

Abstract
Objectives:
To study the relationship between the change in gut microbial communities in HIV-infected individuals on suppressive antiretroviral therapy (cART), and the peripheral HIV-Gag-specific CD8 T cell responses before and after ex-vivo immune checkpoint blockade (ICB).
Design:
Thirty-four HIV-seropositive, 10 HIV-seronegative and 12 HIV-seropositive receiving fecal microbiota transplant (FMT) participants were included. Gut microbial communities and peripheral and gut negative checkpoint receptors were assessed.
Methods:
Bacterial 16s rRNA sequencing for gut microbiome study and flow-based assays for peripheral and gut negative checkpoint receptor (NCR) and their cognate ligand expression, including peripheral HIV-Gag-specific CD8 T cell responses before and after ex-vivo ICB were performed.
Results:
Fusobacteria abundance was significantly higher in HIV-infected donors compared to uninfected controls. In HIV-infected participants receiving Fusobacteria-free fecal microbiota transplant (FMT), Fusobacteria persisted up to 24 weeks in stool post FMT. PD-1 and TIGIT and their ligands were expanded in mucosal versus peripheral T cells and dendritic cells, respectively. PD-L1 and TIGIT blockade significantly increased the magnitude of peripheral anti-HIV-Gag-specific CD8 T cell responses. Higher gut Fusobacteria abundance was associated with lower magnitude of peripheral IFN-γ+ HIV-Gag-specific CD8 T cell responses following ICB.
Conclusions:
Fusobacteria gut colonization in HIV infection is persistent and may influence anti-HIV T cell immunity to PD-1 or TIGIT blockade. Strategies modulating Fusobacteria colonization may elicit a favorable mucosal immune landscape to enhance the efficacy of ICB for HIV cure.
Keywords: HIV, immune checkpoint blockade, Fusobacteria, TIGIT, PD-L1
Introduction
Reversing T cell exhaustion by targeting negative immune checkpoint receptor (NCR) pathways to improve anti-HIV T cell function is currently under investigation as part of potential “Shock and Kill” HIV cure strategy.[1] We previously reported that blockade of PD-1 and TIGIT pathways synergistically enhances HIV-specific CD8 T cell effector functions.[2] In a humanized mouse model, disrupting the receptor-ligand mediated suppression of PD-1 and Programmed Death-Ligand 1 (PD-L1) reduced HIV viral loads in vivo.[3] In addition, administration of anti-PD-L1 monoclonal antibody (mAb) in a small human clinical trial led to enhanced HIV-specific immunity in a subset of participants.[4] These data are complemented by a report in an HIV-infected individual with lung cancer receiving multiple doses of anti-PD-1 immunotherapy with sustained reduction in the CD4 HIV reservoir and a concomitant increase in HIV-specific CD8 T cell effector capacities.[5] Targeting the NCR pathways has evolved as a promising strategy to enhance pre-existing anti-HIV CD8 T cell responses and viral persistence in HIV remission strategies.
Immune checkpoint blockade (ICB) has altered the management of certain cancers resulting in long-term and durable remission, however, not all individuals are responsive to immunotherapy. Several factors have been reported to impact the efficacy of ICB in cancer, including the levels monocyte HLA-DR expression,[6] impaired antigen processing and presentation,[7] and the epigenetic stability of exhausted T cells [8]. Emerging evidence revealed that the gut microbiome may determine the success of ICB in malignany.[9, 10] Gut Bacteroides was reported to enhance the efficacy of anti-CTLA-4 blockade resulting in the elimination of metastatic melanoma in mice, [11] and in a similar study, tumor-specific CD8 T cells restoration upon PD-L1 blockade and tumor clearance relied on gut Bifidobacterium colonization.[12] The oral commensal, Fusobacteria, was shown to associate with decreased T cell responses in several gut-associated malignancies. Indeed the overabundance of Fusobacteria in the gut has been shown to inhibit immune function through the interaction of its membrane protein, Fap2, with the TIGIT receptor on the surface of the effector immune cells.[13, 14] Additionally, in a subset of cancer patients initially resistant to ICB immunotherapy, tumor growth was halted after receiving a fecal material transplant (FMT) from ICB response donors who were responsive to immunotherapy.[15] Cumulatively these finding provide important insights on the influence of the gut microbiome on ICB to improve anti-HIV CD8 T cells immune responses.
Here we profiled NCR and their ligands in the gut and peripheral compartment in HIV infected donors, assessed mucosal gut microbiomes and quantified peripheral effector HIV-specific CD8 T cell immune responses after ex-vivo ICB to understand the role of the gut microbiome on HIV immunity.
Materials and methods
Human Subjects
For this study, fresh biopsies acquired by colonoscopy or rectosigmoidoscopy and PBMCs were obtained from 2 cohorts: Hawaii (UH) cohort and Washington (UW)cohort through either the University of Washington Center for AIDS Research, University of California San Francisco (UCSF) SCOPE cohort, or the University of Washington AIDS Clinical Trials Unit. Additionally, stool samples were obtained from study participants recruited through the UCSF Fecal Microbiota Transplantation in HIV (FMT-HIV) study before (weeks −4, −2), at the time of (week 0), and after FMT (weeks 1, 2, 4, 8, 24). All study participants gave written consent to participate in the study, and the appropriate Institutional Review Board (IRB) approved all protocols.
Fecal microbiota transplantation
Study participants underwent clinical and stool pre-screening prior to joining the study. Donor screen and supplied was performed by OpenBiome (Somerville, MA). Usage of the donor samples was approved by the FDA. Participants underwent standard bowel purge and stool suspension was introduced via colonoscopy. Stool were collected at timepoints described above. Detailed protocol can be found in Supplementary Methods, Supplemental Digital Content 1.
Isolation of bacterial DNA
For DNA isolation from tissue biopsies, two fresh tissue fragments (50–100mg) were re-suspended and lysed in lysis buffer. Genomic DNA was isolated using QIAmp PowerFecal DNA Kit (Qiagen), followed by DNA yield analysis using Qubit 2.0 fluorometer (Invitrogen). For DNA isolation from stool sample, DNA was extracted using a protocol optimized for the isolation of bacterial DNA from feces.[16]
Bacterial 16s rRNA sequencing
Hypervariable regions in the bacterial 16s rRNA gene were targeted with specific primers followed by appropriate library preparations and subsequent sequencing using either Ion Torrent PGM (Ion Torrent, Life Technologies) or MiSeq (Illumina) platform. Additional details can be found in Supplementary Methods, Supplemental Digital Content 1.
Sequence reads from all cohorts were consolidated followed by metagenomic study. FastQC (Babraham Institute) was used to explore the raw reads and Prinseq, a perl script,[17] was used to filter out reads having mean Phred quality score less than 25. High quality reads were then aligned using Kraken,[18] an ultrafast metagenomics sequence classification tool within Partek Flow Software (Partek Inc.) to assign into operational taxonomy units (OTUs). All FastQ files from all three cohorts were analyzed by the same pipeline.
Isolation of rectal mucosal mononuclear cells from fresh gut biopsy
Nine to twelve endoscope obtained tissue fragments were washed thoroughly with cRPMI (RPMI 1640 medium; (Hyclone, Logan, Utah) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 1% penicillin-streptomycin (Hyclone), 10 mM HEPES (Hyclone), 2 mM L-glutamine (Hyclone) and digested with collagenase II (Sigma-Aldrich, Dorset, United Kingdom) solution, and filtered through a 70μm nylon mesh. Leftover undigested tissue was re-digested with collagenase II solution. RMMCs were washed PBS + 2% FBS (Hyclone) and re-suspended for flow cytometry analysis.
Flow cytometric analysis and ex vivo antigen stimulation with antibody blockade assay
Details about antibodies, flow cytometry analysis, and ex vivo antigen stimulation with antibody blockade assay have been previously described [2], and a summary can be found in the Supplementary Methods, Supplemental Digital Content 1.
Statistical analysis
Measures of central tendency are expressed as medians and interquartile ranges (IQRs; given in the form 25th percentile, 75th percentile). Repeated-measures one-way ANOVA followed by Tukey post hoc test was used for comparing multiple treatment groups. Mann-Whitney U Tests were applied to compare between two independent groups while Wilcoxon signed-rank tests were applied to matched groups, and the Spearman’s rank correlation tests were used to discover the association between two variables. All the analyses were performed using Graphpad release 8.0.2 (Graphpad Software) or SPSS 22.0 (IBM) and a p-value of less than 0.05 were considered statistically significant.
Results
Study participants
We assessed biological samples from two cross-sectional study groups: (i) University of Hawaii (UH) cohort (7 HIV+ and 5 HIV-); (ii) the University of Washington (UW) cohort (27 HIV+ and 5 HIV-), and a longitudinal cohort from the University of California San Francisco (UCSF) (n=12) who received a fecal microbiota transplant (FMT) from one of two HIV-uninfected donors (Table 1). All HIV-infected participants are on stable combination antiretroviral therapy (cART). None of the participants were on immunomodulators, probiotics, and have any malignancies 6 months prior to and at baseline visits. No participant was on antibiotics in the 3 months prior to study enrollment, with the exception of 9 study participants from the FMT study who received antibiotics 5 days prior to FMT (Table 1).
Table 1.
Clinical characteristics
| UH and UW Cohorts | UCSF/FMT Cohort | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | HIV+ | HIV− | HIV+ Vs. HIV− p value | Recipient (n=12) | Donor (n=2) | |||||||||
| UH n = 7 | WA n = 27 | Total n = 34 | UH Vs. WA P value | UH n = 5 | WA n = 5 | Total n = 10 | UH Vs. WA P value | Abx+ n = 9 | Abx− n = 3 | Total n = 12 | Abx + Vs. - p value | |||
| Age, Median (min, max) |
57 (51,78) | 53 (24,64) | 53.5 (24,78) | 0.307 | 53 (50,63) | 52 (51,61) | 52.5 (50,63) | 0.881 | 0.741 | 57 (46,63) | 61 (31,70) | 59 (31,70) | 0.95 | N/A |
| Sex, Male, n (%) | 6 (85.7) | 22 (81.5) | 28 (82.3) | 0.798 | 4 (80) | 4 (80) | 8 (80) | 1.00 | 0.87 | 9 (100) | 3(100) | 12 (100) | - | N/A |
| Sexual Orientation | ||||||||||||||
| MSM, n (%) | 6 (85.7) | 22 (81.5) | 28 (82.3) | 0.798 | 0 (0) | 1 (20) | 1 (10) | 0.317 | <0.0001 | 9 (100) | 3 (100) | 12 (100) | - | N/A |
| WSM, n (%) | 0 (0) | 4 (14.8) | 5 (14.7) | 0.286 | 1 (20) | 1 (20) | 2 (20) | 1.00 | 0.690 | 0 (0) | 0 (0) | 0 (0) | - | N/A |
| MSW, n (%) | 1 (14.3) | 1 (3.7) | 1 (2.9) | 0.295 | 4 (80) | 3 (60) | 7 (70) | 0.513 | <0.0001 | 0 (0) | 0 (0) | 0 (0) | - | N/A |
| Viral load, Undetectable, n (%) | 7 (100) | 27 (100) | 34 (100) | - | N/A | N/A | N/A | - | - | 9 (100) | 3 (100) | 12 (100) | - | N/A |
| CD4 Count, Median (min, max) | 560 (304,938) | 645.5 (179,931) | 635 (179.938) | 0.78 | N/A | N/A | N/A | - | - | 505 (257,714) | 463 (357,835) | 490.5 (257,835) | 0.86 | N/A |
UH: University of Hawaii Cohort
WA: University of Washington Cohort
Abx+: received antibiotics treatment 5 days prior to FMT
Abx-: no antibiotics treatment prior to FMT
Gut Fusobacteria abundance of HIV-infected individuals on cART
Microbial taxonomic distributions between individuals in both HIV-infected and HIV-uninfected groups from both cohorts were diverse, however, we observed a significantly higher abundance in only the Fusobacteria phylum in the HIV-infected group when compared to the HIV-uninfected controls (0.18% vs. 0.006% respectively, p = 0.0011) (Fig 1A; Table 2; see Figure A Supplemental Digital Content 2). No significant difference was observed when study participants were stratified based on age and gender (data not shown). Additionally, we did not observe any difference in the alpha diversity or microbial distribution in the other phyla evaluated (see Figure B-C Supplemental Digital Content 2).
Figure 1.
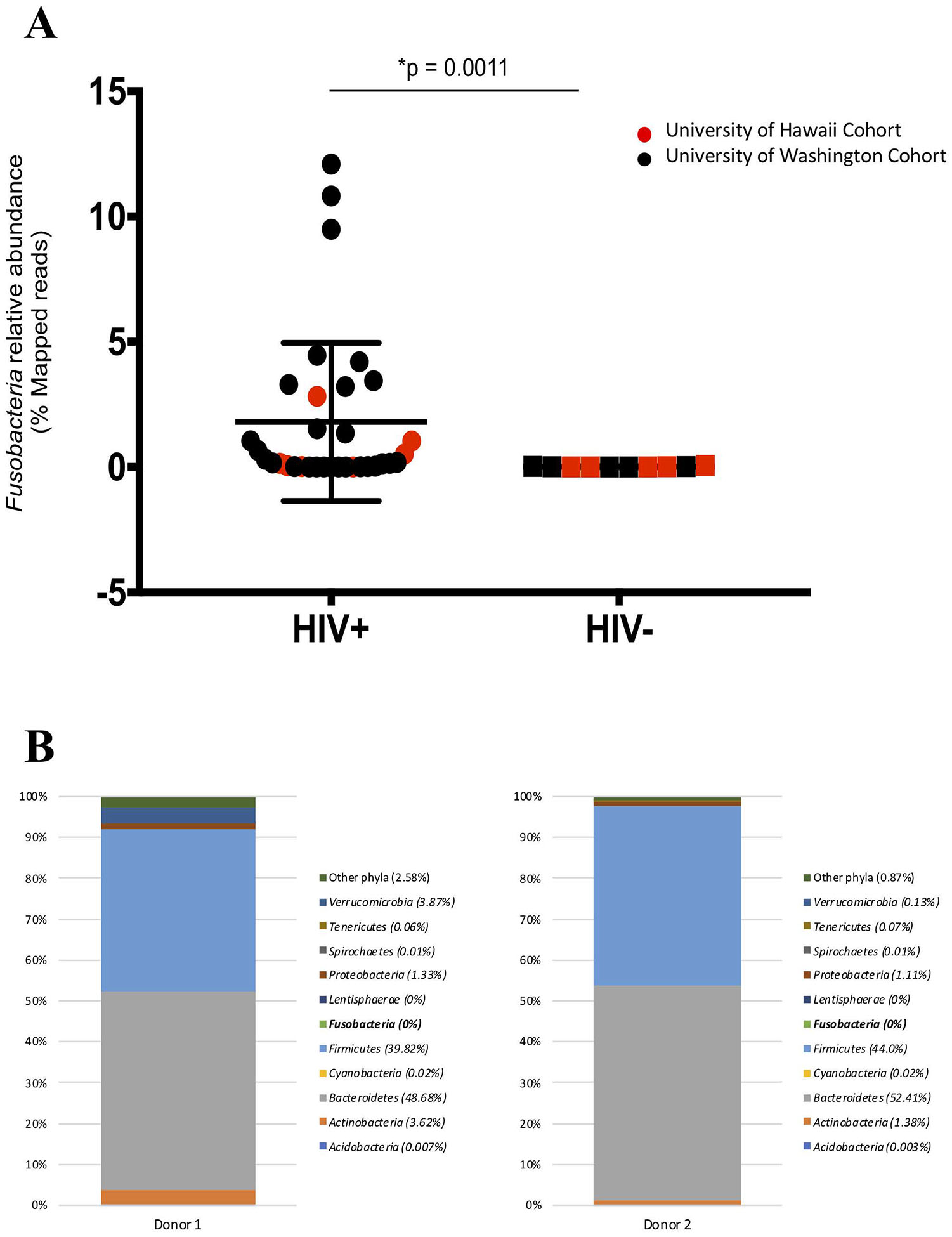
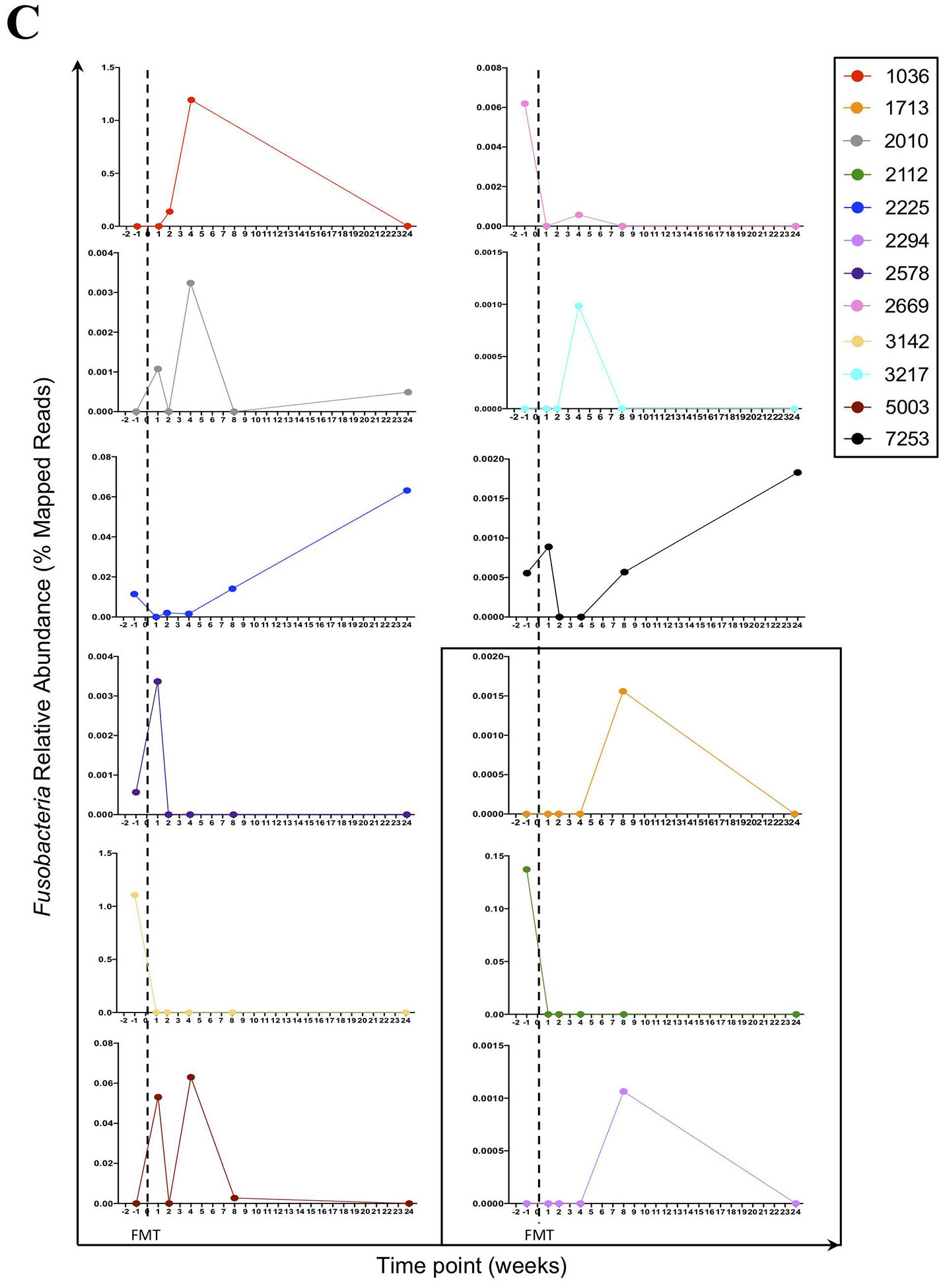
(A) The relative abundance of Fusobacteria is higher in HIV-infected compared to HIV-uninfected individuals. Red represent University of Hawaii (UH) cohort and black represent University of Washington (UW) cohort. Graph show compiled data of Fusobacteria relative abundance in HIV-infected group (n = 34) compared to HIV-uninfected group (n = 10). The p-values were calculated using Wilcoxon matched-pairs signed ranked test for matched pairs and Mann-Whitney U test for group comparison. (B) HIV-uninfected FMT donor Bacterial phyla relative abundance. The distribution of bacterial phyla relative abundance from FMT donor 1 (left) and 2 (right). (C) Fusobacteria was present in the gut of HIV-individuals receiving FMT. Fusobacteria relative abundance in each individual receiving FMT at entry and at week 1, 2, 4, 8 and 24 post FMT. Dashed lines represent the FMT time point. All individuals received antibiotics therapy pre-FMT, except for PID 1713, 2112, and 2294 (boxed).
Table 2.
Gut microbiome phyla relative abundance
| Microbiome | HIV+ | HIV− | p value | ||||
|---|---|---|---|---|---|---|---|
| Median | Min | Max | Median | Min | Max | ||
| Acidobacteria | 0.0000 | 0.0000 | 1.0726 | 0.0025 | 0.0000 | 1.9334 | 0.1815 |
| Actinobacteria | 1.2220 | 0.1938 | 23.8308 | 1.8605 | 0.1631 | 10.2672 | 0.8795 |
| Bacteroidetes | 28.0263 | 3.6643 | 76.4847 | 30.1025 | 2.8395 | 57.6995 | 0.9890 |
| Cyanobacteria | 0.1103 | 0.0000 | 1.9422 | 0.0216 | 0.0000 | 0.4685 | 0.3391 |
| Firmicutes | 40.3029 | 10.4745 | 79.3306 | 45.1707 | 11.7207 | 63.2901 | 0.8578 |
| Fusobacteria | 0.1798 | 0.0008 | 12.0914 | 0.0061 | 0.0007 | 0.0698 | 0.0011 |
| Lentisphaerae | 0.0166 | 0.0000 | 0.9635 | 0.0003 | 0.0000 | 0.5214 | 0.0775 |
| Proteobacteria | 6.8867 | 0.9832 | 67.3723 | 13.1094 | 1.4708 | 72.5461 | 0.6097 |
| Spirochaetes | 0.3573 | 0.0000 | 30.1833 | 0.0768 | 0.0000 | 14.5274 | 0.1417 |
| Tenericutes | 0.0265 | 0.0000 | 1.8604 | 0.0078 | 0.0000 | 2.9573 | 0.8577 |
| Verrucomicrobia | 0.0082 | 0.0003 | 2.5196 | 0.0239 | 0.0006 | 25.8266 | 0.5345 |
| Other phyla | 0.1361 | 0.0000 | 2.1349 | 0.0598 | 0.0142 | 0.4370 | 0.6653 |
| Unclassified | 0.0000 | 0.0000 | 0.2764 | 0.0029 | 0.0000 | 0.3870 | 0.6897 |
In the longitudinal cohort, we observed fluctuations of all relative bacterial abundances pre-FMT and up to 24 weeks post-FMT, and principle component analysis did not show any clear clustering between pre- and post-FMT microbial abundance (see Figure, Supplemental Digital Content 3). Interestingly, while no Fusobacteria was detected from the preparation of fecal bacteria from FMT HIV-uninfected donors (Fig 1B), Fusobacteria was detected in most of the HIV-infected recipients at entry (7 of 12 participants), week 1 (5 of 12 participants), week 2 (3 of 12 participants), week 4 (6 of 12 participants), week 8 (6 of 12 participants), and week 24 (4 of 11 participants) (Fig 1C). Furthermore, some of the recipients received antibiotic therapy yet retained the abundance of Fusobacteria (Fig 1C, Table 1) in the gut.
Expansion of TIGIT and PD-1 T cells and their ligands in periphery and GALT in cART-suppressed HIV-infected individuals
We profiled TIGIT and PD-1 expression on CD8 (Figs 2A–C) and CD4 (Figs 2D–F) T cells obtained from rectal mucosal mononuclear cells (RMMCs) and peripheral blood mononuclear cells (PBMCs) from the UH cohort. We observed significantly higher frequencies of TIGIT+PD-1+ co-expressing CD8 T cells from RMMCs compared to PBMCs from HIV-infected individuals (Median: 33% vs. 14.2% respectively, p = 0.0486) (Fig 2B, left panel). We also observed a significantly lower frequency of RMMC CD8 T cells expressing PD-1+ in HIV-infected compared to HIV-uninfected individuals (46% vs. 61.6% respectively, p = 0.048) (Fig 2C, middle panel). In the HIV-infected group, we observed a higher frequency of RMMC CD4 T cells expressing TIGIT+ (39.7% vs 18% respectively, p = 0.015), PD-1+ (71.8% vs. 36.9% respectively, p = 0.015) and TIGIT+PD-1+ cells (35.3% vs. 6.8% respectively, p = 0.015) (Fig 2E, left panel). A higher frequency of RMMC CD4 T cells expressing TIGIT+ (39.7% vs. 28.8% respectively, p = 0.048) in HIV-infected compared to HIV-uninfected individuals (Fig 2F, left panel) was noted.
Figure 2. TIGIT and PD-1 are upregulated on mucosal mononuclear T cells derived from rectosigmoid biopsies.
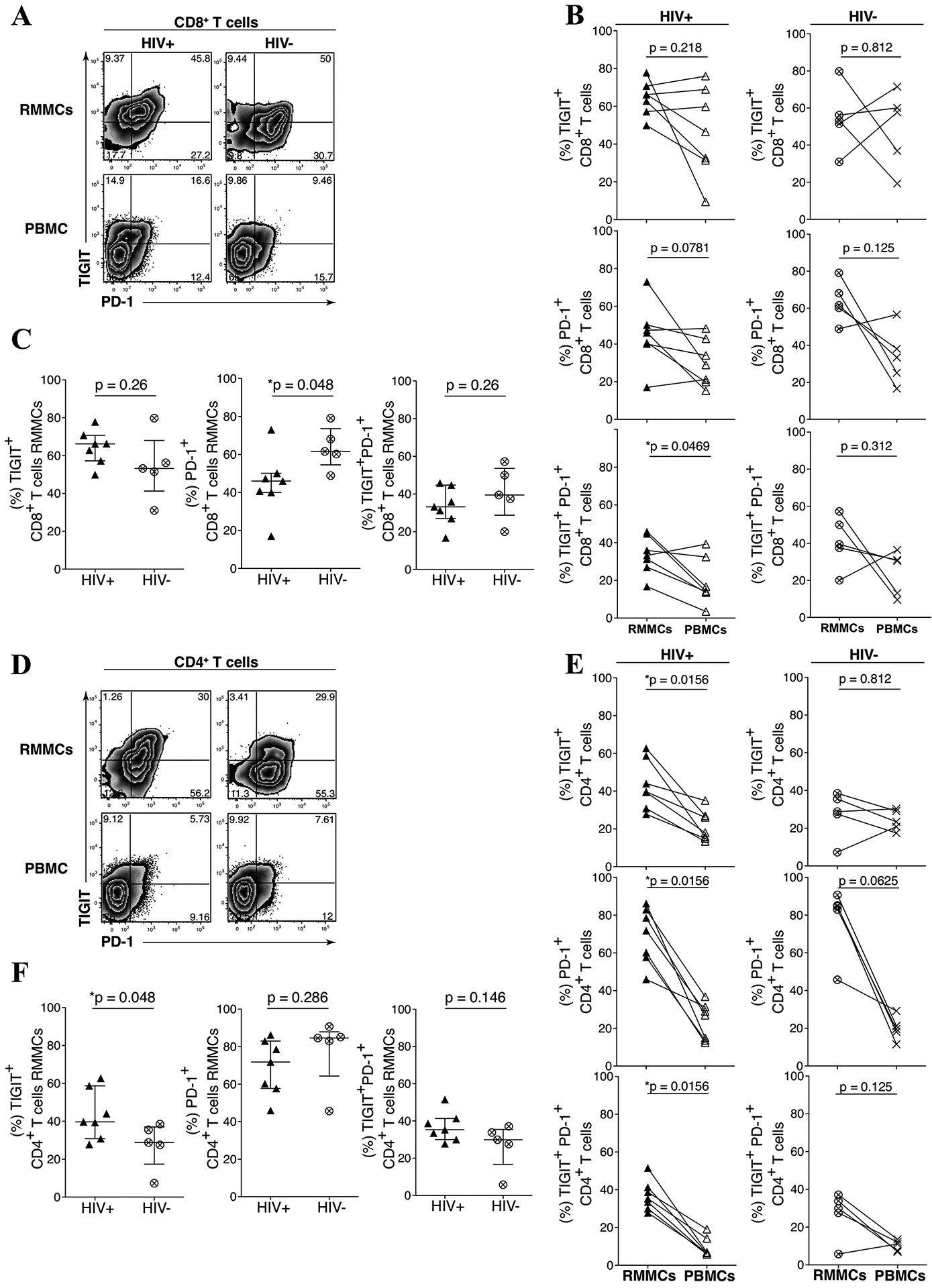
Rectosigmoid mucosal mononuclear cells (RMMCs) and peripheral blood mononuclear cells (PBMCs) from cART-suppressed UH HIV-infected and HIV-uninfected individuals were stained for viability and antibodies against CD3, CD4, CD8, TIGIT and PD-1. Representative flow cytometry plots of matched RMMCs and PBMCs gated on live CD3+ lymphocytes, from representative UH HIV-infected (A) and HIV-uninfected (D) individual. (B) Compiled data comparing the differences of TIGIT and PD-1 expression on CD8+ T cells from RMMCs and matched PBMCs of UH HIV-infected (n = 7) and HIV-uninfected (n = 5) individuals. (C) Compiled data of PD-1+, TIGIT+, and PD-1+ TIGIT+ CD8+ T cells from RMMCs from UH HIV-infected and HIV-uninfected individuals. (E) Compiled data comparing the differences of TIGIT and PD-1 expression on CD4+ T cells from RMMCs and matched PBMCs of UH HIV-infected (n = 7) and HIV-uninfected (n = 5) individuals. (F) Compiled data of PD-1+, TIGIT+, and PD-1+ TIGIT+ CD4+ T cells from RMMCs from UH HIV-infected and HIV-uninfected individuals. The p-values were calculated using Wilcoxon matched-pairs signed ranked test for matched pairs and Mann-Whitney U test for group comparison.
We assessed the TIGIT and PD-1 ligands, poliovirus receptor (PVR; CD155) and PD-L1 expression on dendritic cell subsets (monocyte-derived dendritic cells (mDC) and plasmacytoid dendritic cells (pDC)) respectively from donors in the UH cohort (see Figure A-G, Supplemental Digital Content 4). Compared to PBMCs, RMMCs had significantly higher frequency of PD-L1+ expressing mDCs (80.3% vs. 28.9%, p = 0.015), lower frequency of PVR+ expressing mDCs (78.6% vs. 93.4%, p = 0.015) and higher frequency of dual PVR+PD-L1+ co-expressing mDCs (87.5% vs. 28.7%, p = 0.015) (see Figure B, Supplemental Digital Content 4). We also observed in the pDCs, a significantly higher frequency of PVR+ expressing pDCs (93.5% vs. 54.6%, p = 0.015), PDL1+ expressing pDCs (66.4% vs. 24.2%, p = 0.015), and PVR+PD-L1+ co-expressing pDCs (50.3% vs. 20.8%, p = 0.031) (see Figure C, Supplemental Digital Content 4). No significant differences were observed in RMMCs vs. PBMCs in uninfected controls (see Figure D-F, Supplemental Digital Content 4). Lastly, we observed a significantly higher frequency of PD-L1+ expressing mDCs (80.3% vs. 62.75%, p = 0.0101) and PVR+PD-L1+ co-expressing mDCs (87.5% vs. 64.1%, p = 0.025) in HIV infected group (see Figure G, Supplemental Digital Content 4).
Magnitude of peripheral anti-HIV CD8 T cell responses to ICB and gut microbial phyla abundance
On the basis of the phenotype results, we next assessed the effects of peripheral anti-HIV CD8 T cells responses to TIGIT and PD-L1 blockade (Fig 3A). We observed an increased trend in the fold change of interferon gamma (IFN-γ), CD107a, and tumor necrosis factor alpha (TNF-α) producing anti-HIV Gag-specific CD8 T cell responses to either single TIGIT or PD-L1 blockade, and a significant increase fold change in responses following the combination of TIGIT and PD-L1 blockade when compared to an IgG1 isotype control mAb (Fig 3B).
Figure 3. Peripheral CD8 T cell immune restoration by anti-TIGIT and anti-PD-L1 blockade varied and is associated with microbiome abundance.
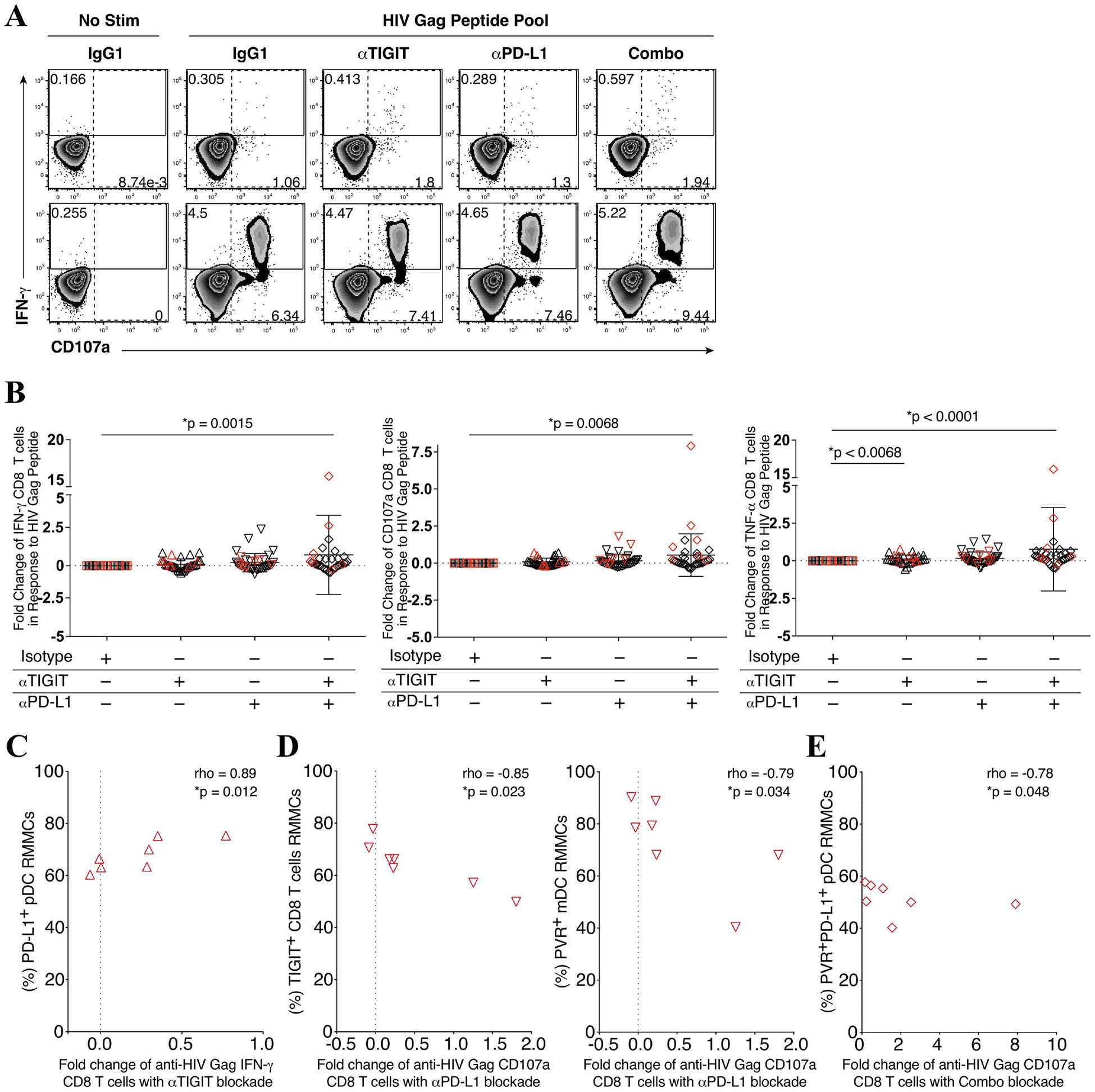
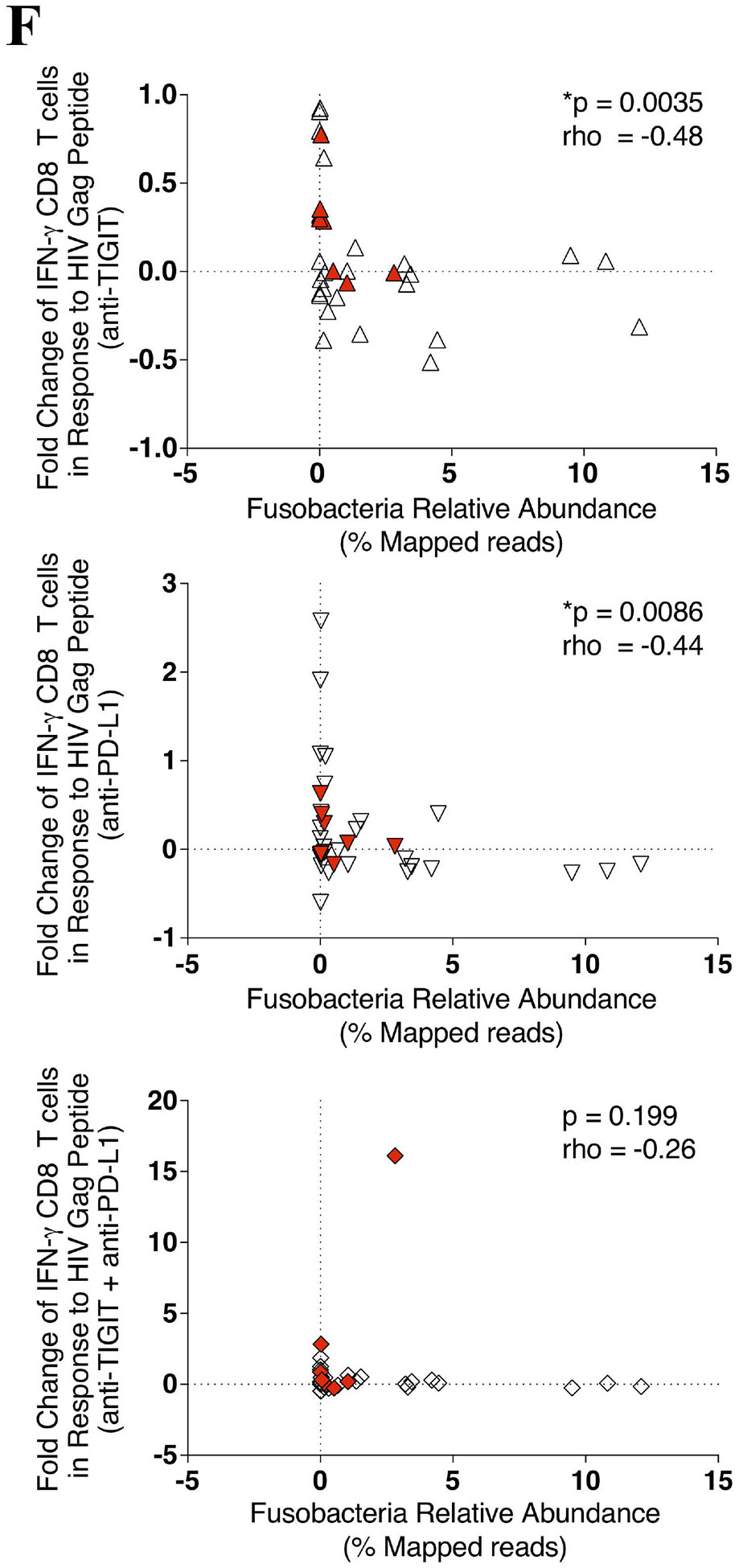
PBMCs were stimulated with a HIV Gag peptide pool in the presence, absence or combination of anti-TIGIT and anti-PD-L1 monoclonal blocking antibodies and assessed for the production of interferon gamma (IFN-γ), degranulation (CD107a), and inflammation (TNF-α). (A) Representative flow cytometry plots gated on Live CD3+ CD8 T cells, showing IFN-γ (solid gate) and CD107a (dashed gate) responses from two cART-suppressed HIV-infected individuals. (B) Graphs show compiled data of the fold change of IFN-γ (right panel), CD107a (middle panel), and TNFα (right panel) in the presence of blocking mAbs normalized to HIV Gag stimulation with IgG1 isotype. Red represents UH cohort (n = 7) and black represent UW cohort (n = 27). Matched phenotypes from RMMCs were compared with the restoration of PBMC CD8 T cells effector functions after mAb blockade. (C) Graph shows correlation of the frequency (%) of PD-L1+ pDCs from RMMCs against the fold change of anti-HIV Gag IFN-γ CD8 T cells with anti-TIGIT blockade (n = 7). (D) Graph shows correlation of the frequency (%) of TIGIT+ CD8 T cells (left panel) and PVR+ mDCs (right panel) from RMMCs against the fold change of anti-HIV Gag CD107a CD8 T cells with anti-PD-L1 blockade (n = 7). (E) Graph shows correlation of the frequency (%) of PVR+PDL1+ pDCs from RMMCs against the fold change of anti-HIV Gag CD107a CD8 T cells with combination blockade (anti-TIGIT + anti-PD-L1) (n = 7). (F) Graph shows correlation of the relative abundance of Fusobacteria from rectosigmoid biopsies against the fold change of anti-HIV Gag IFN-γ CD8 T cells with anti-TIGIT (upper panel), anti-PD-L1 (middle panel) and combination of anti-TIGIT and anti-PD-L1 (lower panel) blockade (n = 34). The p-values were calculated using Spearman’s rho test for correlations.
In the UH cohort we observed a correlation between higher rectal mucosal PD-L1+ expressing pDCs and increased fold change of IFN-γ+ CD8 T cell Gag response to TIGIT blockade (p = 0.012, rho = 0.89) (Fig 3C). Higher TIGIT+ RMMC CD8 T cells and PVR+ RMMC mDCs were associated with lower fold change of CD107a+ CD8 T cell Gag responses to PD-L1 blockade (p = 0.023, rho = −0.85; p = 0.034, rho = −0.79, respectively) (Fig 3D). Furthermore, higher RMMC PVR+PD-L1+ pDCs was associated with a lower fold change of CD107a+ CD8 T cell Gag responses to combination TIGIT and PD-L1 blockade (p = 0.048, rho = −0.78) (Fig 3E).
To gain insights into the variation in the magnitude of the Gag CD8 T cell responses to ICB blockade we performed a regression analysis of the responses relative to gut microbial abundance. A decrease in the fold change of IFN-γ+ CD8 T cell anti-HIV Gag responses after anti-TIGIT and anti-PD-L1 blockade was correlated with higher Fusobacteria abundance (p = 0.0035, rho = −0.486, and p = 0.0086, rho = −0.444, respectively) (Fig 3F). Interestingly, although no significant correlation was found between IFN-γ+ CD8 T cell anti-HIV Gag responses with the relative abundance of Firmicutes, a non-pathogenic commensal, a trend in IFN-γ+ CD8 T cell anti-HIV Gag responses after single anti-TIGIT, anti-PD-L1 and a combination of both anti-TIGIT and anti-PD-L1 blockade correlated with higher abundance of Firmicutes (see Figure, Supplemental Digital Content 5).
Discussion
Our study reveals several novel findings relevant to understanding the host-pathogen interactions and potential mechanisms affecting the efficacy of anti-HIV CD8 T cell immunity essential for viral clearance. First, the only significant difference in gut microbial diversity between virally suppressed HIV-infected and age-matched uninfected individuals in our cohorts, was a higher gut abundance of pathogenic Fusobacteria phyla. Among HIV infected donors receiving FMT that were devoid of Fusobacteria, no appreciable impact on the colonization of Fusobacteria was evident. Second, we observed increased co-expression of the PD-1 and TIGIT receptors on the rectal mucosal resident CD8 T cells compared to the periphery in HIV-infected donors, suggesting that these cells may be sensitive to immune checkpoint blockade. Third, ex vivo studies using an anti-TIGIT or anti-PD-L1 mAb revealed that a reduction in the magnitude of peripheral polyfunctional anti-HIV CD8 T cells responses to ICB was associated with increased abundance of Fusobacteria in the gut mucosa. We propose a model in which an oral commensal bacterium, expands in the gut mucosa in persons with HIV and may significantly impair not only pre-existing HIV-specific CD8 T cell responses, but may restrain the efficacy of ICB immunotherapies in blood and tissues. We believe these finding provide important insights into mechanisms of ICB resistance as we pursue effective strategies to eradicate HIV using immunotherapy.
It was notable that we observed limited gut microbial dysbiosis in our cART-suppressed HIV-infected study population. Longer duration of cART, as opposed to short term therapy, may shift the gut microbiota composition towards an HIV-uninfected profile.[19] Furthermore, partial restoration of intestinal mucosal barrier defects is seen in HIV-infected individuals on long-term cART [20]. While these reports complement our findings, more extensive longitudinal studies would clarify the extent of gut dysbiosis during cART-suppressed HIV-infection.
The persistence of distinct fecal bacterial communities may be influenced by different anti-retroviral drugs such as efavirenz (EFV)-containing regimens. [21] In our study, only four participants received EFV-based regimens (see Table, Supplemental Digital Content 6) and no significant microbial abundance differences were seen between the EFV and non EFV-containing regimens making it unlikely that cART was responsible for the Fusobacteria expansion. While gut microbiome alpha diversity has been reported to be lower in untreated HIV-infected individuals and those on short term cART [22], other studies have found HIV-infected individuals on cART had similar gut microbial species richness to uninfected donors.[23] Our study participants were virally suppressed on cART for ≥ 5 years and the higher abundance of Fusobacteria suggests an HIV-directed mucosal immune environment driving this specific gut colonization,
Fusobacteria are anaerobic gram-negative bacilli that are frequently isolated from the oral cavity of both healthy and from individuals with periodontitis [24, 25] and known to associate with colonic tumor formation in preclinical models.[14] The hypothesis for a causal role of Fusobacteria ectopic colonization in the gut is still unclear. A study by Atarashi et al., demonstrated that in mice, the intestinal tract had the ability to expand specific oral microbial colonies, including Fusobacteria. Moreover, the study also showed that salivary microbiota are strong inducers of T helper 1 (Th1) response that may be responsible for eliciting severe gut inflammation in genetically susceptible hosts.[26] Furthermore, during HIV infection, the gastric acidity is altered, potentially allowing gastric colonization of oral flora.[27] In HIV infection, greater abundance of Fusobacteria was found in subgingival plaque [25] and in anal swabs [28] collected from HIV-infected individuals compared to their matched HIV-uninfected group, increasing the risk of Fusobacteria colonization that may lead to local and systemic dysbiosis.
During acute HIV infection, CD4 T-cell depletion causing epithelial injury leads to a disturbance in intestinal homeostasis resulting in systemic immune activation that persists in the chronic stage but is only partially tempered by early initiation of cART.[29] Combined with the ability of Fusobacteria to colonize the intestinal tract in susceptible hosts to intestinal damage,[26] our current study support the hypothesis that HIV infected persons with Fusobacteria colonization are prone to a cascade of local and systemic immune activation and a continuous cycle of gut dysbiosis, disrupted gut hemostasis and peripheral immune activation and immune dysfunction that persists despite viral suppression.
We didn’t find significant difference in peripheral TIGIT+ CD8 T cells in HIV-infected individuals compared to control, contrary to our previous results. [2] The inconsistent result could be caused by lower number of study participants and higher participants’ median age in the current cohort. Recent study showed that TIGIT was upregulated in CD8+ T cells of elderly adults suggesting TIGIT role as critical immune regulator in aging.[30] A recent study showed a preferential and persistence expansion of TIGIT+ CD8 and CD4 T cells in the gut of cART-treated HIV infected donors over time.[31] Memory CD4 T cells expressing multiple NCRs, including PD-1 and TIGIT, are also enriched in cells harboring inducible proviral HIV DNA.[2, 32, 33] Combined with our results, these data suggest viral persistence in the gut may be related to the degree of NCR expression and be sensitive to immune targeting with ICB. Whether the gut microbiome remains a key regulatory of these anti-reservoirs responses remains to be investigated.
PD-L1 expressing APCs have been implicated in several histolytic and DC disorders [34] and TIGIT and PVR interact bi-directionally to induce T cell dysfunction while promoting mature immunoregulatory DCs.[35] The expansion of RMMC PD-L1+ and PVR+PD-L1+ mDCs during HIV infection could selectively support the persistence of dysfunctional TIGIT+ and PD-1+ T cells in gut and leaving them amenable to ICB. It is notable that we observed relationships between mucosal and not peripheral TIGIT expressing CD8 T cells [2], and the efficacy of ICB and both PVR and PD-L1 DC expression. A previous study reported that colonic mDC from HIV-infected donors had greater responses to stimulation with gut pathobionts compared to those derived from HIV-uninfected control.[36] While we did not perform functional DC studies, this report raises the possibility that activation of colonic mDC by gut bacteria may contribute to systemic immune activation and ICB may dampen the inflammatory microenvironment in the gut. Collectively, these finding raise the importance that targeting these receptor-ligand pairs may have effects beyond the periphery but also in the tissue compartments such as the gut.
Recent studies demonstrate that the human intestinal microbiome is an important mediator that can affect the efficacy of cancer immunotherapy by ICB.[11–14] Experiments using specific pathogen-free mice further demonstrate how intestinal commensal microbiome modulate host immune priming and systemic activation.[37] The Fusobacteria membrane protein, Fap2, engages the TIGIT in the gut mucosa and appears to reduce the ability of T and NK cells to eliminate tumor cells.[13, 35] We postulate that the interaction between Fap2 and TIGIT in the gut may impact the efficacy of TIGIT blockade. Furthermore, this interaction may induce metabolic reprograming of the T cells rendering them insensitive to TIGIT blockade that will require additional interventions to overcome this resistance. Investigating the metabolic demands of effector T cells in the presence of Fap2 may aid in our mechanistic understanding of this interaction.
Monitoring and managing gut Fusobacteria colonization may be a prerequisite for predicting the efficacy of TIGIT inhibitor treatment. Dietary supplements with probiotics [38] or FMT [39] previously may have shown as a favorable adjunctive therapy to “reboot” the gut microbial ecosystem prior to ICB. However, our data showed that FMT may be insufficient. Identifying and targeting an immunogenic epitope of the Fap2 protein,[40] may serve as an alternate and complimentary adjuvant therapy to improve TIGIT blockade.
In summary, our data provide a synopsis of the complex inter-play between human gut microbial communities and how they modulate the anti-HIV effector responses to both PD-1 and TIGIT blockade. As we attempt to understand the validity of ICB for anti-HIV reservoir studies or other anti-viral CD8 T cell dependent responses, characterization of the gut microbiome may serve as an important predictive tool but also aid in the development of more effective immune-based or targeted anti-bacterial therapies.
Supplementary Material
Supplemental Digital Content 2. Figures describing gut microbiome distribution in HIV-infected and uninfected participants .doc
Supplemental Digital Content 3. Figures describing gut microbiome distribution in HIV-infected participants receiving FMT .doc
Supplemental Digital Content 4. Figures describing NCR ligands expression on rectosigmoid dendritic and mucosal mononuclear T cells .doc
Supplemental Digital Content 5. Figures describing peripheral anti TIGIT and anti PD-L1 blockade immune restoration and Firmicutes abundance .doc
Supplemental Digital Content 6. Table describing cART regiments in study participants .doc
Supplemental Digital Content 1. Supplementary material and methods .doc
Acknowledgments
Conceived and designed the experiments: INS GMC LCN NRK MS. Performed the experiments: INS GMC MJC APSP THM IVC. Analyzed the data: INS GMC VSK THM IVC. Contributed materials/analysis tools: NT CMS DC NH NRK MS Wrote and edit the paper: INS GMC IVC MS LCN.
We would like to extend our profound appreciation to the study participants for their participation in this study. We would like to thank the nurses and staff of the Hawaii Center for AIDS for their help in recruiting, organizing and maintaining the cohort. We acknowledge the National Institute of Health (NIH) AIDS Reagent Program for providing HIV-1 Gag peptides and Bristol-Myers Squibb for the donation of the monoclonal antibodies.
Funding: National Institute of General Medical Sciences (NIGMS) 1U54GM104944-01A1 (INS), National Institutes of Health award numbers NIH/NIDDK RO1DK112254 and NIH/NIDA 1DP13A037979 (NRK) and National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) (MS), and University of Washington Center for AIDS Research funded by the National Institutes of Health AI027757.
Source of funding:
The project described was supported in part by several funding agencies: National Institute of General Medical Sciences (NIGMS) 1U54GM104944-01A1 (IS), National Institutes of Health award numbers NIH/NIDDK RO1DK112254 and NIH/NIDA 1DP13A037979 (NRK) and National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) (MS). Additional support was provided by the University of Washington Center for AIDS Research, which is funded by the National Institutes of Health under award number AI027757.
Footnotes
Conflict of interest
None of the authors have any relevant conflict of interest to disclose with the exception of Dr. Nichole Klatt who is on the Editorial Board of AIDS.
References
- 1.Deeks SG. HIV: Shock and kill. Nature. 2012;487(7408):439–40. [DOI] [PubMed] [Google Scholar]
- 2.Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS pathogens. 2016;12(1):e1005349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seung E, Dudek TE, Allen TM, Freeman GJ, Luster AD, Tager AM. PD-1 blockade in chronically HIV-1-infected humanized mice suppresses viral loads. PloS one. 2013;8(10):e77780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical Trial of the Anti-PD-L1 Antibody BMS-936559 in HIV-1 Infected Participants on Suppressive Antiretroviral Therapy. J Infect Dis. 2017;215(11):1725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guihot A, Marcelin AG, Massiani MA, Samri A, Soulie C, Autran B, et al. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2018;29(2):517–8. [DOI] [PubMed] [Google Scholar]
- 6.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–53. [DOI] [PubMed] [Google Scholar]
- 7.Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44(6):1255–69. [DOI] [PubMed] [Google Scholar]
- 8.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (New York, NY). 2016;354(6316):1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (New York, NY). 2017. [DOI] [PubMed] [Google Scholar]
- 10.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (New York, NY). 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (New York, NY). 2015;350(6264):1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, NY). 2015;350(6264):1084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42(2):344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abed J, Emgard JE, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. 2016;20(2):215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaiser J Fecal transplants could help patients on cancer immunotherapy drugs. Science (New York, NY). 2019. [Google Scholar]
- 16.Vujkovic-Cvijin I, Swainson LA, Chu SN, Ortiz AM, Santee CA, Petriello A, et al. Gut-Resident Lactobacillus Abundance Associates with IDO1 Inhibition and Th17 Dynamics in SIV-Infected Macaques. Cell Rep. 2015;13(8):1589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics (Oxford, England). 2011;27(6):863–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology. 2014;15(3):R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14(3):329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epple HJ, Zeitz M. HIV infection and the intestinal mucosal barrier. Ann N Y Acad Sci. 2012;1258:19–24. [DOI] [PubMed] [Google Scholar]
- 21.Pinto-Cardoso S, Lozupone C, Briceno O, Alva-Hernandez S, Tellez N, Adriana A, et al. Fecal Bacterial Communities in treated HIV infected individuals on two antiretroviral regimens. Scientific reports. 2017;7:43741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nowak P, Troseid M, Avershina E, Barqasho B, Neogi U, Holm K, et al. Gut microbiota diversity predicts immune status in HIV-1 infection. AIDS. 2015;29(18):2409–18. [DOI] [PubMed] [Google Scholar]
- 23.McHardy IH, Li X, Tong M, Ruegger P, Jacobs J, Borneman J, et al. HIV Infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome. 2013;1(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bui FQ, Johnson L, Roberts J, Hung SC, Lee J, Atanasova KR, et al. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1beta and the danger signals ASC and HMGB1. Cell Microbiol. 2016;18(7):970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goncalves LS, Goncalves BM, Fontes TV. Periodontal disease in HIV-infected adults in the HAART era: Clinical, immunological, and microbiological aspects. Arch Oral Biol. 2013;58(10):1385–96. [DOI] [PubMed] [Google Scholar]
- 26.Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science (New York, NY). 2017;358(6361):359–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welage LS, Carver PL, Revankar S, Pierson C, Kauffman CA. Alterations in gastric acidity in patients infected with human immunodeficiency virus. Clin Infect Dis. 1995;21(6):1431–8. [DOI] [PubMed] [Google Scholar]
- 28.Yu G, Fadrosh D, Ma B, Ravel J, Goedert JJ. Anal microbiota profiles in HIV-positive and HIV-negative MSM. Aids. 2014;28(5):753–60. [DOI] [PubMed] [Google Scholar]
- 29.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10(9):655–66. [DOI] [PubMed] [Google Scholar]
- 30.Song Y, Wang B, Song R, Hao Y, Wang D, Li Y, et al. T-cell Immunoglobulin and ITIM Domain Contributes to CD8(+) T-cell Immunosenescence. Aging Cell. 2018;17(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS pathogens. 2016;12(7):e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niessl J, Baxter AE, Mendoza P, Jankovic M, Cohen YZ, Butler AL, et al. Combination anti-HIV-1 antibody therapy is associated with increased virus-specific T cell immunity. Nat Med. 2020;26(2):222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Sun HH, Fletcher CD, Hornick JL, Morgan EA, Freeman GJ, et al. Expression of Programmed Cell Death 1 Ligands (PD-L1 and PD-L2) in Histiocytic and Dendritic Cell Disorders. Am J Surg Pathol. 2016;40(4):443–53. [DOI] [PubMed] [Google Scholar]
- 35.Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57. [DOI] [PubMed] [Google Scholar]
- 36.Dillon SM, Lee EJ, Kotter CV, Austin GL, Gianella S, Siewe B, et al. Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T-cell activation in untreated HIV-1 infection. Mucosal Immunol. 2016;9(1):24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison OJ, Powrie FM. Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol. 2013;5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee A, Bhattacharya H, Kandwal A. Probiotics in periodontal health and disease. J Indian Soc Periodontol. 2011;15(1):23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seekatz AM, Aas J, Gessert CE, Rubin TA, Saman DM, Bakken JS, et al. Recovery of the gut microbiome following fecal microbiota transplantation. MBio. 2014;5(3):e00893–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guevarra LA Jr., Afable ACF, Belza PJO, Dy KJS, Lee SJQ, Sy-Ortin TT, et al. Immunogenicity of a Fap2 peptide mimotope of Fusobacterium nucleatum and its potential use in the diagnosis of colorectal cancer. Infectious agents and cancer. 2018;13:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 2. Figures describing gut microbiome distribution in HIV-infected and uninfected participants .doc
Supplemental Digital Content 3. Figures describing gut microbiome distribution in HIV-infected participants receiving FMT .doc
Supplemental Digital Content 4. Figures describing NCR ligands expression on rectosigmoid dendritic and mucosal mononuclear T cells .doc
Supplemental Digital Content 5. Figures describing peripheral anti TIGIT and anti PD-L1 blockade immune restoration and Firmicutes abundance .doc
Supplemental Digital Content 6. Table describing cART regiments in study participants .doc
Supplemental Digital Content 1. Supplementary material and methods .doc