

Abstract
Long-range sequencing information is required for haplotype phasing, de novo assembly, and structural variation detection. Current long-read sequencing technologies can provide valuable long-range information but at a high cost with low accuracy and high DNA input requirements. We have developed a single-tube Transposase Enzyme Linked Long-read Sequencing (TELL-seq) technology, which enables a low-cost, high-accuracy, and high-throughput short-read second-generation sequencer to generate over 100 kb of long-range sequencing information with as little as 0.1 ng input material. In a PCR tube, millions of clonally barcoded beads are used to uniquely barcode long DNA molecules in an open bulk reaction without dilution and compartmentation. The barcoded linked-reads are used to successfully assemble genomes ranging from microbes to human. These linked-reads also generate megabase-long phased blocks and provide a cost-effective tool for detecting structural variants in a genome, which are important to identify compound heterozygosity in recessive Mendelian diseases and discover genetic drivers and diagnostic biomarkers in cancers.
Many second-generation sequencing technologies, which sequence thousands to millions of templates at once, have been developed since 2005 (Margulies et al. 2005; Bentley et al. 2008; Valouev et al. 2008; Rothberg et al. 2011). They can generate terabases of highly accurate sequencing output in a run and are used widely in laboratories today. However, their short read length (150–600 bp) limits their ability to resolve haplotypes, assemble complex genomes, and detect structural variants. Third-generation sequencing platforms that use single-molecule sequencing and promise long-read sequencing capability have also been on the market for nearly a decade, such as SMRT-sequencing (Eid et al. 2009) and nanopore sequencing (Loman et al. 2015). However, they still yield lower sequencing accuracy at higher sequencing cost than second-generation platforms and require micrograms of DNA for library construction, which is very challenging to obtain for many real-world samples or applications.
One (sequencing) system for all (applications) is the desire of the customer but requires a significant breakthrough of an enabling technology on either second- or third-generation sequencing platforms. In the past decade, numerous methods have been developed to capture long-range information with short sequencing reads, including mate-pair (Korbel et al. 2007; Levy et al. 2007), clonal-barcoding methods (e.g., synthetic long reads [Peters et al. 2012; Voskoboynik et al. 2013; Bankevich and Pevzner 2016] and linked-reads [Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019]), and Hi-C (Burton et al. 2013). Of these, clonal-barcoding library technologies (Peters et al. 2012; Voskoboynik et al. 2013; Bankevich and Pevzner 2016; Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019) showed the most promising results to bring routine long-read capability to second-generation platforms. The general concept underlining these clonal-barcoding technologies is to uniquely label subfragments of a long genomic DNA fragment with a common barcode sequence when breaking the long fragment into small subfragments, which later become the inserts of a sequencing library. These methods can be classified into two categories: synthetic long reads (SLRs) and linked-reads. SLR methods, for example, LFR (Peters et al. 2012), LRseq (Voskoboynik et al. 2013), and TSLR (Bankevich and Pevzner 2016), in principle have the ability to assemble those small subfragments back into their original long fragment with the barcode information but only work for limited fragment lengths (up to 10 kb) and face technical challenges and expensive operational costs to scale up for large samples. Linked-reads, sometimes called cloud reads or sparse SLR, have limited potential to re-assemble barcode reads from subfragments back into their original long fragment completely but have been demonstrated to successfully phase the haplotypes, assemble de novo genomes, and detect structural variants with significantly easier sample preparation (Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019). Adoption of current linked-read methods is constrained by their dependency on costly instruments for droplet partition (Zheng et al. 2016), complicated workflows (Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019), limitations on genome size (Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019), or incompatibility with widely used sequencing platforms (Wang et al. 2019).
To enable a routine linked-read sequencing protocol on a second-generation sequencer in all laboratories for all users, we developed an ultralow-input single-tube linked-read library method, Transposase Enzyme Linked Long-read Sequencing (TELL-seq). The TELL-seq method enables barcoding of as little as 0.1 ng of genomic DNA in a single PCR tube with 3-h library construction, without any dedicated specialized instrument.
Results
Clonal barcoding with simultaneous capture and strand transfer reactions
Tn5 and MuA transpososomes have been previously used to simultaneously fragment DNA and introduce adaptors in vitro, creating libraries for second-generation DNA sequencing (Adey et al. 2010; Caruccio 2011). These protocols remove any long-range information when the long DNA molecules are broken into untraceable small fragments. However, when transpososomes attack DNA targets, they form strand transfer complexes (STCs) which are very stable under natural conditions (Surette et al. 1987; Mizuuchi et al. 1992; Savilahti et al. 1995; Au et al. 2004; Amini et al. 2014). Only under harsh conditions, such as heat, protease, or SDS treatment in vitro, will these STCs be disassembled, breaking DNA targets into tagged fragments. This unique feature of the transposition reaction has been used to clonally barcode tagging fragments using the Tn5 system (Zhang et al. 2017; Wang et al. 2019). One method is to anchor the Tn5 transposon and transposase on a barcoded solid bead surface, then react with genomic DNA (Zhang et al. 2017); another method is to have the Tn5 transposon and transposase react with genomic DNA first, then ligate them to barcoded beads (Wang et al. 2019). We tested both methods using a MuA transposition system. However, neither method was efficient (Supplemental Fig. S1). We speculated that when both transposon and transposase were immobilized on the bead surface in the first method, their fixed location and spatial arrangement would restrain the efficiency of capturing the free-floating DNA targets by strand transfer reaction only. The limitation for the latter method was that a DNA target full of STCs after a strand transfer reaction would create significant steric hindrance from the tertiary structure of protein and DNA complex and reduce their chance of being captured onto the bead surface. The TELL-seq method overcame the limitations of both methods with simultaneous strand transfer and transpososome capture reactions in the same solution (Fig. 1A). The barcoding reaction efficiency was significantly improved when both strand transfer and hybridization reactions were dynamically used to capture the DNA targets (Supplemental Fig. S1B). In order to keep the barcoding reaction clonal, we also used nonbarcoded beads as a spacer between clonal barcoded beads and increased the solution viscosity to slow DNA diffusion and keep the beads suspended. The TELL-seq molecular barcode located at the index 1 position in the TELL-seq library (Fig. 1B) was comprised of 18 degenerate nucleotides with a maximum homopolymer length of six bases and over 2.4 billion unique barcodes. This vast barcoding capability enabled the TELL-seq library method to assign one unique barcode to a single DNA target for maximum barcoding resolution.
Figure 1.

Overview of TELL-seq library workflow and structure. (A) Diagram of TELL-seq library preparation procedure. In a 0.2-mL PCR tube, 0.1 ng to 5 ng genomic DNA was mixed with 3–10 million barcoded TELL beads and transpososomes for the clonal barcoding reaction. Genomic DNA fragments were captured on the barcoded TELL beads via connecting strand transfer complexes (STCs) to barcode oligos on the bead surface. A tagging between STCs by a second transpososome introduced a second priming site for library amplification. After breaking the STCs and washing the magnetic TELL beads, sequencing library molecules were amplified off beads with P5 and P7 adaptor sequences incorporated at the same time. The total library procedure took ∼3 h. (B) TELL-seq library structure for Illumina sequencing systems. Index 1 comprises 18-base TELL-seq molecular barcode; Index 2 comprises 8-base barcode for sample indexing.
De novo microbial genome assembly with ultralow input material
We first evaluated the TELL-seq library method for de novo sequencing of microbes. One challenge for microbial sequencing was that the amount of genomic DNA material was often low for fastidious organisms and environmental samples. We developed an ultralow-input TELL-seq protocol, which used 0.1 ng–0.5 ng of genomic DNA from 1-Mb to 50-Mb size microbial genomes for library construction.
In order to effectively use TELL-seq linked-read data for microbial de novo assembly, we also developed a new de novo genome assembler, TuringAssembler, a de Bruijn graph-based assembler that uses linked-read information to perform local assembly and scaffolding in order to produce high-quality assemblies. TuringAssembler worked very well for small genome assemblies under optimal k-mer condition. We compared de novo assembly results of an Escherichia coli K12 MG1655 sample using sequencing data from a standard Illumina fragment library, a TELL-seq Illumina library, and an Oxford Nanopore R10.3 chemistry (Table 1). Three different assemblers, Athena (Bishara et al. 2018), cloudSPAdes (Tolstoganov et al. 2019), and TuringAssembler were used to analyze the same TELL-seq data. Both TuringAssembler and cloudSPAdes were able to assemble the TELL-seq data into one scaffold for the entire E. coli genome. TuringAssembler results from the TELL-seq data were better than the Flye assembly results from the Nanopore R10 sequencing data, with much fewer mismatches and less indel errors. In addition, 1000 ng of genomic DNA was used to construct the library for nanopore sequencing, whereas only 0.5 ng of genomic DNA was used for the TELL-seq library preparation. Athena assembler, which was capable of assembling 10x Genomics linked-reads, did not perform well on the TELL-seq linked-reads with its default setting. Further optimization of the Athena assembler for TELL-seq data might be necessary. The SPAdes assembly from a standard Illumina 2 × 100 paired-end fragment library resulted in short contigs and dozens of scaffolds, which was expected due to its short sequencing read length.
Table 1.
Comparison of de novo assembly results of E. coli K12 MG1655 using sequencing data from Illumina standard fragment library, TELL-seq Illumina library (with different assemblers), and Oxford Nanopore R10.3 chemistry

Escherichia coli DH10B, which contained a 113,260-bp tandem duplication and many insertion sequences not found in E. coli K12 MG1655 (Durfee et al. 2008), was used to further evaluate TELL-seq performance on different amounts of a low-input microbial sample. Either 0.1 ng or 0.5 ng of Escherichia coli DH10B genomic DNA was used with three million barcoded TELL beads for the DNA barcoding reaction, and approximately one million or 0.2 million reacted TELL beads were used for amplification to generate a paired-end library for 2 × 146 paired-end sequencing on an Illumina sequencing system, respectively. With the optimal k-mer condition, we achieved excellent assembly results for both the 0.1-ng- and 0.5-ng-input E. coli DH10B samples (Table 2). Assembly results from the 0.1-ng-input were even better than those from the 0.5-ng-input based on the largest alignment length and the number of misassemblies. This could be due to a lower genomic DNA molecule to barcoded bead ratio in the 0.1-ng-input condition, which in turn decreased the number of different genomic DNA inputs sharing the same barcode and reduced the ambiguity of linked-read clonality during the assembly process.
Table 2.
Summary of de novo assembly results using TuringAssembler on bacterial samples

We sequenced additional bacteria, including those with different GC content in the genome, such as Campylobacter jejuni and Rhodobacter sphaeroides (Table 2). R. sphaeroides, with a 68.8% GC content, was a more challenging genome to sequence and assemble. Its assembly results showed many small contigs with length <5000 bp. The genomic DNA of C. jejuni and R. sphaeroides were purchased and extracted with a standard method having an average length of ∼20 kb, whereas genomic DNA from both strains of E. coli was prepared with a high molecular weight-specific protocol and averaged over 40 kb in length. Calculated DNA molecule length based on the sequencing data (Fig. 2A) confirmed the genomic DNA length differences among these microbial samples. The R. sphaeroides sample had <15% of molecules whose length was >50 kb, whereas all E. coli samples and the C. jejuni sample had more than 54% and 41% of molecules over 50 kb long, respectively. The shorter genomic DNA length of R. sphaeroides may have contributed to the lower assembly performance compared with the other microbial samples. We also calculated per molecule sequencing coverage by TELL-seq linked-reads in these samples. Its average coverage ranged from 10% to 24% among these low-input microbial TELL-seq samples (Fig. 2C).
Figure 2.
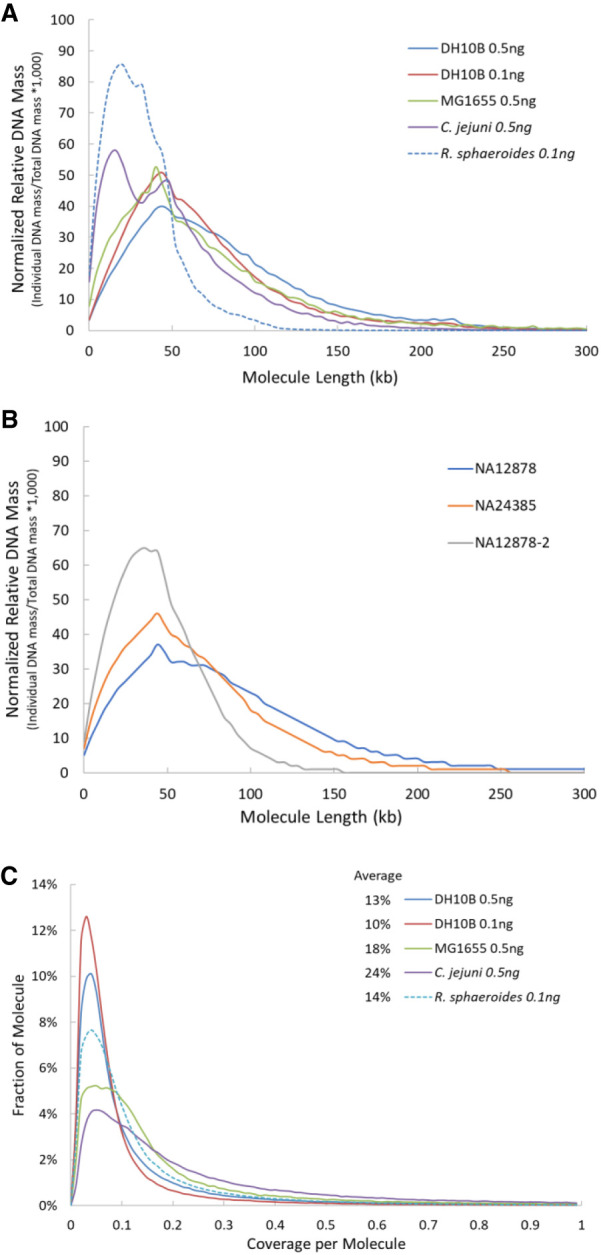
TELL-seq linked-read molecule analyses. (A) Calculated molecule length based on the TELL-seq sequencing data from microbial samples. To compare results from different microbial samples, the calculated DNA mass was normalized as follows: (Individual DNA mass at specified molecule length/Total DNA mass of the microbial sample) × 1000. (B) Calculated molecule length based on the TELL-seq sequencing data from human cell line samples. To compare results from different samples, the calculated DNA mass was normalized as follows: (Individual DNA mass at specified molecule length/Total DNA mass of the cell line sample) × 1000. (C) Distribution of linked-read sequencing coverage per molecule. Average sequencing coverage per molecule was 13%, 10%, 18%, 24%, and 14% for E. coli DH10B (0.5 ng genomic DNA input for library prep), E. coli DH10B (0.1 ng), E. coli K12 MG1655 (0.5 ng), C. jejuni (0.5 ng), and R. sphaeroides (0.1 ng), respectively.
Furthermore, we sequenced and assembled the genome of a human gut microbial isolate, Coprobacillus cateniformis DSM-15921 (Firmicutes), using 0.1 ng genomic DNA. There were seven assemblies previously deposited for this organism in the NCBI database. As expected, most of the contigs generated from our assemblies aligned well to these publicly available assemblies. The best assembly among them was AKCB01, which was a hybrid assembly from Illumina short reads and Pacific Biosciences (PacBio) long reads. In addition, the whole genome shotgun (WGS) assembly CABKQT01 was identical to AKCB01 when comparing their assembly results and was excluded from further evaluation. Our assembly results for DSM-15921 (Supplemental Tables S1, S2) exhibited the longest N50 contig length (3607 kb) among all previously deposited assemblies for this organism and had the highest ratio of largest contig length to total length, 98.4%. To further assess the assembly quality, we ran a BUSCO analysis using Firmicutes as the bacterial lineage (Simão et al. 2015) and identified 221 complete and single-copy BUSCO groups out of 232 expected BUSCO groups with 11 BUSCO groups missing in this lineage (Supplemental Table S3). This 95% complete rate of the DSM-15921 assembly was among the highest of all assemblies. We also noted that the 11 BUSCO groups missing in our assembly were absent in all six deposited assemblies (Supplemental Table S3).
Haplotype phasing human genomes in a single tube
TELL-seq is the first linked-read technology demonstrated for efficient microbial genome sequencing and the first long-range sequencing technology enabling sub-nanogram-input whole genome sequencing. Other linked-read methods have been effectively applied to whole genome haplotype phasing and structural variation detection for human samples (Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019). With over two billion unique barcodes, the TELL-seq technology can be easily used for such applications as well. As a demonstration for such human applications, we applied TELL-seq to two well-characterized Genome in a Bottle (GIAB) consortium samples NA12878 and NA24385. Five nanograms of each input DNA were processed to construct TELL-seq libraries with approximately eight million barcoded TELL beads and sequenced using an S1 flowcell on a NovaSeq 6000 with 2 × 146 paired-end reads. Data were analyzed as described in the Methods section, and results were reported in Table 3. The NovaSeq run generated 1024 million and 959 million cluster reads for NA12878 and NA24385 samples (totaling 1983 million) from the single S1 flowcell run, respectively (Table 3). More than 96% of reads from both samples were mapped to the GRCh38 reference. There were approximately 7.7 million and 7.4 million effective barcodes identified from the two samples, respectively. Over 90% of linked-read molecules had lengths >20 kb, with 20%–30% of linked-read molecules having lengths >100 kb (Fig. 2B). The HapCUT2 tool was used for analysis (Edge et al. 2017), phasing over 99.8% of the heterozygous single nucleotide variants (SNVs) in each sample with low switch error rates and N50 phasing block size >16 Mb (Table 3). The manufacturer's sequencing throughput specification for an S1 flowcell is 1300 million to 1600 million cluster reads with a 200-cycle and a 300-cycle sequencing chemistry available for this flowcell. We subsampled our sequencing reads for each sample down to under 700 million with 2 × 95 read length to simulate a lower loading density on the S1 flowcell run for a 2 × 96 paired-end workflow using a 200-cycle kit. Even with the subsampled read number and shorter read length, we were still able to phase 99.7% heterozygous SNVs for each sample with N50 phasing blocks larger than 7.7 Mb while keeping switch error rates low (Table 3). Phasing results from HapCUT2 outperformed those from Long Ranger, which is also a widely used phasing tool specifically designed for 10x linked-reads. For the same complete NA12878 TELL-seq data set, Long Ranger analysis showed that ∼98.9% of heterozygous SNVs were phased with N50 phasing block size at only 4.2 Mb.
Table 3.
Summary of TELL-seq phasing results on NA12878 and NA24385 samples

Genomic DNA quality played a key role in the phasing application. A two-year-old prep of a NA12878 DNA sample with some noticeable degradation, NA12878-2, was processed and sequenced in the same way as NA12878 and NA24385 samples. Aside from elevated amounts of non-linked-reads, only 4.2% of linked-read molecules were >100 kb (Fig. 2B). With approximately 35× unique sequencing coverage depth, 99.2% of heterozygous SNVs in the NA12878-2 sample were phased with N50 phasing blocks at 1.4 Mb, which clearly underperformed compared with the results of the NA12878 sample. We further examined the coverage uniformity along the GC content for the NA12878 and NA24385 samples (Supplemental Fig. S2). Relatively good coverage uniformity was observed from regions between 20% and 65% GC content. Coverage uniformity declined for very high AT-rich regions (<20% GC) but showed some fluctuation in these two samples. Even more coverage drop was seen in the very high GC-rich regions (>70% GC).
We also checked the variant calling results without incorporating any barcode information on the NA12878 sequencing data and obtained 99.1% recall rate and 98.9% precision rate on SNVs and 89.8% recall rate and 89.3% precision rate for indel variants against the GIAB high-confidence benchmark variant calls with filtering conditions described in the Methods section (Supplemental Table S4).
Phasing major histocompatibility complex (MHC) locus completely
Many diseases have been associated with the MHC region, also referred to as the human leukocyte antigen (HLA) locus, including some autoimmune disorders and infectious diseases (Shiina et al. 2009). In addition, HLA plays a critical role in organ transplantation, where matched HLA alleles of the donor and recipient are required to avoid graft rejection (Choo 2007). However, the MHC region is a particularly challenging genomic region to analyze using standard short-read sequencing technology because it harbors many SNVs, copy number variations (CNVs) and structural variants (Shiina et al. 2009).
We examined the TELL-seq phasing results of the NA12878 sample for the MHC region and identified two complete phasing blocks covering maternal and paternal copies of the entire MHC region. We further analyzed nine well-characterized HLA genes, HLA-A, HLA-B, HLA-C, HLA-DRA, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1, within the MHC region. Six digits allele type for these genes were known for the NA12878 sample. In order to identify the exact nucleotide sequence for comparison with the TELL-seq data, we assigned 01 as the seventh and eighth digits to these genes (Supplemental Table S5) and used the corresponding sequences for these allele types as the “reference data.” All nine genes were phased into a single block for each parental haplotype except for few SNV switch errors in the HLA-A, HLA-DRB1, and HLA-DQA1 genes when compared to the reference (Fig. 3; Table 4). SNVs identified from TELL-seq data against the GRCh38 human genome assembly using the BLASTN program showed good concordance with SNVs identified from the reference data against the GRCh38 assembly except for HLA-DRA and HLA-DRB1 (Table 4). Approximately 90% of SNVs in HLA-DRA and HLA-DRB1 were in noncoding regions. Because there is no precisely validated reference for these two genes, we used 01 allele type as the seventh and eighth digits in the reference of these genes arbitrarily. These two digits defined the variants in the noncoding region specifically. The reference quality may contribute to the low concordance rate of these two genes. Additional optimization on the SNV detection parameters should also improve the variant calling sensitivity and accuracy. Increasing sequencing depth of TELL-seq data should further improve the concordance rate.
Figure 3.

Diagram of phased heterozygous SNVs on nine HLA genes. The major histocompatibility complex region in the NA12878 sample was phased into two complete phasing blocks: one for the maternal haplotype (orange), another for paternal haplotype (blue). Compared with HLA reference on nine well-characterized genes, SNVs with switch error were shown in the opposite color on each haplotype in the HLA-A, HLA-DRB1, and HLA-DQA1 gene.
Table 4.
Summary of TELL-seq phasing results on nine HLA genes in comparison with the reference data

Resolving structural variant call discrepancies
Previous studies have reported 10 structural variants (i.e., deletions) in NA12878 revealed by other linked-read methods (Table 5; Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019). However, there are discrepancies regarding two of these SV calls between the 10x linked-read method (Zheng et al. 2016) and the stLFR method (Wang et al. 2019). With respect to the deletion at Chr 3: 162,512,134–162,626,335, Zheng et al. (10x) reported it as a 114-kb heterozygous deletion (Zheng et al. 2016), whereas Wang et al. (stLFR) claimed it as a 19-kb homozygous deletion (Wang et al. 2019). We used Long Ranger for structural variation detection and Loupe for visualization of TELL-seq data. When looking at these variants manually, our data (Fig. 4A,B) clearly resolved this SV as a small 19-kb homozygous deletion within a larger 114-kb heterozygous deletion, which could explain why other linked-read methods called it differently. Furthermore, we used Loupe to manually check 10x sequencing data of this NA12878 sample and clearly identified the 19-kb homozygous deletion, although it was not automatically called by Long Ranger software. With respect to the deletion at Chr 5: 104,431,113–104,503,673, Zheng et al. identified it as a heterozygous deletion (Zheng et al. 2016), whereas Wang et al. claimed it as a homozygous deletion (Wang et al. 2019). Our data provided support to classify it as a heterozygous deletion (Supplemental Fig. S3A). For this case, a heterozygous deletion call was more reliable than a homozygous deletion call which could be due to the lack of sequencing coverage rather than an actual deletion. We also manually checked and confirmed the other eight SVs in the TELL-seq data as heterozygous deletions (Supplemental Figs. S3–S5), as previously reported by others. However, only five of these eight SVs were automatically called by Long Ranger software, although all deletions were clearly visible when we manually examined the phased reads graph and heat map using the Loupe software.
Table 5.
Comparison of 10 large deletion calls reported by different linked-read methods from 10x, Illumina's CPTv2-seq, and stLFR

Figure 4.

Detection of structural variations in NA12878. (A) Phased read graph from TELL-seq data showed a 19-kb homozygous deletion (Hom) within a 114-kb heterozygous deletion (Het) on Chromosome 3: 162,512,134–162,626,335. For the same location, a 10x study (Zheng et al. 2016) only reported a 114-kb heterozygous deletion, whereas an stLFR study (Wang et al. 2019) only identified a 19-kb homozygous deletion. However, for 10x data, we confirmed the presence of the 19-kb homozygous deletion when we manually examined the data. GRCh38 (hg38) coordinates were used for visualization data. (B) Heat map of the same region from TELL-seq data clearly showed the presence of both the small homozygous deletion and the large heterozygous deletion.
Altogether, for the NA12878 TELL-seq sample, Long Ranger reported 5342 deletions, 338 duplications, and 656 inversions automatically (Supplemental Table S6), of which 1545 deletions, 64 duplications, and 95 inversions overlapped with the set of SVs called by the 1000 Genome Project Consortium for the NA12878 sample, which called 1824 deletions, 211 duplications, and 190 inversions (The 1000 Genomes Project Consortium 2015; Sudmant et al. 2015). Due to different boundary definitions for SV calls between these two data sets, 1600 deletions, 211 duplications, and 190 inversions called by the 1000 Genomes Project Consortium overlapped with the TELL-seq SVs called by Long Ranger. We also used Long Ranger to analyze structural variation in the NA24385 sample, identifying 5198 deletions, 260 duplications, and 450 inversions (Supplemental Table S6), of which 3985 deletions overlapped with deletions calls (total 29,799) made by the GIAB consortium for this NA24385 sample (Zook et al. 2019). For both NA12878 and NA24385 samples, Long Ranger identified over 5000 deletions for each sample; approximately 1800 deletions were reported by the 1000 Genomes Project Consortium for NA12878 and approximately 30,000 deletions were reported by the GIAB consortium for NA24385. As we knew that Long Ranger might significantly undercall the number of SVs in the TELL-seq data at its current default setting, it was very likely that TELL-seq SVs called by Long Ranger for NA12878 and NA24385, and the 1000 Genomes Project Consortium SV calls for NA12878 both underestimated the number of SVs in these samples, although it is not known how many SVs calls for NA24385 in the GIAB data are true positives.
Promising de novo assembly of the human genome
We generated a de novo assembly of NA12878 with the TELL-seq data using Supernova 2.1.1. Although the TELL-seq library insert size averaged 200 bp and was much shorter than the optimal condition required for Supernova (350–400 bp), we used default parameters for the Supernova analysis on reads longer than 125 bp for both R1 and R2. N50 and NA50 scaffold length were 31.5 Mb and 4.3 Mb, respectively. Largest contig and largest alignment lengths were 109.2 Mb and 23.6 Mb, respectively (Supplemental Table S7). In comparison to assembly results using other linked-read methods (Zheng et al. 2016; Zhang et al. 2017; Wang et al. 2019) and nanopore long-read sequencing (Jain et al. 2018), the TELL-seq derived assembly showed longer aligned contig length and at least 28% and 71% fewer misassemblies than other linked-read or nanopore methods, respectively (Supplemental Table S8).
Discussion
Here, we demonstrated TELL-seq as a streamlined linked-read technology for whole genome sequencing of a variety of genomes with different sizes. A major advantage of the TELL-seq method is its low input requirement for library construction which enables long-range sequencing for the first time for many samples with very limited available genomic DNA material. TELL-seq expands linked-read sequencing into small genomes, which was not previously served by the 10x Genomics linked-read method. TELL-seq can assemble a variety of individual microbes. However, many biological microbial samples are not individual microbial isolates. These kinds of metagenomic samples bring new challenges for sequencing technology. It demands long sequencing read length, low-input library construction, and optimized assemblers with minimized chimeric rate across different species. We are exploring TELL-seq for metagenomic applications and expect it to improve gene and genome annotation efficiency over current short-read sequencing methods.
As shown by other existing linked-read technologies, long-range sequencing information generated from a low-input TELL-seq library could be used to effectively phase the whole human genome with bulk genomic DNA isolated from somatic cells directly. TELL-seq library construction in a PCR tube without the expensive instrument required by the droplet-based barcoding method could further increase the adoption of the linked-read method for genome-wide haplotype phasing application. More encouragingly, TELL-seq data could be used to phase the complicated MHC region into completely phased maternal and paternal blocks, as we demonstrated with NA12878 whole genome sequencing data here. Ideally, more sequencing depth for this region would be appreciated to further improve the sensitivity and accuracy of the variant detection. The apparently increased cost for high-depth whole genome sequencing is a bottleneck for its broad implementation. Recently, 10x linked-reads were used for diplotyping of CRISPR-Cas9 captured targeted gene loci, ranging from the 200-kb BRCA1 gene to the 4-Mb MHC locus (Shin et al. 2019). These target sizes fit well with our TELL-seq technology, as our ultralow-input protocol for small bacterial genome sequencing has demonstrated. One of the current challenges for sequencing long contiguous targeted regions is the sample preparation of these ultralong targeted regions. Existing enrichment or capture methods have an on-target capture rate of only 1%–3% (Shin et al. 2019). Significant improvement on the capture rate of these ultralong targets is critical for adoption of targeted diplotyping in the biomedical community.
Long-read and/or long-range sequencing information are essential for SV detection. We demonstrated that TELL-seq linked-read data could be used to detect SVs using analysis tools developed for the 10x linked-read method. However, the current Long Ranger tool could not automatically identify all the SVs in the linked-read data from either 10x or TELL-seq. Among the 11 total deletions including the smaller homozygous deletion within the larger heterozygous deletion region in the NA12878 described earlier, only seven were automatically called out from TELL-seq data by Long Ranger. We could easily confirm the presence of the other four uncalled deletions by manual inspection of phased read data using the Loupe visualization tool. In addition, Long Ranger was developed for 10x linked-reads specifically and has not been optimized for TELL-seq data at all. Due to the short library insert length and different barcoding chemistry of TELL-seq, further fine tuning and data training of Long Ranger for TELL-seq will be necessary. The same should also apply to using Supernova for de novo assembly of TELL-seq data. We are working to optimize these tools internally. At the same time, we are encouraging the sequencing community to develop and optimize other linked-read analysis tools to improve the sensitivity and accuracy of SV detection using linked-read data.
In a standard TELL-seq barcoding reaction, there were, on average, three to five genomic DNA fragments captured on a TELL bead for a microbial sample and six to 10 genomic DNA fragments per TELL bead for a human sample. TELL-seq has a capacity of 2.4 billion unique barcodes. By adjusting the amount of input DNA and the number of TELL beads in a barcoding reaction, the ratio of genomic DNA molecule per TELL bead can be modified easily. When necessary, the TELL-seq technology can assign one unique barcode to each input DNA molecule, which will be critical for application of phasing small targeted genes.
We present an easy-to-use and easy-to-automate single-tube linked-read library method that can cost-effectively generate long-range information from short-read NGS systems. The lengths of many linked molecules generated from sub-nanogram to nanogram input material are over 100 kb. Such long-range information generated from such low input is very difficult to achieve by current commercially available long-read sequencing technologies. With the TELL-seq library technology, a routine linked-read library for whole genome and contiguous targets will become a reality for accurate haplotype-resolved sequencing and de novo sequencing.
Methods
Genomic DNA
Genomic DNA of C. jejuni and R. sphaeroides were purchased from ATCC and used directly without any size selection. Average genomic DNA size of C. jejuni and R. sphaeroides were 28 kb and 20 kb, respectively.
Genomic DNA of E. coli DH10B and K12 MG1655 were extracted using a modified salting-out protocol (Miller et al. 1988) described below. NA12878 and NA24385 DNA were extracted from harvested immortalized human lymphocyte cells GM12878 and GM20847 (Coriell Institute) using the same salting-out protocol, respectively. Briefly, 5 × 106 human/bacterial cells were resuspended in 3 mL of 10 mM Tris, 400 mM NaCl, and 2 mM EDTA at pH 8.0 and lysed by the addition of 0.2 mL 10% SDS and 0.5 mL Proteinase K solution (1 mg/mL Proteinase K [Ambion], 1% SDS, and 2 mM EDTA at pH 8.0). After an overnight incubation at 37°C (12–18 h), the cell lysate was mixed with 1.2 mL of 5 M NaCl and centrifuged at 1100g for 15 min at 4°C. The supernatant was transferred and mixed with 8 mL of 100% ethanol and centrifuged at 8000g for 15 min at 4°C to precipitate the DNA. The pellet was air-dried and resuspended in 50 μL TE. Following an incubation with 20 μg RNase A (Thermo Fisher Scientific) at room temperature for 30 min, genomic DNA was stored at 4°C in a DNA low-bind tube.
C. cateniformis DSM-15921 was obtained from the DSMZ culture collection and grown inside of a vinyl anaerobic chamber (Coy Laboratory Products) with an atmosphere of 3% hydrogen, 10% CO2 and nitrogen as balance. Liquid pure cultures were grown in anoxic BHI broth supplemented with hemin, vitamin K, and L-cysteine. Genomic DNA of strain DSM-15921 was extracted from pure culture using the MagMAX-96 DNA Multi-Sample kit (Applied Biosystems). We followed the manufacturer's instructions for isolating genomic DNA from cultured cells but extracted in 1.5-mL microcentrifuge tubes in place of a 96-well plate and used a Hula Mixer (Thermo Fisher Scientific) in place of a titer plate shaker. For the elution step, we followed instructions for the nonheated shaking option. After extraction, no vortexing was applied and only wide-boar tips were used to facilitate recovery of high molecular weight DNA fragments. We then performed a 0.4× AMPure size-selection and bead clean-up (Beckman Coulter), followed by further size selection with a Short-Read Eliminator (SRE) kit (Circulomics), which removes all fragments <10 kb. Size-selected genomic DNA was stored at 4°C in a DNA low-bind tube.
TELL-seq library construction and sequencing
TELL-seq libraries were constructed using a TELL-seq WGS Library Prep kit (Universal Sequencing Technology). Briefly, 0.1 ng or 0.5 ng genomic DNA from microbial samples or 5 ng genomic DNA from human samples were used with approximately 3 million or 8 million TELL beads for the barcoding reaction in a 0.2 mL PCR tube according to the manufacturer's protocol, respectively. TELL beads are 3-μm magnetic beads, each having at least one unique barcode sequence conjugated on the surface. There are approximately 50,000 copies of total barcode templates on each TELL bead. Among them, ∼1% barcode templates share at least one common barcode sequence with other TELL beads. After the barcoding reaction, TELL beads with captured barcoded DNA were amplified for 13–14 cycles for microbial samples and eight cycles for human samples to produce final sequencing libraries. The high-throughput nature of the reaction allows for construction of multiple libraries in a 96-well format by one person in 4 h. The TELL-seq libraries were quantified by Qubit dsDNA BR Assay kit (Thermo Fisher Scientific) and pooled for sequencing on a MiSeq/NextSeq for microbial samples or a NovaSeq for human samples with 2 × 146 paired-end reads, 18-cycle Index 1 reads and 8-cycle Index 2 reads based on the manufacturer's protocols.
Primary sequencing data processing
The sequenced raw data were first processed by the TELL-Read analysis pipeline software for barcode correction and filtering before proceeding to downstream analysis, such as phasing, variant calling, SV detection, and de novo genome assembly.
After sequencing, raw read BCL files were converted to FASTQ files using bcl2fastq, and adaptor sequences were removed. The FASTQ files were then demultiplexed into Index I1 reads, R1 reads, and R2 reads for each sample based on the Index 2 reads. I1 reads are the TELL-seq barcode sequences. For each sequencing library construction, a set of unique barcode sequences was randomly chosen from a 2.4 billion-barcode pool. After sequencing, all unique barcodes were identified along with the count for the number of reads they were associated with. The unique barcodes associated with only one read were error-corrected if they were 1-base mismatched with one of the barcodes associated with multiple reads. Barcodes with errors after this step were filtered out. The erroneous barcodes along with their associated reads were removed and excluded from the rest of analyses.
De novo assembly of microbial samples
E. coli K12 MG1655 sequencing data from a standard Illumina fragment library were subsampled from a public database (https://www.ebi.ac.uk/ena/data/view/ERA000206&display=html). Out of a total 14,214,324, 5,585,398 read-pairs were used for SPAdes assembly (Bankevich et al. 2012). E. coli K12 MG1655 sequencing data from an Oxford Nanopore R10.3 chemistry was from a public resource (https://figshare.com/articles/Ecoli_K12_MG1655_R10_3_HAC/11823087) and was assembled using Flye assembler v2.5 (Kolmogorov et al. 2019).
TuringAssembler was developed and used for microbial genome assembly of TELL-seq data. It combines paired-end read and linked-read barcode information to perform local assembly in order to resolve local complex regions caused by tandem duplications; linked-read information is also applied for scaffolding the contigs resulting from early assembly steps.
The process that makes TuringAssembler unique is local assembly. For two contigs that are predicted to be consecutive on the genome based on both paired-end reads and barcode information, finding the path between them on the assembly graph is not straightforward because they usually are stitched into a complex region, together with other pairs of contigs. This region usually comprises similar k-mer compositions from many copies of a repetitive sequence in the genome. Rather than ignoring it and filling ambiguous characters between the two consecutive contigs, TuringAssembler attempts to de novo assemble the repeat region between them locally using linked-read information.
First, reads that originated from the start and the end of those contigs along with reads within the gap region between the contigs are isolated using barcode information. We denote the barcode tagged on a read r as b(r). R(x) denotes the set of reads that can be aligned on the region x. The set of barcodes that “span” a region x is denoted as B(x) = {b(r) | r ∈ R(x)}. A long contig C (larger than 4 kb) has two bounded regions Ch, at the beginning (head) of C, and Ct, at the end (tail) of C. Each region Ch/t has the length |Ch/t| = min((|C|/2), 3000). For example, a pair of long contigs C1 and C2 has the representation on the genome as (C1t, C1h, C2t, C2h). The set of barcodes that span C1hor C2t is denoted as Bunion = B(C1h) ∪ B(C2t). The set of barcodes that span both C1hand C2t is denoted as Bshare = B(C1h) ∩ B(C2t). The set of reads that is used to construct the local de Bruijn graph for the region (C1h, C2t) is denoted as Runion = {r | b(r) ∈ Bunion}. Since this approach is still unable to capture short molecules that are located completely in the gap region, in order to keep the graph connected, we use a smaller k-mer size to construct the “local” de Bruijn graph. We next identify a pair of edges E1, E2 in the assembly graph Glocal that represent the sequences of C1h, C2t. The nucleotide sequence that “bridges” C1h and C2t can be represented by a path that starts at E1 and ends at E2 in the assembly graph Glocal. There might be multiple paths like that in Glocal, but the true path must represent the true multiplicity of its edges in the local region (C1h, C2t) and maximize the mapping capability of the local set of reads Rshare = {r | b(r) ∈ Bshare}. We first use Rshare to estimate the coverage of all edges in Glocal, then use the average coverages of E1 and E2 to compute the unit coverage. The multiplicity of each edge in Glocal is the ratio of its coverage to the unit coverage. We use the total number of concordance aligned read-pairs as a score to sort all paths that start at E1 and end at E2, then choose the nucleotide sequence of the best path as the sequence in the local region (C1h, C2t). In case the local region is very complex and we cannot enumerate all paths between E1 and E2, we fill a series of ‘N’ characters in the region (C1h, C2t) where the length of the series equals the shortest path that connects C1hand C2t.
TuringAssembler uses two different k-mer sizes during assembly: a global k-mer size to construct the de Bruijn graph from all input data, and a local k-mer size to construct the de Bruijn graphs in the local regions. The global k-mer size can be inferred from the estimated genome coverage and the read length. It is an odd number close to the estimated genome coverage but not larger than the average read length. The local k-mer size should be smaller than the global k-mer size and may vary between data sets. Usually, multiple k-mer sizes should be tested in order to identify an optimal k-mer combination for a data set.
For assembly completeness evaluation, BUSCO (Simão et al. 2015) was run using the “-m geno” option to indicate that the input was a genome assembly, using the firmicutes_odb9 lineage as a reference.
Variant calling and filtering
Paired-end reads with corrected barcode information were mapped to the reference genome using BWA-MEM (0.7.17-r1188) (Li 2013), and the mapped BAM file was sorted by chromosome coordinates. After duplicate reads were marked and removed with Picard (v2.18.7, http://broadinstitute.github.io/picard) and read group information were added to the BAM file, the germline variants including single nucleotide polymorphisms (SNPs) and small indels between the sample and the reference genome were called using HaplotypeCaller in the GATK tool package (GATK4-4.1.2.0-1) (Van der Auwera et al. 2013). Variants were filtered using the following VCF parameters: QUAL > 15, QUAL < 50, and MQRankSum < 6. SNPs were filtered as those with AD ≥ 2 and AF > 0.15, whereas short indels had AD ≥ 4 and AF > 0.25. These parameters ensure that the reads were mapped to a unique place in the assembly with high quality, that the reads carrying the alleles were sufficient in terms of frequency, depth, and mapping quality (AD, AF, MQRankSum), and that the actual variants were called with high quality (QUAL). For the NA12878 sample, the called and filtered variants were compared with GIAB variant calls (ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/latest/GRCh38/) using the Illumina haplotype VCF comparison tool, hap.py (https://github.com/Illumina/hap.py.git).
Phasing linked-reads using HapCUT2
Linked-reads were phased with HapCUT2 (https://github.com/vibansal/HapCUT2) using heterozygous SNVs that involved two alleles of the same length. The BAM file of diploid variants with duplicates removed was used as input to “extractHAIRS” in the HapCUT2 tool to create the compact fragment file containing only haplotype-relevant information. Linked fragments were generated using the “LinkFragments.py” program in the package. The linked fragments and variants in VCF format were then used as input to “HapCUT2” for phasing.
For assessing the accuracy of the linked-read haplotypes, we used the high-quality phased genotypes for the two individuals, NA12878 and NA24385, from the GIAB project. For NA12878, 99% of the variants were phased using the Platinum Genome pedigree analysis, whereas for the NA24385 genome, 87.0% of the calls were phased using trio analysis. The haplotypes used for benchmarking phasing accuracy should have very few errors (or very high accuracy) since errors in these haplotypes would inflate the long and short switch errors of an independent set of haplotypes assembled using sequence reads and make it difficult to assess the true accuracy. Previous work has shown that using a consensus of haplotypes inferred from two different sequencing technologies improved phasing accuracy (Edge et al. 2017; Chaisson et al. 2019). Therefore, we assembled a consensus of the GIAB haplotypes and 10x Genomics linked-read haplotypes (VCF files downloaded from the GIAB ftp site) by discarding the small fraction of variants that were inconsistently phased between the two sets of haplotypes (Edge and Bansal 2019). These consensus haplotypes were used for calculating short and long switch error rates using scripts in the HapCUT2 software package.
MHC phasing analysis
In order to evaluate the phasing result on highly polymorphic MHC region (28.5 M to 33.5 M on Chromosome 6 in the GRCh38 human genome assembly), a comparison analysis on nine relatively well-characterized HLA genes, HLA-A, HLA-C, HLA-B, HLA-DRA, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1, were performed for the NA12878 sample. Six digits allele type of these genes were known for the NA12878 sample. In order to get the exact nucleotide sequence for comparison with TELL-seq data, we assigned 01 as the seventh and eighth digit to these genes (Supplemental Table S5) and used the corresponding sequences on these allele types as the “reference data.” Briefly, the haplotype allele sequences in FASTA format for these nine genes in the NA12878 sample were downloaded and extracted from ftp://ftp.ebi.ac.uk/pub/databases/ipd/imgt/hla/fasta/ based on the allele type in the Supplemental Table S5. These sequences were aligned to human Chromosome 6 from the GRCh38 assembly by the standalone BLASTN program https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/. The blast result was parsed so that the alignment pieces, which were most likely the origins to hub the alleles, have been determined. Those ambiguous alignments, either weak identities, short alignments, or in different locations and/or strand, were ignored for the process to avoid collecting too much noise for the downstream analyses. Paternal and maternal alleles from NA12878 were mapped to each other using GRCh38 coordinates as the references. This mapping result was used to extract out the heterozygous SNVs in NA12878, which then served as the reference for our comparison process. The heterozygous SNVs detected in TELL-seq from the phasing analysis were compared to these references. The recall and precision rates were calculated based on how many SNVs were overlapped between TELL-seq data and reference data. TELL-seq phasing analysis did not provide any paternal and maternal information on the phased haplotype. During the comparison, the parental origin of each haplotype from TELL-seq was determined based on the parental information from the reference alleles.
Structural variant detection and de novo assembly of the human genome
Structural variation detection and visualization were performed using Long Ranger (v2.2.2) and Loupe (v2.1.1) tools (10x Genomics), respectively. First, TELL-seq data were converted into a 10x-compatible data format. Briefly, all unique TELL-seq 18-base barcodes used in each sequencing library were identified and converted to a whitelist of 10x-compatible (i.e., 16-base) unique barcodes. The mapping was also done in such a way that any two of the resulting barcodes were 2+ Hamming distance away from each other to avoid error correction step in the Long Ranger process. The converted barcodes followed by seven ‘N’ characters were then added to the beginning of each corresponding R1 read in FASTQ format. These converted R1 reads, along with R2 reads, were used as the input for Long Ranger. In addition, Long Ranger's barcode whitelist file, for example, /longranger-2.2.2/longranger-cs/2.2.2/tenkit/lib/python/tenkit/barcodes/4M-with-alts-february-2016.txt, was replaced with a newly created TELL-seq converted barcode whitelist. All subsequent analyses were done following the standard Long Ranger procedure. For the human NA12878 sample, to keep the total unique barcode count <16 million which was the upper limit for the Loupe program, barcodes associated with only one read were removed and Long Ranger v2.2.2 was run with default parameters on the remaining set of 1014 million cluster reads using the GRCh38-2.1.0 reference and the GATK-3.8-0 variant caller. Results of Long Ranger were visualized with Loupe program v2.1.1. Genome coordinate conversion between GRCh38 (hg38) and GRCh37 (hg19) references was done with Lift Genome Annotations (https://genome.ucsc.edu/cgi-bin/hgLiftOver).
The human genome de novo assembly was performed using Supernova (v2.1.1) with default parameters on all reads longer than 125 bp on both PE ends.
Data access
The sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA591637.
TuringAssembler is embedded in the TELL-Link v1.0.2 assembly pipeline. The TELL-Link v1.0.2 assembly pipeline and TELL-seq data to 10x compatible data conversion tool are available freely at https://www.universalsequencing.com/protocol-gate and as Supplemental Code.
Competing interest statement
Z.C., L.P., T.-C.W., G.M., Y.X., P.L.C., D.P., Y.W., and M.L. declare competing financial interests in the form of stock ownership, patent application, or employment through Universal Sequencing Technology Corporation; T.P., H.C., H.T., and S.P. declare competing financial interests in the form of stock ownership or employment through Bioturing, Inc.
Supplementary Material
Acknowledgments
We thank Ivan Tolstoganov for E. coli assembly analysis using SPAdes and cloudSPAdes; Feng Pan for testing data analysis software; Dennis Woods, Kyle LeBlanc, and Alice Tagle for supporting TELL-seq kit production; and Michelle Zhang for assistance with optimizing DNA extraction from DSM-15921. We thank Dr. Wei Wang and the Princeton University Genomics Core Facility for NovaSeq sequencing.
Author contributions: Z.C. conceived the study; Z.C. and M.L. oversaw the technology development; and Z.C. and Y.W. oversaw the analysis pipeline and software development. Z.C., L.P., T.-C.W., and G.M. developed the assays; D.P. helped with the assay development; L.P., T.-C.W., and G.M. performed the experiments; and J.S., P.B.-F., and G.H. prepared the microbial sample. Y.X., P.L.C., and Y.W. developed the data analysis pipeline; Y.X., P.L.C., P.P., Y.W., and Z.C. analyzed the data; T.P., H.C., H.T., and S.P. developed the TuringAssembler; and V.B. performed the phasing analysis. Z.C., R.K., and P.P. led the project coordination. Z.C., T.P., Y.X., P.L.C., Y.W., and R.K. cowrote the paper. All authors contributed to the revision and review of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.260380.119.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119 10.1186/gb-2010-11-12-r119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini S, Pushkarev D, Christiansen L, Kostem E, Royce T, Turk C, Pignatelli N, Adey A, Kitzman JO, Vijayan K, et al. 2014. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat Genetics 46: 1343–1349. 10.1038/ng.3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au TK, Pathania S, Harshey RM. 2004. True reversal of Mu integration. EMBO J 23: 3408–3420. 10.1038/sj.emboj.7600344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Pevzner PA. 2016. TruSPAdes: barcode assembly of TruSeq synthetic long reads. Nat Methods 13: 248–250. 10.1038/nmeth.3737 [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov AS, Lesin V, Nikolenko S, Pham S, Prjibelski A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59. 10.1038/nature07517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishara A, Moss EL, Kolmogorov M, Parada AE, Weng Z, Sidow A, Dekas AE, Batzoglou S, Bhatt AS. 2018. High-quality genome sequences of uncultured microbes by assembly of read clouds. Nat Biotechnol 36: 1067–1075. 10.1038/nbt.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J. 2013. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31: 1119–1125. 10.1038/nbt.2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruccio N. 2011. Preparation of next-generation sequencing libraries using Nextera™ technology: simultaneous DNA fragmentation and adaptor tagging by in vitro transposition. Methods Mol Biol 733: 241–255. 10.1007/978-1-61779-089-8_17 [DOI] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 1784 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo SY. 2007. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med J 48: 11–23. 10.3349/ymj.2007.48.1.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T, Nelson R, Baldwin S, Plunkett G 3rd, Burland V, Mau B, Petrosino JF, Qin X, Muzny DM, Ayele M, et al. 2008. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J of Bacteriology 190: 2597–2606. 10.1128/JB.01695-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge P, Bansal V. 2019. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat Commun 10: 4660 10.1038/s41467-019-12493-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge P, Bafna V, Bansal V. 2017. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27: 801–812. 10.1101/gr.213462.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36: 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner P. 2019. Assembly of long error-prone reads using repeat graphs. Nature Biotech 37: 540–546. 10.1038/s41587-019-0072-8 [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426. 10.1126/science.1149504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254 10.1371/journal.pbio.0050254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]. [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380. 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215 10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi M, Baker TA, Mizuuchi K. 1992. Assembly of the active form of the transposase-Mu DNA complex: a critical control point in Mu transposition. Cell 70: 303–311. 10.1016/0092-8674(92)90104-K [DOI] [PubMed] [Google Scholar]
- Peters BA, Kermani BG, Sparks AB, Alferov O, Hong P, Alexeev A, Jiang Y, Dahl F, Tang YT, Haas J, et al. 2012. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487: 190–195. 10.1038/nature11236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, et al. 2011. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475: 348–352. 10.1038/nature10242 [DOI] [PubMed] [Google Scholar]
- Savilahti H, Rice PA, MiZuuchi K. 1995. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J 14: 4893–4903. 10.1002/j.1460-2075.1995.tb00170.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiina T, Hosomichi K, Inoko H, Kulski JK. 2009. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 54: 15–39. 10.1038/jhg.2008.5 [DOI] [PubMed] [Google Scholar]
- Shin G, Greer SU, Xia LC, Lee H, Zhou J, Boles TC, Ji HP. 2019. Targeted short read sequencing and assembly of re-arrangements and candidate gene loci provide megabase diplotypes. Nucleic Acids Res 47: e115 10.1093/nar/gkz661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Frit MH, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette M, Buch SJ, Chaconas G. 1987. Transpososomes: stable protein–DNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell 49: 253–262. 10.1016/0092-8674(87)90566-6 [DOI] [PubMed] [Google Scholar]
- Tolstoganov I, Bankevich A, Chen Z, Pevzner PA. 2019. cloudSPAdes: assembly of synthetic long reads using de Bruijn graphs. Bioinformatics 35: i61–i70. 10.1093/bioinformatics/btz349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. 2008. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18: 1051–1063. 10.1101/gr.076463.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro M, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit Best Practices pipeline. Curr Protoc Bioinformatics 43: 11.10.1–11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, Passarelli B, Fan HC, Mantalas GL, Palmeri KJ, et al. 2013. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2: e00569 10.7554/eLife.00569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang O, Chin R, Cheng X, Wu MKY, Mao Q, Tang J, Sun Y, Anderson E, Lam HK, Chen D, et al. 2019. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res 29: 798–808. 10.1101/gr.245126.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Christiansen L, Thomas J, Pokholok D, Jackson R, Morrell N, Zhao Y, Wiley M, Welch E, Jaeger E, et al. 2017. Haplotype phasing of whole human genomes using bead-based barcode partitioning in a single tube. Nat Biotechnol 35: 852–857. 10.1038/nbt.3897 [DOI] [PubMed] [Google Scholar]
- Zheng GX, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. 2016. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34: 303–311. 10.1038/nbt.3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, McDaniel J, Olson ND, Wagner J, Parikh H, Heaton H, Irvine SA, Trigg L, Truty R, McLean CY, et al. 2019. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol 37: 561–566. 10.1038/s41587-019-0074-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.