosphorylation in a SH-SY5Y whole-cell lysate kinase assay. Cell lysate aliquots were fortified with 0.25% (w/w) GSK-3β, and incubated with 2–50 μM of 9, 17, 21, 30, or 5% DMSO vehicle in a kinase buffer at 37 °C for 2 h. 10 μM TDZD-8 and 100 μM isoorientin (1) were used as reference controls. ELISA analysis was performed with specific antibody against Tau pS396 to quantify tau phosphorylation levels. Fold changes were calculated relative to the control with ± SEM (n = 4). Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ####p < 0.0001 relative to inactive GSK-3β fortified control; ****p < 0.0001 relative to the active GSK-3β fortified control; •p < 0.05, ••p < 0.01, and ••••p < 0.0001 relative to the TDZD-8 reference control.
GSK-3β Kinetic Studies on the Inhibition Mode of 30
To determine the GSK-3β inhibitory mechanism, the most potent analogue 30 (Figure 4A) was assayed to competitively replace ATP or the GSK-3β substrate GS2 (a peptide derived from human muscle glycogen synthase). Under a constant concentration of the substrate GS2 (17 μM), ATP concentrations varied from 2 to 50 μM and 30 concentrations varied from 0 to 5 μM. The Lineweaver–Burk plots show a convergence of intersecting lines on the x-axis, indicating an unaltered Michaelis–Menten constant (Km) but a reduced GSK-3β activity (increased 1/Vmax) when the concentration of 30 increased (Figure 4B(a)). This suggested no competition between ATP and 30. In the second set of experiments under a constant concentration of ATP (10 μM), substrate GS2 concentrations varied from 8 to 66 μM and 30 concentrations varied from 0 to 5 μM. The Lineweaver–Burk plots show that all lines intersected at the same point on the y-axis, suggesting an unchanged 1/Vmax but an increase of Km when the concentration of 30 increased (Figure 4B(b)). The data demonstrated that 30 competed with the substrate GS2. The enzyme inhibitory behaviors of 30 are similar to that of the parent compound 123 and therefore confirm that the new analogues are indeed reversible and substrate-competitive inhibitors of GSK-3β.
Figure 4.

Compound 30 selectively inhibits GSK-3β via a substrate-competitive mechanism. (A) Structure of 30 and inhibition curves of 30 with an IC50 of 0.59 μM and 1 with an IC50 of 184.9 μM. The results were presented as the percentage of the kinase activity relative to control (5% DMSO vehicle). Inhibition curves were analyzed by four-parameter regression. (B) Lineweaver–Burk plots of GSK-3β kinetic data at increasing concentrations of 30 from 0 to 5 μM. (a) The lines are linear regression plotting of 1/V against 1/ATP at a given concentration of 30. ATP concentrations varied from 2 to 50 μM, while the concentration of the GSK-3β substrate GS2 was kept constant at 17 μM. Intersecting at the same point on the x-axis indicates noncompetitive inhibition with respect to ATP. (b) The lines are linear regression plotting of 1/V against 1/GS2 at a given concentration of 30. Substrate GS2 concentrations varied from 8 to 66 μM, while the ATP concentration was kept constant at 10 μM. Intersecting at the same point on the y-axis indicates competitive inhibition with respect to the substrate GS2. (C) Inhibitory effects of 30 on the activities of 41 kinases. Kinases were assayed in the presence of 5 μM 30 or control (5% DMSO vehicle). Data were the mean of quadruplicate of each of two independent experiments with ± SEM. The data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 relative to the control.
Kinase Selectivity Profile of 30
It is prudent to evaluate kinase selectivity in the early phases of drug discovery. Compound 30 was screened against a panel of 41 human protein kinases for selectivity relevant to AD and other CNS disorders.9 30 at 5 μM showed an overall good selectivity as it effectively inhibited GSK-3β by decreasing 92.3% kinase activity (p < 0.0001) in comparison to the control (100% kinase activity), whereas it showed only marginal or weak inhibition against 40 out of 41 kinases in the test panel (Figure 4C). Notably, between the two GSK-3 isoforms (GSK-3α and GSK-3β), 30 at 5 μM displayed a 12.3-fold selectivity to GSK-3β (92.3% inhibition) versus GSK-3α (7.5% inhibition). The exceptional selectivity of 30 to GSK-3β could minimize risk of off-target effects.
Evaluation of 30 on Cytotoxicity and Anti-Amyloid Neurotoxicity
To investigate whether semisynthetic 6-C-glycosylflavone analogues exert neuroprotection against Aβ-induced neurotoxicity, 30 was assayed in an AD cellular model where Aβ42 oligomers were administrated in human SH-SY5Y neuroblastomas.23,35 Compound 30 displayed a good tolerability profile similar to 1 as no observable cytotoxicity up to a dose of 1000 μM (Figure 5A).23 On the other hand, treatment of 10 μM Aβ42 oligomers decreased cell viability to 40% compared with the controls (Figure 5B). However, such Aβ42 neurotoxicity was greatly relieved, as pretreatment of SH-SY5Y cells with 30 at concentrations from 1.25 to 20 μM for 1 h followed by coincubation with 10 μM Aβ42 for 72 h recovered cell viability from 40% to 100% in a dose-dependent manner. The neuroprotective potency of 30 (EC50, 8.7 μM) (Figure 5C) was a 5.4-fold increase in comparison to its parent compound 1 (EC50, 47 μM).23
Figure 5.

Compound 30 alleviates Aβ42 induced neurotoxicity in SH-SY5Y cells. (A) Cytotoxicity assessment of 30 in SH-SY5Y cells. Cells were treated with varying concentrations of 30 or 0.2% DMSO vehicle and incubated for 72 h. Cell viability was determined with the MTX assay. (B) Cells were pretreated with varying concentrations of 30 or 0.2% DMSO vehicle for 1 h followed by 10 μM Aβ42 treatment and incubated for 72 h. TDZD-8 at 10 μM was used as a reference control. (C) 30 inhibited neurotoxicity induced by 10 μM Aβ42 with an EC50 value of 8.7 μM. The results were presented as the percentage of the neuroprotective activity relative to control (100%) and 10 μM Aβ42 treatment (0%). Neuroprotection curve was analyzed by four-parameter regression. Data were the mean of triplicate of each of two independent experiments with ± SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ####p < 0.0001 relative to vehicle control; ****p < 0.0001 relative to the 10 μM Aβ42 treatment. (D) Morphological changes of SH-SY5Y cells after treatment for 72 h. (a) 0.2% DMSO treatment as vehicle control. Differentiated cells with extended axons and dendrites. (b) 10 μM Aβ42 treatment. Dying and impaired cells with retracted neurites. (c) Pretreatment of 10 μM 30 followed by 10 μM Aβ42 treatment. Protected differentiated cells with extended axons and dendrites. Micrographs represent the average morphologic characteristics of cell cultures under a given condition of 4–6 experimental replicates. Scale bar = 100 μm.
Morphological observations also illustrated that pretreatment by 30 at 10 μM effectively protected the SH-SY5Y cells from Aβ42 intoxication, as the neuronal cells were healthy and well differentiated with extended axons and dendrites (Figure 5D). Although a similar neuroprotective activity was observed to 1, 30 exhibited approximately 20-fold improvement of effectiveness in comparison to its parent compound (effective dose: 30 in 10 μM versus 1 in 200 μM).23 In good agreement with our previous findings, 30 and the other 6-C-glycosylflavone analogues exerted anti-Aβ neurotoxicity through protecting neurite outgrowth and neuronal differentiation, whose functions are constitutively regulated by GSK-3β and tau protein within axonal microtubules.7
Evaluations of 1, 4, 9, 17, 21, and 30 on Passive Membrane Permeability
Oral administration is noninvasive and is preferred for chronical diseases such as AD and other CNS disorders. It is advantageous to discover passive membrane permeable AD drugs targeting GSK-3β upon oral administration. The parallel artificial membrane permeability assay (PAMPA) is used to assess passive and transcellular permeability, which is well correlated with in vivo oral absorption rates for drugs that cross the gastrointestinal barrier.36 In the PAMPA model, the amount of target compounds diffused from a donor compartment to an acceptor compartment through a trilayer phospholipid precoated membrane was measured to assess oral-absorption potential of drug candidates. In the present study, 1, 4, 9, 17, 21, and 30 were evaluated with the PAMPA assay. Theophylline and atenolol known for their low permeability were used as negative controls. Desipramine known for its high permeability was used as a positive control. The measured Pe values were compared with the literature data.36
The new analogues 9, 17, 21, and 30 demonstrated a significant increase of permeability upon 5 h of incubation in comparison with their parent compound 1 (Table 3). Methylation of phenolic hydroxyls and transformation of the primary hydroxyl to hydrophobic amides on the C-glycone of 1 increased the PAMPA permeability (Pe) ranging from 2.6 to 4.4-fold relative to 1. Such Pe changes agreed well with the CLogP changes (Table 3). The best PAMPA permeability of 30 is probably attributed to the CF3 group. Multiple hydroxyl groups in C-glycone and flavone moieties make 1 poorly permeable, which is consistent with the literature reports.37 For comparison, luteolin (4), an aglycone flavone, had a medium PAMPA permeability similar to 9, 17, and 21, indicating the liability of the polar C-glycone of 1 in respect to passive diffusion. Regardless, it is indispensable to assess pharmacokinetics of the new GSK-3β inhibitors for their in vivo bioavailability which involves the complexity of active and facilitated transports in addition to passive diffusion.
Table 3.
PAMPA Permeability and CLogP Values of Compounds Tested
| PAMPA permeability |
||||
|---|---|---|---|---|
| cmpd no. | Pe (×10−6 cm/s)a | R (%)b | permeability classificationc | CLogP |
| isoorientin (1) | 0.51 ± 0.01 | 48 | low | 0.21 |
| luteolin (4) | 1.42 ± 0.07 | 43 | medium | 2.31 |
| 9 | 1.55 ± 0.06 | 46 | high | 1.35 |
| 17 | 1.77 ± 0.07 | 20 | high | 2.55 |
| 21 | 1.35 ± 0.08 | 30 | medium | 2.34 |
| 30 | 2.23 ± 0.21 | 30 | high | 1.62 |
| theophylline | 0.04 ± 0.01 | n/a | low | −0.03 |
| atenolold | 0.29 ± 0.09 | 40 | low | −0.11 |
| desipramined | 13.43 ± 0.91 | 96 | high | 4.47 |
Pe values were the mean of PAMPA measurements (n = 3–4) with ± SD.
Percent recovery (R%) measures mass retention of compounds trapped inside the PAMPA membrane.
PAMPA permeability classification: high (Pe > 1.5 × 10−6 cm/s), medium (1.0 × 10−6cm/s ≤ Pe ≤ 1.5 × 10−6 cm/s), low (Pe < 1.0 × 10−6 cm/s).
The measured Pe values are comparable to the data in ref 36.
■ COMPUTATIONAL MODELING
Key Molecular Interactions between New C-Glycosylflavones and the Substrate Site of GSK-3β
To elucidate the molecular mechanisms by which the new 6-C-glycosylflavones increase the binding affinity and selectivity to GSK-3β relative to 1, compounds 5–31 were docked into GSK-3β enzyme. The AutoDock Vina program was used in the present study because it has shown good accuracies for binding pose prediction and scoring function.38–40 This docking tool has been widely used in drug design and applied in many cases of GSK-3β inhibitor discovery.17,41
Two X-ray crystallographic structures of GSK-3β (PDB codes 1PYX42 and 1H8F43) were chosen to take into account for potential induced fit effects upon ligand binding. The GSK-3β in 1PYX contains two Mg2+ ions and a ligand ANP, a nonhydrolyzed ATP derivative, at the ATP site. Upon binding of Mg2+ ions and an ATP-mimic ligand, it is postulated that GSK-3β undergoes subtle conformational changes in both the ATP and substrate sites for an optimal kinase reaction, a phenomenon known as induced fit effects.44,45 The GSK-3β structure in 1PYX hence adopts a conformation that resembles the native enzyme ready for substrate recognition. Docking 5–31 into the substrate site of GSK-3β using 1PYX would give more reliable binding poses. On the other hand, the GSK-3β in 1H8F is by far the only available X-ray crystallographic structure containing the ligand HEPES in the substrate site. HEPES in 1H8F may cause induced fit changes of GSK-3β conformation, particularly for the substrate site. Given the new 6-C-glycosylflavones are substrate competitive, we used 1PYX and 1H8F in molecular docking. The docking method was validated by redocking and cross-docking experiments using both ATP-competitive and substrate-competitive inhibitors (Supporting Information S1).
The synthetic 6-C-glycosylflavones (5–31) and their parent 1 were thus docked into 1PYX and 1H8F. Docking scores of 1PYX show a better linear fit to the experimental pIC50 values (R2 = 0.7844) than that of 1H8F (R2 = 0.6999), suggesting more accurate predictions of binding poses using 1PYX than 1H8F (Supporting Information S2). The 1PYX docking data set was therefore used in the remainder of the study to analyze the molecular interactions responsible for the improved potency and selectivity to GSK-3β.
The docking results of 1PYX showed that the C-glycone moiety of 5–31 forms hydrogen bonds with GSK-3β residues Gln89, Asn95, Arg96, and Glu97 within the substrate pocket, which is similar to 1 (Supporting Information S3). Moreover, the newly introduced hydrophobic groups indeed exhibit a favorable ligand pose to the concave surface comprised of Phe67, Val87, Leu88, and Phe93 on GSK-3β. The most potent compounds 30 and 31 (IC50, 0.59 and 2.3 μM, respectively) contain a CF3 moiety, which may form orthogonal multipolar interactions for protein binding.46,47 To visualize such molecular interactions, we conducted flexible-residue docking40,48 to refine binding poses of 9, 30, and 31 in 1PYX. To prepare GSK-3β structure, side chains of Ser66, Phe67, Leu88, Gln89, Asp90, Phe93, Asn95 and Lys183 within the substrate site were treated flexible. The docking results of 30 suggested formation of orthogonal multipolar interactions. The (S)-CF3 in 30 shows a favorable geometry within typical F···C distances to Leu88 (backbone amide carbonyl carbon, 3.3 Å), Gln89 (backbone amide carbonyl carbon, 3.6 Å) and Asp90 (side chain carbonyl carbon, 3.3 and 3.8 Å) (Figure 6C). It also forms a polar interaction with the backbone NH of Asp90 (F···H distance, 2.6 Å). In contrast, the (R)-CF3 group of 31 is absent from these interactions probably owing to the orientation and steric hindrance by switching CH3 and CF3 positions at the stereocenter (Figure 6B,C). Therefore, only hydrophobic interactions with Phe67, Val87 and Leu88 of GSK-3β exist in 31 (improper configuration of CF3 for orthogonal multipolar interactions) as well as 9 (lack of CF3) (Figure 6A,B).
Figure 6.

Predicted docking poses of 9 (A), 31 (B), and 30 (C) within the substrate site of GSK-3β (PDB code 1PYX). Dotted lines represent interactions via hydrogen bonding, π–cation interaction, or orthogonal multipolar interactions with key amino acid residues of GSK-3β. Key molecular interaction distances are highlighted.
Interestingly, the docking results showed that the catechol Bring of the flavone core in 9, 30, and 31 appears to have a π–cation interaction with Lys183.49,50 The distance between the catechol B-ring center and ε-NH3+ cation of Lys183 is 4.1 Å at an angle of 71.5° (Figure 6C and Supporting Information S4). In conjunction, the side chain ε-NH3+ of Lys183 forms weak hydrogen bonds with two methoxy oxygen atoms of catechol Bring (distances of 3.1 and 3.2 Å). The orthogonal multipolar and π–cation interactions potentially enhance the binding affinity to GSK-3β.
Potential Causes for the Isoform-Selectivity of 30 between GSK-3α and GSK-3β
Isoform-selectivity of 30 to GSK-3β over GSK-3α (Figure 4C) was assessed with comparative molecular modeling. Yet no X-ray crystallographic structures of GSK-3α available in PDB, a homology model of GSK-3α was built with the SWISS-MODEL server51 (Supporting Information S5). A full human GSK-3α amino acid sequence (UniProt code: P49840) was searched against protein databases in BLAST and HHblits and overall 4470 templates were found. A GSK-3β template (PDB code 1PYX) resulted in a top ranking with an overall sequence identity of 82.97%, thereby selected for homology modeling of GSK-3α. A sequence alignment between the two GSK-3 isoforms indicated that GSK-3α has extra amino acid portions flanking the N- and C-termini of GSK-3β (Supporting Information S6). Within the matched sequence portion, most amino acid differences occur in the N- and C-terminal regions. In contrast, both the ATP and substrate domains of GSK-3α/β isoforms are conserved as high as 92.37% identical, in which only few amino acid residues are different in these regions. The superposition and comparison of GSK-3β structure (PDB code 1PYX) with the GSK-3α homology model (Figure 7A) implied that those subtle differences of amino acid residues in the kinase catalytic domain (both ATP and substrate pockets) may affect substrate recognition as well as ligand binding. Intriguingly, docking experiment using the GSK-3α homology model showed that 30 resides a similar location in the substrate site in comparison to GSK-3β docking data (Figures 7B,C). However, the resulting binding pose of 30 in GSK-3α favors neither a π-cation interaction of catechol B-ring nor the orthogonal multipolar interactions of the (S)-CF3 group. It is conceivable that the isoform-selectivity of 30 to GSK-3β might be in part due to the lack of these critical molecular interactions in GSK-3α. Instead, the (S)-trifluoroisopropyl group of 30 simply exerts hydrophobic affinity with the homologous residues Phe130, Val150, and Leu151 in the substrate site of GSK-3α (Figure 7C).
Figure 7.

Molecular models of GSK-3β and GSK-3α and the docking complexes with 30. (A) Superposition of GSK-3β structure (PDB code 1PYX) and GSK-3α homology model (UniProt code P49840). The ATP and substrate sites are highlighted in circles, and the N- and C-termini are depicted. The ATP and substrate sites of GSK-3α/β isoforms are highly conserved and most of amino acid differences occur in the N- and C-terminal regions. (B) Predicted docking pose of 30 into the substrate site of GSK-3β structure. (C) Predicted docking pose of 30 into the substrate site of GSK-3α homology model. GSK-3 isoforms are shown as gray cartoons. In both isoforms, selected conserved residues are displayed as green sticks. In GSK-3β, nonconserved residues are displayed as blue sticks. In GSK-3α, nonconserved residues are displayed as magenta sticks. The dotted lines represent key molecular interactions.
■ DISCUSSION
GSK-3β plays a central role in signaling pathways of AD. Accumulating evidence has demonstrated that GSK-3β is activated abnormally by Aβ and in turn hyperphosphorylates tau proteins in neurons. This eventually triggers the tauopathic cascade in AD progression.1,7,8 Development of GSK-3β inhibitors as a disease-modifying therapy for AD, therefore, is highly attractive. In recent years, discovery of selective inhibitors targeting the substrate site of GSK-3β has emerged as a rational and feasible strategy in AD drug design.11 Unlike the ATP-site directed GSK-3β inhibitors that frequently bind to many off-target kinases, inhibitors targeting the substrate site on GSK-3β plausibly overcome this limitation. GSK-3β phosphorylates its specific substrates for biological processes such as glucose homeostasis, immune response, neurogenesis, cell proliferation and apoptosis, and circadian rhythm, which has a plethora of normal functions for human health.52 However, GSK-3β is aberrantly overactive in AD. Subtle modulation of GSK-3β activity at a normal level rather than complete shut-off to prevent disruption of its normal cellular functions will be highly appreciated in AD therapy.8 Targeting the substrate site to intervene the GSK-3β–substrate recognition would be a feasible means to find selective inhibitors.14 Until now, substrate-competitive inhibitors of GSK-3β are limited in the structural classes of marine alkaloid manzamines,15 labdane diterpenoid andrographolide,17 peptidomimetic inhibitors,18 and synthetic compound ITDZ series.16 These inhibitors were in the low micromolar to submicromolar range of potency, but exhibited a good selectivity to GSK-3β under in vitro or in vivo evaluations. Such findings substantiate the rationale of developing substrate-competitive inhibitors of GSK-3β for AD by which the agents can attain an utmost therapeutic advantage—a high selectivity to avoid potential side effects.
Herein, we reported a new structural class of substratecompetitive inhibitors of GSK-3β, inspired by isoorientin (1), containing a 6-C-glycosylflavone scaffold. It is a valuable addition to the known substrate-competitive inhibitors and offers new lead compounds for AD. The inhibitors are new chemical probes allowing to elucidate the underling mechanisms of GSK-3β inhibition and selectivity.
The SAR analysis, in vitro enzymatic kinetics, and in silico docking studies indicate that both the presence and position of C-glycone on the flavone core are essential features binding to the substrate site on GSK-3β. A flavone core (4) alone is promiscuous and tends to sneak in the ATP site and thus lose the required selectivity (Supporting Information S1). The presence of a C-glycone at 8-position (3, IC50, >5 mM) rather than 6-position (1, IC50, 185 μM) results in an unfavorable binding pose to the substrate site plausibly due to the loss of key hydrogen bonds with Gln89, Asp90, Asn95, and Arg96 (Supporting Information S3). Such unique SAR features inspired us to semisynthesize a series of novel analogues of 1 that selectively inhibit GSK-3β at the substrate site. Data suggest that the new lipophilic amide analogues (8–31) increase the potency against GSK-3β for 3–310-fold (Table 2) and passive membrane permeability for 2.6–4.4-fold relative to 1 (Table 3). Systematic modifications of the carbon-chain length and ring size and bioisosteric replacement in the R2-group on the C-glycone of 1 (Table 1) confer a dramatic GSK-3β potency improvement. The structural modifications contribute hydrophobic affinities with Phe67, Val87 and Leu88 in the substrate site of GSK-3β (Supporting Information S3), which is concurrently supported by the LiPE analysis (LiPE highlights potency changes to the net of lipophilicity). Compounds 9, 30, and 31 containing an isopropyl moiety have IC50 values of 5.4, 0.59, and 2.3 μM, respectively, which are most potent among the analogues. It suggests that they adopt a suitable carbon-chain length and topological size within the hydrophobic cleft of the substrate site. LiPE analysis also indicates that 9, 30, and 31 are quality ligands (LiPE values ≥ 4, Figure 2B). In addition, the new analogues alleviate tau hyperphosphorylation and Aβ neurotoxicity through GSK-3β inhibition in the human SH-SY5Y neuronal model of AD (Figures 3 and 5).
With respect to the GSK-3β selectivity of 30, π–cation interaction and orthogonal multipolar interactions appear to play a critical role. In protein structures, the aromatic side chain of Phe or Tyr usually involves a favorable π–cation interaction with the cationic side chain of Lys or Arg.49 In analogy, if a small aromatic ligand binds to a protein, potential π–cation interactions would be an important contribution to the binding affinity. Such phenomena have been exemplified in our previous study50 as well as other investigations.53,54 Flavonoids seem to be the case: interactions between the aromatic A/B-rings with the cationic Lys or Arg residues.41,55 In the docking experiments, we found that the catechol B-ring of the flavone core in 30 forms a π–cation interaction with Lys183 of GSK-3β (distance, 4.1 Å; angle, 71.5°). To a certain extent, this might stabilize the binding conformation and orientation within the substrate site.
It is known that a fluorine atom has distinct chemical properties contributing to the molecular recognition.46 Fluorinated compounds commonly form orthogonal multipolar interactions with the backbone and side chain carbonyl carbons (Asp/Glu/Asn/Gln) or the guanidinium carbon (Arg) of amino acid residues in protein structures.46,47 Introduction of a CF3 group is a common tactic in drug design. It is estimated that substitution of CH3 for CF3 may improve 5–10-fold ligand binding affinity.47,56 In the present study, 27 (IC50, 19.3 μM) and 29 (IC50, 17.2 μM) containing a 2,2,2-trifluoroethyl and 3,3,3-trifluoropropyl, respectively, show an improved potency relative to their analogue 8 (IC50, 29.2 μM) without a CF3 group. In addition, 30 and 31 containing a 1,1,1-trifluoroisopropyl are more potent than their analogue 9 without a CF3 group. Docking experiments suggest that the isopropyl (S)-CF3 group of 30 interacts with the backbone and side chain carbonyls of Leu88, Gln89, and Asp90 (Figure 6C) within typical interaction distances (F···C, 3.0–3.7 Å).47 Such orthogonal multipolar interactions might in part give rise to an increased potency of 30 (IC50, 0.59 μM) against GSK-3β approximately 9-fold and 4-fold compared to 9 (IC50, 5.4 μM) and 31 (IC50, 2.3 μM), respectively. Conversely, 31 with a (R)-CF3 group may be lacking these critical molecular interactions (Figure 6B,C) and thus has weaker binding affinity than 30. The differential and stereospecific binding of the CF3 group between 30 and 31 plausibly explains the high inhibitory selectivity to GSK-3β (Figures 2 and 4).
Interestingly, 30 exhibits isoform-selectivity to GSK-3β over GSK-3α, which deserves discussion. In humans, GSK-3α (51 kDa) and GSK-3β (47 kDa) are derived from different genes and have distinct functions.52 Both isoforms share an overall sequence identity of 83%, especially at the ATP catalytic domain with 93% identity.57 However, they differ substantially in the N- and C-terminal lobes that are thought to cause protein conformational dynamics and differential cellular localization.52,57 Subtle amino acid differences in the substrate site between two isoforms (Supporting Information S6) putatively affect substrate recognition and preference as noted in the literature.58,59 Wang et al. reported that GSK-3β phosphorylates a substrate (phosphatase inhibitor-2) 10-time faster than GSK-3α,58 indicating the differential substrate recognition of GSK-3 isoforms. Soutar et al. demonstrated that the specific sites T231, T235, and S396 on tau proteins are solely phosphorylated by GSK-3β but not GSK-3α,59 giving an evidence of substrate specificity between two isoforms. Those studies pose a possibility to discover isoform-selective inhibitors via targeting the substrate site of GSK-3β. Compound 30 attains GSK-3β isoform-selectivity probably because of both the π-cation interaction with Lys183 and the orthogonal multipolar interactions with Leu88, Gln89, and Asp90 in GSK-3β on the basis of the comparative docking of GSK-3α/β (Figures 6C and 7). In contrast, 30 is absent from those key molecular interactions within the substrate site of GSK-3α (Figure 7C). In addition, the highly nonconserved regions in GSK-3α, such as the N- and C-terminal domains (Figure 7A), can modulate enzyme conformations and thereby may affect the binding of 30. Notwithstanding, the molecular mechanism suggested by computational modeling requires further verification by conclusive experimental evidence.
■ CONCLUSIONS
In summary, we described a new class of substrate-competitive inhibitors of GSK-3β focusing on modifications of the primary hydroxyl group in the C-glycone of isoorientin (1). The results demonstrated that the 6-C-glycone moiety of the new inhibitors defines the specific binding at the substrate site rather than the ATP site on GSK-3β. The data also help explore topological requirements in the substrate site of GSK-3β and highlight the critical SAR. Those inhibitors effectively attenuate tau hyperphosphorylation and amyloid neurotoxicity in molecular and cellular AD models by which the mechanism is involved in GSK-3β inhibition via a non-ATP competitive but substrate competitive manner. The inhibitors not only significantly increased the potency, kinase selectivity, and isoform-selectivity, but also improved passive membrane permeability in comparison with the natural product counterparts. Among them, 30 (IC50, 0.59 μM) showed a potency improvement by 310-fold and a promising profile in various biological assessments, which warrants further exploration. SAR analyses and in silico mechanistic investigations suggested that the hydrophobic, π-cation and orthogonal multipolar interactions of 30 with the substrate site lead to selective inhibition against GSK-3β, but neither GSK-3α nor a broad panel of kinases tested. Nevertheless, additional SAR knowledge and physiochemical property optimization of the GSK-3β inhibitors based on the 6-C-glycosylflavone scaffold are essential to the CNS drug discovery campaign. Pharmacokinetics and in vivo animal studies of the new GSK-3β inhibitors will help understand drug delivery, target engagement and efficacy, which would suggest the therapeutic potential of these agents for AD and other GSK-3β relevant neuropsychiatric and neurodegenerative diseases.
■ METHODS
Chemicals and Reagents
All solvents and reagents were from commercial sources and were used without further purification. Natural flavones (isoorientin, orientin, isovitexin, and luteolin), staurosporine, TDZD-8, theophylline, atenolol, desipramine, TMSCHN2, [TEMPO]+[BF4]−, HCTU, organic amines, and protease inhibitor cocktail were from Sigma-Aldrich (Saint Louis, MO). β-Amyloid fragment peptide 1–42 (Aβ42) was from AnaSpec (Fremont, CA). Kinase Selectivity Profiling Assay Kit, ADP-Glo Kinase Assay Kit, and CellTiter 96 AQueous One Solution Cell Proliferation MTX Assay Kit were from Promega (Madison, WI). Human Tau pS396 ELISA Kit and Cell Extraction Buffer were from Invitrogen (Camarillo, CA). Precoated PAMPA plate system was from Corning (Tewksbury, MA).
General Experimental Procedures
High-resolution mass spectrometric data were obtained on a Bruker maXis Impact nanoLC-QTOF-MS spectrometer in ESI positive mode. Accurate masses of all analytes were obtained from the pseudomolecule [M + H]+ and were within 5 ppm mass error. 1H, 13C and 2D NMR data were recorded with a Varian Unity Inova 500 MHz spectrometer. NMR spectra were referenced to the appropriate residual solvent signal (δH 2.50, δC 39.5 for DMSO-d6) with chemical shifts reported in δ units (ppm). Resonance multiplicities are denoted s, d, t, q, m, and br for singlet, doublet, triplet, quartet, multiplet, and broad, respectively.
All reactions were monitored by LC-MS (Bruker nanoLC-QTOF-MS). Compounds in crude reaction mixtures were separated by flash column chromatography on HyperSep C18 (40–63 μm), and purified by semipreparative reverse phase Agilent HPLC with a diode array detector (Waters XSELECT CSH Phenyl-Hexyl column, 150 × 10 mm, 5 μm, a linear gradient over 30 min from 10 to 50% aqueous acetonitrile containing 0.1% formic acid, flow rate 2.5 mL/min).
The purity of each compound was determined by analytical reverse phase Agilent HPLC with a diode array detector (Waters XSELECT CSH Fluoro-Phenyl column, 150 × 4.6 mm, 3.5 μm, isocratic elution with 35% aqueous acetonitrile containing 0.1% formic acid, detection at 210, 254, and 340 nm, flow rate of 0.8 mL/min). All the tested compounds were over 95% purity by HPLC-UV at 210, 254, and 340 nm.
Luminescent measurement was performed on an Agilent Cary Eclipse fluorescence spectrophotometer. Optical absorbance was measured on a Multiskan GO Microplate spectrophotometer. Microscopic images were observed under a Nikon Diaphot inverted tissue culture microscope with Optronics MicroFire microscope camera.
General Procedure for the Semisynthesis of C-Glycosylflavone Analogues
Preparation of Compound 5
The chemoselective methylation proceeded as described.28 To a stirred solution of 1 (45 mg, 0.1 mmol) in a mixture of toluene (6 mL) and methanol (4 mL) was added TMSCHN2 (2 M in hexane, 0.5 mL, 1 mmol). The reaction solution was stirred at room temperature for 8 h and the solvent was evaporated. The residue was purified by RP-HPLC (Waters XSELECT CSH Fluoro-Phenyl column, 150 × 4.6 mm, 3.5 μm, isocratic elution with 35% aqueous acetonitrile containing 0.1% formic acid, detection at 210, 254, and 340 nm, flow rate of 0.8 mL/min) to afford 5.
2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-6-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-4H-chromen-4-one (5).
Light yellow solid (80% yield). 1H NMR (DMSO-d6) δ 7.65 (dd, J = 8.6, 2.2 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.09 (br s, 1H), 6.72 (br s, 1H), 4.65 (br d, J = 9.7 Hz, 1H), 4.05 (dd, J = 12.7, 9.2 Hz, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.81 (s, 3H), 3.77 (s, 3H), 3.72 (dd, J = 12.6, 12.5 Hz, 1H), 3.34–3.28 (m, 1H), 3.27–3.12 (m, 3H). 13C NMR (DMSO-d6) δ 175.5, 163.4, 160.2, 158.9, 158.5, 151.8, 149.1, 123.2, 119.7, 111.9, 111.3, 109.4, 107.1, 107.0, 97.0, 82.0, 79.2, 73.1, 71.3, 70.9, 62.6, 62.0, 61.8, 56.4, 55.8. HRESI-TOFMS m/z [M + H]+ 505.1710 (calcd for C25H29O11+, 505.1704, −1.1 ppm error). HPLC purity: 97.1% (210 nm).
Preparation of Compound 6
To a stirred solution of 5 (45 mg, 0.09 mmol) in a mixture of dichloromethane (6 mL) and pyridine (3 mL) was added [TEMPO]+[BF4]− (oxoammonium salt, 60 mg, 0.2 mmol). The reaction mixture was stirred at room temperature for 5 h. The reaction was quenched by adding drops of methanol and then evaporated to dryness. The residue was reconstituted in 5% MeOH/H2O and then eluted on a HyperSep C18 column using the same solvents to remove the red-orange nitroxide. Elution was continued with 90% MeOH/H2O and the eluate was collected as the crude product, which was further purified by RP-HPLC (Waters XSELECT CSH Fluoro-Phenyl column, 150 × 4.6 mm, 3.5 μm, isocratic elution with 35% aqueous acetonitrile containing 0.1% formic acid, detection at 210, 254, and 340 nm, flow rate of 0.8 mL/min) to afford 6.
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic Acid (6)
Light yellow solid (95% yield). 1H NMR (DMSO-d6) δ 7.65 (dd, J = 8.5, 2.0 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.12 (d, J = 8.5 Hz, 1H), 7.09 (br s, 1H), 6.76 (d, J = 6.6 Hz, 1H), 4.60 (d, J = 10.1 Hz, 1H), 4.02 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.30–3.16 (m, 3H). 13C NMR (DMSO-d6) δ 175.5, 173.0, 162.2, 160.4, 158.9, 158.5, 151.9, 149.2, 123.2, 119.7, 111.9, 111.4, 109.4, 107.1, 107.0, 97.1, 79.0, 78.5, 74.1, 72.5, 71.3, 63.5, 62.6, 56.6, 56.1. HRESI-TOFMS m/z [M + H]+ 519.1502 (calcd for C25H27O12+, 519.1497, −0.9 ppm error). HPLC purity: 97.8% (210 nm).
Preparation of Compound 7
To a stirred solution of 6 (1 mg, 2 μmol) in a mixture of toluene (1.5 mL) and methanol (1 mL) was added TMSCHN2 (2 M in hexane, 2 μL, 4 μmol). The reaction solution was stirred at room temperature for 1 h and the solvent was evaporated. The residue was purified by RP-HPLC (Waters XSELECT CSH Fluoro-Phenyl column, 150 × 4.6 mm, 3.5 μm, isocratic elution with 35% aqueous acetonitrile containing 0.1% formic acid, detection at 210, 254, and 340 nm, flow rate of 0.8 mL/min) to afford 7.
Methyl (2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylate (7)
Light yellow solid (87% yield). 1H NMR (DMSO-d6) δ 7.69 (dd, J = 8.5, 2.1 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.15 (br s, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.80 (d, J = 5.5 Hz, 1H), 4.57 (d, J = 9.6 Hz, 1H), 4.00 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H), 3.63 (s, 3H), 3.29–3.13 (m, 3H). 13C NMR (DMSO-d6) δ 175.6, 172.9, 162.1, 160.1, 159.0, 158.4, 151.8, 149.1, 123.2, 119.7, 111.9, 111.4, 109.4, 107.1, 107.0, 97.0, 79.0, 78.5, 74.1, 72.5, 71.3, 63.4, 62.6, 56.6, 56.0, 51.8. HRESI-TOFMS m/z [M + H]+ 533.1656 (calcd for C26H29O12+, 533.1654, −0.5 ppm error). HPLC purity: 97.1% (210 nm).
Preparation of Compounds 8–31
To a stirred solution of 6 (5 mg, 9.7 μmol) in a mixture of dimethylformamide (1 mL) and DIPEA (0.5 mL) was added HCTU (10 mg, 24 μmol) and then stirred for 10 min at room temperature. To this solution was added corresponding organic amines (each 50 μmol) and stirred at room temperature for 5 h.
The reaction was quenched by adding 1 N HCl followed by evaporation of solvents to dryness. The residue was purified by RPHPLC (Waters XSELECT CSH Fluoro-Phenyl column, 150 × 4.6 mm, 3.5 μm, isocratic elution with 35% aqueous acetonitrile containing 0.1% formic acid, detection at 210, 254, and 340 nm, flow rate of 0.8 mL/min) to afford the final products.
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-propyltetrahydro-2H-pyran-2-carboxamide (8)
Light yellow solid (90% yield). 1H NMR (DMSO-d6) δ 7.67 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 2.2 Hz, 1H), 7.13 (s, 1H), 7.12 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 5.9 Hz, 1H), 4.67 (br d, J = 10.1 Hz, 1H), 4.06 (q, J = 10.3 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.77 (s, 3H), 3.71 (s, 3H), 3.60 (m, 1H), 3.50 (m, 1H), 3.23 (m, 1H), 2.99 (m, 2H), 1.36 (m, 2H), 0.78 (td, J = 7.4, 2.2 Hz, 3H). 13C NMR (DMSO-d6) δ 175.7, 168.7, 163.4, 160.5, 160.2, 158.4, 151.6, 149.3, 123.1, 120.4, 112.2, 111.9, 109.6, 107.2, 107.0, 96.8, 80.6, 79.0, 74.4, 71.6, 69.6, 63.9, 63.1, 56.7, 56.1, 40.8, 22.6, 11.5. HRESI-TOFMS m/z [M + H]+ 560.2135 (calcd for C28H34NO11+, 560.2126, −1.6 ppm error). HPLC purity: 98.6% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-isopropyltetrahydro-2H-pyran-2-carboxamide (9)
Light yellow solid (90% yield). 1H NMR (DMSO-d6) δ 7.67 (d, J = 8.1 Hz, 1H), 7.55 (d, J = 2.2 Hz, 1H), 7.14 (br s, 1H), 7.12 (d, J = 9.7 Hz, 1H), 6.77 (d, J = 5.1 Hz, 1H), 4.67 (d, J = 9.7 Hz, 1H), 4.05 (m, 1H), 3.92 (s, 3H), 3.89 (s, 3H), 3.85 (s, 3H), 3.83 (m, 1H), 3.81 (s, 3H), 3.59 (m, 1H), 3.57 (m, 1H), 3.23 (m, 1H), 1.04 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H). 13C NMR (DMSO-d6) δ 175.5, 168.7, 163.3, 161.7, 160.1, 158.7, 152.1, 149.2, 123.1, 119.5, 111.9, 111.3, 109.4, 107.1, 107.0, 97.0, 79.8, 78.5, 73.8, 71.4, 70.3, 63.4, 62.5, 56.6, 55.8, 40.3, 22.2, 22.2. HRESI-TOFMS m/z [M + H]+ 560.2117 (calcd for C28H34NO11+, 560.2126, 1.7 ppm error). HPLC purity: 97.2% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-butyltetrahydro-2H-pyran-2-carboxamide (10)
Light yellow solid (90% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 2.2 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 6.7 Hz, 1H), 4.67 (br d, J = 10.2 Hz, 1H), 4.06 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.58 (m, 1H), 3.50 (m, 1H), 3.23 (m, 1H), 3.02 (m, 2H), 1.34 (m, 2H), 1.23 (m, 2H), 0.83 (m, 3H). 13C NMR (DMSO-d6) δ 175.9, 168.8, 163.2, 160.3, 160.2, 158.3, 151.8, 149.1, 123.3, 119.7, 111.7, 111.5, 109.2, 107.2, 106.9, 96.5, 80.1, 78.8, 74.0, 71.5, 70.3, 63.8, 62.9, 56.7, 56.1, 38.2, 31.2, 19.6, 13.8. HRESI-TOFMS m/z [M + H]+ 574.2289 (calcd for C29H36NO11+, 574.2283, −1.1 ppm error). HPLC purity: 97.4% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-isobutyltetrahydro-2H-pyran-2-carboxamide (11)
Light yellow solid (88% yield). 1H NMR (DMSO-d6) δ 7.62 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 1.9 Hz, 1H), 7.14 (s, 1H), 7.11 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 6.9 Hz, 1H), 4.68 (br d, J = 10.2 Hz, 1H), 4.07 (m, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H), 3.62 (m, 1H), 3.49 (m, 1H), 3.24 (m, 1H), 2.87 (t, J = 6.4 Hz, 2H), 1.66 (m, 1H), 0.79 (d, J = 6.7 Hz, 6H). 13C NMR (DMSO-d6) δ 176.2, 167.3, 163.5, 160.6, 160.7, 158.8, 153.1, 149.5, 123.7, 120.7, 113.0, 112.7, 112.4, 108.3, 107.4, 97.7, 81.0, 79.9, 75.2, 72.8, 71.5, 64.6, 64.1, 57.8, 56.9, 47.2, 29.4, 21.3, 21.3. HRESI-TOFMS m/z [M + H]+ 574.2292 (calcd for C29H36NO11+, 574.2283, −1.6 ppm error). HPLC purity: 99.1% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-pentyltetrahydro-2H-pyran-2-carboxamide (12)
Light yellow solid (89% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.68 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 6.3 Hz, 1H), 4.67 (br d, J = 10.2 Hz, 1H), 4.06 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.58 (m, 1H), 3.50 (m, 1H), 3.22 (m, 1H), 3.03 (m, 2H), 1.35 (m, 2H), 1.23 (m, 2H), 1.20 (m, 2H), 0.83 (m, 3H). 13C NMR (DMSO-d6) δ 175.9, 168.9, 163.2, 160.5, 160.3, 158.5, 151.9, 149.2, 123.4, 119.7, 111.8, 111.6, 109.3, 107.0, 106.8, 96.4, 80.1, 78.8, 73.5, 71.6, 70.4, 63.5, 62.7, 56.1, 55.9, 38.4, 28.7, 28.6, 21.9, 14.0. HRESI-TOFMS m/z [M + H]+ 588.2456 (calcd for C30H38NO11+, 588.2439, −2.8 ppm error). HPLC purity: 98.2% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-hexyltetrahydro-2H-pyran-2-carboxamide (13)
Light yellow solid (90% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 2.3 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.81 (d, J = 5.9 Hz, 1H), 4.66 (br d, J = 10.5 Hz, 1H), 4.06 (q, J = 8.6 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.60 (m, 1H), 3.50 (m, 1H), 3.23 (m, 1H), 3.01 (m, 2H), 1.40–1.15 (m, 8H), 0.82 (m, 3H). 13C NMR (DMSO-d6) δ 175.6, 168.8, 163.4, 160.3, 160.2, 158.6, 151.8, 149.1, 123.1, 119.6, 112.1, 111.8, 109.3, 107.1, 107.0, 97.1, 79.9, 78.7, 74.5, 71.5, 69.4, 63.5, 62.7, 56.6, 56.0, 38.5, 31.0, 28.9, 26.1, 22.1, 13.9. HRESI-TOFMS m/z [M + H]+ 602.2572 (calcd for C31H40NO11+, 602.2596, 3.9 ppm error). HPLC purity: 98.5% (210 nm).
(2S,3S,4R,5R,6S)-N-Cyclopropyl-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxamide (14)
Light yellow solid (82% yield). 1H NMR (DMSO-d6) δ 7.67 (dt, J = 8.3, 2.2 Hz, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.16 (s, 1H), 7.12 (d, J = 8.3 Hz, 1H), 6.78 (d, J = 5.7 Hz, 1H), 4.64 (br d, J = 9.7 Hz, 1H), 4.06 (q, J = 8.9 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.79 (s, 3H), 3.73 (s, 3H), 3.51 (m, 1H), 3.50 (m, 1H), 3.19 (m, 1H), 2.62 (m, 1H), 0.79 (m, 2H), 0.58 (m, 2H). 13C NMR (DMSO-d6) δ 175.3, 167.6, 163.4, 161.5, 160.3, 159.6, 152.5, 149.3, 123.4, 119.8, 111.8, 111.5, 109.4, 107.7, 107.0, 97.0, 80.2, 78.9, 74.1, 71.3, 70.3, 63.5, 62.8, 56.1, 55.8, 23.1, 10.8, 10.8. HRESI-TOFMS m/z [M + H]+ 558.1967 (calcd for C28H32NO11+, 558.1970, 0.5 ppm error). HPLC purity: 98.2% (210 nm).
(2S,3S,4R,5R,6S)-N-Cyclobutyl-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxamide (15)
Light yellow solid (85% yield). 1H NMR (DMSO-d6) δ 7.67 (dt, J = 8.6, 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.16 (s, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.77 (d, J = 7.2 Hz, 1H), 4.66 (br d, J = 9.7 Hz, 1H), 4.16 (q, J = 7.8 Hz, 1H), 4.05 (td, J = 9.3, 5.7 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.78 (s, 3H), 3.74 (s, 3H), 3.54 (m, 1H), 3.51 (m, 1H), 3.21 (m, 1H), 2.09 (m, 2H), 1.86 (m, 2H), 1.58 (m, 2H). 13C NMR (DMSO-d6) δ 175.5, 167.4, 163.1, 161.4, 160.5, 159.3, 152.2, 149.1, 123.2, 119.6, 111.9, 111.6, 109.1, 107.5, 107.0, 97.1, 80.4, 78.5, 74.2, 71.2, 70.4, 63.3, 62.7, 55.9, 55.7, 43.6, 29.9, 29.9, 14.3. HRESI-TOFMS m/z [M + H]+ 572.2120 (calcd for C29H34NO11+, 572.2126, 1.1 ppm error). HPLC purity: 98.3% (210 nm).
(2S,3S,4R,5R,6S)-N-Cyclopentyl-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxamide (16)
Light yellow solid (86% yield). 1H NMR (DMSO-d6) δ 7.66 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 7.16 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 6.5 Hz, 1H), 4.64 (br d, J = 10.2 Hz, 1H), 3.95 (m, 1H), 3.87 (s, 3H), 3.82 (s, 3H), 3.79 (m, 1H), 3.77 (s, 3H), 3.71 (s, 3H), 3.53 (m, 1H), 3.51 (m, 1H), 3.21 (m, 1H), 1.79–1.40 (m, 8H). 13C NMR (DMSO-d6) δ 175.4, 167.1, 163.6, 161.4, 160.3, 158.8, 152.1, 149.1, 123.0, 119.8, 112.1, 111.8, 109.2, 107.7, 107.1, 97.3, 80.2, 78.5, 74.2, 71.2, 68.6, 63.6, 62.8, 56.3, 55.9, 50.5, 32.1, 32.1, 23.6, 23.6. HRESI-TOFMS m/z [M + H]+ 586.2291 (calcd for C30H36NO11+, 586.2283, −1.4 ppm error). HPLC purity: 96.6% (210 nm).
(2S,3S,4R,5R,6S)-N-Cyclohexyl-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxamide (17)
Light yellow solid (88% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 6.5 Hz, 1H), 4.65 (br d, J = 10.2 Hz, 1H), 4.04 (q, J = 8.4 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.79 (s, 3H), 3.74 (s, 3H), 3.58 (m, 1H), 3.51 (m, 1H), 3.49 (m, 1H), 3.21 (m, 1H), 1.74–1.47 (m, 6H), 1.27–0.99 (m, 4H). 13C NMR (DMSO-d6) δ 175.5, 167.2, 163.2, 161.3, 160.3, 158.8, 152.0, 149.1, 123.1, 119.8, 112.0, 111.4, 109.4, 107.2, 107.0, 96.9, 80.5, 79.1, 74.1, 71.6, 70.6, 63.9, 62.7, 56.4, 56.0, 47.8, 32.6, 32.6, 29.1, 24.6, 24.6. HRESI-TOFMS m/z [M + H]+ 600.2446 (calcd for C31H38NO11+, 600.2439, −1.1 ppm error). HPLC purity: 98.6% (210 nm).
(2S,3S,4R,5R,6S)-N-(Cyclohexylmethyl)-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxamide (18)
Light yellow solid (88% yield). 1H NMR (DMSO-d6) δ 7.66 (d, J = 8.7 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.13 (s, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 6.2 Hz, 1H), 4.66 (br d, J = 9.9 Hz, 1H), 4.05 (q, J = 9.9 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.78 (s, 3H), 3.73 (s, 3H), 3.61 (m, 1H), 3.48 (m, 1H), 3.23 (m, 1H), 2.87 (m, 2H), 1.69–1.47 (m, 6H), 1.33 (m, 1H), 1.08 (m, 2H), 0.80 (m, 2H). 13C NMR (DMSO-d6) δ 176.0, 167.0, 163.7, 160.8, 160.6, 158.9, 152.0, 149.3, 123.4, 119.7, 112.0, 111.7, 109.3, 107.2, 107.1, 96.2, 80.3, 78.7, 74.7, 71.3, 69.9, 63.8, 63.1, 56.9, 56.2, 45.1, 37.1, 30.6, 30.6, 26.3, 25.7, 25.7. HRESI-TOFMS m/z [M + H]+ 614.2597 (calcd for C32H40NO11+, 614.2596, −0.1 ppm error). HPLC purity: 98.3% (210 nm).
(2S,3S,4R,5R,6S)-N-(Adamantan-1-yl)-6-(2-(3,4-dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxytetrahy-dro-2H-pyran-2-carboxamide (19)
Light yellow solid (81% yield). 1H NMR (DMSO-d6) δ 7.67 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.13 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 6.8 Hz, 1H), 4.65 (br d, J = 10.2 Hz, 1H), 4.03 (m, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.57 (m, 1H), 3.52 (m, 1H), 3.49 (m, 1H), 1.96 (m, 3H), 1.88–1.57 (m, 12H). 13C NMR (DMSO-d6) δ 175.3, 167.2, 163.3, 161.1, 160.2, 158.9, 152.1, 149.2, 123.2, 119.8, 112.1, 111.6, 109.5, 107.2, 107.0, 97.0, 80.7, 79.2, 74.2, 71.7, 70.5, 63.9, 62.5, 56.4, 56.0, 51.2, 41.2, 41.2, 41.2, 36.1, 36.1, 36.1, 28.9, 28.9, 28.9. HRESI-TOFMS m/z [M + H]+ 652.2764 (calcd for C35H42NO11+, 652.2752, −1.8 ppm error). HPLC purity: 97.7% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-phenyltetrahydro-2H-pyran-2-carboxamide (20)
Light yellow solid (88% yield). 1H NMR (DMSO-d6) δ 7.66 (dd, J = 8.6, 2.2 Hz, 1H), 7.62 (dd, J = 8.2, 2.5 Hz, 2H), 7.55 (d, J = 2.2 Hz, 1H), 7.28 (td, J = 8.1, 2.2 Hz, 2H), 7.16–7.10 (m, 2H), 7.03 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 6.5 Hz, 1H), 4.75 (br d, J = 9.7 Hz, 1H), 4.12 (td, J = 9.7, 2.4 Hz, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.85 (s, 3H), 3.84 (m, 1H), 3.79 (s, 3H), 3.66 (m, 1H), 3.29 (m, 1H). 13C NMR (DMSO-d6) δ 175.4, 167.3, 163.4, 161.9, 160.3, 158.8, 151.8, 149.1, 138.8, 128.6, 128.6, 123.4, 123.1, 119.5, 119.3, 119.3, 112.0, 111.8, 109.4, 107.1, 106.9, 97.1, 81.0, 78.6, 73.7, 71.2, 69.6, 63.4, 62.7, 56.5, 56.0. HRESI-TOFMS m/z [M + H]+ 594.1986 (calcd for C31H32NO11+, 594.1970, −2.8 ppm error). HPLC purity: 96.5% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-benzyltetrahydro-2H-pyran-2-carboxamide (21)
Light yellow solid (92% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.67 (d, J = 10.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.29–7.16 (m, 5H), 7.16–7.08 (m, 2H), 6.80 (d, J = 6.7 Hz, 1H), 4.68 (dd, J = 9.8, 6.7 Hz, 1H), 4.27 (m, 2H), 4.10 (m, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.79 (s, 3H), 3.75 (s, 3H), 3.71 (m, 1H), 3.57 (m, 1H), 3.26 (m, 1H). 13C NMR (DMSO-d6) δ 175.6, 169.1, 163.6, 162.0, 160.3, 158.9, 151.8, 149.1, 139.2, 128.2, 128.2, 127.3, 127.3, 126.7, 123.1, 119.6, 112.1, 111.7, 109.2, 107.1, 107.0, 97.1, 80.2, 78.8, 73.7, 71.6, 71.0, 63.5, 62.8, 56.6, 56.0, 42.0. HRESI-TOFMS m/z [M + H]+ 608.2130 (calcd for C32H34NO11+, 608.2126, −0.5 ppm error). HPLC purity: 97.1% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(pyridin-4-yl)tetrahydro-2H-pyran-2-carboxamide (22)
Light yellow solid (81% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.40 (d, J = 5.0 Hz, 2H), 7.68 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 5.0 Hz, 2H), 7.55 (s, 1H), 7.15 (s, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.81 (d, J = 6.9 Hz, 1H), 4.72 (br d, J = 9.7 Hz, 1H), 4.11 (td, J = 9.2, 4.1 Hz, 1H), 3.94 (m, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.76 (s, 3H), 3.62 (m, 1H), 3.29 (m, 1H). 13C NMR (DMSO-d6) δ 175.6, 167.5, 163.3, 161.7, 160.2, 158.7, 152.0, 150.9, 150.9, 149.2, 145.3, 123.1, 119.9, 112.1, 113.9, 113.9, 111.5, 109.8, 107.5, 106.8, 97.1, 81.2, 78.8, 74.4, 71.5, 69.8, 63.9, 63.0, 56.5, 56.1. HRESI-TOFMS m/z [M + H]+ 595.1921 (calcd for C30H31N2O11+, 595.1922, 0.1 ppm error). HPLC purity: 97.8% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(thiazol-2-yl)tetrahydro-2H-pyran-2-carboxamide (23)
Light yellow solid (80% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.18 (d, J = 1.7 Hz, 1H), 7.17 (s, 1H), 7.12 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 6.8 Hz, 1H), 4.67 (br d, J = 10.5 Hz, 1H), 4.03 (m, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.76 (s, 3H), 3.60 (m, 1H), 3.55 (m, 1H), 3.23 (m, 1H). 13C NMR (DMSO-d6) δ 175.4, 168.5, 163.2, 162.3, 161.6, 160.5, 158.9, 151.9, 149.2, 137.2, 123.3, 119.8, 114.1, 111.9, 111.5, 109.5, 107.3, 105.8, 97.3, 80.0, 79.2, 74.3, 72.1, 70.3, 63.7, 62.9, 56.2, 56.0. HRESI-TOFMS m/z [M + H]+ 601.1493 (calcd for C28H29N2O11S+, 601.1487, −1.1 ppm error). HPLC purity: 96.8% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(4-fluorophenyl)tetrahydro-2H-pyran-2-carboxamide (24)
Light yellow solid (88% yield). 1H NMR (DMSO-d6) δ 7.70–7.61 (m, 3H), 7.54 (d, J = 2.1 Hz, 1H), 7.18–7.08 (m, 4H), 6.78 (d, J = 7.1 Hz, 1H), 4.72 (dd, J = 12.0, 9.7 Hz, 1H), 4.11 (q, J = 8.7 Hz, 1H), 3.92 (s, 3H), 3.88 (s, 3H), 3.84 (s, 3H), 3.83 (m, 1H), 3.76 (s, 3H), 3.61 (m, 1H), 3.28 (m, 1H). 13C NMR (DMSO-d6) δ 175.6, 167.4, 162.8, 160.4, 160.1, 158.6, 156.9, 150.7, 149.2, 136.5, 121.2, 121.2, 123.5, 119.8, 115.5, 115.5, 113.5, 111.8, 109.3, 107.0, 105.7, 97.2, 81.2, 78.5, 74.7, 71.7, 70.4, 63.7, 63.0, 56.1, 55.9. HRESI-TOFMS m/z [M + H]+ 612.1892 (calcd for C31H31FNO11+, 612.1876, −2.7 ppm error). HPLC purity: 97.1% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(4-fluorobenzyl)tetrahydro-2H-pyran-2-carboxamide (25)
Light yellow solid (89% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.68 (d, J = 8.5 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.27 (ddd, J = 8.1, 5.4, 2.2 Hz, 2H), 7.18–7.06 (m, 4H), 6.80 (d, J = 6.8 Hz, 1H), 4.67 (t, J = 9.3 Hz, 1H), 4.24 (dd, J = 9.9, 9.0 Hz, 2H), 4.07 (m, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.78 (s, 3H), 3.74 (s, 3H), 3.69 (m, 1H), 3.54 (m, 1H), 3.24 (m, 1H). 13C NMR (DMSO-d6) δ 175.7, 169.1, 163.5, 162.0, 161.2, 160.4, 159.0, 151.9, 149.1, 135.4, 129.3, 129.3, 126.5, 119.7, 115.1, 115.1, 111.8, 111.3, 109.3, 107.1, 105.5, 97.2, 80.2, 78.8, 73.7, 71.9, 71.0, 63.6, 62.9, 56.6, 55.9, 41.3. HRESI-TOFMS m/z [M + H]+ 626.2043 (calcd for C32H33FNO11+, 626.2032, −1.7 ppm error). HPLC purity: 97.4% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(2-fluoroethyl)tetrahydro-2H-pyran-2-carboxamide (26)
Light yellow solid (84% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.17 (s, 1H), 7.12 (d, J = 8.7 Hz, 1H), 6.82 (d, J = 6.6 Hz, 1H), 4.67 (br d, J = 10.5 Hz, 1H), 4.45 (m, 1H), 4.35 (m, 1H), 4.07 (t, J = 9.2 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H), 3.60 (m, 1H), 3.50 (m, 1H), 3.23 (m, 1H), 3.37 (m, 2H). 13C NMR (DMSO-d6) δ 175.1, 168.6, 163.5, 161.6, 160.5, 158.9, 151.9, 149.2, 123.6, 119.7, 111.8, 111.4, 109.3, 107.2, 105.5, 97.3, 81.7, 79.9, 79.4, 74.3, 72.3, 70.3, 63.5, 62.9, 56.1, 55.9, 39.1. HRESI-TOFMS m/z [M + H]+ 564.1892 (calcd for C27H31FNO11+, 564.1876, −2.9 ppm error). HPLC purity: 97.5% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(2,2,2-trifluoroethyl)tetrahydro-2H-pyran-2-carboxamide (27)
Light yellow solid (85% yield). 1H NMR (DMSO-d6) δ 7.68 (dd, J = 8.5, 2.0 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 7.17 (s, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.81 (d, J = 7.7 Hz, 1H), 4.68 (br d, J = 9.7 Hz, 1H), 4.07 (q, J = 9.1 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.69 (m, 1H), 3.48 (m, 1H), 3.67 (m, 2H), 3.21 (m, 1H). 13C NMR (DMSO-d6) δ 174.5, 169.5, 163.4, 161.3, 160.4, 158.9, 151.8, 149.1, 125.0, 123.4, 119.7, 112.1, 111.2, 109.4, 107.1, 105.4, 97.2, 80.4, 78.5, 74.8, 71.6, 69.5, 63.6, 62.7, 56.3, 55.9, 41.6. HRESI-TOFMS m/z [M + H]+ 600.1700 (calcd for C27H29F3NO11+, 600.1687, −2.2 ppm error). HPLC purity: 96.8% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(3-fluoropropyl)tetrahydro-2H-pyran-2-carboxamide (28)
Light yellow solid (86% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.17 (s, 1H), 7.12 (d, J = 8.7 Hz, 1H), 6.82 (d, J = 6.4 Hz, 1H), 4.66 (br d, J = 10.6 Hz, 1H), 4.46 (m, 1H), 4.37 (m, 1H), 4.07 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.59 (m, 1H), 3.50 (m, 1H), 3.22 (m, 1H), 3.12 (m, 2H), 1.83–1.68 (m, 2H). 13C NMR (DMSO-d6) δ 175.1, 168.5, 163.4, 161.6, 160.6, 158.8, 151.9, 149.3, 123.7, 119.8, 111.9, 111.5, 109.6, 107.3, 105.5, 97.3, 81.9, 80.2, 78.8, 74.2, 71.8, 70.6, 63.6, 62.8, 56.2, 55.8, 34.9, 30.0. HRESI-TOFMS m/z [M + H]+ 578.2041 (calcd for C28H33FNO11+, 578.2032, −1.6 ppm error). HPLC purity: 98.3% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-(3,3,3-trifluoropropyl)tetrahydro-2H-pyran-2-carboxamide (29)
Light yellow solid (85% yield). 1H NMR (DMSO-d6) δ 7.68 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 2.2 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.81 (d, J = 5.5 Hz, 1H), 4.68 (br d, J = 10.0 Hz, 1H), 4.07 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.74 (s, 3H), 3.61 (m, 1H), 3.49 (m, 1H), 3.28 (m, 2H), 3.21 (m, 1H), 2.40 (m, 2H). 13C NMR (DMSO-d6) δ 174.6, 169.3, 163.4, 161.1, 160.3, 158.7, 151.8, 149.1, 125.7, 123.1, 119.6, 111.7, 110.9, 109.2, 107.0, 105.2, 97.4, 79.6, 78.3, 73.6, 71.6, 69.5, 63.4, 62.7, 56.0, 55.8, 32.8, 32.1. HRESI-TOFMS m/z [M + H]+ 614.1857 (calcd for C28H31F3NO11+, 614.1844, −2.2 ppm error). HPLC purity: 97.2% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-((S)-1,1,1-trifluoropropan-2-yl)tetrahydro-2H-pyran-2-carboxamide (30)
Light yellow solid (82% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.67 (dd, J = 8.5, 2.1 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H), 7.15 (br s, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.78 (d, J = 6.2 Hz, 1H), 4.69 (d, J = 9.6 Hz, 1H), 4.55 (m, 1H), 4.07 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.79 (s, 3H), 3.76 (s, 3H), 3.71 (m, 1H), 3.56 (m, 1H), 3.24 (m, 1H), 1.23 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 175.5, 168.8, 163.5, 161.7, 160.3, 158.6, 152.1, 149.1, 124.4, 123.1, 119.5, 111.8, 111.3, 109.4, 107.7, 106.9, 96.2, 79.8, 78.3, 73.8, 71.3, 70.3, 62.9, 62.7, 55.9, 55.6, 45.1, 13.3. HRESI-TOFMS m/z [M + H]+ 614.1843 (calcd for C28H31F3NO11+, 614.1844, 0.1 ppm error). HPLC purity: 98.1% (210 nm).
(2S,3S,4R,5R,6S)-6-(2-(3,4-Dimethoxyphenyl)-5,7-dimethoxy-4-oxo-4H-chromen-6-yl)-3,4,5-trihydroxy-N-((R)-1,1,1-trifluoropropan-2-yl)tetrahydro-2H-pyran-2-carboxamide (31)
Light yellow solid (86% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.66 (dd, J = 8.5, 2.0 Hz, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.14 (br s, 1H), 7.12 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 6.4 Hz, 1H), 4.68 (d, J = 9.8 Hz, 1H), 4.54 (m, 1H), 4.06 (m, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.79 (s, 3H), 3.75 (s, 3H), 3.71 (m, 1H), 3.55 (m, 1H), 3.24 (m, 1H), 1.23 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 175.3, 168.6, 163.4, 161.5, 160.2, 158.5, 152.1, 149.0, 124.2, 123.0, 119.4, 111.6, 111.1, 109.3, 107.6, 106.8, 96.4, 79.7, 78.5, 73.9, 71.2, 70.4, 63.1, 62.5, 55.7, 55.4, 45.1, 13.3. HRESI-TOFMS m/z [M + H]+ 614.1847 (calcd for C28H31F3NO11+, 614.1844, −0.5 ppm error). HPLC purity: 96.5% (210 nm).
Kinase Luminescent Assay
Kinase inhibition was assessed with the ADP-Glo Kinase Assay. For screening, 5 ng/μL kinase was assayed in a reaction containing 50 ng/μL substrate, 40 mM Tris, pH 7.5, 20 mM MgCl2, 0.1 mg/mL bovine serum albumin, 50 μM dithiothreitol (DTT), 25 μM ATP, varying concentrations of test samples, or 5% DMSO as vehicle. The reaction mixture was incubated for 1 h at room temperature followed by the addition of the ADP-Glo reagents according to the manufacturer’s protocol. The kinase inhibitor staurosporine was used at 1 μM as a reference control. Each data point was collected in quadruplicate of two independent experiments. All new analogues were not promiscuous or pan-assay interference compounds as determined with a detergent-based assay.23,31
To study the GSK-3β kinetics, a reaction solution contained 5 ng/μL kinase, 40 mM Tris, pH 7.5, 20 mM MgCl2, 0.1 mg/mL BSA, 50 μM DTT, and varying concentrations of ATP or substrate GS2 (peptide YRRAAVPPSPSLSRHSSPHQ(pS)EDEEE that is derived from human muscle glycogen synthase) versus test samples. The mixture was incubated for 5, 15, 30, and 60 min at room temperature followed by the addition of the ADP-Glo reagents according to the manufacturer’s protocol. The Lineweaver–Burk representation is derived from the double reciprocal plotting of the enzyme kinetic data.
Cell Culture
Human neuroblastoma SH-SY5Y cell line (Sigma-Aldrich, Saint Louis, MO) was cultured in DMEM/F12 (v/v 1:1) media supplemented with 2 mM glutamine, 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics including penicillin and streptomycin. After reaching 70–80% confluence, cells were then subcultured on poly-L-lysine plates with 10 μM retinoic acid in a reduced serum media (1% FBS) to promote neuronal maturation and differentiation as described.35 Cell cultures were incubated at 37 °C in a fully humidified atmosphere containing 5% CO2.
Whole-Cell Lysate GSK-3β Assay
The assay procedure was followed as described.23 SH-SY5Y cells were washed with phosphate buffered saline (PBS) and lysed with cell extraction buffer containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1 mM phenylmethanesulfonyl fluoride (PMSF) and a protease inhibitor cocktail. Lysate was diluted with kinase buffer (40 mM Tris, pH 7.5, 20 mM MgCl2, 50 μM DTT, 400 μM ATP) to afford a concentration of 5 μg/μL of total protein, and split into aliquots. Recombinant human GSK-3β was fortified into lysate aliquots to a final concentration of 0.25% (w/w) of total protein. A lysate aliquot fortified with heat-inactivated GSK-3β was used as a negative control. The fortified lysate aliquots were incubated with test sample or 5% DMSO vehicle at 37 °C for 2 h followed by ELISA analysis. The GSK-3β inhibitor TDZD-8 was used at 10 μM as a reference control.
Human Tau pS396 ELISA
The quantitative determination of phosphorylated human tau at GSK-3β specific pS396 site was conducted by taking 50 μL diluted cell lysate and using a specific antibody against human tau [pS396] in a sandwich ELISA according to the manufacturer’s protocol. Tau phosphorylation was quantified by measuring the absorbance at 450 nm in a microtiter plate reader. The analysis was collected in quadruplicate of two independent experiments.
Aβ42 Oligomer Preparation
The toxic oligomers of Aβ42 were prepared as described.23 Briefly, lyophilized Aβ42 peptide was dissolved in hexafluoroisopropanol, dried under vacuum, and stored at −20 °C. Immediately prior to use, the peptide residue was reconstituted in DMEM/F12 media to make a stock solution at 0.1 mM and incubated at 4 °C for 24 h to form diffusible oligomers. Aβ42 oligomers at a final concentration of 10 μM were assayed for cell viability
Anti-Aβ42 Neurotoxicity Assay
SH-SY5Y cells were seeded at a density of 3 × 105 cells/mL in a 96-well plate in DMEM/F12 media containing 10 μM retinoic acid and 1% FBS to suppress cell proliferation. Cells were incubated under regular culture conditions for attachment. After 24 h of plating, the cells were pretreated with different concentrations of test samples or the 0.2% DMSO as a vehicle control for 1 h and then coincubated with 10 μMAβ42 for 72 h. After the experimental treatment, the cells were subject to a CellTiter 96 AQueous One Solution Cell Proliferation MTX Assay according to the manufacturer’s instruction. Staurosporine at 1 μM was used as a reference control for cytotoxicity, while 10 μM TDZD-8 was used as a reference control for GSK-3β inhibition. Each data point was collected in triplicate of two independent experiments.
PAMPA Studies
A 96-well filter plate with 0.45 μm polyvinylidene fluoride (PVDF) membrane was precoated with trilayer phospholipids. Then 300 μL of sample solutions (20 μM) in 5% DMSO-PBS at pH 7.4 were added to the donor wells. The acceptor plate containing 200 μL of 5% DMSO-PBS was then placed on top of the donor plate so that the artificial membrane was in contact with the solution below. The PAMPA system was covered with a lid and incubated for 5 h at room temperature. The concentration of compound in the donor and acceptor wells was quantified by LC-ESI-QTOF-MS. Theophylline and atenolol known for their low permeability were used as negative controls, and desipramine known for its high permeability was used as a positive control. Samples were run in quadruplicate. Pe values and R % were calculated according to the manufacturer’s instruction.
Docking Studies
Compounds of interest were docked with AutoDock Vina 1.1.238,40 using the X-ray crystallographic structures of GSK-3β (PDB codes 1PYX42 and 1H8F43). To streamline the docking process, the PDB crystallographic structures were treated without water molecules according to the published GSK-3β docking protocols.17,40 Proteins were prepared by adding polar hydrogens and Gasteiger charges using AutoDockTools.40 Ligands were optimized for their energy and geometry using MMFF94 and AM1 force fields prior to docking as described.60 All bonds of ligands were treated as rotatable except for the aromatic, alkenyl, carbonyl bonds and rings. The dimensions of the grid map were 30 × 30 × 30 points with a grid-point spacing of 1 Å. Docking was proceeded with an exhaustiveness value of 500 and a maximum output of 100 structures. Redocking experiments were conducted using the ligands ANP and HEPES for 1PYX and 1H8F, respectively. ANP showed a binding pose at the ATP site of GSK-3β (PDB code 1PYX) with a RMSD of 0.33 Å as compared to its original crystal structure. HEPES showed a binding pose at the substrate site of GSK-3β (PDB code 1H8F) with a RMSD of 0.62 Å as compared to its original crystal structure. AutoDock-Tools40 was used to analyze the docking data of compounds of interest in molecular interactions including hydrogen bonds, hydrophobic contact, π–cation interactions, π–π interactions, and multipolar interactions.
Homology Modeling
The GSK-3α homology model was built with the SWISS-MODEL server.51 The full sequence of human GSK-3α (UniProt code P49840) was obtained from the Universal Protein Resource. The target sequence was searched against BLAST and HHblits databases for evolutionary related protein structures. A total of 4470 templates were found. For each identified template, the template’s quality was predicted from features of the target-template alignment. A template of the GSK-3β structure (PDB code 1PYX) showing the highest quality (sequence identity, 82.97%) in the template ranking was selected for model building. The model was built based on the target-template alignment using ProMod3. Coordinates that are conserved between the target and the template were copied from the template to the model. Insertions and deletions were remodeled using a fragment library. Side chains were then rebuilt. Finally, the geometry and energy minimization of the resulting model was performed using the OpenMM molecular mechanics force field. The model quality assessment was performed based on the global and per-residue model quality using the QMEAN scoring function. A detailed homology modeling method was elaborated in Supporting Information S5.
Statistical Analysis
Data were presented as the mean ± SEM or ± SD. The data were analyzed by one-way ANOVA with Tukey’s multiple comparison posthoc test and Student’s t test. The p values less than 0.05 were considered statistically significant. Analyses were performed using Excel and GraphPad Prism.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Philip Williams, Dr. Robert Nichols, and Dr. Jon-Paul Bingham, UH-Manoa, for helpful discussions; Dr. Wei Wen Su, UH-Manoa, for the assistance of bioluminescence measurement; Dr. Yong-Soo Kim and Dr. Jinzeng Yang, UH-Manoa, for the assistance of cell culture; and Wesley Yoshida, UH-Manoa NMR facility, for acquiring the NMR spectra.
Funding
This work was supported in part by the NIH National Institute on Minority Health and Health Disparities grant 8G12MD007601, and by the USDA National Institute of Food and Agriculture, Hatch Project HAW5032-R, managed by the College of Tropical Agriculture and Human Resources, University of Hawaii.
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ42
β-amyloid fragment peptide 1–42
- NFT
neurofibrillary tangle
- GSK-3
glycogen synthase kinase-3
- ERK2
extracellular signal regulated kinase 2
- JNK1
c-Jun N-terminal kinase 1
- JNK3
c-Jun N-terminal kinase 3
- p38α
p38 mitogen-activated protein kinase 14
- p38β
mitogenactivated protein kinase 14B
- p38γ
mitogen-activated protein kinase 12
- p38δ
p38 mitogen-activated protein kinase 13
- CDK1/CyclinA
cyclin-dependent kinase 1 with subunit cyclin A
- CDK2/CyclinE
cyclin-dependent kinase 2 with subunit cyclin E
- CDK3/CyclinE
cyclin-dependent kinase 3 with subunit cyclin E
- CDK5/p25
cyclin-dependent kinase 5 with subunit p25
- CDK5/p35
cyclin-dependent kinase 5 with subunit p35
- CDK6/CyclinD
cyclin-dependent kinase 6 with subunit cyclin D
- CDK9/CyclinK
cyclin-dependent kinase 9 with subunit cyclin K
- CLK1
dual specificity protein kinase 1
- AKT1
v-akt murine thymoma viral oncogene homologue 1
- p70S6Kβ
p70 ribosomal protein S6 kinase beta
- PDK1
phosphoinositide-dependent kinase 1
- PKA
protein kinase A
- PKC
protein kinase C
- PRKG1
cGMP-dependent protein kinase 1
- ROCK1
Rho-associated, coiled-coil containing protein kinase 1
- RSK2
ribosomal protein S6 kinase 2
- AMPK
AMP-activated protein kinase with subunits
- CAM-KIIα
Ca2+/calmodulin-dependent protein kinase Iiα
- CAM-KIIγ
Ca2+/calmodulin-dependent protein kinase IIγ
- CAM-KIV
Ca2+/calmodulin-dependent protein kinase IV
- DAPK1
death-associated protein kinase 1
- STK33
serine/threonineprotein kinase 33
- CK2α1
casein kinase 2α1
- DNA-PK
DNA-dependent protein kinase
- CK1α1
casein kinase 1α1
- CK1ε
casein kinase 1ε
- CK1γ1
casein kinase 1γ1
- VRK2
vaccinia related kinase 2
- ANP
adenylyl imidodiphosphate
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- TMSCHN2
trimethylsilyldiazomethane
- HCTU
2-(6-chloro-1-H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- [TEMPO]+[BF4]−
4-acetamido-2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate
- TDZDs
thiadiazolidinones
- CLogP
calculated logarithm of partition coefficient
- LiPE
ligand-lipophilic efficiency
- SAR
structure–activity relationship
- ELISA
enzyme-linked immunosorbent assay
- PAMPA
parallel artificial membrane permeability assay
- HPLC
high performance liquid chromatography
- LC-ESI-QTOF-MS
liquid chromatography-electrospray ionization-quadrupole time-of-flight-mass spectrometry
- HRMS
high resolution mass spectrometry
- NMR
nuclear magnetic resonance
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00010.
Docking method validation, tabulated docking scores, predicted docking poses of selected C-glycosylflavones with GSK-3β, summary of molecular interaction distances of compound 30 with GSK3β, GSK-3α homology modeling method, protein sequence alignment of GSK-3α/β isoforms, HPLC analysis data, detail of the kinase selectivity screening, LC-MS method for the PAMPA studies, and structures of the reference compounds (PDF)
■ REFERENCES
- (1).Querfurth HW, and LaFerla FM (2010) Alzheimer’s disease. N. Engl. J. Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- (2).McDade E, and Bateman RJ (2017) Stop Alzheimer’s before it starts. Nature 547, 153–155. [DOI] [PubMed] [Google Scholar]
- (3).Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, Ashe KH, and Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Khanna MR, Kovalevich J, Lee VMY, Trojanowski JQ, and Brunden KR (2016) Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimer’s Dementia 12, 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Reardon S (2015) Alzheimer antibody drugs show questionable potential. Nat. Rev. Drug Discovery 14, 591–592. [DOI] [PubMed] [Google Scholar]
- (6).Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Hoffmann VP, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, and Siemers E (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med 378, 321–330. [DOI] [PubMed] [Google Scholar]
- (7).Iqbal K, Liu F, and Gong C-X (2016) Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol 12, 15–27. [DOI] [PubMed] [Google Scholar]
- (8).Hooper C, Killick R, and Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem 104, 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Yardin C, and Terro F (2013) Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev 12, 289–309. [DOI] [PubMed] [Google Scholar]
- (10).Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, Mantua V, Mecocci P, Pani L, Winblad B, and Kivipelto M (2014) Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med 275, 251–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pandey MK, and DeGrado TR (2016) Glycogen synthase kinase-3 (GSK-3) – targeted therapy and imaging. Theranostics 6, 571–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, and Medina M (2012) Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J. Biol. Chem 287, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Swinney ZT, Haubrich BA, Xia S, Ramesha C, Gomez SR, Guyett P, Mensa-Wilmot K, and Swinney DC (2016) A four-point screening method for assessing molecular mechanism of action (MMOA) identifies tideglusib as a time-dependent inhibitor of Trypanosoma brucei GSK3β. PLoS Neglected Trop. Dis 10, e0004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huggins DJ, Sherman W, and Tidor B (2012) Rational approaches to improving selectivity in drug design. J. Med. Chem 55, 1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Peng J, Kudrimoti S, Prasanna S, Odde S, Doerksen RJ, Pennaka HK, Choo Y-M, Rao KV, Tekwani BL, Madgula V, Khan SI, Wang B, Mayer AMS, Jacob MR, Tu LC, Gertsch J, and Hamann MT (2010) Structure-activity relationship and mechanism of action studies of manzamine analogues for the control of neuroinflammation and cerebral infections. J. Med. Chem 53, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Palomo V, Perez DI, Perez C, Morales-Garcia JA, Soteras I, Alonso-Gil S, Encinas A, Castro A, Campillo NE, Perez-Castillo A, Gil C, and Martinez A (2012) 5-Imino-1,2,4-thiadiazoles: First small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J. Med. Chem 55, 1645–1661. [DOI] [PubMed] [Google Scholar]
- (17).Tapia-Rojas C, Schüller A, Lindsay CB, Ureta RC, Mejías-Reyes C, Hancke J, Melo F, and Inestrosa NC (2015) Andrographolide activates the canonical Wnt signalling pathway by a mechanism that implicates the non-ATP competitive inhibition of GSK-3β: Autoregulation of GSK-3β in vivo. Biochem. J 466, 415–430. [DOI] [PubMed] [Google Scholar]
- (18).Licht-Murava A, Paz R, Vaks L, Avrahami L, Plotkin B, Eisenstein M, and Eldar-Finkelman H (2016) A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signaling 9, ra110. [DOI] [PubMed] [Google Scholar]
- (19).Rodrigues T, Reker D, Schneider P, and Schneider G (2016) Counting on natural products for drug design. Nat. Chem 8, 531–541. [DOI] [PubMed] [Google Scholar]
- (20).Baptista FI, Henriques AG, Silva AMS, Wiltfang J, and da Cruz e Silva OAB (2014) Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer’s disease. ACS Chem. Neurosci 5, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao J, Capanoglu E, Jassbi AR, and Miron A (2016) Advance on the flavonoid C-glycosides and health benefits. Crit. Rev. Food Sci. Nutr 56, S29–S45. [DOI] [PubMed] [Google Scholar]
- (22).Sciacca MFM, Romanucci V, Zarrelli A, Monaco I, Lolicato F, Spinella N, Galati C, Grasso G, D’Urso L, Romeo M, Diomede L, Salmona M, Bongiorno C, Di Fabio G, La Rosa C, and Milardi D (2017) Inhibition of Aβ amyloid growth and toxicity by silybins: The crucial role of stereochemistry. ACS Chem. Neurosci 8, 1767–1778. [DOI] [PubMed] [Google Scholar]
- (23).Liang Z, Zhang B, Su WW, Williams PG, and Li QX (2016) C-Glycosylflavones alleviate tau phosphorylation and amyloid neurotoxicity through GSK3β inhibition. ACS Chem. Neurosci 7, 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Song Y, Kim H-D, Lee M-K, Hong I-H, Won C-K, Bai H-W, Lee SS, Lee S, Chung BY, and Cho J-H (2017) Maysin and its flavonoid derivative from centipedegrass attenuates amyloid plaques by inducting humoral immune response with Th2 skewed cytokine response in the Tg (APPswe, PS1dE9) Alzheimer’s mouse model. PLoS One 12, e0169509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Peng K-Z, Zhang S-Y, and Zhou H-L (2016) Toxicological evaluation of the flavonoid-rich extract from Maydis stigma: Subchronic toxicity and genotoxicity studies in mice. J. Ethnopharmacol 192, 161–169. [DOI] [PubMed] [Google Scholar]
- (26).Ilouz R, Kowalsman N, Eisenstein M, and Eldar-Finkelman H (2006) Identification of novel glycogen synthase kinase-3β substrate-interacting residues suggests a common mechanism for substrate recognition. J. Biol. Chem 281, 30621–30630. [DOI] [PubMed] [Google Scholar]
- (27).Walle T (2007) Methylation of dietary flavones greatly improves their hepatic metabolic stability and intestinal absorption. Mol. Pharmaceutics 4, 826–832. [DOI] [PubMed] [Google Scholar]
- (28).Liang Z, Sorribas A, Sulzmaier FJ, Jiménez JI, Wang X, Sauvage T, Yoshida WY, Wang G, Ramos JW, and Williams PG (2011) Stictamides A–C, MMP12 inhibitors containing 4-amino-3-hydroxy-5-phenylpentanoic acid subunits. J. Org. Chem. 76, 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bobbitt JM, Bartelson AL, Bailey WF, Hamlin TA, and Kelly CB (2014) Oxoammonium salt oxidations of alcohols in the presence of pyridine bases. J. Org. Chem 79, 1055–1067. [DOI] [PubMed] [Google Scholar]
- (30).Baell JB (2016) Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod 79, 616–628. [DOI] [PubMed] [Google Scholar]
- (31).Feng BY, and Shoichet BK (2006) A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc 1, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wager TT, Hou X, Verhoest PR, and Villalobos A (2016) Central nervous system multiparameter optimization desirability: Application in drug discovery. ACS Chem. Neurosci 7, 767–775. [DOI] [PubMed] [Google Scholar]
- (33).Freeman-Cook KD, Hoffman RL, and Johnson TW (2013) Lipophilic efficiency: The most important efficiency metric in medicinal chemistry. Future Med. Chem 5, 113–115. [DOI] [PubMed] [Google Scholar]
- (34).Chou JT, and Jurs PC (1979) Computer-assisted computation of partition coefficients from molecular structures using fragment constants. J. Chem. Inf. Model 19, 172–178. [Google Scholar]
- (35).Agholme L, Lindström T, Kågedal K, Marcusson J, and Hallbeck M (2010) An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimer’s Dis 20, 1069–1082. [DOI] [PubMed] [Google Scholar]
- (36).Chen X, Murawski A, Patel K, Crespi CL, and Balimane PV (2008) A novel design of artificial membrane for improving the PAMPA model. Pharm. Res 25, 1511–1520. [DOI] [PubMed] [Google Scholar]
- (37).Courts FL, and Williamson G (2015) The occurrence, fate and biological activities of C-glycosyl flavonoids in the human diet. Crit. Rev. Food Sci. Nutr 55, 1352–1367. [DOI] [PubMed] [Google Scholar]
- (38).Trott O, and Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang Z, Sun H, Yao X, Li D, Xu L, Li Y, Tian S, and Hou T (2016) Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys 18, 12964–12975. [DOI] [PubMed] [Google Scholar]
- (40).Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, and Olson AJ (2016) Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hassandarvish P, Rothan HA, Rezaei S, Yusof R, Abubakar S, and Zandi K (2016) In silico study on baicalein and baicalin as inhibitors of dengue virus replication. RSC Adv. 6, 31235–31247. [Google Scholar]
- (42).Bertrand JA, Thieffine S, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, and Flocco M (2003) Structural characterization of the GSK-3β active site using selective and nonselective ATP-mimetic inhibitors. J. Mol. Biol 333, 393–407. [DOI] [PubMed] [Google Scholar]
- (43).Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, and Pearl LH (2001) Crystal structure of glycogen synthase kinase 3β: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732. [DOI] [PubMed] [Google Scholar]
- (44).Gadakar PK, Phukan S, and Balaji VN (2007) Pose prediction accuracy in docking studies and enrichment of actives in the active site of GSK-3β. J. Chem. Inf. Model 47, 1446–1459. [DOI] [PubMed] [Google Scholar]
- (45).Fu G, Sivaprakasam P, Dale OR, Manly SP, Cutler SJ, and Doerksen RJ (2014) Pharmacophore modeling, ensemble docking, virtual screening, and biological evaluation on glycogen synthase kinase-3β. Mol. Inf 33, 610–626. [DOI] [PubMed] [Google Scholar]
- (46).Müller K, Faeh C, and Diederich F (2007) Fluorine in pharmaceuticals: Looking beyond intuition. Science 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (47).Bissantz C, Kuhn B, and Stahl M (2010) A medicinal chemist’s guide to molecular interactions. J. Med. Chem 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Abreu RMV, Froufe HJC, Queiroz M-JRP, and Ferreira ICFR (2012) Selective flexibility of side-chain residues improves VEGFR-2 docking score using AutoDock Vina. Chem. Biol. Drug Des 79, 530–534. [DOI] [PubMed] [Google Scholar]
- (49).Gallivan JP, and Dougherty DA (1999) Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U. S. A 96, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pellequer J-L, Zhao B, Kao H-I, Bell CW, Li K, Li QX, Karu AE, and Roberts VA (2000) Stabilization of bound polycyclic aromatic hydrocarbons by a π-cation interaction. J. Mol. Biol 302, 691–699. [DOI] [PubMed] [Google Scholar]
- (51).Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, and Schwede T (2014) SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Beurel E, Grieco SF, and Jope RS (2015) Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Liang SH, Chen JM, Normandin MD, Chang JS, Chang GC, Taylor CK, Trapa P, Plummer MS, Para KS, Conn EL, Lopresti-Morrow L, Lanyon LF, Cook JM, Richter KEG, Nolan CE, Schachter JB, Janat F, Che Y, Shanmugasundaram V, Lefker BA, Enerson BE, Livni E, Wang L, Guehl NJ, Patnaik D, Wagner FF, Perlis R, Holson EB, Haggarty SJ, El Fakhri G, Kurumbail RG, and Vasdev N (2016) Discovery of a highly selective glycogen synthase kinase-3 inhibitor (PF-04802367) that modulates tau phosphorylation in the brain: Translation for PET neuroimaging. Angew. Chem., Int. Ed 55, 9601–9605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Koehler MFT, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, Salphati L, Schmidt S, Schneider EV, Wu J, and Beresini MH (2016) Development of a potent, specific CDK8 kinase inhibitor which phenocopies CDK8/19 knockout cells. ACS Med. Chem. Lett 7, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wei J, Jin F, Wu Q, Jiang Y, Gao D, and Liu H (2014) Molecular interaction study of flavonoid derivative 3d with human serum albumin using multispectroscopic and molecular modeling approach. Talanta 126, 116–121. [DOI] [PubMed] [Google Scholar]
- (56).Pollock J, Borkin D, Lund G, Purohit T, Dyguda-Kazimierowicz E, Grembecka J, and Cierpicki T (2015) Rational design of orthogonal multipolar interactions with fluorine in protein–ligand complexes. J. Med. Chem 58, 7465–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ali A, Hoeflich KP, and Woodgett JR (2001) Glycogen synthase kinase-3: Properties, functions, and regulation. Chem. Rev 101, 2527–2540. [DOI] [PubMed] [Google Scholar]
- (58).Wang QM, Park IK, Fiol CJ, Roach PJ, and DePaoli-Roach AA (1994) Isoform differences in substrate recognition by glycogen synthase kinases 3.alpha. and 3.beta. in the phosphorylation of phosphatase inhibitor 2. Biochemistry 33, 143–147. [DOI] [PubMed] [Google Scholar]
- (59).Soutar MPM, Kim W-Y, Williamson R, Peggie M, Hastie CJ, McLauchlan H, Snider WD, Gordon-Weeks PR, and Sutherland C (2010) Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem 115, 974–983. [DOI] [PubMed] [Google Scholar]
- (60).Liang Z, Sulzmaier FJ, Yoshida WY, Kelly M, Ramos JW, and Williams PG (2015) Neopetrocyclamines A and B, polycyclic diamine alkaloids from the sponge Neopetrosia cf exigua. J. Nat. Prod 78, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.