

Abstract
Monobodies, built with the scaffold of the fibronectin type III domain, are among the most well-established synthetic binding proteins. They promote crystallization of challenging molecular systems. They have strong tendency to bind to functional sites and thus serve as drug-like molecules that perturb the biological functions of their targets. Monobodies lack disulfide bonds and thus they are particularly suited as genetically encoded reagents to be used intracellularly. This article reviews recent monobody-enabled studies that reveal new structures, molecular mechanisms and potential therapeutic opportunities. A systematic analysis of the crystal structures of monobody-target complexes suggest important attributes that make monobodies effective crystallization chaperones.
Introduction
Target-binding proteins, including antibodies and synthetic binding proteins, are powerful and integral tools in biology and medicine. These binding proteins, or “binders” for brevity, are particularly useful in two ways: First, as tools for perturbing biological function; and second, as chaperones that promote productive crystallization. Structural biology and binder development are strongly synergistic. Binders help harvesting ‘high-hanging fruit’ systems for structure determination. In addition, atomic structures of the binder-target complexes help elucidate molecular mechanisms underlying target recognition and inform further development of binder technology [1]. Since one of us published an early review of recombinant crystallization chaperones ten years ago [2], numerous crystal structures of binder-target complexes have been reported. We will focus this review on structural and mechanistic studies enabled by monobodies.
Monobodies are synthetic proteins built using the tenth fibronectin type III (FN3) domain of human fibronectin as the molecular scaffold. Since the initial report [3], multiple monobody library designs [4–8] and analogous systems (e.g. adnectins [9], centyrins [10], tenascins [11]) have been developed in academia and industry [12]. In terms of structural applications, the monobody system dominates among FN3-based binders: there are 47 PDB entries for monobody-target complexes, whereas only six structures have been reported for the other homologous FN3-based systems.
Recent monobodies are developed from two types of combinatorial phage-display libraries with different diversification patterns (Fig. 1a): one concentrates diversity in three loops that are structurally equivalent to antibody CDRs; the other uses two loops on opposite ends of the FN3 scaffold and the β-sheet surface in between [3,5,6,13,14]. monobodies have produced the most diverse topography among well-established binder systems [12], ranging from convex to concave surfaces, which in turn increases the types of sites (epitopes) to which monobodies can bind. monobodies intended as crystallization chaperons are generally produced without imposing selection bias toward a particular epitope. Additional negative selection steps with a blocking ligand or a mutant decoy can be incorporated to enrich monobodies that bind to a specific site [15–17]. Such site-directed monobodies are particularly effective tools for addressing mechanistic questions. Ultimately monobodies are produced in E. coli and purified, or alternatively can be expressed for functional assays in cultured cells and in animals [18–21].
Figure 1:
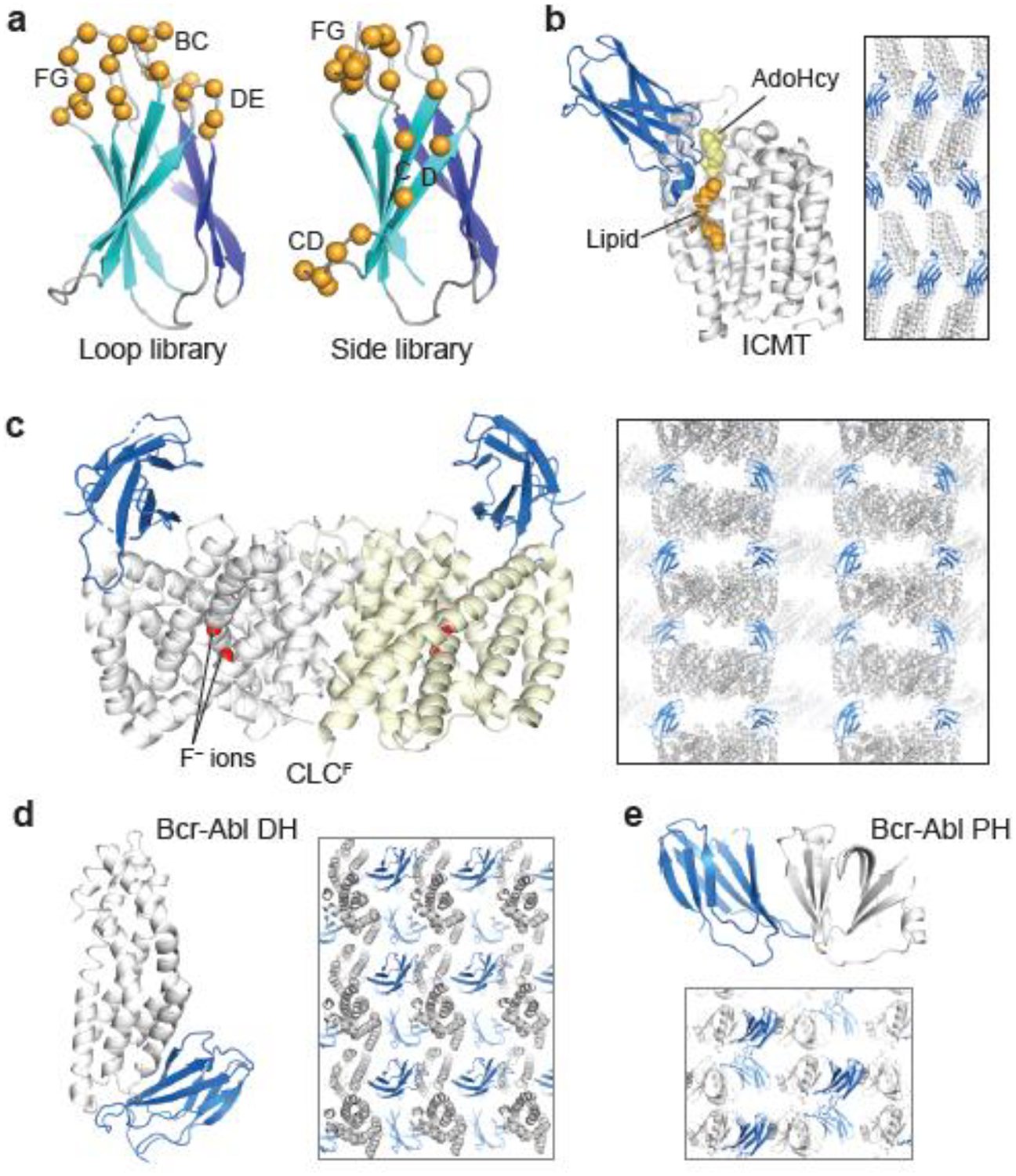
De novo crystal structures enabled with monobody chaperones. (a) Designs of monobody combinatorial libraries. The diversified positions in the Loop and Side libraries are shown with spheres. (b–e) Crystal structures of isoprenylcysteine carboxyl methyltransferase (ICMT), CLCF transporter, Bcr-Abl Dbl-homology (DH) and Pleckstrin-homology (PH) domains. monobodies are shown in blue. Crystal packing is also shown.
Novel structures enabled by monobody chaperones
In the past two years, monobodies served as effective crystallization chaperones for a wide variety of targets and have helped determine several novel structures [20,22–24].
Isoprenylcysteine carboxyl methyltransferase (ICMT) is an integral membrane protein with eight transmembrane α-helices and resides in the endoplasmic reticulum membrane. It is a member of the CAAX family of methyltransferases [25]. After conjugation to a prenyl lipid to the Cys residue of the CAAX motif and removal of the AAX motif by proteolytic cleavage, ICMT methylates the C-terminal prenyl cysteine of RAS and other CAAX motif-containing proteins. This modification is critical for membrane localization and downstream activity of RAS [26]. The monobody-ICMT complex structure was determined in the lipidic-cubic phase at 2.3 Å resolution [27]. ICMT is comprised of an 8-helix bundle, with a pronounced central cleft that binds both the cofactor and substrate, bringing them into close proximity for methylation to occur (Fig. 1b). A 4.0 Å resolution structure of detergent-solubilized ICMT in the absence of a monobody revealed a nearly identical conformation, including the large active site cleft, indicating that monobody binding did not substantially alter the enzyme conformation. The monobody inhibitor occludes substrate binding, thereby helping reveal the catalytic mechanism. In the crystal structure, each monobody molecule contacts three ICMT molecules (the biologically-relevant target and two symmetry mates), bridging between the solvent-exposed loops of ICMT (Fig. 1b).
CLCF transporters are members of the ubiquitous CLC family of anion-transport proteins whose primary function is to export F− from bacterial cells and prevent this ion from reaching toxic levels [28]. Although CLCF is homologous to the extensively studied CLC Cl− transporters, the mechanisms of F− selectivity and ion transport were unknown due to low sequence homology at key positions. The crystal structures of the wild-type and two mutants were determined in complex with the same monobody chaperone (Fig. 1c) at 3.0 Å [29]. These structures revealed both similarities and differences between CLCF and Cl− transporters, and identified the basis for ion selectivity. In particular, one mutant structure captured an unprecedented, intracellular-facing conformation of the critical, proton-transferring Glu residue. From these structures and accompanying mutational studies, the authors propose a ‘windmill’ model for antiporter function in which the Glu residue cycles through alternate conformers that are coupled with both the occupancy of F− ions at two sites and Glu protonation. The monobody does not perturb F− transport, and it appears to serve exclusively as a crystallization chaperone that enhances stacking of CLCF homodimers by bridging their solvent-exposed surfaces, resulting in the formation of loosely associated columns in the crystals (Fig. 1c).
Bcr-Abl is a large multi-domain tyrosine kinase whose two isoforms, p210 and p190, are associated with different types of leukemia [30]. The presence or absence of the Dbl-homology (DH) and Pleckstrin-homology (PH) domains constitute the only structural differences between the isoforms. The Bcr-Abl DH domain crystallized only in complex with monobodies, and the structure was determined at 1.65 Å (Fig. 1d) [31]. This study determined the solution NMR structure in parallel, and the two structures together revealed an identical overall fold with only minor variations in helix orientation. This is a rare case in which target structure was determined both from the crystals of a monobody complex and in isolation by NMR spectroscopy. Both models show close agreement of the structures, underlining the notion that monobodies stabilize a low-energy state among the native conformational ensemble [2]. In the same study, the structure of the Bcr-Abl PH domain was determined in complex with a monobody. The authors deleted an unstructured 59-amino acid segment (almost a third of the domain) and employed a monobody to obtain crystals that diffracted to 1.65 Å [31]. The structures of the Bcr-Abl DH and PH domains allowed a thorough functional analysis of these domains, providing a better understand of functional differences between p210 and p190. These structures and monobodies will enable studies on higher order structural organization of full-length Bcr-Abl.
Monobodies promote crystallization by reducing disorder
Crystallization chaperones are thought to promote crystallization by two complementary mechanisms: one that reduces the conformational heterogeneity of the target molecule and the other that increases surfaces that can contribute to productive crystal contacts [2]. The availability of nearly 50 monobody-target complex structures amassed in the last ten years (Supplementary Table 1) allows us to better examine the validity of this view.
Binding of a ligand (e.g. a monobody) generally leads to immobilization of a flexible region in the target into a conformation suitable for binding. A series of monobody-mediated crystal structures of the Fluc F− channel offers a rare direct support for this immobilization effect [24,32]. Fluc is a small integral membrane protein consisting of two subunits that are arranged in an anti-parallel manner, and thus the two solvent-accessible surfaces of the dimer are structurally equivalent. Most of Fluc structures have been determined with two monobody molecules bound to both sides of the Fluc dimer, which show that the monobody-bound Fluc is fairly rigid, with low B factors observed near the binding interface (Fig. 2a). In contrast, a structure in which a single monobody is bound to one subunit of the Fluc dimer [32] shows that solvent-exposed regions distal to the bound monobody have conspicuously high B factors as compared with the monobody-bound side (Fig. 2b). This observation supports the role of monobodies in reducing the conformational heterogeneity of a target molecule, which in turn promotes crystallization. It is also notable that although the use of diverse monobodies resulted in diverse crystal packing modes, the Fluc channel in these structures are very similar.
Figure 2: Monobody-mediated packing interactions.
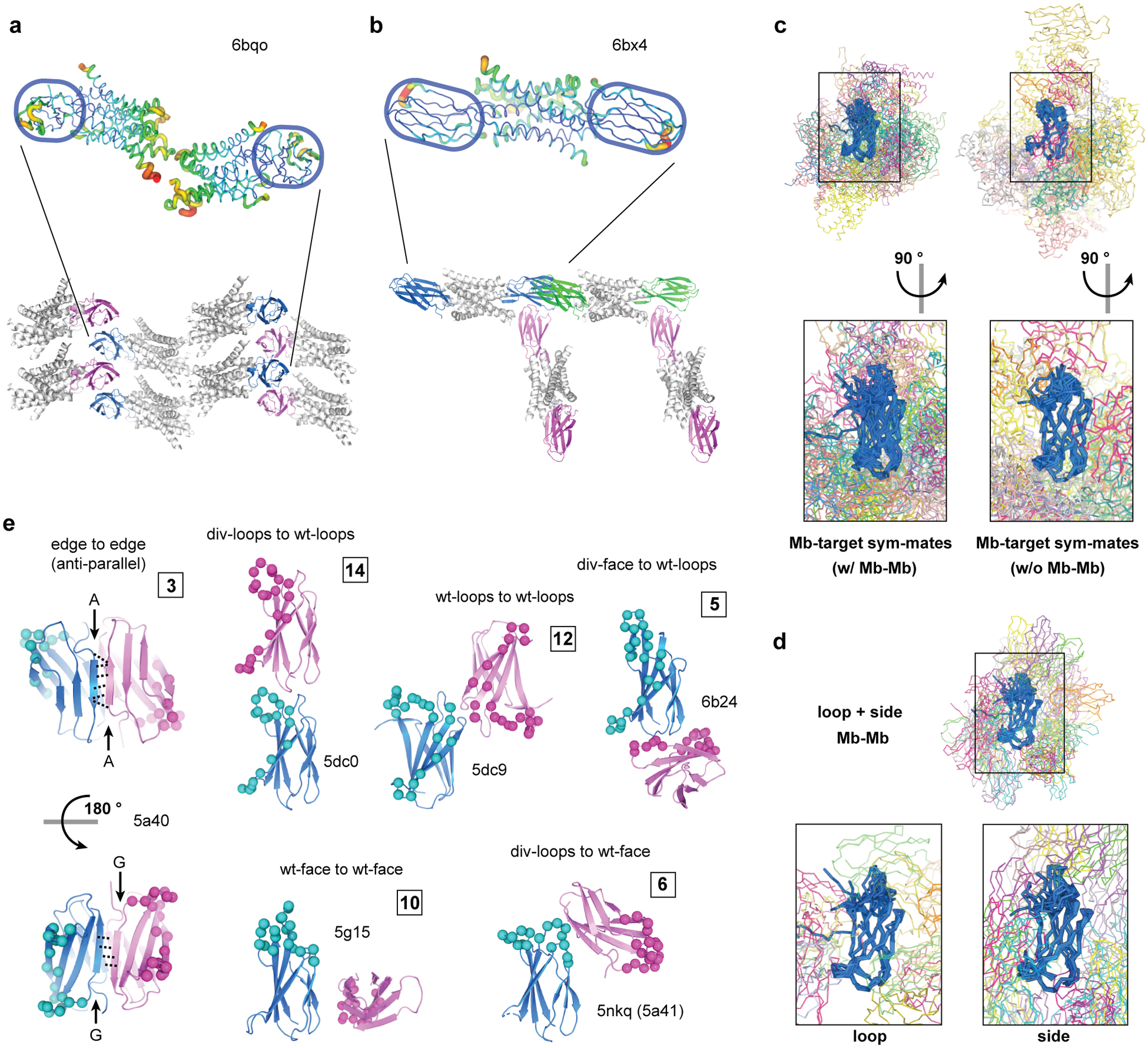
(a and b). Monobody-mediated reduction of disorder in target molecules. The Fluc F− channel Ec2 dimer bound with one monobody chaperone (a) and that bound by two copies of a different monobody (b). The cartoons are colored based on the B factor (top) and the packing modes are shown with monobodies in blue, magenta, and green (bottom). (c) Crystal packing interactions between monobodies and their target molecule. Alignment of all monobody-target complexes, subdivided into structures with monobody-monobody contacts and those with no monobody-monobody contacts. The aggregate alignments are shown in the upper panel, with a magnified view rotated 90° from the upper panel shown below. (d) Alignment of monobody-monobody interactions observed in crystal structures of the monobody-target complexes. The aggregate alignment is shown in the upper panel, and a magnified view of these interactions subdivided into the loop and side libraries are shown below. (e) Representative modes of monobody-monobody interactions observed in crystal structures. The reference monobody is shown in blue, and the interacting monobody is shown in magenta. The diversified positions are indicated as spheres. The numbers in the boxes indicate the number of instances in which the indicated modes are observed. Symmetry-mates were generated and contacts were mapped by selecting any residue within 5 Å of the reference monobody in PyMOL. Protein molecules are considered interacting if three or more residues are contacting.
Monobodies binding to Aurora A (AurA) kinase demonstrate their ability to capture and stabilize different states within the conformational ensemble of the target molecule [17]. The primary goal of this study was to develop monobodies that allosterically modulate AurA kinase. A selection strategy was developed to direct monobodies to the AurA allosteric effector site, which included negative selection using a decoy target harboring a mutation in this site. Functional analysis identified activating, inhibiting, and functionally neutral monobodies, although no attempts were made to shift the conformational equilibrium toward the active or inactive states of AurA during monobody library selection and thus to enrich a particular type of allosteric modulators. The crystal structure of AurA in complex with an activating monobody revealed a canonical active kinase conformation similar to that with a natural allosteric activator, TPX2, including the binding of the YxY motif in the allosteric effector site (Fig. 3a). In contrast, AurA in complex with an inhibitory monobody revealed a distorted conformation similar to a previously reported one without an allosteric ligand (Fig. 3a). These findings are consistent with solution studies indicating that both active and inactive states of AurA are substantially populated in the absence of an allosteric modulator [33]. This work furthers the view that monobodies are capable of binding selectively to distinct low energy conformers and thereby reducing the conformational heterogeneity of the target.
Fig. 3. Monobodies that modulate biological functions of their respective targets and their crystal structures.
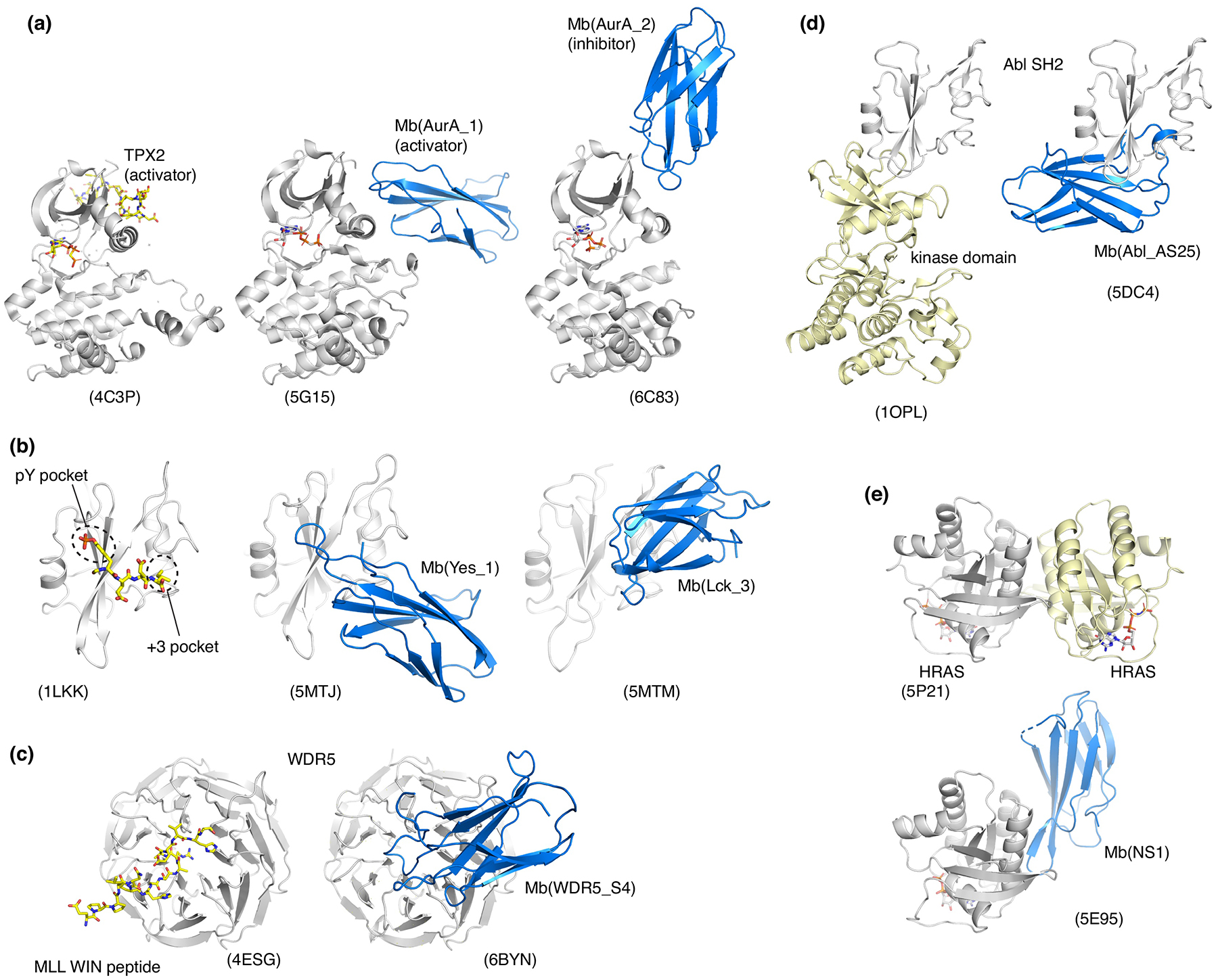
(a) Allosteric activator and inhibitor of Aurora A kinase. (b) Competitive inhibitors of the interaction between SH2 domains and pY peptides. (c) Competitive inhibitor of the interaction between WDR5 and a fragment of MLL1. (d) Allosteric inhibitor targeting the interface between the Bcr-Abl SH2 and kinase domains. (e) Allosteric inhibitor targeting the self-association surface of HRAS.
A previously developed monobody to maltose-binding protein (MBP) [34] was used as a crystallization chaperone of an MBP fusion with DUSP1 phosphatase (the intended target for structure determination). The monobody appears to restrict the flexibility of MBP and provides contacts between MBP and DUSP that reduce the overall flexibility of the fusion protein [35].
Monobodies produce diverse crystal contacts
There are currently 47 structures of monobody-target complexes in the PDB, of which 35 can be considered nonredundant after eliminating virtually identical structures of highly similar complexes. These structures give an opportunity to systematically survey how monobodies promote crystal packing. The nonredundant structures can be further divided in two classes: one in which each monobody contacts only its target through the cognate (engineered) interaction and fortuitous (crystal) contacts (15 entries), and the other in which monobody-monobody contacts are present (20 entries). These crystal structures show a diverse set of packing interactions between monobodies and their targets (Fig. 2c), as well as between adjacent monobody molecules (Fig. 2d, Supplementary Fig. 1 and 2). As expected, the diversified residues of the monobodies, particularly the FG loop that is both the longest and the most extensively diversified, are largely sequestered within the monobody-target interface and do not extensively engage in crystal contacts (Fig. 2c and d). Consequently, invariant positions of the monobody framework dominantly contribute to forming crystal contacts. Each monobody contacts up to three adjacent monobodies in the crystal lattice. There are 12 potential modes of monobody-monobody interactions designated by which portion(s) of the monobody molecules are involved in crystal packing, as tabulated in Supplementary Fig. 1b, of which we observed nine modes (Fig. 2e, Supplementary Fig. 1, and Supplementary Table 1). Contacts between monobody loops and/or β-sheet surfaces distant from the target-binding interface predominate in crystal contacts, as expected. Although the β-sandwich structure of the FN3 scaffold has a total of four exposed β-sheet edges, strand-strand pairings resulting in the formation of an intermolecular β-sheet are observed only in three structures (Supplementary Fig. 1 and 2d). The paucity of these strand-strand interactions may reflect the distortion of β-sheet edges, a negative design feature that prevents self-association and is commonly observed among natural β-sheet proteins including FN3 [36]. In summary, the monobody scaffold has the capacity to form highly diverse types of crystal contacts, thereby promoting crystal formation.
In addition to the use of diverse monobody surfaces for crystal packing, the diversity in target engagement seems to contribute to the formation of diverse crystal contacts. Src homology 2 (SH2) domains are the largest class of proteins for which monobodies have been developed. SH2 domains are modular protein-protein interaction domains that recognize tyrosine-phosphorylated peptides through two adjacent binding pockets: one binds the phosphotyrosine (pY) side chain, and the other recognizes the third residue downstream of the pY residue that dictates sequence selectivity [37,38]. Monobodies targeting nine human SH2 domains have been developed [6,16,21,39]. Strikingly, most of these monobodies robustly compete with pY peptide binding to the SH2 domain, although no selective pressure favoring binding to the peptide-binding cleft was applied. This observation highlights the strong tendency of monobodies to target functional surfaces involved in protein-protein or protein-ligand interactions [12]. A total of 6 monobody-SH2 complexes revealed at least three different modes by which monobodies engage an SH2 domain and inhibit pY-ligand binding: (i) the FG loop mimics the canonical mode of the pY-SH2 interaction with a tyrosine residue and an inorganic sulfate or phosphate in the pY pocket (PDB entries 3K2M and 5MTJ) [6,39], (ii) their CD loops block the +3 specificity pocket, but with otherwise little contacts to the pY peptide binding groove (5MTM and 5MTN) [39] (Fig. 3b), (iii) a β-strand of monobodies runs in opposite orientation from a canonical pY-peptide (4JE4 and 4JEG) [21]. These distinct binding modes of SH2-targeting monobodies further supports their ability to present diverse topography for target recognition [12] and clearly eliminate the possibility that the monobody scaffold is predisposed to binding to SH2 domains. Different monobodies can produce distinct monobody-target orientations, which in turn present distinct monobody surfaces available for crystal contacts, as also seen for the Fluc F− channel [24,32].
Monobodies as tools for advancing mechanistic insights and drug discovery
Monobodies are increasingly used as ligands for controlling biological functions. Several attributes make monobodies ideally suited as a platform to develop drug-like molecules that can also be used as genetically encoded tools. As discussed above, monobodies have strong tendency to bind to a functional site in a target [12]. The ability of monobodies to achieve high specificity suitable for cellular studies has been consistently demonstrated. For instance, despite the presence of closely related 122 SH2 domains in the human proteome, SH2-binding monobodies are highly selective to a single or a few members, as validated in proteome-wide binding assays [6,21,39]. The overall larger interaction surface as compared with pY-peptide ligands and small molecule inhibitors enable monobodies to recognize regions outside the highly conserved binding groove, thus rationalizing their outstanding selectivity. Because monobodies have no Cys residues and their integrity does not rely on disulfide bonds, monobodies can readily be expressed in intracellular compartments with a reducing environment. Moreover, the capability of monobodies as crystallization chaperones facilitates structural studies, which in turn advances both a mechanistic understanding of the biological process being studied and further improvement via structure-guided design.
Distinct modes of action have been observed among monobodies that modulate functions of their targets. The first mode is competitive inhibition. This class included monobody inhibitors of pY-SH2 interactions in oncogenic tyrosine kinases Abl and Src, as well as the SHP-2 tyrosine phosphatase [6,21,39] (Fig. 3b), monobody targeting the peptide-binding pocket of WDR5 essential for the formation of the mixed lineage leukemia (MLL) methyltranferase complex [18] (Fig 3c), and monobody that target the protein-protein interaction interface of PRDM14, an epigenetic regulator important for stem cell development [20].
The second class is allosteric modulation by targeting domain interfaces, an approach pioneered by monobodies that disrupt the interface between the SH2 and kinase domains in Bcr-Abl. The SH2-kinase interaction is necessary for leukemogenesis, and intracellular expression of these monobodies suppressed leukemia cell survival and oncogenic transformation (Fig. 3d) [15,16]. A monobody targeting RAS also falls in this class. RAS and its oncogenic mutants remain among the most challenging drug targets [40]. Unbiased selection identified a monobody termed NS1 that binds to KRAS and HRAS in an unprecedented manner in that the monobody is insensitive to the activation (GTP/GDP)-state of RAS [41]. The crystal structure revealed that NS1 bound to a previously uncharacterized ‘α4/ α5’ region located on the opposite side of RAS from the well-known switch regions important for signaling (Fig. 3e). NS1 does not inhibit known interactors of RAS, including GAP, GEF and other downstream effectors. Surprisingly, NS1 is nevertheless a potent inhibitor of RAS-mediated signaling and oncogenesis when expressed intracellularly in both cell-based and mouse xenograft studies [19,41]. These and additional results support a model that NS1 disrupts a signaling complex consisting of two RAS molecules and two RAF kinase molecules by inhibiting RAS self-association.
Although both Bcr-Abl and RAS are among the most intensely studied oncogenes, monobody-enabled studies of these systems reveal novel vulnerabilities that could be therapeutically exploited and that provide important insights into their cellular functions.
The third class is allosteric modulation of the target conformation in a classical sense, as seen in AurA kinase, described above (Fig. 3a).
Conclusions
Recent studies further highlight the effectiveness of integrated utilization of monobodies both as crystallization chaperones and as drug-like molecules for functional studies in cultured cells and animal models. This approach advances mechanistic studies on diverse proteins, while also identifying and validating drug binding sites on prospective therapeutic targets. We expect that future studies will further increase the impact of monobody-enabled investigations.
Supplementary Material
Acknowledgements
This work was supported by the European Research Council (Grant ERC-2016-CoG 682311-ONCOINTRABODY) and within the scope of the National Research Programme National Center of Competence in Research (NCCR) in Chemical Biology, which was funded by the Swiss National Science Foundation (OH) and by the National Institutes of Health Grants R01 CA194864 and R01 CA212608 (SK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Gilbreth RN, Koide S: Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr Opin Struct Biol 2012, 22:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koide S: Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol 2009, 19:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koide A, Bailey CW, Huang X, Koide S: The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol 1998, 284:1141–1151. [DOI] [PubMed] [Google Scholar]
- 4.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S: High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A 2007, 104:6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S: Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. Journal of molecular biology 2012, 415:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S: A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat Struct Mol Biol 2010, 17:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hackel BJ, Kapila A, Wittrup KD: Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol 2008, 381:1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackel BJ, Wittrup KD: The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein engineering, design & selection : PEDS 2010, 23:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipovsek D: Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel 2011, 24:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diem MD, Hyun L, Yi F, Hippensteel R, Kuhar E, Lowenstein C, Swift EJ, O’Neil KT, Jacobs SA: Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng Des Sel 2014, 27:419–429. [DOI] [PubMed] [Google Scholar]
- 11.Oganesyan V, Ferguson A, Grinberg L, Wang L, Phipps S, Chacko B, Drabic S, Thisted T, Baca M: Fibronectin type III domains engineered to bind CD40L: cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of two complexes. Acta Crystallogr Sect F Struct Biol Cryst Commun 2013, 69:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sha F, Salzman G, Gupta A, Koide S: Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci 2017, 26:910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S: Teaching an Old Scaffold New Tricks: Monobodies Constructed Using Alternative Surfaces of the FN3 Scaffold. J Mol Biol 2012, 415:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koide S, Koide A, Lipovsek D: Target-Binding Proteins Based on the 10th Human Fibronectin Type III Domain ((10)Fn3). Methods Enzymol 2012, 503:135–156. [DOI] [PubMed] [Google Scholar]
- 15.Grebien F, Hantschel O, Wojcik J, Kaupe I, Kovacic B, Wyrzucki AM, Gish GD, Cerny-Reiterer S, Koide A, Beug H, et al. : Targeting the SH2-Kinase Interface in Bcr-Abl Inhibits Leukemogenesis. Cell 2011, 147:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wojcik J, Lamontanara AJ, Grabe G, Koide A, Akin L, Gerig B, Hantschel O, Koide S: Allosteric Inhibition of Bcr-Abl Kinase by High Affinity Monobody Inhibitors Directed to the Src Homology 2 (SH2)-Kinase Interface. J Biol Chem 2016, 291:8836–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zorba A, Nguyen V, Koide A, Hoemberger M, Zheng Y, Kutter S, Kim C, Koide S, Kern D: Allosteric modulation of a human protein kinase with monobodies. Proc Natl Acad Sci U S A 2019, 116:13937–13942. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors identified activating, inhibiting, and functionally neutral monobodies that bind to the allosteric effector site of Aurora A kinase. Together, these monobodies modulate kinase activity over 3,000-fold between activated and inhibited states. The crystal structures provide starting points for the design of selective modulators of this important kinase.
- 18.Gupta A, Xu J, Lee S, Tsai ST, Zhou B, Kurosawa K, Werner MS, Koide A, Ruthenburg AJ, Dou Y, et al. : Facile target validation in an animal model with intracellularly expressed monobodies. Nat Chem Biol 2018, 14:895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The first characterization of a monobody targeting WDR5, a core component of the mixed lineage leukemia 1 (MLL1) methyltransferase complex whose mutations are associated with acute leukemia. The authors demonstrate the utility of genetically encoded monobodies as target validation tools in animal models (see also ref 19).
- 19.Khan I, Spencer-Smith R, O’Bryan JP: Targeting the alpha4-alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 2019, 38:2984–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nady N, Gupta A, Ma Z, Swigut T, Koide A, Koide S, Wysocka J: ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife 2015, 4:e10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sha F, Gencer EB, Georgeon S, Koide A, Yasui N, Koide S, Hantschel O: Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proceedings of the National Academy of Sciences of the United States of America 2013, 110:14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu M, Symersky J, Radchenko M, Koide A, Guo Y, Nie R, Koide S: Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci U S A 2013, 110:2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salzman GS, Ackerman SD, Ding C, Koide A, Leon K, Luo R, Stoveken HM, Fernandez CG, Tall GG, Piao X, et al. : Structural Basis for Regulation of GPR56/ADGRG1 by Its Alternatively Spliced Extracellular Domains. Neuron 2016, 91:1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stockbridge RB, Kolmakova-Partensky L, Shane T, Koide A, Koide S, Miller C, Newstead S: Crystal structures of a double-barrelled fluoride ion channel. Nature 2015, 525:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S, Philips MR: Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem 1998, 273:15030–15034. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Casey PJ: Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 2016, 17:110–122. [DOI] [PubMed] [Google Scholar]
- 27.Diver MM, Pedi L, Koide A, Koide S, Long SB: Atomic structure of the eukaryotic intramembrane RAS methyltransferase ICMT. Nature 2018, 553:526–529. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This paper provides the first structural insight into the integral membrane enzyme critical for post-translational modification of Ras and other CAAX proteins. A monobody served as a crystallization chaperone as well as an inhibitor of enzyme activity.
- 28.Stockbridge RB, Lim HH, Otten R, Williams C, Shane T, Weinberg Z, Miller C: Fluoride resistance and transport by riboswitch-controlled CLC antiporters. Proc Natl Acad Sci U S A 2012, 109:15289–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Last NB, Stockbridge RB, Wilson AE, Shane T, Kolmakova-Partensky L, Koide A, Koide S, Miller C: A CLC-type F(−)/H(+) antiporter in ion-swapped conformations. Nat Struct Mol Biol 2018, 25:601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reckel S, Hamelin R, Georgeon S, Armand F, Jolliet Q, Chiappe D, Moniatte M, Hantschel O: Differential signaling networks of Bcr-Abl p210 and p190 kinases in leukemia cells defined by functional proteomics. Leukemia 2017, 31:1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reckel S, Gehin C, Tardivon D, Georgeon S, Kukenshoner T, Lohr F, Koide A, Buchner L, Panjkovich A, Reynaud A, et al. : Structural and functional dissection of the DH and PH domains of oncogenic Bcr-Abl tyrosine kinase. Nat Commun 2017, 8:2101. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This paper reports the structure determination of the two previously uncharacterized domains of the Bcr-Abl oncoprotein in complex with monobodies, which together provide important functional insights into Bcr-Abl localization and signaling.
- 32.McIlwain BC, Newstead S, Stockbridge RB: Cork-in-Bottle Occlusion of Fluoride Ion Channels by Crystallization Chaperones. Structure 2018, 26:635–639 e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pitsawong W, Buosi V, Otten R, Agafonov RV, Zorba A, Kern N, Kutter S, Kern G, Padua RA, Meniche X, et al. : Dynamics of human protein kinase Aurora A linked to drug selectivity. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S: A dominant conformational role for amino acid diversity in minimalist protein-protein interfaces. J Mol Biol 2008, 381:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gumpena R, Lountos GT, Raran-Kurussi S, Tropea JE, Cherry S, Waugh DS: Crystal structure of the human dual specificity phosphatase 1 catalytic domain. Protein Sci 2018, 27:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richardson JS, Richardson DC: Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A 2002, 99:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu BA, Jablonowski K, Raina M, Arce M, Pawson T, Nash PD: The human and mouse complement of SH2 domain proteins-establishing the boundaries of phosphotyrosine signaling. Mol Cell 2006, 22:851–868. [DOI] [PubMed] [Google Scholar]
- 38.Waksman G, Kuriyan J: Structure and specificity of the SH2 domain. Cell 2004, 116:S45–48, 43 p following S48. [DOI] [PubMed] [Google Scholar]
- 39.Kukenshoner T, Schmit NE, Bouda E, Sha F, Pojer F, Koide A, Seeliger M, Koide S, Hantschel O: Selective Targeting of SH2 Domain-Phosphotyrosine Interactions of Src Family Tyrosine Kinases with Monobodies. J Mol Biol 2017, 429:1364–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Detailed structural and functional characterization of monobodies targeting the SH2 domains of six Src kinases in a highly selective manner, allowing perturbation studies of active and inactive Src kinase conformations.
- 40.Simanshu DK, Nissley DV, McCormick F: RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, et al. : Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol 2017, 13:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This seminal paper describes the development of the NS1 monobody that binds to a previously uncharacterized α4-α5 surface of H- and K-RAS, resulting in allosteric inhibition of RAS-mediated signaling and transformation in cell lines. These results strengthen the importance of RAS self-association through the α4-α5 surface as a critical step in RAS-mediated signaling.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.