

Summary
Determining the off-target cleavage profile of programmable nucleases is an important consideration for any genome editing experiment, and a number of Cas9 variants have been reported that improve specificity. We describe here Tagmentation-based Tag Integration Site Sequencing (TTISS), an efficient, scalable method for analyzing double-strand breaks that we apply in parallel to eight Cas9 variants across 59 targets. Additionally, we generated thousands of other Cas9 variants and screened for variants with enhanced specificity and activity, identifying LZ3 Cas9, a high specificity variant with a unique +1 insertion profile. This comprehensive comparison reveals a general trade-off between Cas9 activity and specificity and provides information about the frequency of generation of +1 insertions, which has implications for correcting frameshift mutations.
eTOC Blurb
Schmid-Burgk et al. develop Tagmentation-based Tag Integration Site Sequencing (TTISS), a rapid, streamlined protocol for analyzing double-strand breaks, such as those created by CRISPR nucleases. Using TTISS, they comprehensively assess Cas9 variants, revealing a trade-off between specificity and activity and identifying LZ3 Cas9, a variant with a unique +1 insertion profile.
Graphical Abstract
Introduction
CRISPR-Cas9 technology is widely used for genome editing and is currently being tested in clinical trials as a therapeutic. Many applications of this technology rely on Cas9 from Streptococcus pyogenes (SpCas9), and a number of engineered or evolved SpCas9 variants have been reported that impact Cas9 specificity. It is known that Cas9 activity and editing outcome vary depending on both the protein and the guide RNA, and thus empirically determining optimal enzyme-guide combinations may be helpful, particularly for clinical applications. Although a number of techniques have been developed that assess off-target cleavage (Tsai and Joung, 2016), these techniques are relatively low-throughput—limited to one guide per barcoded sample. We therefore developed Tagmentation-based Tag Integration Site Sequencing (TTISS), an efficient, rapid, scalable method to assess editing outcomes.
Design
Our method builds on the GUIDE-seq (Tsai et al., 2015) approach of tagging double-strand breaks (DSBs) induced by nuclease cleavage through integration of a double-stranded donor DNA, but makes use of guide multiplexing and bulk tagmentation by Tn5 (Picelli et al., 2014), which can be performed directly in lysed cells, leading to an efficient, rapid protocol (Fig. 1A). Following tagmentation, DNA is quickly purified using a spin column. Integration sites are enriched using two nested PCRs, which provide sufficient specificity to allow direct sequencing of the final product without further enrichment. Assigning the sequenced integration sites to guides by sequence similarity generates a list of off-target sites for each guide in parallel.
Figure 1 |. TTISS allows multiplexed assessment of nuclease off-targets.

(A) Schematic of TTISS off-target detection method. (B) TTISS results for 59 guides from the GeCKO library tested across eight SpCas9 specificity variants and WT SpCas9. (C) Specificity and activity scores for all tested SpCas9 variants. See also Figures S1, S2 and Tables S1 – S3.
Results
The sensitivity of TTISS is comparable to GUIDE-seq (Supplemental Table 1, note GUIDE-seq data is from U-2 OS cells using matched single guides) and DISCOVER-Seq (Supplemental Table 1, using matched single guides) (Wienert et al., 2019). TTISS is scalable to at least 60 guides per transfection in HEK 293T cells (Supplemental Fig. 1A), while retaining 71.4% of off-target sites detected in a single guide experiment, and is compatible with multiple cell types (Supplemental Fig. 1B). Additionally, TTISS can be extended to profiling of prime editing-mediated donor integration (Anzalone et al., 2019), which showed no off-target integration events for three integration sites tested (Supplemental Fig. 1C).
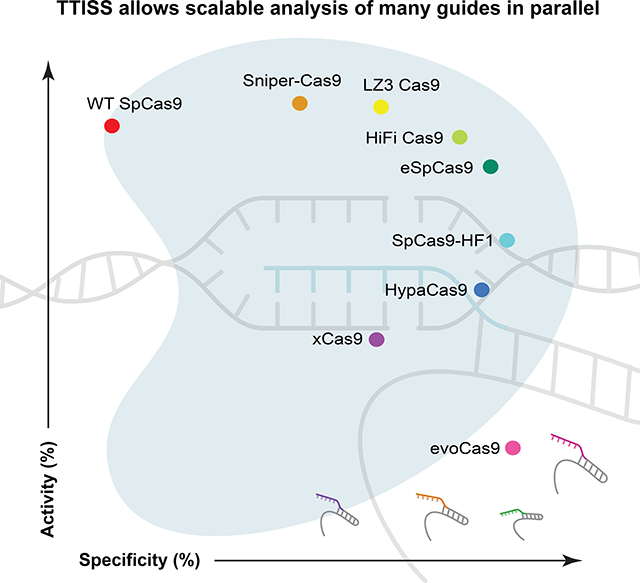
We used TTISS to assess the specificity of WT SpCas9 and eight SpCas9 specificity variants – eSpCas9(1.1) (Slaymaker et al., 2015), SpCas9-HF1 (Kleinstiver et al., 2016), HypaCas9 (Chen et al., 2017), evoCas9 (Casini et al., 2018), xCas9(3.7) (Hu et al., 2018), Sniper-Cas9 (Lee et al., 2018), HiFi Cas9 (Vakulskas et al., 2018) – and one newly generated specificity variant, LZ3 Cas9 (see Methods, Fig. 2) in parallel using 59 guides in two pools randomly selected from the GeCKO library (Shalem et al., 2014) that all start with a guanine to improve U6 transcription (Fig. 1B). For WT SpCas9, TTISS detected 607 total off-target sites across two technical replicates, with individual guides contributing 0–225 off-target sites (Supplemental Fig. 1D, Supplemental Table 2). Although each specificity variant showed improvement relative to WT SpCas9, a systematic comparison of these variants has not been reported. Using TTISS, we found that, although each specificity variant eliminated at least half of the WT SpCas9 off-targets, there was a wide range of specificities among variants, with evoCas9 being most specific (4 detected off-targets) and SniperCas9 being least specific (287 detected off-targets) (Fig. 1B).
Figure 2 |. High-throughput profiling of SpCas9 mutant fitness in human cells.

(A) Crystal structure of SpCas9 (PDB ID: 5F9R) showing the positions of 157 residues (magenta) selected for mutagenesis. (B) Sequences of target sites used for screening. (C) Approach for pooled lentiviral screening of SpCas9 variants in HEK 293FT cells. (D) Scatter plots of on-target vs. off-target activity scores for 2,420 SpCas9 single amino acid variants. The dashed box in each subplot contains all variants with ≥80% of the median wild-type on-target activity and ≤50% of the median wild-type off-target activity; activities were calculated after subtracting the median background activity of stop codon variants. The percentage within each box represents the percentage of all variants that lie within the box. (E) On-target and off-target activity of 254 SpCas9 single amino acid variants, quantified by targeted deep sequencing of individually transfected constructs. See also Figure S2.
Measuring on-target indel frequencies by targeted sequencing revealed that evoCas9 and xCas9(3.7) have the lowest on-target activity, while LZ3 Cas9, HiFi Cas9 and Sniper-Cas9 have on-target activity comparable to WT SpCas9 (Supplemental Fig. 2A, B). To compare specificity variants more broadly, we calculated an activity and a specificity score for each variant (Fig. 1C), revealing a general trade-off between activity and specificity among all variants.
To assess whether this observed trade-off between activity and specificity is a general feature of the SpCas9 mutation space, we performed a high-throughput pooled lentiviral screen to comprehensively profile variant activity in human cells. We selected 157 residues for mutagenesis (Fig. 2A), focusing on the HNH and RuvC nuclease domains, as well as the L1 and L2 linkers connecting them, as these regions play a key role in the conformational activation of Cas9 to license target cleavage (Palermo et al., 2016). We selected four diverse target sites to assay the variants on: a putative ‘permissive’ guide (g1) known to be highly active for eSpCas9(1.1) and SpCas9-HF1; a ‘difficult’ guide (g2) with no activity for eSpCas9(1.1) and SpCas9-HF1; and two simulated off-targets (g3 and g4) bearing two mismatches each (Fig. 2B). Barcoded variants were cloned into a lentiviral vector and transduced into HEK 293FT cells (Fig. 2C), along with a guide RNA cassette and cognate target site. A total of 2,420 single amino acids variants exceeded the minimum read threshold for all four targets, representing 9.2% of all possible single amino acid variants of SpCas9. The activity of these variants was highly guide-dependent: over 20% of the variants improved specificity (≤50% activity at mismatched off-target; ≥80% activity on-target) when comparing g1 vs. g3, while <1% of variants met these criteria when comparing g2 vs. g4 (Fig. 2D). We validated the performance of 254 variants on a broader range of targets (including three targets known to have low activity for eSpCas9(1.1) and SpCas9-HF1) by individual transfections and targeted deep sequencing (Fig. 2E). Overall, these results suggest that a simple guide-dependent trade-off describes the performance of a broad range of Cas9 variants.
A number of algorithms have been developed that aim to predict editing outcomes, including specificity and, more recently, indel distributions. Comparison of TTISS specificity data to two published computational tools that provide specificity scores for guides – GuideScan (guidescan.com) (Perez et al., 2017) and CRISPR ML (crispr.ml) (Listgarten et al., 2018) showed a weak correlation (GuideScan, n = 59, R = 0.408, CRISPR ML, n = 47, R = 0.111) between the predicted metric and empirical observation (Supplemental Fig. 1E, F). Although the predominant outcome of Cas9 cleavage is a blunt DSB created by the concerted effort of the two nuclease domains, HNH and RuvC, the RuvC domain is not as rigidly positioned and it can slide one base upstream (distal to the PAM), giving rise to a staggered cut that is filled in by the cellular repair machinery and leads to duplication of a single base (+1 insertion) (Fig. 3A) (Zuo and Liu, 2016). This property is particularly useful in the genome engineering context because +1 insertions in protein-coding regions guarantee frameshifts, which has utility either for knocking out a gene or for the correction of a genetic variant. We therefore examined whether we could predict the relative frequencies of +1 insertions in the indel distribution for a given on-target site from multiplex TTISS data. Because TTISS relies on integration of a donor, we cannot directly observe +1 insertions, so we developed an algorithm to predict +1 insertions based on the distribution of the position of the donor relative to the cut site. To obtain the distribution for each cut site, we compiled the number of donor integrations at each nucleotide position relative to the cut site for both ends of the donor. We then used a convolution operation to merge these two distributions to model the situation in which no donor is integrated, allowing us to predict +1 frequencies (Fig. 3B). To validate our approach, we compared the +1 frequencies obtained by TTISS for WT SpCas9 for 58 guides to those measured by targeted indel sequencing (Supplemental Fig. 3A) and found a high correlation (r = 0.829), suggesting TTISS can be used to predict +1 frequency of a given guide. Prediction tools for Cas9-induced indel length distributions performed heterogeneously in predicting +1 frequencies compared to our empirical data (FORECasT (Allen et al., 2018), R = 0.782; inDelphi (Shen et al., 2018), R = −0.075; Lindel (Chen et al., 2019), R = 0.839)(Supplemental Fig. 3A).
Figure 3 |. Multiplexed assessment of +1 indel frequencies using TTISS.

(a) Editing outcomes of nuclease-induced blunt or staggered cuts in the human genome. As a simplified model, blunt or staggered cuts can either be resected prior to re-ligation, creating random deletions (top panel) or re-ligated without resection (middle panel). Staggered 5’-overhangs can be filled in before re-ligation, causing duplication of base −4 respective to the PAM motif (bottom panel). (b) Schematic for convolution operation used to predict indel distributions by TTISS. (c) Representative examples of TTISS-predicted +1 insertion frequencies compared between specificity variants versus WT SpCas9 for 58 gRNAs. (d) Differential +1 indel frequencies between LZ3 Cas9 and WT SpCas9 +1 insertion frequencies from targeted indel sequencing, grouped by the nucleotide identity at the −2 position relative to the PAM. Results from two-tailed t-test for significant divergence from zero are indicated by ** (p < 0.01), *** (p < 0.001), n.s. (not significant). See also Figure S3.
Given that many of the Cas9 variants contain mutations impacting DNA binding, which could potentially affect RuvC positioning, we compared the indel patterns of Cas9 specificity variants across a set of 58 guides. While most variants closely mirrored +1 frequencies of WT SpCas9 across on-target sites by TTISS (Supplemental Fig. 3B), the variant LZ3 Cas9 exhibited a markedly different +1 frequency profile relative to WT SpCas9 (Fig. 3C), which was confirmed by targeted sequencing data (Supplemental Fig. 3D). Exploring sequence determinants for +1 frequencies of LZ3 Cas9 and WT SpCas9 revealed that for both enzymes, the presence of a thymidine or a guanine in the −4 position with respect to the PAM led to the highest and lowest rates of +1 insertion respectively (Supplemental Fig. 3C). However, when comparing LZ3 Cas9 to WT SpCas9, LZ3 Cas9 showed elevated +1 frequency given a guanine at position −2 (Fig. 3D). In contrast, overall indel profiles were not found to be altered for any of the Cas9 variants tested (Supplemental Fig. 3E).
Discussion
Here we have shown that TTISS is a scalable, accessible, and cost-effective method for examining off-targets and +1 insertion frequencies of programmable nucleases. Beyond these applications, TTISS has been successfully applied to detect off-targets in other genome editing contexts, including editing by Cas enzymes creating overhanging, rather than blunt, ends (Strecker et al., 2019a), Cas enzymes delivered as ribonucleoprotein complexes, and ShCAST-mediated genome insertions (Strecker et al., 2019b). Multiplex TTISS enables the creation of substantially larger sets of empirical data that could contribute to improved predictive algorithms or identify high-specificity guides suitable for clinical applications. Applying TTISS across a panel of SpCas9 variants revealed a tradeoff between activity and specificity, which is also supported by our Cas9 mutational screening results. We also showed that the newly evolved LZ3 Cas9 variant exhibits high activity, increased specificity, and a differential +1 insertion profile as compared to WT SpCas9. Further rational engineering of LZ3 Cas9 might provide an avenue for non-templated correction of disease-causing frameshift mutations in the human population.
Limitations
Among published off-target detection methods, TTISS requires the least hands-on time with very few enzymatic steps, and is the only method demonstrated to be compatible with multiplexing >50 guides in a single experiment. For instances where in vitro rather than in vivo specificity profiles are desired, Digenome-Seq, CIRCLE-seq, or SITE-seq should be used (Cameron et al., 2017; Kim et al., 2015; Tsai et al., 2017), all of which can potentially be adapted to guide multiplexing using our analysis pipeline. Whereas GUIDE-seq and TTISS capture relevant in vivo biases from cleavage and NHEJ repair processes, DISCOVER-Seq instead captures binding of the MRN repair complex, adding potentially relevant information for assessing the safety of Cas enzymes (and other programmable nucleases) for clinical application (Wienert et al., 2019).
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Feng Zhang (zhang@broadinstitute.org). Plasmids generated in this study have been deposited to Addgene (catalog numbers pending).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK 293T cells
HEK 293T cells were maintained at 37C, 5% CO2 in DMEM-GlutaMAX (Gibco) supplemented with 10% FBS (Seradigm) and 10 μg/ml Ciprofloxacin (Sigma-Aldrich). HEK 293T cells were originally derived from a female human embryo. Cells were obtained from the lab of Veit Hornung. Cell line authentication was not performed.
U-2 OS cells
U-2 OS cells were maintained at 37C, 5% CO2 in DMEM-GlutaMAX (Gibco) supplemented with 10% FBS (Seradigm) and 10 μg/ml Ciprofloxacin (Sigma-Aldrich). U-2 OS were originally established from the osteosarcoma of female patient. Cells were obtained from ATCC. Cell line authentication was performed by the vendor.
K562 cells
K562 cells were maintained at 37C, 5% CO2 in RPMI-GlutaMAX (Gibco) supplemented with 10% FBS and 10 μg/ml Ciprofloxacin (Sigma-Aldrich). K562 cells were originally established from the chronic myelogenous leukemia of a female patient. Cells were obtained from Sigma-Aldrich. Cell line authentication was performed by the vendor.
E. coli strains
STBL3 E. coli cells (ThermoFisher) were grown in LB media at 37C overnight. Chemocompetent cells were generated using the Mix&Go kit (Zymo).
METHOD DETAILS
Tn5 purification
Tn5 was purified as previously described (Picelli et al., 2014). E. coli cells (NEB C3013) harboring pTBX1-Tn5 were grown in terrific broth to an OD of 0.65 before addition of IPTG at 0.25 mM. Protein expression was induced at 23°C overnight, and cells were harvested and stored at −80°C until purification. 20 g of E. coli pellet was lysed in 200 mL HEGX buffer (20 mM HEPES-KOH pH 7.2, 800 mM NaCl, 1 mM EDTA, 0.2% Triton, 10% glycerol) with cOmplete protease inhibitor (Roche) and 10 uL of benzonase (Sigma-Aldrich). Cells were lysed using a LM20 microfluidizer device (Microfluidics) and cleared by centrifugation at max speed for 30 min. 5.25 mL of 10% PEI (pH 7) was added dropwise to a stirring solution to remove E. coli DNA and the resulting precipitation removed after centrifugation for 10 min. Cleared supernatant was added to 30 mL of equilibrated chitin resin (NEB), mixed end-over-end for 30 min, added to column, washed with 1L HEGX buffer. 75 mL HEGX buffer with 100 mM DTT was added to column, 30 mL drawn through the resin before sealing the column and storing at 4°C for 48h to allow for intein cleavage and elution of free Tn5. Eluted Tn5 was dialyzed into 2xTn5 dialysis buffer (100 HEPES, 200 NaCl, 2 EDTA, 0.2 Triton, 20% glycerol), with two exchanges of 1L of buffer. The final solution was concentrated to 50 mg/mL as determined by A280 absorbance (A280 = 1 = 0.616 mg/mL = 11.56 mM) and flash frozen in liquid nitrogen before storage at −80°C.
Tn5 loading with single handle
Oligonucleotides Transposon ME and Transposon read 2 were annealed at a concentration of 42 μM each in annealing buffer (1.5 mM Tris-HCl pH 8.0, 150 μM EDTA, 30 mM NaCl) by heating to 95°C for 3 minutes, and subsequently ramping the temperature from 70C to 25°C at a rate of 1°C per minute. 1 ml of purified Tn5 (50 mg/ml) were incubated with 355 μl of annealed oligonucleotides for 1 hour at room temperature. Of note, loaded Tn5 can crash out as white precipitate, but retains activity. Loaded Tn5 is stored at −20C and ready to be thawed on ice for later use.
Cas9 variant cloning
Cas9 variants were cloned by site-directed mutagenesis into pX165 (Addgene #48137), which encodes a CBh promoter-driven SpCas9 containing a 3xFLAG tag and SV40 NLS on the N terminus and a nucleoplasmin NLS on the C terminus.
Cell transfection
HEK 293T cells were seeded in poly-D-lysine coated 96-well plates (Corning) at a density of 25,000 cells in 100 μl medium per well. The next day, 250 μl OptiMEM (Thermo) were mixed with 1 μg of oligonucleotide donor (TTISS donor sense and TTISS donor antisense, annealed in 0.1x IDT Nuclease-Free Duplex Buffer by ramping the temperature from 95°C to 25°C at a rate of 1°C per minute), 750 ng Cas9 expression plasmid, and a total of 250 ng of 1–60 different gRNA expression plasmids (sequences in Supplemental Table 3). In parallel, 250 μl OptiMEM were mixed with 5 μl GeneJuice (Millipore) and incubated at room temperature for 5 minutes. After mixing all components and incubating them for 20 minutes, 50 μl were added drop-wise per 96-well of cells in a total of ten wells per condition. For prime editing, the same transfection protocol was used with 1.5 μg pCMV-PE2 plasmid and 500 ng pU6-pegRNA. For TTISS in K562 and U-2 OS cells, one million cells were nucleofected with pulse code FF-120 (K562) or CM-104 (U-2 OS) using a Lonza 4D-Nucleofector X unit in 100 μl buffer SF (K562) or SE (U-2 OS) with the same amounts of Cas9, gRNA, and donor as listed above.
Cell lysis and genome tagmentation
Three days after transfection, cells were washed with PBS, trypsinized, and washed again in a 1.5 ml tube. Pelleted cells were lysed by re-suspending one million cells in 100 μl lysis buffer (1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10 mM Tris pH 7.5, 8 units/ml Proteinase K (NEB)) and heating to 65° C for 10 minutes. For tagmentation, 80 μl crude lysate were mixed with 25 μl 5x TAPS buffer (50 mM TAPS-NaOH pH 8.5 at room temperature, 25 mM MgCl2) and 20 μl hyperactive loaded Tn5 transposase and were heated to 55° C for 10 minutes. Reactions were mixed with 625 μl PB buffer (Qiagen) and purified on a mini-prep silica spin column according to the protocol (Qiagen). DNA was eluted in 50 μl water (typical concentration: 200–300 ng/μl).
PCR amplification
Total eluates were denatured at 95° C for 5 minutes, snap-cooled on ice, and amplified in 200μl PCR reactions using KOD Hot Start polymerase (Millipore) according to the manufacturer’s protocol (12 cycles, Ta = 60° C, one minute elongation, primers: TTISS PCR fwd 1, Transposon read 2). For each sample, a secondary 50 μl KOD PCR was templated with 3 μl of the first PCR reaction and a unique barcoding primer (20 cycles, Ta = 65° C, one minute elongation, primers: TTISS PCR fwd 2, TTISS PCR rev BC1–24). For mapping prime-mediated insertions, primers TTISS PCR prime +24 fwd a, b or TTISS PCR prime +38 fwd a1, a2, b1, b2 were used instead.
Deep sequencing
PCRs were pooled, column-purified, and 250–1,000 bp fragments were enriched using a 2% agarose gel. After two consecutive column purifications, the library was quantified using a NanoDrop spectrometer (Thermo) and sequenced using an Illumina NextSeq 500 sequencer with a 75-cycle high-output v2 kit (cycle numbers: read 1 = 59, index 1 = 8, read 2 = 25, no index 2).
Read mapping
Reads were mapped to human genome version hg38 using BrowserGenome.org (Schmid-Burgk and Hornung, 2015) with mapping parameters: read filter = NNNNNNNNNNNNNNNNNNNNNNNAAC, forward mapping start = 26 bp, forward mapping length = 25 bp, reverse mapping length = 15 bp, max forward/reverse span = 1000 bp. For mapping prime-mediated insertions, read filters CTTATCGTCGTCATCCTTGTAATC (+24 a, forward mapping start = 25), GATTACAAGGATGACGACGATAAG (+24 b, forward mapping start = 25), GACGGCGGTCTCCGTCGTCAGGATCAT (+38 a, forward mapping start = 28), or GACGGAGACCGCCGTCGTCGACAAGCC (+38 b, forward mapping start = 28) were used instead. Mapped read pairs spanning fewer than 37 genome bases were discarded in order to omit signal from the pegRNA expression plasmid.
Integration site detection
Common break sites, common mispriming sites and reads mapping to the human U6 promoter were filtered out. These were detected by TTISS in the absence of a nuclease, donor, and/or gRNA plasmid. Following removal of non-overlapping single-read noise, putative break sites were identified by the presence of two or more unique reads mapping to the reference sequence within a window of 20 nucleotides. For all sites passing filters, TTISS read counts mapping to a 60-nucleotide window were tabulated and stored for downstream analysis.
gRNA assignment
For each 60-nucleotide window, peaks were identified in both the sense and antisense reads, and each peak was grouped with all gRNA sequences used in the respective experiment whose spacers had an edit distance less than or equal to 6 mismatches for any 20-mer in a window of 25 nucleotides on either side of the detected peak site. If a given peak site had at least one such gRNA, then a cut site score was calculated for each putative gRNA match. The cut site score was defined as the distance between the expected cut site of the spacer and the peak. Each remaining peak site was then assigned to gRNA with the lowest cut site score and all peak sites with a cut site score of between −3 and 3 were retained and reported for each individual gRNA. This allows for the possibility of multiple cut sites within the same window, as well as for the removal of false hits where the apparent cut site does not line up with the expected cut site from the spacer sequence.
Prediction of indel length distributions
Genomic positions of TTISS-detected donor integration events were tabulated for each gRNA target site with more than 50 reads mapping in each orientation. Obtained distributions were normalized to their total number of reads in order to obtain two frequency distributions per target site. TTISS-predicted indel length distributions were calculated by numerically convolving the two directional distributions for each target site. From each indel length distribution, relative +1 frequencies were calculated as the ratio of +1 frequency to the sum of all non-+0 repair frequencies.
Variant Scoring
Specificity scores were calculated by subtracting from 100 the percent of TTISS reads that corresponds to off-targets. Activity scores were calculated as the mean indel percentage across all 59 on-target sites, normalized to WT SpCas9.
Cas9 variant library construction
SpCas9 variants were screened using a pool of self-targeting lentiviral vectors in which each lentiviral insert contained a Cas9 variant and a constant target site, allowing indel formation at the target site to be coupled to its corresponding Cas9 variant. For the variant pool, >150 residue positions, concentrated in the HNH and RuvC nuclease domains, were selected for single amino acid saturation mutagenesis. For each residue, a mutagenic insert was synthesized as short complementary oligonucleotides, with the mutated codon replaced by a degenerate NNK mixture of bases, as previously described in (Gao et al., 2017). Furthermore, variants were barcoded with a random 24-nt sequence placed in close proximity to the target site in order to allow direct variant-to-indel association by short-read paired-end sequencing. Barcode-to-variant associations were determined by targeted deep sequencing prior to performing the screen.
Lentiviral Cas9 variant library screen
HEK 293FT cells were transduced with the variant library at MOI <0.1 and selected with puromycin at 1 μg/mL over several passages to eliminate non-transduced cells. Variant library-transduced cells were subsequently transduced with a second lentivirus containing an U6-sgRNA expression cassette at MOI >> 1 and >1000 cells/variant, in order to initiate indel formation at the target site. After approximately 4 days, genomic DNA from cells were isolated, and the target site and corresponding barcodes were PCR-amplified and paired-end sequenced with a 150-cycle NextSeq 500/550 High Output Kit v2 (Illumina). This procedure was repeated for four different sgRNAs: Two fully matched sgRNAs, to assess on-target efficiency of the variants; and two sgRNA bearing double base mismatches, to assess specificity (all guide sequences in Supplemental Table 3). Highly abundant barcodes (above 50 reads; comprising 5%, 2%, 3% and 3% of all barcodes for g1, g2, g3 and g4, respectively) were discarded to reduce noise. For each guide, the score of a variant was calculated as 100 * (number of reads containing an indel) / (total number of reads pooled across all retained barcodes for that variant). Variants with fewer than 100 reads for any of the four target sites were discarded, resulting in a final set of 130 wild-type, 112 stop codons, and 2,420 single amino acid variants.
Cas9 variant validation and combinatorial mutagenesis
Top hits from the pooled variant screen that exhibited both high on-target efficiency and high specificity were individually cloned into pX165 (Ran et al., 2013) and tested at additional target sites in HEK 293T cells, including sites that were previously observed to have substantially reduced activity with eSpCas9, SpCas9-HF1, and HypaCas9. Top-performing variants were combined to produce combination mutants, including LZ3 Cas9, which were re-tested as described and refined over 10 subsequent rounds of mutagenesis.
Prime editing constructs
The following pegRNA sequences were cloned into pU6-pegRNA-GG-acceptor according to the protocol described in Anzalone et al., 2019 (Supplemental Table 3).
Targeted indel sequencing
Indel frequencies were quantified by targeted deep sequencing (Illumina) as previously described in (Gao et al., 2017). Indel distribution profiles were analyzed using OutKnocker.org (Schmid-Burgk et al., 2014).
Indel distribution and specificity predictors
Elevation scores (Listgarten et al., 2018) and GuideScan (Perez et al., 2017) scores were calculated by inputting the gene into the online interfaces (crispr.ml and guidescan.com) and storing the Elevation aggregate value and specificity value for the correct gRNA respectively. Predicted +1 insertion frequencies from FORECasT (Allen et al., 2018) and inDelphi (Shen et al., 2018) were evaluated by inputting the genomic locus (FORECasT) or 30 bp on either side of the cut site (inDelphi) into the correct online interface (partslab.sanger.ac.uk/FORECasT and the HEK 293 predictor on indelphi.giffordlab.mit.edu/single) and recording the total predicted % of 1-bp insertions Lindel-predicted values (Chen et al., 2019) were calculated similarly to inDelphi using the Python library (github.com/shendurelab/Lindel).
QUANTIFICATION AND STATISTICAL ANALYSIS
The code used for sequencing data mapping used in this study is available at www.BrowserGenome.org. No data were excluded from analysis. Statistical tests and significance thresholds are indicated in the legends to Fig. 3.
ADDITIONAL RESOURCES
Detailed protocol
A detailed bench protocol (Method S1) describes the experimental details of the TTISS method.
DATA AND CODE AVAILABILITY
The sequencing data generated during this study are available at SRA (BioProject PRJNA602092). The code used for read post-processing used in this study is available at GitHub (schmidburgk/TTISS).
Bench Protocol
Step 1: Tn5 purification
Grow E. coli cells (NEB C3013) harboring the plasmid pTBX1-Tn5 in terrific broth to an OD of 0.65
Add IPTG to a concentration of 0.25 mM and shake at 23°C overnight
Harvest cells by centrifugation and store at −80°C until purification
Lyse 20 g of E. coli pellet in 200 mL HEGX buffer (20 mM HEPES-KOH pH 7.2, 800 mM NaCl, 1 mM EDTA, 0.2% Triton, 10% glycerol) with cOmplete protease inhibitor (Roche) and 10 μL of Benzonase (Sigma-Aldrich), using an LM20 microfluidizer device (Microfluidics)
Clear the lysate by centrifugation at max speed for 30 min
Add 5.25 mL of 10% PEI (pH 7) dropwise to a stirring solution to remove E. coli DNA. for 10 min
Add cleared supernatant to 30 mL of equilibrated chitin resin (NEB) and mix end-over-end for 30 min • Add mixture to column, wash with 1L HEGX buffer
Add 75 mL HEGX buffer with 100 mM DTT to column, draw 30 mL through the resin before sealing the column and storing at 4°C for 48h to allow for intein cleavage and elution of free Tn5
Dialyze eluted Tn5 into 2xTn5 dialysis buffer (100 HEPES, 200 NaCl, 2 EDTA, 0.2 Triton, 20% glycerol), with two exchanges of 1L of buffer
Concentrate the final solution to 50 mg/mL as determined by A280 absorbance (A280 = 1 = 0.616 mg/mL = 11.56 mM) • Flash-freeze in liquid nitrogen before storage at −80°C Step 2: Tn5 loading with single handle
Anneal oligonucleotides Transposon ME and Transposon read 2 at a concentration of 42 μM each in annealing buffer (1.5 mM Tris-HCl pH 8.0, 150 μM EDTA, 30 mM NaCl) by heating to 95C for 3 minutes, and subsequently ramping the temperature from 70C to 25C at a rate of 1C per minute
Incubate 1 ml of purified Tn5 (50 mg/ml) with 355 μl of annealed oligonucleotides for 1 hour at room temperature. Of note, loaded Tn5 can crash out as white precipitate, but retains activity.
Store loaded Tn5 at −20C, ready to be thawed on ice for later use. Resuspend before use.
Step 3: Cell transfection
Seed HEK293T cells in poly-D-lysine coated 96-well plates (Corning) at a density of 25,000 cells in 100 μl medium per well
Anneal TTISS donor sense and TTISS donor antisense in 0.1x IDT Nuclease-Free Duplex Buffer by ramping the temperature from 95°C to 25°C at a rate of 1°C per minute
The next day, mix 250 μl OptiMEM (Thermo) with 1 μg of annealed oligonucleotide donor, 750 ng Cas9 expression plasmid, and a total of 250 ng of 1–60 different gRNA expression plasmids for each condition
In parallel, mix 250 μl OptiMEM with 5 μl GeneJuice (Millipore) and incubate at room temperature for 5 minutes for each condition
Mix all components for each condition and incubate them for 20 minutes
Add 50 μl drop-wise per 96-well of cells in a total of ten wells per condition
Step 4: Cell lysis and genome tagmentation
Two to three days after transfection, wash cells with PBS, trypsinize, and wash again with PBS in a 1.5 ml tube
Lyse pelleted cells by re-suspending one million cells in 100 μl lysis buffer (1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10 mM Tris pH 7.5, 8 units/ml Proteinase K (NEB))
Heat lysates to 65° C for 10 minutes, then keep on ice
For tagmentation, mix 80 μl crude lysate with 25 μl 5x TAPS buffer (50 mM TAPS-NaOH pH 8.5 at room temperature, 25 mM MgCl2) and 20 μl hyperactive loaded Tn5 transposase. Heat to 55° C for 10 minutes.
Mix reactions with 625 μl PB buffer (Qiagen) and bind to a mini-prep silica spin column. Wash with 750 μl buffer PE (Qiagen), spin dry, and elute DNA in 50 μl water (typical concentration: 200–300 ng/μl).
Run 3μl of the eluate on a 2% Agarose gel to check size range • If size range is outside the range of 300 to 1,000 bp, repeat with adjusted amounts of Tn5 and note adjustments for future use of the Tn5 batch. Alternatively, you can perform a titration of loaded Tn5 at the start using extra cell lysate to determine optimal tagmentation conditions.
Step 5: PCR amplification
Denature total eluates at 95° C for 5 minutes, then snap-cool on ice
Amplify in 200μl PCR reactions using KOD Hot Start polymerase (Millipore) according to the manufacturer’s protocol (12 cycles, Ta = 60° C, one minute elongation, primers: TTISS PCR fwd 1, Transposon read 2)
For each sample, perform a secondary 50 μl KOD PCR templated with 3 μl of the first PCR reaction and a unique barcoding primer (20 cycles, Ta = 65° C, one minute elongation, primers: TTISS PCR fwd 2, TTISS PCR rev BC1–24)
Step 6: Deep sequencing
Pool PCRs on ice, column-purify on a mini-prep silica gel column, and purify fragments within a size range of 250–1,000 bp using a 2% agarose gel
Perform two consecutive column purifications (first with buffer QG (Qiagen) and isopropanol added to the gel slice before loading, second with buffer PB and the eluate from the previous column) • Quantify the library using a NanoDrop spectrometer (Thermo)
Sequence using an Illumina NextSeq 500 sequencer with a 75-cycle high-output v2 kit (cycle numbers: read 1 = 59, index 1 = 8, read 2 = 25, no index 2)
Step 7: Read mapping
Open in a web browser the site www.BrowserGenome.org
Click the “Map deep sequencing data” tab
Under point 2 click “Browse” to choose the human genome file “hg38.2bit” on your hard drive (download from http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.2bit)
Under point 3 click “Browse” to choose all un-compressed FASTQ files to be analyzed
Under point 4, enter the filter values 0 bp, NNNNNNNNNNNNNNNNNNNNNNNAAC
Under point 5 enter forward mapping start = 26 bp
Under point 6 enter forward mapping length = 25 bp
Under point 7 enter reverse mapping length = 15 bp
Under point 8 enter max forward/reverse span = 1000 bp
Click “Start mapping”, which takes about one hour per ten million reads
When all data has been processed, click “Save all” on bottom right to save mapping data files
Click on the “Process” tab, then “Remove single read noise” and “Enforce antisense-overlap reads” for basic noise reduction and off-target site identification
Click “Export peak list” to save a list of detected cleavage sites, which can be opened in a text or spreadsheet editor for further analysis
For more complex analyses (such as gRNA multiplexing or indel distribution prediction), refer to the Read Me on the Github repository available at https://github.com/schmidburgk/ttiss
Supplementary Material
Supplemental Table 1 | Comparison of TTISS to GUIDE-Seq and DISCOVER-Seq, Related to Figure 1. (First Tab) List of target sites detected for the EMX1 and VEGFA 3 gRNAs from single-guide TTISS runs in HEK 293T cells. TTISS reads and published GUIDE-seq read counts from an experiment using the same gRNAs in U2OS cells are listed. (Second Tab) List of target sites detected for the RNF2 and VEGFA gRNAs from single-guide TTISS runs in K562 cells. TTISS reads and published DISCOVER-seq read counts from an experiment using the same gRNAs in K562 cells are listed.
Supplemental Table 2 | TTISS-detected target sites across 59 guides and Cas9 variants used, Related to Figure 1.
Supplemental Table 3 | Sequences of guide RNAs and pegRNAs used, Related to STAR Methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| STBL3 | ThermoFisher | C737303 |
| T7 Express lysY/Iq Competent E. coli (High Efficiency) | NEB | C3013 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FBS, USA, Seradigm Premium | VWR | 97068-085 |
| KOD Hot Start DNA Polymerase | Millipore Sigma | 71086-3 |
| Proteinase K | NEB | P8107S |
| Tn5 | F. Zhang Lab | - |
| Qiaprep spin miniprep kit | Qiagen | 27106 |
| IPTG | Millipore Sigma | I6758 |
| cOmplete protease inhibitor | Millipore Sigma | 11697498001 |
| Benzonase | Millipore Sigma | E1014-25KU |
| Chitin resin | NEB | S6651L |
| OptiMEM | ThermoFisher | 31985070 |
| E-Gel™ EX Agarose Gels, 2% | ThermoFisher | G402002 |
| GeneJuice | Millipore Sigma | 70967-3 |
| SF Cell Line 4D-Nucleofector® X Kit | Lonza | V4XC-2012 |
| SE Cell Line 4D-Nucleofector® X Kit | Lonza | V4XC-1012 |
| Puromycin | ThermoFisher | A1113802 |
| NextSeq 500/550 High Output Kit v2, 75 cycles | Illumina | FC-404-2005 |
| NextSeq 500/550 High Output Kit v2, 150 cycles | Illumina | FC-404-2002 |
| Nuclease-Free Duplex Buffer | IDT | 11-01-03-01 |
| Deposited Data | ||
| Deep Sequencing data | SRA | PRJNA602092 |
| Experimental Models: Cell Lines | ||
| HEK 293T | Gift from Veit Hornung | - |
| U-2 OS | ATCC | HTB-96 |
| K562 | Millipore Sigma | 89121407-1VL |
| Oligonucleotides | ||
| /5Phos/CTGTCTCTTATACA/3ddC/ | IDT | Transposon ME |
| GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG | IDT | Transposon read 2 |
| /5phos/G*T*TGTGAGCAAGGGCGAGGAGGATAACGCCTCTCTCCCAGCGACT*A*T | IDT | TTISS donor sense |
| /5phos/A*T*AGTCGCTGGGAGAGAGGCGTTATCCTCCTCGCCCTTGCTCACA*A*C | IDT | TTISS donor antisense |
| GTCGCTGGGAGAGAGGCGTTATC | IDT | TTISS PCR fwd 1 |
| AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTTATCCTCCTCGCCCTTGCTCAC | IDT | TTISS PCR fwd 2 |
| CAAGCAGAAGACGGCATACGAGATCGAGTAATGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC1 |
| CAAGCAGAAGACGGCATACGAGATTCTCCGGAGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC2 |
| CAAGCAGAAGACGGCATACGAGATAATGAGCGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC3 |
| CAAGCAGAAGACGGCATACGAGATGGAATCTCGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC4 |
| CAAGCAGAAGACGGCATACGAGATTTCTGAATGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC5 |
| CAAGCAGAAGACGGCATACGAGATACGAATTCGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC6 |
| CAAGCAGAAGACGGCATACGAGATAGCTTCAGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC7 |
| CAAGCAGAAGACGGCATACGAGATGCGCATTAGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC8 |
| CAAGCAGAAGACGGCATACGAGATCATAGCCGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC9 |
| CAAGCAGAAGACGGCATACGAGATTTCGCGGAGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC10 |
| CAAGCAGAAGACGGCATACGAGATGCGCGAGAGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC11 |
| CAAGCAGAAGACGGCATACGAGATCTATCGCTGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC12 |
| CAAGCAGAAGACGGCATACGAGATTGTAGTGCGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC13 |
| CAAGCAGAAGACGGCATACGAGATGCGTCGACGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC14 |
| CAAGCAGAAGACGGCATACGAGATGGTCTTCTGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC15 |
| CAAGCAGAAGACGGCATACGAGATAAATGTCCGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC16 |
| CAAGCAGAAGACGGCATACGAGATGTTGAAACGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC17 |
| CAAGCAGAAGACGGCATACGAGATTCTTTACGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC18 |
| CAAGCAGAAGACGGCATACGAGATATGCCTGGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC19 |
| CAAGCAGAAGACGGCATACGAGATCAATAAGGGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC20 |
| CAAGCAGAAGACGGCATACGAGATCGCCGTAAGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC21 |
| CAAGCAGAAGACGGCATACGAGATTAAGGCTTGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC22 |
| CAAGCAGAAGACGGCATACGAGATTTGCTGCCGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC23 |
| CAAGCAGAAGACGGCATACGAGATCTCAATGTGTCTCGTGGGCTCGGAGATGTGT | IDT | TTISS PCR rev BC24 |
| AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGctcttccgatctCTTATCGTCGTCATCCTTGT | IDT | TTISS PCR prime +24 fwd a |
| AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGctcttccgatctGATTACAAGGATGACGACGA | IDT | TTISS PCR prime +24 fwd b |
| GGCTTGTCGACGACGGCGGTC | IDT | TTISS PCR prime +38 fwd a1 |
| AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGctcttccgatctGACGGCGGTCTCCGTCGTCAG | IDT | TTISS PCR prime +38 fwd a2 |
| ATGATCCTGACGACGGAGACCG | IDT | TTISS PCR prime +38 fwd b1 |
| AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGctcttccgatctGACGGAGACCGCCGTCGTCGA | IDT | TTISS PCR prime +38 fwd b2 |
| Recombinant DNA | ||
| pTBX1-Tn5 | Addgene | #60240 |
| pX165 | Addgene | #48137 |
| pCMV-PE2 | Addgene | #132775 |
| pU6-pegRNA-GG-acceptor | Addgene | #132777 |
| pX165-Sniper-Cas9 | This study | - |
| pX165-LZ3 Cas9 | This study | - |
| pX165-HiFi Cas9 | This study | - |
| pX165-eSpCas9 | This study | - |
| pX165-Cas9-HF1 | This study | - |
| pX165-HypaCas9 | This study | - |
| pX165-xCas9 | This study | - |
| pX165-evoCas9 | This study | - |
| Software and Algorithms | ||
| BrowserGenome | BrowserGenome.org | - |
| Elevation scoring | crispr.ml | - |
| GuideScan | guidescan.com | - |
| FORECasT | partslab.sanger.ac.uk/FORECasT | - |
| inDelphi | indelphi.giffordlab.mit.edu/single | - |
| Lindel | github.com/shendurelab/Lindel | - |
| Other | ||
| Bench Protocol | (this paper) | Methods S1 |
Highlights.
Tagmentation-based Tag Integration Site Sequencing (TTISS) scalably detects DSBs
TTISS is a rapid and streamlined protocol compatible with multiplexing
Application of TTISS highlights trade-off in Cas9 variant specificity and activity
LZ3 Cas9 variant exhibits a unique +1 insertion profile
Acknowledgements
We thank Rhiannon Macrae for helpful discussions. J.L.S.-B. is supported by an EMBO Long-Term Fellowship (ALTF 199-2017). J.S. is supported by the Human Frontier Science Program. F.Z. is supported by NIH grants (1R01-HG009761, 1R01-MH110049, 1DP1-HL141201, and 5R-M1HG006193-09); the Howard Hughes Medical Institute; the Harold G. and Leila Mathers and Edward Mallinckrodt, Jr. Foundations; the Poitras Center for Psychiatric Disorders Research at MIT; the Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT; and by the Phillips family and J. and P. Poitras. Deep sequencing data is made available on SRA (BioProject PRJNA602092, Submission SUB6858869). Reagents will be available to the academic community through Addgene and additional information on using these reagents can be obtained via the Zhang lab website (https://zlab.bio/resources).
Footnotes
Declaration of Interests
The Broad Institute has filed patent applications related to the work presented here. F.Z. is a co-founder of Editas Medicine, Beam Therapeutics, Pairwise Plants, Arbor Biotechnologies, and Sherlock Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen F, Crepaldi L, Alsinet C, Strong AJ, Kleshchevnikov V, De Angeli P, Páleníková P, Khodak A, Kiselev V, Kosicki M, et al. (2018). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nature Biotechnology 37, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron P, Fuller CK, Donohoue PD, Jones BN, Thompson MS, Carter MM, Gradia S, Vidal B, Garner E, Slorach EM, et al. (2017). Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Meth 14, 600–606. [DOI] [PubMed] [Google Scholar]
- Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, et al. (2018). A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nature Biotechnology 36, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, and Doudna JA (2017). Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, McKenna A, Schreiber J, Haeussler M, Yin Y, Agarwal V, Noble WS, and Shendure J (2019). Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucl. Acids Res. 47, 7989–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, and Zhang F (2017). Engineered Cpf1 variants with altered PAM specificities. Nature Biotechnology 163, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim J-I, and Kim J-S (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Meth 12, 237–243. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, and Joung JK (2016). High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim Y-H, Lee K, Jung I, Kim D, Kim S, et al. (2018). Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications 9, 3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J, Weinstein M, Kleinstiver BP, Sousa AA, Joung JK, Crawford J, Gao K, Hoang L, Elibol M, Doench JG, et al. (2018). Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nature Biomedical Engineering 2018 2:7 2, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G, Miao Y, Walker RC, Jinek M, and McCammon JA (2016). Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Cent Sci 2, 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, and Ventura A (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nature Biotechnology 35, 347–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, and Hornung V (2015). BrowserGenome.org: web-based RNA-seq data analysis and visualization. Nat Meth 12, 1001–1001. [DOI] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Schmidt T, Gaidt MM, Pelka K, Latz E, Ebert TS, and Hornung V (2014). OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 24, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, and Zhang F (2015). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Jones S, Koopal B, Schmid-Burgk J, Zetsche B, Gao L, Makarova KS, Koonin EV, and Zhang F (2019a). Engineering of CRISPR-Cas12b for human genome editing. Nature Communications 10, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, and Zhang F (2019b). RNA-guided DNA insertion with CRISPR-associated transposases. Science eaax9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, and Joung JK (2016). Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Publishing Group 17, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, and Joung JK (2017). CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Meth 14, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, et al. (2018). A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 24, 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienert B, Wyman SK, Richardson CD, Yeh CD, Akcakaya P, Porritt MJ, Morlock M, Vu JT, Kazane KR, Watry HL, et al. (2019). Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z, and Liu J (2016). Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Scientific Reports 6, 37584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 | Comparison of TTISS to GUIDE-Seq and DISCOVER-Seq, Related to Figure 1. (First Tab) List of target sites detected for the EMX1 and VEGFA 3 gRNAs from single-guide TTISS runs in HEK 293T cells. TTISS reads and published GUIDE-seq read counts from an experiment using the same gRNAs in U2OS cells are listed. (Second Tab) List of target sites detected for the RNF2 and VEGFA gRNAs from single-guide TTISS runs in K562 cells. TTISS reads and published DISCOVER-seq read counts from an experiment using the same gRNAs in K562 cells are listed.
Supplemental Table 2 | TTISS-detected target sites across 59 guides and Cas9 variants used, Related to Figure 1.
Supplemental Table 3 | Sequences of guide RNAs and pegRNAs used, Related to STAR Methods.
Data Availability Statement
The sequencing data generated during this study are available at SRA (BioProject PRJNA602092). The code used for read post-processing used in this study is available at GitHub (schmidburgk/TTISS).
Bench Protocol
Step 1: Tn5 purification
Grow E. coli cells (NEB C3013) harboring the plasmid pTBX1-Tn5 in terrific broth to an OD of 0.65
Add IPTG to a concentration of 0.25 mM and shake at 23°C overnight
Harvest cells by centrifugation and store at −80°C until purification
Lyse 20 g of E. coli pellet in 200 mL HEGX buffer (20 mM HEPES-KOH pH 7.2, 800 mM NaCl, 1 mM EDTA, 0.2% Triton, 10% glycerol) with cOmplete protease inhibitor (Roche) and 10 μL of Benzonase (Sigma-Aldrich), using an LM20 microfluidizer device (Microfluidics)
Clear the lysate by centrifugation at max speed for 30 min
Add 5.25 mL of 10% PEI (pH 7) dropwise to a stirring solution to remove E. coli DNA. for 10 min
Add cleared supernatant to 30 mL of equilibrated chitin resin (NEB) and mix end-over-end for 30 min • Add mixture to column, wash with 1L HEGX buffer
Add 75 mL HEGX buffer with 100 mM DTT to column, draw 30 mL through the resin before sealing the column and storing at 4°C for 48h to allow for intein cleavage and elution of free Tn5
Dialyze eluted Tn5 into 2xTn5 dialysis buffer (100 HEPES, 200 NaCl, 2 EDTA, 0.2 Triton, 20% glycerol), with two exchanges of 1L of buffer
Concentrate the final solution to 50 mg/mL as determined by A280 absorbance (A280 = 1 = 0.616 mg/mL = 11.56 mM) • Flash-freeze in liquid nitrogen before storage at −80°C Step 2: Tn5 loading with single handle
Anneal oligonucleotides Transposon ME and Transposon read 2 at a concentration of 42 μM each in annealing buffer (1.5 mM Tris-HCl pH 8.0, 150 μM EDTA, 30 mM NaCl) by heating to 95C for 3 minutes, and subsequently ramping the temperature from 70C to 25C at a rate of 1C per minute
Incubate 1 ml of purified Tn5 (50 mg/ml) with 355 μl of annealed oligonucleotides for 1 hour at room temperature. Of note, loaded Tn5 can crash out as white precipitate, but retains activity.
Store loaded Tn5 at −20C, ready to be thawed on ice for later use. Resuspend before use.
Step 3: Cell transfection
Seed HEK293T cells in poly-D-lysine coated 96-well plates (Corning) at a density of 25,000 cells in 100 μl medium per well
Anneal TTISS donor sense and TTISS donor antisense in 0.1x IDT Nuclease-Free Duplex Buffer by ramping the temperature from 95°C to 25°C at a rate of 1°C per minute
The next day, mix 250 μl OptiMEM (Thermo) with 1 μg of annealed oligonucleotide donor, 750 ng Cas9 expression plasmid, and a total of 250 ng of 1–60 different gRNA expression plasmids for each condition
In parallel, mix 250 μl OptiMEM with 5 μl GeneJuice (Millipore) and incubate at room temperature for 5 minutes for each condition
Mix all components for each condition and incubate them for 20 minutes
Add 50 μl drop-wise per 96-well of cells in a total of ten wells per condition
Step 4: Cell lysis and genome tagmentation
Two to three days after transfection, wash cells with PBS, trypsinize, and wash again with PBS in a 1.5 ml tube
Lyse pelleted cells by re-suspending one million cells in 100 μl lysis buffer (1 mM CaCl2, 3 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10 mM Tris pH 7.5, 8 units/ml Proteinase K (NEB))
Heat lysates to 65° C for 10 minutes, then keep on ice
For tagmentation, mix 80 μl crude lysate with 25 μl 5x TAPS buffer (50 mM TAPS-NaOH pH 8.5 at room temperature, 25 mM MgCl2) and 20 μl hyperactive loaded Tn5 transposase. Heat to 55° C for 10 minutes.
Mix reactions with 625 μl PB buffer (Qiagen) and bind to a mini-prep silica spin column. Wash with 750 μl buffer PE (Qiagen), spin dry, and elute DNA in 50 μl water (typical concentration: 200–300 ng/μl).
Run 3μl of the eluate on a 2% Agarose gel to check size range • If size range is outside the range of 300 to 1,000 bp, repeat with adjusted amounts of Tn5 and note adjustments for future use of the Tn5 batch. Alternatively, you can perform a titration of loaded Tn5 at the start using extra cell lysate to determine optimal tagmentation conditions.
Step 5: PCR amplification
Denature total eluates at 95° C for 5 minutes, then snap-cool on ice
Amplify in 200μl PCR reactions using KOD Hot Start polymerase (Millipore) according to the manufacturer’s protocol (12 cycles, Ta = 60° C, one minute elongation, primers: TTISS PCR fwd 1, Transposon read 2)
For each sample, perform a secondary 50 μl KOD PCR templated with 3 μl of the first PCR reaction and a unique barcoding primer (20 cycles, Ta = 65° C, one minute elongation, primers: TTISS PCR fwd 2, TTISS PCR rev BC1–24)
Step 6: Deep sequencing
Pool PCRs on ice, column-purify on a mini-prep silica gel column, and purify fragments within a size range of 250–1,000 bp using a 2% agarose gel
Perform two consecutive column purifications (first with buffer QG (Qiagen) and isopropanol added to the gel slice before loading, second with buffer PB and the eluate from the previous column) • Quantify the library using a NanoDrop spectrometer (Thermo)
Sequence using an Illumina NextSeq 500 sequencer with a 75-cycle high-output v2 kit (cycle numbers: read 1 = 59, index 1 = 8, read 2 = 25, no index 2)
Step 7: Read mapping
Open in a web browser the site www.BrowserGenome.org
Click the “Map deep sequencing data” tab
Under point 2 click “Browse” to choose the human genome file “hg38.2bit” on your hard drive (download from http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.2bit)
Under point 3 click “Browse” to choose all un-compressed FASTQ files to be analyzed
Under point 4, enter the filter values 0 bp, NNNNNNNNNNNNNNNNNNNNNNNAAC
Under point 5 enter forward mapping start = 26 bp
Under point 6 enter forward mapping length = 25 bp
Under point 7 enter reverse mapping length = 15 bp
Under point 8 enter max forward/reverse span = 1000 bp
Click “Start mapping”, which takes about one hour per ten million reads
When all data has been processed, click “Save all” on bottom right to save mapping data files
Click on the “Process” tab, then “Remove single read noise” and “Enforce antisense-overlap reads” for basic noise reduction and off-target site identification
Click “Export peak list” to save a list of detected cleavage sites, which can be opened in a text or spreadsheet editor for further analysis
For more complex analyses (such as gRNA multiplexing or indel distribution prediction), refer to the Read Me on the Github repository available at https://github.com/schmidburgk/ttiss