

Abstract
Therapeutic targeting of advanced or metastatic non-small-cell lung cancer (NSCLC) represents a major goal of clinical treatment. Polo-like kinase 1 (PLK1) is an essential mitotic kinase in cell cycle progression and is associated with oncogenesis in a large spectrum of cancer types, including NSCLC. Volasertib (BI 6727) is a potent, selective, PLK1 inhibitor that is currently under phase 2 clinical trials with modest antitumor activity against solid tumors. As the combination of volasertib with pemetrexed does not improve efficacy for NSCLC treatment, it is crucial to identify compounds that could enhance efficacy with volasertib. Immunomodulatory drugs (IMiDs) bind to E3 ligase CRBN and repurposes it to ubiquitinate other proteins as neo-substrates, representing an effective treatment for hematologic malignancies. In this study, by screening IMiDs, we found that a novel CRBN modulator, CC-885, can synergistically inhibit NSCLC with volasertib both in vitro and in vivo. This synergistic effect overcomes volasertib resistance caused by PLK1 mutations and is compromised in CRBN-or p97-depleted cells. Mechanistically, CC-885 selectively promotes CRBN- and p97-dependent PLK1 ubiquitination and degradation, thereby enhancing the sensitivity of NSCLC to volasertib. In conclusion, our findings reveal that PLK1 is a neo-substrate of CUL4-CRBN induced by CC-885 and represent a combinational approach for treating NSCLC.
Keywords: immunomodulatory drugs, IMiDs, CC-885, PLK1, volasertib, NSCLC, protein stability, ubiquitination
Graphical Abstract
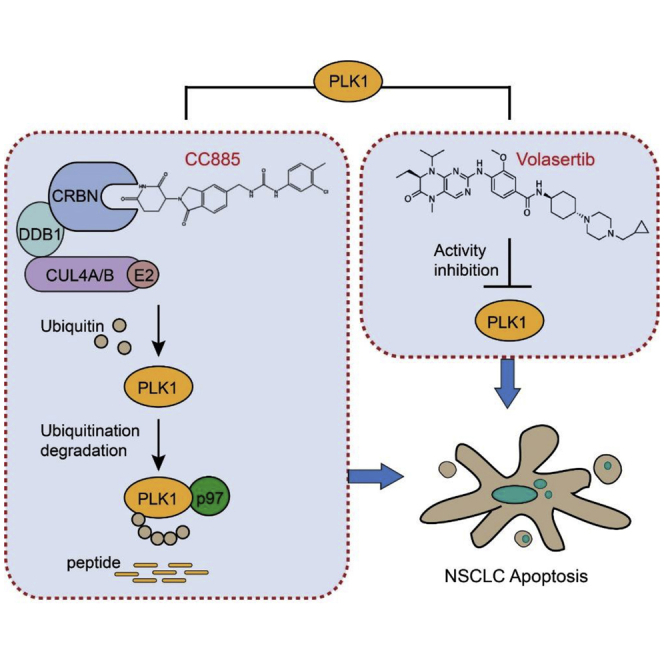
Therapeutic targeting of advanced non-small-cell lung cancer (NSCLC) represents a major goal of clinical treatment. In this study, we show that a novel CRBN modulator, CC-885, selectively promotes CRBN-and p97-dependent PLK1 ubiquitination and degradation, thereby enhancing the sensitivity of NSCLC to a PLK1 inhibitor, volasertib, both in vitro and in vivo.
Introduction
Lung cancer is the leading cause of cancer-related mortality and morbidity worldwide.1 There are two types of lung cancer according to histological classification: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC).2 Approximately 85% of lung cancer cases are NSCLC, which has a poor prognosis and is associated with locally advanced or metastatic disease.3 The expected median survival with NSCLC is about 6 months, and the expected 5-year survival rate is extremely low.4 Targeted therapy based on small-molecule inhibitors is an effective treatment for NSCLC, which can improve the survival rate of patients. However, drug tolerance often occurs in patients, which impairs the therapeutic potential.3,5 Therefore, finding alternative or complementary therapies to reduce drug tolerance and improve the efficacy of targeted therapy is a crucial issue.
The polo-like kinase (PLK) family members are serine/threonine protein kinases, which regulate many steps of the cell cycle.6 This family has five members: PLK1, PLK2, PLK3, PLK4, and PLK5. Among these members, PLK1 plays a crucial role in cell mitosis.7 PLK1 has been considered to be an intriguing drug target for cancer treatment, as its overexpression has been found in multiple human malignancies.8 Volasertib (BI 6727) is a highly selective and potent inhibitor of PLK1, which can disrupt spindle assembly, leading to mitotic arrest and the apoptosis of human cancer cell lines.9,10 Importantly, it has also been validated to efficiently reduce tumor growth in preclinical settings.11 Unfortunately, only modest antitumor activity against solid tumors was observed in phase 1 and 2 clinical trials, suggesting that volasertib alone as a single agent might not be sufficient enough against cancer.12,13 Moreover, a randomized, open-label phase 2 trial of volasertib as monotherapy and in combination with a standard-dose pemetrexed study found that this combination does not improve efficacy compared with single-agent pemetrexed for the treatment of advanced or metastatic NSCLC.14 Thus, it is crucial to identify compounds that could have enhanced efficacy with volasertib to fight against NSCLC.
The development of the immunomodulatory drugs (IMiDs) such as lenalidomide and pomalidomide has been recognized as an effective treatment for myeloma.15 Through affinity-bead technology, CRBN was identified as a common direct and primary target of IMiDs.16 CRBN is an adaptor protein for the E3 ubiquitin ligase complex CUL4-DDB1-RBX1-CRBN (CUL4-CRBN) and determines substrate specificity.17 IMiDs recruit CRBN to promote ubiquitination and proteasome-dependent degradation of diverse proteins as neo-substrates, which is required for the therapeutic effects of the drugs.18 For example, the clinical activity of IMiDs in multiple myeloma is achieved by selective degradation of IKZF1 and IKZF3.19,20 The degradation of casein 1α protein kinase (CK1α) is considered the key mechanism of IMiDs in the treatment of the deletion 5q (del(5q)) subtype of myelodysplastic syndrome (MDS).21 A new CRBN modulator, CC-885, has recently been identified. Unlike other IMiDs, CC-885 has potent anti-solid tumor activity, which is considered to be mediated through the CRBN-dependent ubiquitination and degradation of the translation termination factor GSPT1.22 However, whether CC-885 can recruit neo-substrates other than GSPT1 to CUL4-CRBN is completely unknown.
In this study, by screening IMiDs, we found that CC-885 acted synergistically with volasertib to suppress NSCLC. At the molecular level, CC-885 selectively promotes both CRBN- and p97-dependent PLK1 ubiquitination and sequential proteasomal degradation, thereby enhancing NSCLC cell sensitivity to volasertib.
Results
IMiD Drugs Screen Identified that CC-885 Could Synergistically Enhance Volasertib-Induced Cell Death in NSCLC
To test whether IMiDs have synergistic effects with volasertib against NSCLC cells, we initiated a small molecular inhibitors synergistic screen using thalidomide and thalidomide analogs. We found that among these IMiDs, only CC-885 achieved a significantly synergistic effect with volasertib in A549 cells (Figures 1A and 1B). We further confirmed the screen data using both Cell Counting Kit-8 (CCK-8) and clonogenic survival assays (Figures 1C and 1D). This synergistic effect was also obtained in another NSCLC cell line, NCI-H1299 (Figure S1). We found that subtoxic and low nanomolar CC-885 acts in concert with volasertib-induced cell death in both A549 and NCI-H1299 cells (Figure 1E; Figure S2). It has been reported that volasertib exhibited its anti-tumor activity via G2/M arrest and caspase-3 activation in cells.23 Indeed, CC-885 administration amplified the pro-apoptotic effects of volasertib to further induce G2/M arrest, as evidenced by morphological changes (Figure 1F), cell cycle distribution analysis (Figure 1G), and the expression of mitotic markers including cyclin B1 and phosphorylated H3, as well as activated caspase-3 proteins (Figure 1H), eventually leading to enhanced cell death. Next, we assessed the therapeutic efficacy of this combination in vivo by using nude mice bearing tumors. While volasertib and CC-885 alone inhibited tumor growth, the combination of both small molecular drugs markedly inhibited tumor growth and reduced tumor weights (Figures 1I and IJ). Taken together, these data clearly show that CC-885 synergizes with volasertib against NSCLC cells both in vitro and in vivo.
Figure 1.

CC-885 Synergistically Enhanced Volasertib-Induced Cell Death in NSCLC Identified by IMiD Drug Screen
(A) A549 cells were inoculated in a 96-well plate 24 h before drug administration. Cells were then treated with 50 nM volasertib with or without 1 μM IMiDs for 24 h, and CCK-8 solution was added to each well and incubated for an additional 4 h in an incubator. The absorbance at 450 nm was then measured. Results are representative of three biological replicates. Data are shown as mean ± SD; n = 3. ∗∗∗p < 0.001. (B) Chemical structure of CC-885. (C) A549 cells in a 96-well plate were treated with 50 nM CC-885 and/or indicated doses of volasertib for 24 h, and CCK-8 solution was added to each well and incubated for an additional 4 h. The absorbance at 450 nm was then measured. (D) Clonogenic assay of A549 cells treated with increasing concentrations of volasertib, CC-885, or their combination for a week and the relative cell numbers (as a percentage of the untreated group) were counted. The percentage of cell growth inhibition at each concentration of volasertib, CC-885, or their combination is shown in a heatmap. (E) Flow cytometry demonstrated the effects of 50 nM volasertib and the combination of 10 or 50 nM CC-885 with 50 nM volasertib on A549 cells for 48 h. Error bars indicate the means ± SD. ∗∗∗p < 0.001. (F) Morphological changes of 50 nM volasertib, 50 nM CC-885, or both on A549 cells for 24 h. Cells were imaged using a phase-contrast microscope. (G) Cell cycle distribution of A549 cells treated with 50 nM volasertib, 50 nM CC-885, or both for 24 h. (H) A549 cells were treated with 50 nM volasertib, 50 nM CC-885, or both for 48 h. Cells were harvested and subjected to western blot with the indicated antibodies. (I) 1 × 107 A549 cells were resuspended in serum-free medium and injected subcutaneously into BALB/cA mice. One week after xenograft tumor growth was notable, mice were divided into four groups and treated with volasertib, CC-885, their combination, or placebo, respectively. Tumor growth was measured using a caliper at the indicated times after injection. n = 5 for each group. Error bars indicate the means ± SD. ∗∗∗p < 0.001. (J) The tumor weights of each mice in (I) at day 28 were measured immediately after mice were sacrificed. n = 5 for each group. Error bars indicate the means ± SD. ∗p < 0.05, ∗∗p < 0.01.
CRBN, but Not GSPT1, Is Required for the Synergistic Effect of Volasertib and CC-885
CC-885 is a recently identified novel CRBN modulator.22 Similar to lenalidomide and its analogs, CC-885 contains a glutarimide ring that can directly interact with CRBN (Figure 1B), and cells without CRBN were resistant to CC-885 treatment.22 It is reasonable to predict that this synergistic effect might be invalid in CRBN-depleted cells. To this end, we generated CRBN−/− A549 cells by clustered regularly interspaced short palindromic repeats (CRISPR)-based gene editing. Indeed, CC-885 failed to amplify the cell cycle arrest and cell death-induction ability of volasertib in CRBN−/− cells (Figures 2A and 2B), indicating that CRBN is essential for this drug combination. Interestingly, volasertib alone still induces dose-dependent cell death in CRBN−/− cells (Figures 2C and 2D), suggesting that CRBN is not required for the pharmacological action of volasertib. It has been reported that the anti-tumor activity of CC-885 is mediated through CRBN-dependent ubiquitination and degradation of GSPT1.22 Therefore, we thought that this synergy might be related to GSPT1 protein. To test this, we silenced GSPT1 by three different short hairpin RNAs (shRNAs) in A549 cells (Figure 2E; Figure S3). Although silencing of GSPT1 caused noticeable cell death, it had no further synergistic effect with volasertib (Figure 2F). Furthermore, the combination of volasertib and CC-885 did not further enhance CC-885-induced GSPT1 degradation (Figure 2G). Consistent with this, volasertib alone also induced indistinguishable cell death in cells overexpressing GSPT1 wild-type (WT) or GSPT1 mutant (MUT) (G575N), with the latter being resistant to CC-885-induced GSPT1 degradation as well as CC-885-mediated cell death (Figures 2H and 2I). Taken together, these data suggest that the synergistic effect of CC-885 with volasertib is dependent on CRBN but independent on GSPT1, indicating that the degradation of other unknown proteins might be involved in this process.
Figure 2.

CRBN, but Not GSPT1, Mediated the Synergistic Effect of Volasertib and CC-885
(A) Cell cycle distribution of A549 CRBN−/− cells treated with 50 nM volasertib, 50 nM CC-885, or both for 24 h. (B) Flow cytometry demonstrated the effects of 50 nM volasertib and the combination of 10 or 50 nM CC-885 with 50 nM volasertib on A549 CRBN−/− cells for 48 h. Error bars indicate the means ± SD. (C) A549 CRBN−/− cells were cultured in a 96-well plate treated with indicated doses of volasertib for 24 h, and CCK-8 solution was added to each well and incubated for an additional 4 h. The absorbance at 450 nm was then measured. Error bars indicate the means ± SD. (D) Morphological changes of the indicated doses of volasertib on A549 CRBN−/− cells for 24 h. Cells were imaged using a phase-contrast microscope. (E) A549 cells were infected with lentiviral vectors expressing control shRNA or any of the three GSPT1-specific shRNAs. Four days after infection, cells were collected for immunoblot analysis with the indicated antibodies. (F) A549 cells infected with lentiviral vectors expressing control shRNA or any of the three GSPT1-specific shRNAs for 4 days were cultured in a 96-well plate and treated with either DMSO or 50 nM volasertib for 24 h, and CCK-8 solution was added to each well and incubated for an additional 4 h. The absorbance at 450 nm was then measured. Error bars indicate the means ± SD. (G) A549 cells were treated with 50 nM volasertib, 50 nM CC-885, or both for 24 h. Cells were harvested and subjected to western blot with the indicated antibodies. (H) A549 cells were transfected with Flag-GSPT1 WT or MUT (G575N) for 36 h, and 1μM CC-885 was added for an additional 8 h. Cells were then harvested and subjected to western blot with indicated antibodies. (I) A549 cells transfected with Flag-GSPT1 WT or Flag-GSPT1 MUT (G575N) for 36 h were cultured in a 96-well plate and treated with either DMSO or 50 nM volasertib for 24 h, and CCK-8 solution was added to each well and incubated for an additional 4 h. The absorbance at 450 nm was then measured. Error bars indicate the means ± SD. ∗∗∗p < 0.001.
CC-885 Overcomes Volasertib Resistance in NSCLC Cells by Inducing Rapid Degradation of PLK1
Volasertib is effective against a variety of cancer cell lines by competitively binding to the ATP-binding pocket of the PLK1 protein, but resistance is inevitable.24 One of the mechanisms of volasertib resistance is the mutations in the ATP binding sites of PLK1. The PLK1 mutants evade the inhibitory binding of volasertib but retain the original protein kinase activity, ultimately leading to drug tolerance.25 CC-885 has been shown to induce apoptosis in a wide range of cancer cell lines,22 and thus it is reasonable to speculate that CC-885 alone might be able to overcome volasertib resistance. To test this possibility, we generated A549 cells with stable expression of PLK1 mutants (F183L and L59W) or PLK1 WT (Figure S4). As expected, cells carrying PLK1 mutants were highly resistant to volasertib (Figure 3A). However, CC-885 indistinguishably induced death in both PLK1 WT and PLK1 mutants carrying cells (Figure 3A), suggesting that CC-885 could efficiently overcome volasertib resistance. Unexpectedly, we found that all PLK1 proteins, including both PLK1 mutants, rapidly disappeared after CC-885 administration (Figure 3B), indicating that CC-885 fights against NSCLC cells by inducing rapid depletion of PLK1. Indeed, CC-885 induced both a dose- and time-dependent decrease of endogenous PLK1 protein in A549 cells (Figures 3C and 3D). However, the mRNA levels of PLK1 were unperturbed (Figure 3E), further suggesting the existence of a post-translational regulation mechanism. Indeed, in the presence of CC-885, the half-life of PLK1 was significantly decreased (Figure 3F). In line with this, both proteasome inhibitor MG132 and NEDD8-activating enzyme inhibitor MLN4924 dramatically prevented the decrease of both endogenous and exogenous PLK1 proteins (Figure 3G; Figure S5). In addition, CC-885 increased the ubiquitination of all PLK1 proteins regardless of the variation of their amino acid sequences (Figure 3H). PLK1 protein is composed of a kinase domain and two polo-box domains. To determine which domain is required for CC-885-dependent PLK1 destruction, we generated a series of truncations (Figure 3I). We found that a peptide containing 19 aa at the C-terminal of PLK1 protein is critical for CC-885-dependent PLK1 destruction (Figure 3J). Indeed, PLK1 1–584, which missed those amino acids, was resistant to CC-885-induced ubiquitination-dependent degradation (Figures 3K and 3L). Moreover, cells stably expressing PLK1 1–584 were resistant to CC-885-induced cell death, but sensitive to volasertib treatment (Figure 3M). These findings demonstrated that CC-885 induced the proteasome-dependent degradation of PLK1 proteins, and the combination of CC-885 and volasertib could be applied to NSCLC cells even carrying PLK1 mutations, thus representing a reasonable molecular explanation for CC-885 to overcome volasertib resistance in NSCLC cells.
Figure 3.

CC-885 Overcomes Volasertib Resistance in NSCLC Cells by Inducing Rapid Degradation of PLK1
(A) A549 cells with stable expression of Flag-PLK1 mutants (F183L and L59W) or PLK1 WT were cultured in a 96-well plate and treated with either DMSO, 50 nM volasertib, or 500 nM CC-885 for 24 h, and CCK-8 solution was added to each well and incubated for additional 4 h. The absorbance at 450 nm was then measured. Error bars indicate the means ± SD. ∗∗∗p < 0.001. (B) A549 cells with stable expression of Flag-PLK1 mutants (F183L and L59W) or PLK1 WT were treated with 1 μM CC-885 for 12 h, and cells were then harvested and subjected to western blot with the indicated antibodies. (C) Immunoblot analysis of A549 cells treated with the indicated doses of CC-885 for 12 h. Results are representative of three immunoblot analyses. (D) Time course of PLK1 protein levels in A549 cells treated with DMSO or 2 μM CC-885. (E) Time course of PLK1 mRNA levels in A549 cells treated with DMSO or 2 μM CC-885. Real-time PCR data are presented as the means ± SD (n = 3). (F) Immunoblot analysis of A549 cells treated with 100 μg/mL cycloheximide (CHX) and 100 nM CC-885 for the indicated periods. Cells were pretreated with 100 nM CC-885 for 2 h. Results are representative of three immunoblot analyses. Statistic results of western blotting analysis were obtained by ImageJ software and normalized to actin intensities. Error bars indicate the means ± SD. n = 3. (G) A549 cells were pretreated with 1 μM MLN4924 or 10 μM MG132 before the addition of 1μM CC-885 for 12 h. (H) A549 cells with stable expression of Flag-PLK1 mutants (F183L and L59W) or PLK1 WT were pretreated with 10 μM MG132 before the addition of 1 μM CC-885 for 12 h, and cells were then harvested and subjected to immunoprecipitation with Flag M2 beads followed by immunoblot analyses with the indicated antibodies. Results are representative of three immunoblot analyses. (I) Schematic diagram of the different FLAG-tagged PLK1 deletion mutants. (J) 293T cells transiently expressing either full-length FLAG-PLK1 or various FLAG- PLK1 mutants were treated with 1 μM CC-885 for 12 h, and cells were then harvested and subjected to western blot with the indicated antibodies. (K) A549 cells with stable expression of either full-length FLAG-PLK1 or FLAG- PLK1 1–584 were treated with 100 μg/mL CHX and 100 nM CC-885 for the indicated periods. Cells were then harvested and subjected to western blot with the indicated antibodies. (L) A549 cells with stable expression of Flag-PLK1 WT or PLK1 1–584 were pretreated with 10 μM MG132 before the addition of 1 μM CC-885 for 12 h, and cells were then harvested and subjected to immunoprecipitation with Flag M2 beads followed by immunoblot analyses with the indicated antibodies. (M) A549 cells with stable expression of Flag-PLK1 WT or PLK1 1-584 were cultured in a 96-well plate and treated with either DMSO, 50 nM volasertib, or 500 nM CC-885 for 24 h, and CCK-8 solution was added to each well and incubated for additional 4 h. The absorbance at 450 nm was then measured. Error bars indicate the means ± SD. ∗∗p < 0.01.
PLK1 Is a CRBN-Dependent Neosubstrate of CC-885
We then asked whether CC-885-induced PLK1 degradation is CRBN-dependent. Similar to GSPT1, CC-885 failed to reduce PLK1 in CRBN−/− cells, but not in CRBN+/+ cells (Figure 4A). To avoid the off-target effects, we used two different shRNAs to inhibit CRBN expression in NCI-H1299 cells. Both shRNAs achieved considerable efficient silencing effects (Figure 4B; Figure S6). Consist with previous reports, downregulation of CRBN in NCI-H1299 cells by both shRNAs prevented CC-885-induced degradation of GSPT1, as well as PLK1 (Figure 4B). Similarly, both shRNAs also efficiently blocked the downregulation of Flag-PLK1 by CC-885 treatment in A549 cells (Figure S7). Interestingly, PLK1 protein levels were unaltered in CRBN knockout or knockdown cells regardless of CC-885 treatment (Figures 4A and 4B), suggesting that PLK1 is not an endogenous native substrate of CRBN. Moreover, overexpression of a WT CRBN, but not an IMiD resistant CRBN mutant (YW/AA), into CRBN−/− cells largely restored CC-885-induced PLK1 protein degradation (Figure 4C). IMiDs impart gain-of-function properties to CRL4-CRBN that enable binding, ubiquitination, and degradation of therapeutic targets. Thus, we then tested whether CC-885 could regulate the interaction between PLK1 and CRBN. Notably, we found that PLK1 interacted with CRBN only in the presence of CC-885 in a dose-dependent manner (Figure 4D). In contrast, neither pomalidomide nor CC-122 could promote PLK1 binding to CRBN (Figure 4D). In addition, PLK1 immunoprecipitation followed by western blot with an anti-ubiquitin antibody further confirmed that CC-885-induced PLK1 ubiquitination is dependent on CRBN (Figure 4E). Taken together, these findings demonstrate that CC-885 induces the assembly between PLK1 and CRBN, thereby promoting ubiquitination and proteasomal degradation of PLK1. It has been reported that p97 plays a critical role in IMiD-induced degradation of CUL4-CRBN neo-substrates, including CC-885-mediated degradation of GSPT1.26 We then tested the role of p97 in CC-885-induced degradation of PLK1. CB-5083 is a novel first-in-class, potent selective p97 inhibitor that disrupts cellular protein homeostasis and causes the accumulation of ubiquitinated proteins.27 We found that CB-5083 completely blocked CC-885-induced degradation of PLK1 in both A549 and NCI-H1299 cells (Figure 4F; Figure S8). In line with this, silencing of p97 by two different shRNAs also led to CC-885-induced PLK1 degradation (Figure 4G; Figure S9). To confirm that CB-5083 was indeed blocking PLK1 proteolysis, we performed a cycloheximide chase experiment. Consistent with our observation, CC-885 induced rapid loss of PLK1 in A549 cells, which was completely blocked by CB-5083 administration (Figure 4H). Moreover, CC-885 promoted the binding of endogenous PLK1 to endogenous p97 in CRBN+/+ cells, but not in CRBN−/− cells (Figure 4I), suggesting that the ubiquitinated PLK1 mediated by CRBN, but not the unmodified one, is required for p97 recognition and degradation. Indeed, CB-5083 administration slightly enhanced the ubiquitinated form of PLK1 induced by CC-885 (Figure 4J), indicating that CB-5083 did not decrease the ubiquitinated form of PLK1 to stabilize it. However, CB-5083 alone did not affect PLK1 protein levels (Figure S10). Taken together, these data suggest that p97 is required for CC-885-induced degradation of PLK1, but not for its constitutive degradation.
Figure 4.

PLK1 Is a CRBN-Dependent Neo-Substrate of CC-885
(A) A549 CRBN+/+ cells and CRBN−/− cells were treated with 1 μM CC-885 for 24 h. Cells were then harvested and subjected to western blot with the indicated antibodies. (B) NCI-H1299 cells were infected with lentiviral vectors expressing control shRNA or any of the two CRBN-specific shRNAs. Four days after infection, cells were treated with 1 μM CC-885 for 24 h and subjected to immunoblot analysis with the indicated antibodies. (C) A549 CRBN−/− cells transfected with indicated plasmids for 36 h were treated with 1 μM CC-885 for 24 h and subjected to immunoblot analysis with the indicated antibodies. (D) A549 cells with stable expression of Flag-PLK1 WT were treated with the indicated doses of pomalidomide, CC-885, or CC-122 for 2 h. Cells were then harvested and subjected to immunoprecipitation with Flag M2 beads followed by immunoblot analyses with the indicated antibodies. (E) A549 CRBN+/+ cells and CRBN−/− cells were pretreated with 10 μM MG132 before the addition of 1 μM CC-885 for 12 h. Cells were then harvested and subjected to immunoprecipitation with PLK1 antibody followed by immunoblot analyses with the indicated antibodies. (F) A549 cells were pretreated with 1 μM MLN4924 or 1 μM CB-5083 before the addition of 1 μM CC-885 for 24 h. Cells were then harvested and subjected to western blot with the indicated antibodies. (G) A549 cells were infected with lentiviral vectors expressing control shRNA or any of the two p97-specific shRNAs. Four days after infection, cells were treated with 1 μM CC-885 for 24 h and subjected to immunoblot analysis with the indicated antibodies. (H) Immunoblot analysis of A549 cells treated with 100 μg/mL cycloheximide (CHX) and 100 nM CC-885 with or without 1 μM CB-5083 for the indicated periods. Results are representative of three immunoblot analyses. Statistical results of western blotting analysis were obtained by ImageJ software and normalized to actin intensities. Error bars indicate the means ± SD. n = 3. (I) A549 CRBN+/+ cells and CRBN−/− cells were pretreated with 10 μM MG132 before the addition of 1 μM CC-885 for 12 h. Cells were then harvested and subjected to immunoprecipitation with p97 antibody followed by immunoblot analyses with the indicated antibodies. (J) A549 cells were pretreated with 10 μM MG132 before the addition of 1 μM CC-885 with or without 1 μM CB-5083 for 12 h. Cells were then harvested and subjected to immunoprecipitation with PLK1 antibody followed by immunoblot analyses with the indicated antibodies.
CC-885 Inhibits the Activation of PLK1 Substrates in NSCLC Cell Lines
To investigate the downstream effects of CC-885-induced PLK1 degradation, we measured the phosphorylation levels of PLK1 substrates, including phosphorylated TCTP and phosphorylated Myt1, in NSCLC cells.28,29 As expected, we observed that CC-885 decreased the phosphorylation levels of both TCTP and Myt1 proteins in CRBN+/+ cells, but not in CRBN−/− cells, or cells treated with MLN4924 or MG132 (Figures 5A and 5B), indicating that these effects were highly dependent on the E3 ligase activity of CRBN. Interestingly, the combination of volasertib and CC-885 at a low concentration caused significant decline of both phosphorylated TCTP and Myt1 proteins (Figure 5C). Collectively, these data indicate that CC-885 magnifies the inhibitory effects of volasertib by promoting PLK1 degradation and its downstream substrate inactivation.
Figure 5.

CC-885 Inhibits the Activation of PLK1 Substrates
(A) A549 CRBN+/+ cells and CRBN−/− cells were treated with 1 μM CC-885 for 24 h. Cells were then harvested and subjected to western blot with the indicated antibodies. Results are representative of three immunoblot analyses. (B) A549 cells were pretreated with 1 μM MLN4924 or 10 μM MG132 before the addition of 1 μM CC-885 for 24 h. Cells were then harvested and subjected to western blot with the indicated antibodies. Results are representative of three immunoblot analyses. Results are representative of three independent experiments. (C) A549 cells were treated with 50 nM volasertib, 50 nM CC-885, or both for 24 h. Cells were harvested and subjected to western blot with the indicated antibodies. Results are representative of three independent experiments.
Discussion
Initially, we intended to determine whether a certain IMiD drug has a synergistic effect with volasertib in the treatment of NSCLC, as volasertib was reported to be impotent against NSCLC when used alone.14 Instead of the well-studied IMiDs such as lenalidomide and pomalidomide, we found that a novel CRBN modulator CC-885 has the greatest synergistic effect with volasertib against NSCLC cells, regardless of their sensitivity to volasertib. Our results demonstrate that the synergistic effect of volasertib and CC-885 is primarily driven by CC-885-induced CRBN- and p97-dependent PLK1 ubiquitination and degradation. The hypothesized model of this synergistic effect is depicted in Figure 6.
Figure 6.

Working Model
In this working model, we show that volasertib inhibits the kinase activity of PLK1, whereas CC-885 induces both CRBN- and p97-dependent PLK1 ubiquitination and degradation. The combination targets the same oncoprotein PLK1 by different means, providing a reasonable molecular mechanism explanation for this synergistic effect against NSCLC.
Selectively, CRBN-dependent degradation of GSPT1 has been shown to contribute to the broad-spectrum anti-cancer activity of CC-885.22 Indeed, CC-885 loses its synergistic effect with volasertib in CRBN−/− cells. However, silencing of GSPT1 did not have a synergistic effect with volasertib, suggesting that CRBN-dependent and GSPT1-independent mechanisms existed. We then intended to identify the proteins for which degradation could be required for this synergistic effect. Recent studies have demonstrated that protein inhibition is not always equal to protein degradation, especially for small molecular compound-mediated kinase inhibition. For example, Olson et al.30 revealed that cyclin-dependent kinase 9 (CDK9) degradation can induce distinct pharmacological effects compared to its inhibition. Interestingly, the combination of arsenic trioxide (ATO) and all-trans retinoic acid (ATRA) safely cures fatal acute promyelocytic leukemia (APL) by targeting promyelocytic leukemia (PML)-retinoic acid receptor α (RARα) fusion protein.31 In this case, ATRA associated with RARα to inhibit its transcriptional activity, whereas ATO directly interacts with PML to promote its ubiquitination and degradation.32,33 The combination of ATO and ATRA targets the same oncoprotein by means of both inhibition and degradation, providing an excellent example for treating acute myeloid leukemia (AML).34 Thus, we asked whether CC-885 has some effect on PLK1 protein. Interestingly, CC-885 induced both a dose- and time-dependent decrease of PLK1 protein without affecting its mRNA level, representing a reasonable justification for this combination. However, we still could not exclude the possibility that other unidentified CC-885 substrates might also contribute to this synergistic effect.
p97, also known as valosin-containing protein (VCP), is a member of the AAA family of adenosine triphosphatases (ATPases).35 p97 extracts proteins destined for destruction by the ubiquitin-proteasome system (UPS) and plays a key regulatory role in protein homeostasis by interactions with various E3 ligases and their substrates.36 It has been reported that p97 is required for all IMiD-induced degradation of CUL4-CRBN neosubstrates.26 In agreement, our data indicate that p97 is also indispensable for CC-885-induced PLK1 degradation, further suggesting that PLK1 is a neo-substrate of CUL4-CRBN induced by CC-885.
A recent structural study identified 11 zinc finger-contained transcriptional factors as neo-substrates of IMiDs, which all existed as a Cys2-His2 (C2H2) zinc finger “degrome.”37 However, we think that this zinc finger degrome might not always be necessary for the destruction of IMiDs substrates, as two known neo-substrates, GSPT1 and CK1a, do not contain zinc fingers. Instead, the G-motif degrons of both proteins are formed by a β sheet hairpin.22,38 As PLK1 is not a transcriptional factor and does not contain a C2H2 domain, it will not be easy to predict its degrome. Unexpectedly, we found the 19 aa at the C-terminal of PLK1 protein were critical for CC-885-induced PLK1 destruction, suggesting a potential novel degrome in PLK1. Therefore, in the future the structural basis of CC-885-induced degron recognition of PLK1 by CUL4-CRBN is warranted.
In conclusion, our results demonstrate that PLK1 is a bona fide CC-885-dependent neo-substrate of CUL4-CRBN E3 ligase, providing a reasonable explanation to the synergistic effect of the volasertib and CC-885 combination in the treatment of NSCLC.
Materials and Methods
Cell Culture and Compounds
All cells used in cell culture experiments were bought from ATCC. Hoechst DNA staining was used to make sure that all cells were not contaminated by mycoplasma. A549 and NCI-H1299 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing penicillin-streptomycin solution and 10% fetal bovine serum (FBS) and incubated in 37°C with 5% CO2. Thalidomide, lenalidomide, pomalidomide, and MG132 were purchased from Sigma. Volasertib and CC-122 were purchased from Selleck Chemicals. CC-885, MLN4924, and CB-5083 were purchased from MedChemExpress (MCE).
Animal Studies
BALB/cA nude mice were purchased from National Rodent Laboratory Animal Resources (Shanghai, China). All mice were housed at 21°C ± 1°C with humidity of 55% ± 10%, fed with sterilized food and water, and kept on a 12-h light/12-h dark cycle. 1 × 107 A549 cells were resuspended in serum-free medium and injected subcutaneously into BALB/cA mice. One week later, when tumor growth was visible to the naked eye, mice were randomly selected to receive treatments with volasertib (20 mg/kg, intraperitoneally [i.p.], three times/week, Selleck Chemicals) and/or CC-885 (20 mg/kg, i.p., three times/week, Efebio, Shanghai, China) or placebo. All treatments were administered according to the guidelines of Institutional Animal Care and Use Committee, and all the protocols were approved by The First Affiliated Hospital of Zhengzhou University, Zhengzhou. Mice were treated with the indicated drugs or vehicles for 4 weeks, and tumor sizes were measured by a caliper. Tumor volumes were calculated using the formula (length × width2) × ½. Tumor weights were measured after mice were sacrificed.
Cell Growth and Viability
Viable cells were analyzed by a CCK-8 assay according to the manufacturer’s instructions (Cell Counting Kit C0038, Beyotime Biotechnology, China), as described previously. Briefly, cells were inoculated in a 96-well plate 24 h before drug administration. After treatment, CCK-8 solution was added to each well of the plate, which was then incubated for 4 h in an incubator. The absorbance at 450 nm was measured by using a microplate reader (SpectraMax Paradigm, Molecular Devices, USA).
Lentiviral shRNA
Pairs of synthetic complementary oligonucleotides targeting GSPT1, CRBN, p97, or a control (con-shRNA) were annealed and cloned into a pLKO.1 vector. Targeting sequences are listed as follows: con-shRNA, 5′-CAACAAGATGAAGAGCACCAA-3′; shGSPT1-1, 5′-GATTACCGTTTATTCCATA-3′; shGSPT1-2, 5′-CCGATGATGTAGAGACTGATA-3′; shGSPT1-3, 5′-CCTGCACAATACTGTGAGGAA-3′; shCRBN-1, 5′-GCTGCTTGTCTTCCTATTGAT-3′; shCRBN-2, 5′-GGATAGTAAAGAAGCCAAA-3′; shP97-1, 5′-TGATGTGAAGTACGGCAAA-3′; shP97-2, 5′-TATCAACAGCCATTCTCAA-3′.
RNA Isolation and Real-Time PCR
Total RNA was isolated from cells using TRIzol (Invitrogen) according to the manufacturer’s instructions. Purified RNA was reverse transcribed using PrimeScript RT master mix. The cDNA was quantified with a SYBR Premix Ex Taq II kit on LightCycler 480 (Roche, Switzerland), as described previously. Primers were selected from PrimerBank and are listed as follows: CRBN (forward, 5′-CAGTCTGCCGACATCACATAC-3′, reverse, 5′-GCACCATACTGACTTCTTGAGGG-3′); GSPT1 (forward, 5′-TCAGGACTTACTGGAGCAAATCT-3′, reverse, 5′-AACTTCCACGTTGTGCTTGTT-3′); P97 (forward, 5′-CAAACAGAAGAACCGTCCCAA-3′, reverse, 5′-TCACCTCGGAACAACTGCAAT-3′); β-actin (forward, 5′-CATGTACGTTGCTATCCAGGC-3′, reverse, 5′-CTCCTTAATGTCACGCACGAT-3′). Relative quantitation analysis of gene expression data was conducted according to the 2−ΔΔCt method.
CRISPR-Cas9 System
CRBN guide RNAs (gRNAs) were designed using the online CRISPR design tool (http://CRISPR.mit.edu/). The gRNA sequences were as follows: forward oligonucleotide, 5′-CACCGCACCATACTGACTTCTTGA-3′, reverse oligonucleotide, 5′-AAACTCAAGAAGTCAGTATGGTGC-3′. The CRISPR construction was performed according to the protocol provided by the Zhang Lab (http://genome-engineering.org/gecko/). The gRNA oligonucleotides were cloned into the gRNA cloning vector, pSpCas9 (BB)-2A-Puro (PX459). To obtain single clones of CRBN knockout (KO) cells, cells were transfected with the PX459 plasmids, selected with puromycin for 3 days. The single clones were then picked and validated by immunoblotting analysis and DNA sequencing.
Western Blot
Cells were harvested and lysed with ice-cold lysis buffer (62.5 mM Tris-HCl [pH 6.8], 100 mM DTT, 2% SDS, 10% glycerol). Proteins were separated by SDS-PAGE and transferred to a nitrocellulose (NC) membrane (Amersham Biosciences, Buckinghamshire, UK). Membranes were blocked in 5% non-fat milk in phosphate-buffered saline with Tween 20 (PBST) for 1 h before incubation with primary antibody overnight at 4°C. Membranes were washed with PBST five times and incubated with secondary antibody for 1 h. The following primary antibodies were used as indicated: monoclonal anti-Flag M2 (1:5,000 dilution, F1804, Sigma, USA), monoclonal anti-Flag M2-peroxidase (horseradish peroxidase [HRP]) antibody (1:4,000 dilution, A8592, Sigma, USA), polyclonal anti-PLK1 (1:2,000 dilution, #4535, Cell Signaling Technology, USA), polyclonal anti-TCTP (1:1,000 dilution, #8441, Cell Signaling Technology, USA), polyclonal anti-Myt1 (1:1,000 dilution, #4282, Cell Signaling Technology, USA), polyclonal anti-phosphorylated (phospho-)TCTP (Ser46) (1:1,000 dilution, #5251, Cell Signaling Technology, USA), polyclonal anti-phospho-Myt1 (Ser83) (1:1,000 dilution, #4281, Cell Signaling Technology, USA), polyclonal anti-phospho-histone H3 (Ser10) (1:5,000 dilution, #9701, Cell Signaling Technology, USA), polyclonal anti-cleaved caspase-3 (Asp175) (5A1E) (1:1,000 dilution, #9664, Cell Signaling Technology, USA), polyclonal anti-ubiquitin (P4D1) (1:1,000 dilution, #3936, Cell Signaling Technology, USA), polyclonal anti-CRBN (1:1,000 dilution, #71810, Cell Signaling Technology, USA), polyclonal anti-p97/VCP (1:2,000 dilution, #2648, Cell Signaling Technology, USA), polyclonal anti-GSPT1 (1:500 dilution, 10763-1-AP, Ptglab, Wuhan, Hubei, China), polyclonal anti-cyclin B1 (1:1,000 dilution, sc-4073, Santa Cruz, USA), and polyclonal anti-β-actin (1:10,000 dilution, #3700, Cell Signaling Technology, USA).
Immunoprecipitation (IP)
The whole IP procedure has been described previously.39 Briefly, cells were treated with 10 μM MG132 for an additional 4 h and lysed in lysis buffer for 20 min with gentle rocking at 4°C. Lysates were cleared and subjected to IP with 50 μL of anti-FLAG M2 affinity resin (Sigma) overnight at 4°C. Resin-containing immune complexes were washed with ice-cold lysis buffer followed by Tris buffered saline (TBS) washes. Proteins were eluted with 5% SDS and subjected to immunoblot analysis.
In Vivo Ubiquitination Assay
Cells were transfected with combinations of plasmids. After incubation for 48 h, cells were treated with 10 μM MG132 or 1 μM MLN4924 for an additional 4 h, and lysed in lysis buffer containing 5 0mM Tris-HCl (pH 7.5), 0.15 M NaCl, 1 mM EDTA, 1% Nonidet P-40 (NP-40), and 10% glycerol. Lysates were cleared and subjected to IP with 2 μg of anti-PLK1 antibody and 50 μL of protein A/G affinity resin (Beyotime Biotechnology, China) overnight. The beads were sequentially washed with lysis buffer and PBST. Bound proteins were eluted with 2× SDS loading buffer and subjected to immunoblot analysis.
Clonogenic Assay
Cells were seeded into 12-well plates in complete media and cultured with or without drugs as indicated for a week. Survived cells were fixed with 4% paraformaldehyde, stained with 1% crystal violet, and imaged using a digital scanner. Crystal violet was extracted from the stained cells using 10% of acetic acid and relative growth was then quantified by densitometry. All experiments were performed at least three times.
Flow Cytometric Analysis
For cell cycle analysis, 1 × 105 cells/mL were collected, washed with ice-cold PBS, and fixed with 75% ice-cold ethanol. Cells were washed with ice-cold PBS and suspended in 300 μL of PBS in the presence of 20 μL of RNase A, followed by incubation for 30 min at 37°C. Then, cells were stained with propidium iodide (PI) for 45 min in the dark. Cell cycle analysis at 488 nm was performed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). For apoptosis assays, cells were seeded into six-well plates and cultured in the presence or absence of drugs as indicated. Apoptosis cells were determined using annexin V-fluorescein isothiocyanate (FITC) and a PI apoptosis detection kit according to the manufacturer’s instruction. Cell apoptosis was then analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Statistical Analysis
All in vitro experiments were performed independently three times. All values are shown as mean ± SD. The differences between groups were calculated using the Student’s t test or one-way ANOVA followed by a Tukey’s post hoc test, and p values less than 0.05 were considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). Data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
J.Z. and B.L. conceived the research ideas, supervised the project, and wrote the manuscript. L.L., W.X., Z.S., Y.W., and Y.C. performed animal and cellular experiments and analyzed the data. J.L., M.H., Z.C., H.C., and Y.L. provided technical advice on the animal and cellular studies. B.L. and J.Z. are the guarantors of this work and had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by grants from the National Key Research and Development Program of China (“Precision Medicine Research” key project, grant 2017YFC0909900), the National Natural Science Foundation of China (grants 71673254 and 81773018), the Doctor Research Team Fund from The First Affiliated Hospital of Zhengzhou University for the in-hospital Interdisciplinary Collaboration Research (grant 2016-BSTDJJ-15), the Research Foundation of Hubei Polytechnic University for Talented Scholars (grant 17xjz06A), and by the Henan Province Medical Science and Technology Research Project Joint Construction Project (Grant No. LHGJ20190003, LHGJ20190055).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.013.
Contributor Information
Bin Liu, Email: liubin3575630@163.com.
Jie Zhao, Email: jiezhaoz2016@163.com.
Supplemental Information
References
- 1.Hong Q.Y., Wu G.M., Qian G.S., Hu C.P., Zhou J.Y., Chen L.A., Li W.M., Li S.Y., Wang K., Wang Q., Lung Cancer Group of Chinese Thoracic Society; Chinese Alliance Against Lung Cancer Prevention and management of lung cancer in China. Cancer. 2015;121(Suppl 17):3080–3088. doi: 10.1002/cncr.29584. [DOI] [PubMed] [Google Scholar]
- 2.Gazdar A.F., Bunn P.A., Minna J.D. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat. Rev. Cancer. 2017;17:725–737. doi: 10.1038/nrc.2017.87. [DOI] [PubMed] [Google Scholar]
- 3.Gridelli C., Rossi A., Carbone D.P., Guarize J., Karachaliou N., Mok T., Petrella F., Spaggiari L., Rosell R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers. 2015;1:15009. doi: 10.1038/nrdp.2015.9. [DOI] [PubMed] [Google Scholar]
- 4.Osmani L., Askin F., Gabrielson E., Li Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 2018;52:103–109. doi: 10.1016/j.semcancer.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lategahn J., Keul M., Rauh D. Lessons to be learned: the molecular basis of kinase-targeted therapies and drug resistance in non-small cell lung cancer. Angew. Chem. Int. Ed. Engl. 2018;57:2307–2313. doi: 10.1002/anie.201710398. [DOI] [PubMed] [Google Scholar]
- 6.Lens S.M., Voest E.E., Medema R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 7.Strebhardt K., Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 8.Sebastian M., Reck M., Waller C.F., Kortsik C., Frickhofen N., Schuler M., Fritsch H., Gaschler-Markefski B., Hanft G., Munzert G., von Pawel J. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J. Thorac. Oncol. 2010;5:1060–1067. doi: 10.1097/JTO.0b013e3181d95dd4. [DOI] [PubMed] [Google Scholar]
- 9.Gjertsen B.T., Schöffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11–19. doi: 10.1038/leu.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudolph D., Steegmaier M., Hoffmann M., Grauert M., Baum A., Quant J., Haslinger C., Garin-Chesa P., Adolf G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 11.Gutteridge R.E., Ndiaye M.A., Liu X., Ahmad N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol. Cancer Ther. 2016;15:1427–1435. doi: 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van den Bossche J., Lardon F., Deschoolmeester V., De Pauw I., Vermorken J.B., Specenier P., Pauwels P., Peeters M., Wouters A. Spotlight on volasertib: preclinical and clinical evaluation of a promising Plk1 inhibitor. Med. Res. Rev. 2016;36:749–786. doi: 10.1002/med.21392. [DOI] [PubMed] [Google Scholar]
- 13.Yim H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs. 2013;24:999–1006. doi: 10.1097/CAD.0000000000000007. [DOI] [PubMed] [Google Scholar]
- 14.Ellis P.M., Leighl N.B., Hirsh V., Reaume M.N., Blais N., Wierzbicki R., Sadrolhefazi B., Gu Y., Liu D., Pilz K., Chu Q. A randomized, open-label phase II trial of volasertib as monotherapy and in combination with standard-dose pemetrexed compared with pemetrexed monotherapy in second-line treatment for non-small-cell lung cancer. Clin. Lung Cancer. 2015;16:457–465. doi: 10.1016/j.cllc.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Quach H., Ritchie D., Stewart A.K., Neeson P., Harrison S., Smyth M.J., Prince H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24:22–32. doi: 10.1038/leu.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 17.Fink E.C., Ebert B.L. The novel mechanism of lenalidomide activity. Blood. 2015;126:2366–2369. doi: 10.1182/blood-2015-07-567958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito T., Handa H. Myeloid disease: another action of a thalidomide derivative. Nature. 2015;523:167–168. doi: 10.1038/nature14628. [DOI] [PubMed] [Google Scholar]
- 19.Lu G., Middleton R.E., Sun H., Naniong M., Ott C.J., Mitsiades C.S., Wong K.K., Bradner J.E., Kaelin W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krönke J., Udeshi N.D., Narla A., Grauman P., Hurst S.N., McConkey M., Svinkina T., Heckl D., Comer E., Li X. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krönke J., Fink E.C., Hollenbach P.W., MacBeth K.J., Hurst S.N., Udeshi N.D., Chamberlain P.P., Mani D.R., Man H.W., Gandhi A.K. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matyskiela M.E., Lu G., Ito T., Pagarigan B., Lu C.C., Miller K., Fang W., Wang N.Y., Nguyen D., Houston J. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 23.Hugle M., Belz K., Fulda S. Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs. Cell Death Differ. 2015;22:1946–1956. doi: 10.1038/cdd.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C.P., Hsieh C.H., Hsiao S.H., Luo S.Y., Su C.Y., Li Y.Q., Huang Y.H., Huang C.W., Hsu S.C. Human ATP-binding cassette transporter ABCB1 confers resistance to volasertib (BI 6727), a selective inhibitor of polo-like kinase 1. Mol. Pharm. 2015;12:3885–3895. doi: 10.1021/acs.molpharmaceut.5b00312. [DOI] [PubMed] [Google Scholar]
- 25.Adachi Y., Ishikawa Y., Kiyoi H. Identification of volasertib-resistant mechanism and evaluation of combination effects with volasertib and other agents on acute myeloid leukemia. Oncotarget. 2017;8:78452–78465. doi: 10.18632/oncotarget.19632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen T.V., Li J., Lu C.J., Mamrosh J.L., Lu G., Cathers B.E., Deshaies R.J. p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc. Natl. Acad. Sci. USA. 2017;114:3565–3571. doi: 10.1073/pnas.1700949114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson D.J., Le Moigne R., Djakovic S., Kumar B., Rice J., Wong S., Wang J., Yao B., Valle E., Kiss von Soly S. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cucchi U., Gianellini L.M., De Ponti A., Sola F., Alzani R., Patton V., Pezzoni A., Troiani S., Saccardo M.B., Rizzi S. Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res. 2010;30:4973–4985. [PubMed] [Google Scholar]
- 29.Nakajima H., Toyoshima-Morimoto F., Taniguchi E., Nishida E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003;278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 30.Olson C.M., Jiang B., Erb M.A., Liang Y., Doctor Z.M., Zhang Z., Zhang T., Kwiatkowski N., Boukhali M., Green J.L. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018;14:163–170. doi: 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J., Liu Y.F., Wu C.F., Xu F., Shen Z.X., Zhu Y.M., Li J.M., Tang W., Zhao W.L., Wu W. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 2009;106:3342–3347. doi: 10.1073/pnas.0813280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X.W., Yan X.J., Zhou Z.R., Yang F.F., Wu Z.Y., Sun H.B., Liang W.X., Song A.X., Lallemand-Breitenbach V., Jeanne M. Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science. 2010;328:240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z.Y., Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 34.Burnett A.K., Russell N.H., Hills R.K., Bowen D., Kell J., Knapper S., Morgan Y.G., Lok J., Grech A., Jones G., UK National Cancer Research Institute Acute Myeloid Leukaemia Working Group Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015;16:1295–1305. doi: 10.1016/S1470-2045(15)00193-X. [DOI] [PubMed] [Google Scholar]
- 35.Meyer H., Weihl C.C. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 2014;127:3877–3883. doi: 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Boom J., Meyer H. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell. 2018;69:182–194. doi: 10.1016/j.molcel.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Sievers Q.L., Petzold G., Bunker R.D., Renneville A., Słabicki M., Liddicoat B.J., Abdulrahman W., Mikkelsen T., Ebert B.L., Thomä N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science. 2018;362 doi: 10.1126/science.aat0572. eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petzold G., Fischer E.S., Thomä N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 39.Liu B., Jiang S., Li M., Xiong X., Zhu M., Li D., Zhao L., Qian L., Zhai L., Li J. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018;9:4770. doi: 10.1038/s41467-018-07185-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.