

Abstract
Secondary organic aerosol (SOA) can affect the atmospheric radiation balance through absorbing light at shorter visible and UV wavelengths. However, the composition and optical properties of light-absorbing SOA is poorly understood. In this work, SOA filter samples were collected during individual chamber experiments conducted with three biogenic and eight aromatic volatile organic compound (VOC) precursors in the presence of NOX and H2O2. Compared with the SOA generated using the aromatic precursors, biogenic SOA generally exhibits negligible light absorption above 350 nm; the aromatic SOA generated in the presence of NOX shows stronger light absorption than that generated with H2O2. Fifteen nitroaromatic compound (NAC) chemical formulas were identified and quantified in SOA samples. Their contributions to the light absorption of sample extracts were also estimated. On average, the m-cresol/NOX SOA sample has the highest mass contribution from NACs (10.4 ± 6.74%, w/w), followed by naphthalene/NOX (6.41 ± 2.08%) and benzene/NOX (5.81 ± 3.82%) SOA. The average contributions of NACs to total light absorption were at least two times greater than their average mass contributions at 365 and 400 nm, revealing the potential use of chromophoric NACs as brown carbon (BrC) tracers in source apportionment and air quality modeling studies.
Introduction
The light absorption of atmospheric aerosols significantly impacts the Earth’s radiation balance.(1, 2) Black carbon (BC) and mineral dusts have been recognized as efficient light absorbing agents for decades,(3–6) while global climate models commonly treat organic matter (OM) as a purely light scattering component.(7, 8) Many studies show that OM emitted during open biomass burning can strongly absorb light at shorter visible and UV wavelengths (λs) with strong λ dependency.(9–12) This light-absorbing OM is often referred to as “brown carbon” (BrC). Measurement-based estimates of radiative forcing due to OM have suggested that BrC accounts for ∼20% of the solar absorption of carbonaceous aerosols globally.(13, 14) Feng et al.(15) applied chemical transport and radiative transfer models to estimate the enhanced absorption of solar radiation due to BrC and found that the contribution of BrC to aerosol forcing could account for more than a quarter of the estimated radiative effects of BC globally. However, the available modeling studies on aerosol absorption with the inclusion of BrC only parametrized primary emissions of BrC from biomass or biofuel combustion,(13) and the light absorption of secondary organic aerosol (SOA) is rarely considered or assumed to be less important.(16)
Multiple studies have demonstrated the formation of secondary BrC through laboratory experiments.(9, 17–25) Laboratory-based chamber experiments suggest that a variety of factors impact the light absorption of SOA, including precursor composition (volatile organic compounds, VOC),(17) oxidants (e.g., hydroxyl radical, OH; nitrogen oxides NOX),(20) aging,(9) and seed acidity.(25) These studies conclude that the SOA derived from the photo-oxidation of anthropogenic aromatic VOCs (e.g., toluene) has stronger light absorption than typical biogenic (e.g., α-pinene) SOA reaction products. Additionally, BrC formation is favored in reactions in the presence of gaseous ammonia and amines, high NOX, and highly acidic seed particles. Some nitroaromatic compounds (NACs, e.g., nitrocatechol) were identified in laboratory-generated SOA and accounted for part of the total light absorption at near-UV (300–400 nm) irradiance.(26) However, the molecular structure and composition of NACs from different reaction conditions (VOC/oxidant) and their contributions to light absorption remain uncertain. Most previous studies investigated the light absorption of SOA from only a few biogenic and/or anthropogenic precursors with NOX and/or OH.(17, 19, 20, 26) The potential of different chemical precursors to produce secondary BrC under identical conditions (e.g., oxidant, seed type) is poorly understood.
In the present study, SOA was generated by irradiating three biogenic (α-pinene, isoprene, and β-caryophyllene) and eight aromatic (1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene, m-xylene, toluene, ethylbenzene, benzene, naphthalene, and m-cresol) VOCs individually in the presence of NOX and hydrogen peroxide (H2O2), respectively, in a smog chamber. The SOA was collected on filters, extracted in methanol, and analyzed with a UV/vis spectrometer and a high performance liquid chromatograph/diode array detector (HPLC/DAD)-quadrupole (Q)-time-of-flight mass spectrometer (ToFMS). First, we compared the bulk mass absorption efficiency (MAE, m2 g–1) – a measure of BrC – of SOA from different precursors reacted under NOX or H2O2. Next, the identification and quantification of NACs were conducted, and the mass contributions of NACs to SOA were calculated. Finally, the contribution of identified NACs to the light absorption of SOA was estimated. These study results provide a comprehensive picture about the light-absorbing characteristics of typical biogenic and aromatic SOA at bulk chemical and molecular levels, benefiting chemical source apportionment models and climate models using optical properties.
Methods
Smog Chamber Experiments
All SOA experiments were conducted at the U.S. Environmental Protection Agency, RTP, NC location within a fixed volume 14.5 m3 indoor chamber. Details of the equipment and experimental methods for the indoor chamber system were described elsewhere.(27–29) A summary of experimental conditions for the photo-oxidation of all VOC precursors is provided in Table S1 of the Supporting Information. Each VOC precursor was photo-oxidized under both NOX and H2O2 conditions. For reactions with NOX, nitric oxide (NO) was continuously injected from high pressure cylinders into the smog chamber through a mixing manifold. In the H2O2 experiments, 50 wt % H2O2 solution was vaporized and flushed into the chamber using via a heated glass bulb and photolyzed in the chamber to generate OH radicals in the absence of NOX. Ammonium sulfate [(NH4)2SO4] seed aerosol at a concentration of 0.1–1 μg m–3 was introduced for all experiments to promote SOA formation. All experiments were conducted in dynamic flow mode with a ∼4 h residence time.
During experiments, the VOC precursor concentration in the chamber was determined using a cryogenic trap followed by GC-FID analysis (model 5890, Hewlett-Packard, Palo Alto, CA). NO and NOX were measured using an oxides of nitrogen analyzer (TECO model 42C, Franklin, MA). Ozone was measured using an ozone monitor (Bendix model 8002, Lewisburg, WV). The radiation intensity was continuously monitored with an integrating radiometer (Eppley Laboratory, Inc., Newport, RI). The relative humidity (RH) and temperature were measured with an Omega digital thermohydrometer (model RH411, Omega Engineering, Inc., Stamford, CT). Once steady-state conditions were obtained (24 h), particle samples were collected at 16.7 L min–1 using 47 mm Teflon-impregnated glass fiber filters (Pall Gelman Laboratory, Ann Arbor, MI). A 60 cm XAD4-coated annular denuder (URG, Inc., Chapel Hill, NC) was applied upstream of the filter to remove gas-phase organics in the air stream. Mass concentrations of SOA (μg m–3) in the chamber were determined from gravimetric measurements of the filters. Air volumes and SOA concentrations for the filter samples analyzed in this work are listed in Table S1.
UV/Vis Analysis
Details of the extraction and instrument setup are given in the Supporting Information. Briefly, a 1.5 cm2 punch of each filter sample was extracted ultrasonically with methanol (5 mL) for 15 min, followed by filtration and analysis with a UV/vis spectrometer. To convert the light absorption due to chromophores measured in solution to ambient aerosol chromophore concentrations, the light absorption values of all the filter extracts are converted to light absorption coefficients (Absλ, Mm–1) by(30)
| (1) |
where A700 is referenced to account for systematic baseline drift,(30) absorption was not observed at λ ≥ 700 nm for any sample, Vl (m3) is the volume of methanol (5 mL) used for extraction, Va (m3) is the volume of the sampled air, and L (m) is the optical path length of the quartz cuvette (1 cm) in the UV/vis spectrometer. For ease of analysis, this work focused on Absλ at λ = 300–550 nm, where most of the BrC absorption was observed.(31)
To identify which reaction (VOC/oxidant) generated the strongest light-absorbing SOA, the Absλ value was normalized by the SOA concentration for each sample using eq 2:
| (2) |
in which MAEλ is the bulk mass absorption efficiency (m2 g–1), and OM (μg m–3) is the mass concentration of SOA in each sample. It was assumed that the SOA is 100% extracted by methanol. The solution absorption Ångström exponent (Åa) is a measure of spectral dependence of light absorption and is determined from the linear regression fit of log10(Absλ) vs log10(λ) over the λ range of 300–550 nm. Finally, the imaginary part of the complex refractive index (k), a parameter used to evaluate the light-absorbing characteristics of particulate matter, was calculated for each SOA sample based on the spectroscopic data. See the Supporting Information for calculation method details.
Sample Extraction and HPLC/DAD-Q-ToFMS Analysis
Internal standard (25 μL of 10 ng μL–1 nitrophenol-d4) was spiked onto each filter sample prior to extraction. Approximately 3–5 mL of methanol was used to ultrasonically extract each sample twice (15 min each time). Extract was filtered into a pear-shaped glass flask and rotary evaporated to ∼0.1 mL. Concentrated extracts were then transferred into a solvent-rinsed 2 mL amber vial. The final extract sample volume was ∼500 μL prior to HPLC/DAD-Q-ToFMS analysis. All glassware used during the extraction procedure was prebaked at 550 °C overnight. Sample and extract storage temperatures were ≤ −20 °C.
The identification and quantification of target NACs were conducted with an HPLC (1200 series, Agilent Technologies, Santa Clara, CA) coupled to a UV/vis DAD (G1315C, Agilent Technologies, Santa Clara, CA) followed by a Q-ToFMS (Agilent 6520). The Q-ToFMS used a multimode source operating in electrospray ionization (ESI) and negative ion (−) modes. Details of the HPLC/DAD-Q-ToFMS instrument conditions and methods are provided in the Supporting Information. Briefly, samples were analyzed in full scan mode (40–1000 Da). The combination of HPLC, online light absorbance, and accurate MS detection (2 ppm) permits direct identification of chemical compound formulas responsible for absorbing photons in the near UV and visible range. Selected samples were re-examined using MS-MS mode under identical chromatographic conditions. [M–H]− ions were targeted for collision-induced dissociation (CID) using predetermined accurate mass and retention times. MS/MS data offered structural m/z data and were used to compare standard compounds to those identified in SOA samples. The standard, surrogate assignments, proposed structures, and 15 sample-identified chemical formulas are listed in Table S2. Acceptance criterion for compound identification and quantification was set at ±10 ppm mass accuracy. The extracted ion chromatograms (EICs) and Q-ToF MS/MS spectra for standard compounds are shown in Figure S1, Supporting Information. The EICs, MS/MS spectra, and UV/vis chromatograms (DAD signal at 365 nm) for identified compounds in selected SOA samples are provided in Figures S2–S10. Because the sample extract was analyzed by UV/vis detector before entering the MS, the retention times of the identified compounds in UV/vis chromatogram was ∼0.15 min earlier than the EIC.
Quantification was performed using the internal standard method. Multipoint (n = 9) calibration curves were generated using the relative MS response following standard dilution (∼0.01–2 ng μL–1). The identified NACs (including isomers) represented by each formula were quantified individually using standard or surrogate compounds with similar structure or [M–H]− ion (Table S2) and then added together for the calculation of mass contribution to the total SOA in each sample. Standard recoveries were determined after spiking blank filters with known target compound concentrations and following the extraction and analysis procedures detailed above. Average recoveries of standard compounds ranged from 75.1 to 116% (Table S3). Table S3 also provides the method detection limit (MDL) of each standard compound. Each MDL was calculated as 3σ following multiple injections (n = 7) at the lowest calibration concentration. All reported data were above MDL. Blank filters were extracted and analyzed in the same manner as SOA samples, and no contamination was observed for identified compounds.
Estimation of NACs Contribution to Absλ
The MAE values of individual compound standards in methanol were used to estimate NAC absorption at a given UV/vis λ, similar to the method applied in Zhang et al.(32) Standards were diluted in series (5–11 points) over the concentration ranges in the sample extracts. Concentrations of identified compounds in sample extracts (Cl, ng μL–1) for UV/vis analysis were calculated as
| (3) |
where Ca (ng m–3) is the air concentration of the NAC based on HPLC-ToFMS quantification, Va (m3) is the sample volume corresponding to the filter aliquot (1.5 cm2) used for extraction, and Vl (μL) is the solvent volume (5 mL). The UV/vis spectra of standard compounds are shown in Figure S11. Details of MAE calculations for standard compounds and Absλ attribution for identified NACs are given in the Supporting Information. The MAE values at 365 nm, 400 nm, 450 nm, 500 nm, and 550 nm and the corresponding linear ranges of standard compounds are provided in Table S4.
Results and Discussion
Light-Absorbing Characteristics of SOA Extracts
Average MAE spectra for biogenic and aromatic SOA generated under NOX and H2O2 conditions are shown in Figure 1. The summary of average bulk MAE and k at five representative wavelengths over the λ range of 300–550 nm (λ = 365, 400, 450, 500, 550 nm) and Åa is given in Table S5. In Figure 1a-c, biogenic SOA generated under both NOX and H2O2 conditions shows minimal absorption from 300 to 550 nm, while the MAE spectra for aromatic SOA (Figure 1d-k) reflect typical atmospheric BrC with high UV absorption. Among the eight aromatic VOCs, SOA generated from toluene, ethylbenzene, benzene, naphthalene and m-cresol showed significantly (p < 0.05) higher average MAE365 under NOX conditions as compared to H2O2 conditions (Table S5). In the presence of NOX, the SOA of m-cresol had the highest average MAE at 365 (2.47 ± 1.05 m2 g–1) and 400 nm (1.19 ± 0.37 m2 g–1); naphthalene had the highest average MAE at 450 (0.43 ± 0.034 m2 g–1), 500 (0.25 ± 0.015 m2 g–1), and 550 nm (0.12 ± 0.011 m2 g–1), followed by benzene SOA (Table S5). Light absorption was also observed in the range of 300–550 nm for SOA generated under H2O2 conditions with m-xylene, toluene, ethylbenzene, benzene, naphthalene, and m-cresol. Naphthalene had the strongest light-absorbing SOA in the presence of H2O2 at each of the five representative wavelengths, followed by benzene, ethylbenzene, and m-cresol. The imaginary part of complex refractive index (k) was linearly derived from MAE, so the difference in average k values across different VOCs or between NOX and H2O2 conditions is reflected by the average MAE values. The average k values were summarized in Table S5. For the five aromatic VOCs with relatively strong light-absorbing SOA under NOX conditions (Figure 1g-k), the average Åa values were lower for NOX conditions (range 4.92 ± 0.23 to 6.89 ± 0.63) than H2O2 conditions (6.46 ± 0.25 to 7.75 ± 0.24, Table S5). The Åa values calculated for those SOA extracts (e.g., isoprene) with large percentages of Aλ – A700 (λ = 300–550 nm, >20%) measurements with close to zero or negative values and those with very high standard deviations are subject to large uncertainty because the SOA had weak or no absorption (Figure 1a-d) and thus may not contain BrC. The above results suggest that the photo-oxidation of biogenic VOCs may not generate light-absorbing SOA on neutral seed [(NH4)2SO4]; the aromatic SOA contains BrC, for which the light-absorption is sensitive to differences in molecular structures of SOA precursors and the availability of NOX.
Figure 1.
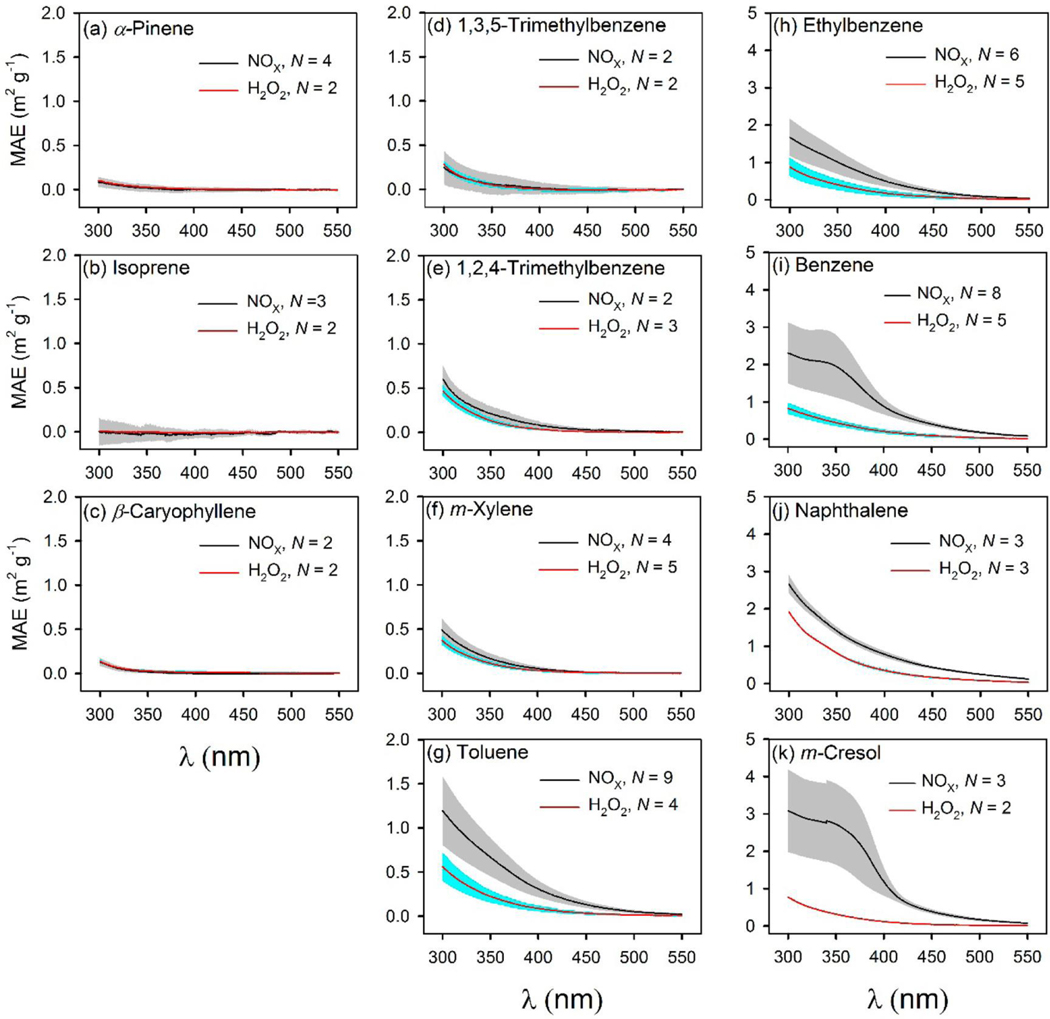
Bulk MAE spectra for SOA formed under NOX and H2O2 conditions, from (a) α-pinene, (b) isoprene, (c) β-caryophyllene, (d) 1,3,5-trimethylbenzene, (e) 1,2,4-trimethylbenzene, (f) m-xylene, (g) toluene, (h) ethylbenzene, (i) benzene, (j) naphthalene, and (k) m-cresol. For sample number N > 2, gray and cyan areas represent ±1 standard deviation; and for sample number N = 2, cyan and gray areas represent ± half the difference between the two measurements.
Liu et al.(20) have investigated the light absorption of SOA generated under NOX and NOX-free conditions for two aromatic (toluene and trimethylbenzene) and two biogenic VOCs (α-pinene and isoprene). The average MAE365 for toluene/NOX SOA (0.55 ± 0.17 m2 g–1) in this study is comparable to that observed by Liu et al.(20) (∼0.8 m2 g–1). However, the MAE365 for the NOX-free condition is 1 order of magnitude lower in Liu et al.(20) (∼0.01 m2 g–1) than here (0.17 ± 0.051 m2 g–1). For naphthalene SOA in the presence of NOX, the MAE400 (0.78 ± 0.074 m2 g–1) obtained here is comparable to the MAE405 obtained by Metcalf et al.(33) (0.81 m2 g–1) but much higher than the MAE405 values in Updyke et al.(24) (0.10 m2 g–1). The k365 values for toluene (with NOX ∼ 0.025, with H2O2 ∼ 0.008) and m-xylene (with NOX ∼ 0.008, with H2O2 ∼ 0.002) SOA in Liu et al.(19) are obtained similarly and are comparable to the corresponding average k365 values in the present study (Table S5). The low absorbance for biogenic SOA materials observed presently is consistent with previous results.(17, 20, 34) The differences in the light absorption of aromatic SOA across studies may be ascribed to the different reaction conditions, including VOC/NOX ratio, irradiation power, reaction time in the chamber, etc., which warrants further study.
Identification and Quantification of NACs and Their Contributions to Bulk Absλ
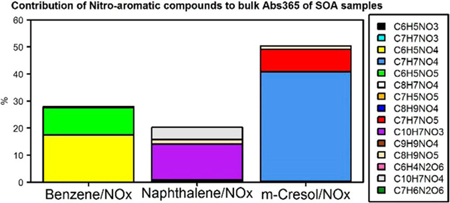
Since NACs have been detected as light-absorbing components in the atmosphere(32, 35) and can be generated from photochemical reactions,(26) the NAC composition and its contribution to bulk absorption of SOA samples are investigated. First, 15 NAC chemical formulas in the SOA samples were determined (Table S2) from the HPLC/DAD-Q-ToF MS analysis. Multiple NAC compound structures were examined using MS/MS data. The relative mass contribution (%) to total SOA of each NAC formula categorized by precursor VOC and oxidant type (NOX or H2O2) is given in Table S6, and the average relative mass contributions (%) of each NAC in SOA samples can be visualized in Figure 2. The m-cresol/NOX SOA has the highest average mass contribution from total NACs (10.4 ± 6.74%), followed by naphthalene/NOX (6.41 ± 2.08%) and benzene/NOX (5.81 ± 3.82%) SOA. The contribution of each identified NAC to the bulk Absλ of SOA samples was estimated from the mass concentration (ng m–3) and MAE (m2 g–1) of individual standards. Average contributions of identified NACs to the light absorption (Absλ) of SOA samples are listed in Tables S7–S11 for λ’s of 365 nm, 400 nm, 450 nm, 500 nm, and 550 nm. For simplicity, the average contributions of identified NACs to Absλ of SOA samples from different experiments at 365 nm, 400 nm, and 450 nm are visualized in Figures 3, S13, and S14, respectively. In Figure 3, the m-cresol/NOX SOA had the highest Abs365 contribution of all identified NACs (average ± standard deviation, 50.5 ± 15.8%), followed by benzene/NOX (28.0 ± 8.86%) and naphthalene/NOX (20.3 ± 8.01%) SOA; for these three SOAs, roughly 36.5 ± 15.8–47.4 ± 20.0% of Abs400 could be attributed to identified NACs (Figure S13). The biogenic SOA samples show weak light absorption (Figure 1) and comprise virtually no NACs under NOX and H2O2 conditions (Table S6 and Figure 2). The lack of aromaticity in the biogenic precursors and secondary reaction products presumably explains this observation.(36) Clearly, light-absorbing conjugated double bonds are retained in aromatic SOA as NACs; thus, NAC compositions and contributions to Absλ in the aromatic SOA samples are the focus henceforth.
Figure 2.
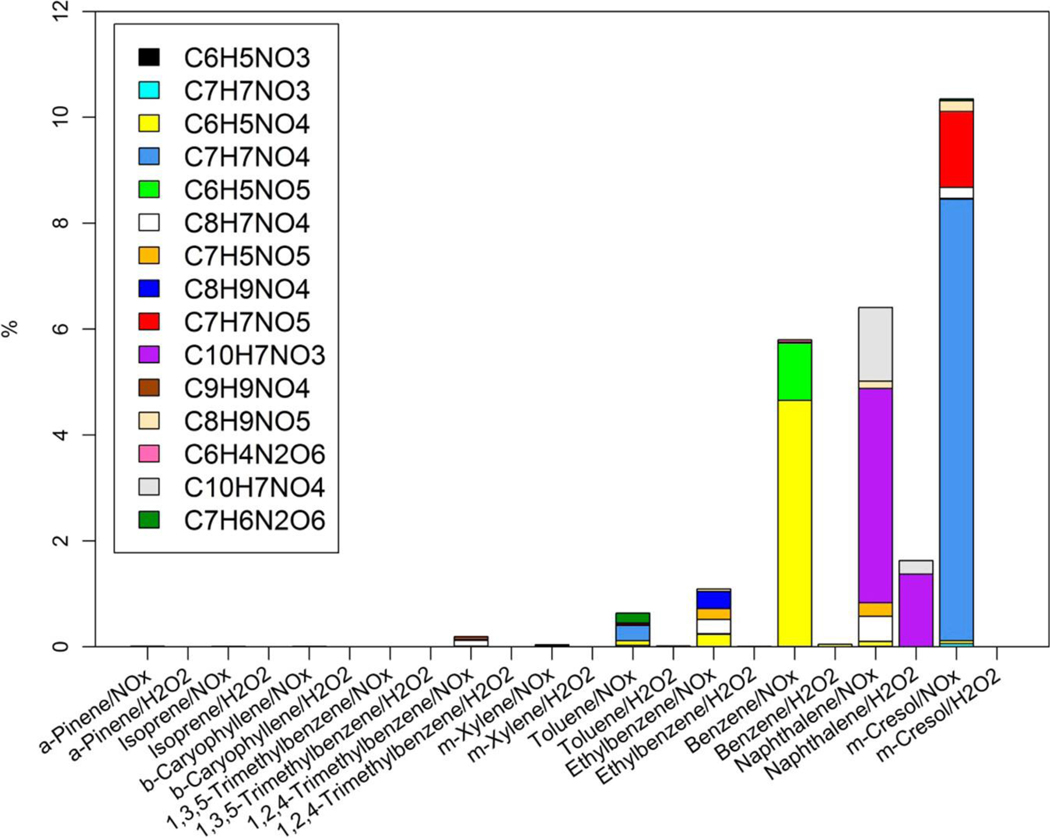
Average mass contributions (%) of nitroaromatic compounds to total SOA generated in different chamber experiments.
Figure 3.
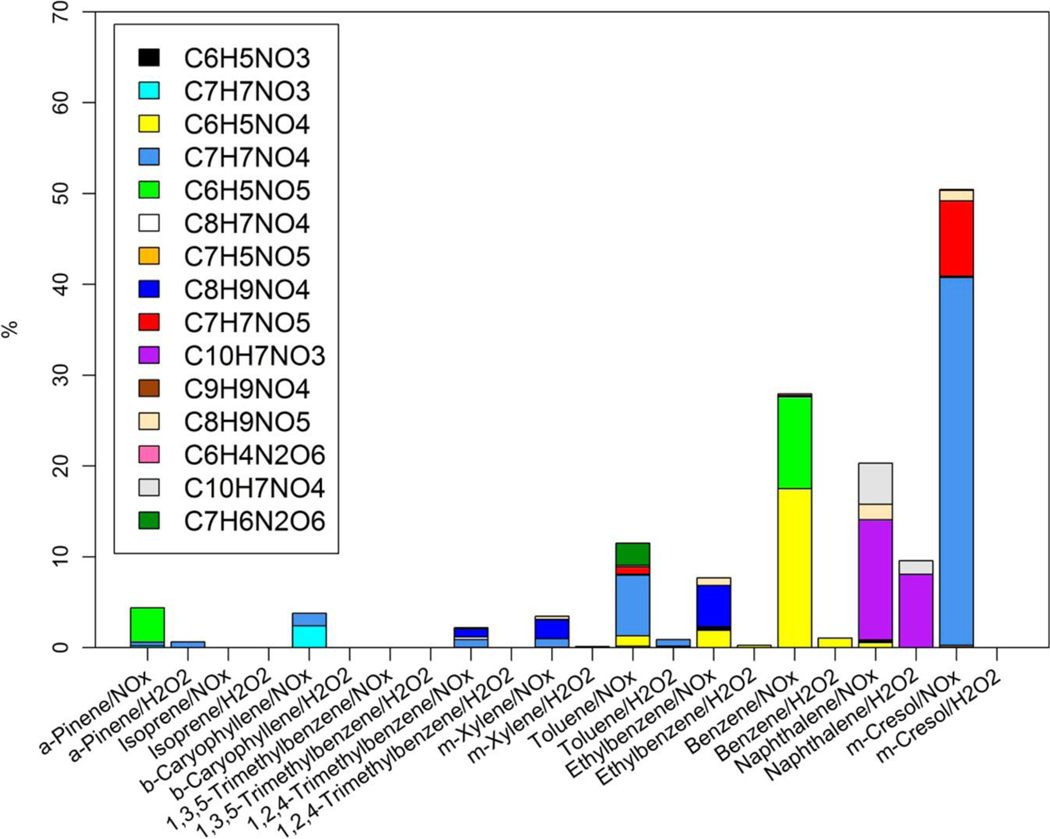
Average contributions (%) of nitroaromatic compounds absorption to the light absorption coefficients of SOA extracts at 365 nm (Abs365) for different chamber experiments.
NAC Composition in Aromatic SOAs
For the m-cresol/NOX experiment, C7H7NO4 products dominate identified NACs (Figure 2), accounting for 8.34 ± 5.87% of the total SOA (Table S6), consistent with the observations (9.9%) of Iinuma et al.(37) Three structural isomers are observed for C7H7NO4 with the strongest EIC signal intensity (∼106) and light absorption (DAD signal at 365 nm; Figure S2a-c). These three compounds show similar MS/MS spectra (Figure S2A-C). In the MS/MS spectra, the m/z 137 ion explains the loss of H and NO ([M–H]−, 168), and the m/z 109 ion explains continuous CO loss possibly due to aromatic ring opening. Iinuma et al.(37) identified three C7H7NO4 isomers using authentic standards in field and m-cresol/NOX photo-oxidation samples. Their separation and analytical methodology is similar to our own, so the elution order of the three C7H7NO4 isomers peaks in the EIC is also presumably similar. According to Iinuma et al.,(37) the C7H7NO4 signals at 8.46, 8.55, and 9.22 min in Figure S2b are 4-methyl-5-nitrocatechol, 3-methyl-5-nitrocatechol, and 3-methyl-6-nitrocatechol, respectively. In our study, three C7H7NO5 isomers were also identified, and their MS/MS spectra showed abundant m/z 125 and 153 ions (Figure S3a, A-C). Comparing to the MS/MS spectra of 4-nitrocatechol standard and C7H7NO4 (Figures S1C, S2A-C), the C7H7NO5 compounds should have a nitrocatechol skeleton with either one methyl and one hydroxyl group or one hydroxymethyl group. The three C7H7NO5 isomers also have discernible light absorption at 365 nm (Figure S3b). Due to the weak EIC signal intensity for the C8H9NO5 compound eluting at 4.14 min (∼103), the MS/MS spectra are only available for C8H9NO5 compounds at 8.51 and 9.27 min (Figure S4). After losing one CH3 and one NO2, the fragmentation patterns of C8H9NO5 compounds are similar as C7H7NO4 with abundant m/z ions of 137 and 109. Thus, the C8H9NO5 compounds may have a methyl nitrocatechol skeleton with either one methyl and one hydroxyl group or one hydroxymethyl group. None of the 15 identified NACs is observed in m-cresol/H2O2 samples.
Nitronaphthol and hydroxyl nitronaphthol were identified in naphthalene SOA under both NOX and H2O2 conditions (Figure 2) from their predicted formula and by comparing their MS/MS spectra (Figure S5A-C) to the reference compound (2-nitro-1-naphthol, Figure S1H). These two compounds accounted for 5.44 ± 1.73% and 1.63 ± 1.83% of the naphthalene SOA under NOX and H2O2 conditions, respectively. However, their light absorption signals at 365 nm could hardly be resolved from background noise in the UV/vis chromatogram (Figure S5c). This might be ascribed to their low concentrations in the sample extract, as reflected by their weak EIC signal intensity (Figure S5a,b). Among all the chamber experiments with H2O2 as the oxidant, only naphthalene SOA contained a significant amount (mass contribution >1%) of NACs. Updyke et al.(24) observed strong light-absorbing SOA at visible wavelengths from naphthalene/H2O2 experiments. They also demonstrated that the exposure of SOA to gaseous NH3 (RH ∼ 100%, 72 h) can increase SOA absorption. In the current work, the strong light absorption and high concentrations of NACs observed for naphthalene/H2O2 samples might be explained as follows: the trace amount of residue NOX (<10 ppb) or nitrogen containing substance in the chamber tends to react with naphthalene in a short-term; the interactions between gas phase NH3 or dissolved NH4+ from seed aerosols and naphthalene/H2O2 SOA lead to NAC generation. However, the latter might be less likely and warrants further study, since the photochemical reactions of naphthalene in the current work take place under dry conditions (RH < 20%) and the residence time of SOA is short (4 h), and trivial NACs were observed in other VOC/H2O2 reactions.
In the benzene/NOX SOA sample, the C6H5NO4 and C6H5NO5 dominated the composition of NACs (Figure 2), contributing 4.65 ± 3.78% and 1.08 ± 0.39% to the total SOA mass on average (Table S6). Referring to the EIC and MS/MS spectrum of 4-nitrocatechol (Figure S1c, C), the C6H5NO4 compound could be identified as 4-nitrocatechol; the C6H5NO5 compound should contain a nitrocatechol skeleton with an extra hydroxyl group on the benzene ring. These two compounds also had strong light absorption from the UV/vis chromatogram (Figure S6c). In Figure S7a, the C6H5NO3 peak at 8.27 min was identified as 4-nitrophenol using standard reagent (Figure S1a, A), and the peak at 8.54 min might be a structural isomer of 4-nitrophenol. The C6H4N2O6 compound contains two nitro groups based on its fragmentation pattern (Figure S7B). Among the eight benzene/NOX samples analyzed in this work, four were collected from the chamber experiments with added methyl nitrite (CH3ONO), and the other four were from the experiment without methyl nitrite. As shown in Figure S12, the presence of methyl nitrite did not change the composition of NACs in SOA products, as compared with the experiment with only benzene, but it might lead to a decrease in SOA yield (Table S1) and the mass contribution of C6H5NO4 to total SOA.
In Figure S8a, three C8H9NO4 isomers are observed in the ethylbenzene/NOX sample. In their MS/MS spectra, the compound eluting at 8.27 min had the dominant ions of m/z 138 and 108 (Figure S8A); the two C8H9NO4 compounds eluting at 9.59 and 10.22 min had the same dominant ions (m/z 109 and 137; Figure S8B, C) as methyl nitrocatechols (Figure S2A-C) and were postulated as ethyl nitrocatechols. In contrast to the latter two C8H9NO4 isomers, the C8H9NO4 compound eluting at 8.27 min might experience ring opening (loss of CO) before the loss of NO during the fragmentation. Three carboxylic acid-like compounds (C8H7NO4, C7H5NO5, and C9H9NO4) were observed for the ethylbenzene/NOX sample (Figure S9). These compounds have low EIC signal intensity and are more fragile than nitrophenolic compounds with the CID fragmentation, so their MS/MS spectra were not available in the current work. For the ethylbenzene/NOX experiment, the C8H9NO4 (0.32 ± 0.26%), C8H7NO4 (0.27 ± 0.21%), C6H5NO4 (0.23 ± 0.10%), and C7H5NO5 (0.21 ± 0.095%) compounds had significant and comparable mass contribution to total SOA products (Table S6).
In Figure S10a, the C7H7NO3 compound eluting at 9.74 min is identified, using standard reagent (Figure S1b, B), as 2-methyl-4-nitrophenol, which dominates the C7H7NO3 isomers from the toluene/NOX sample. This is consistent with the observation of toluene/NOX products from Sato et al.(38) The C7H7NO4 isomers from toluene/NOX SOA eluting during 8.43–9.20 min in Figure S10b are the same as those in Figure S2b and could be identified as methyl nitrocatechols. The MS/MS spectrum for the C7H7NO4 compound eluting at 6.18 min had the same dominant ions (m/z 108 and 138; Figure S10B) as the C8H9NO4 compound from ethylbenze/NOX SOA eluting at 8.27 min (Figure S8A), indicating a common molecular skeleton. The retention times and fragmentation patterns of the C7H6N2O6 compounds (Figure S10c, C) are different from the reference compound (4,6-dinitromethylresorcinol; Figure S1j, J), but the functional groups composition on the benzene ring may be similar, including two nitro, two hydroxyl, and one methyl groups. Among all the NACs identified for toluene/NOX SOA, C7H7NO4 had the highest average mass contribution (0.27 ± 0.17%, Table S6), followed by C7H6N2O6 (0.19 ± 0.093%) and C6H5NO4 (0.088 ± 0.036%). The identified NACs in the SOA products generated with the other aromatic VOCs (m-xylene, 1,2,4-trimethylbenzene, and 1,3,5-trimethylbenzene) in this work had low mass contributions (∼0.2%, Table S6).
Several previous studies have investigated the identification of NACs in aerosols from chamber reactions(26, 37–39) and biomass burning emissions.(40, 41) These studies demonstrated that the NACs could be generated from atmospheric processing of aromatic VOCs and raised the possibility of using the identified NACs as source markers for BrC. However, both biomass burning and fossil fuel combustion could emit a complex mixture of gas-phase aromatic VOCs (e.g., benzene, toluene),(42–47) serving as precursors for NACs. It is impractical to link these NACs to specific pollution sources (e.g., biomass burning), although NACs can be linked with specific aromatic precursors. Moreover, the BrC chromophores (NACs) identified in this work may be subject to aging and photobleaching in the atmosphere. Lin et al.(41) and Forrister et al.(48) examined the light absorption decay of OM from biomass burning and obtained a half-life of ∼15 h. The photobleaching of secondary BrC from reactions of limonene/O3 SOA + NH3 and glyoxal or methyl glyoxal + ammonium sulfate can be quite fast (a few minutes to hours).(49, 50) While the light absorption decay for some NACs (e.g., 4-nitrophenol and 4-nitrocatechol) and naphthalene/NOX SOA in water solutions under UV photolysis (without the addition of H2O2) is much slower, with a half-life comparable to biomass burning OM.(49, 50) Due to the longer half-lives of NACs, several NACs identified in this work (e.g., nitrocatechol, methylnitrocatechol) have been observed in ambient aerosols and cloudwater with the highest concentrations of 1–3 ng m–3 for individual compounds.(32, 35)
The chamber experiments performed here are limited (e.g., light intensity and spectra, temperature and humidity variation, gas and particle composition) in representing the atmosphere, and the identified BrC chromophores are vulnerable to photobleaching. However, the emissions of aromatic VOCs and NOX are prominent in urban areas, and the NACs identified in this work could be generated continuously during the daytime. Moreover, the lifetimes of these NACs are long enough for accumulation and detection in the atmosphere. Thus, the identified NACs in this work can serve as source tracers to represent secondary BrC chromophores. As shown in Figure 2, several aromatic precursors have characteristic NAC compositions, and some NACs are uniquely generated from specific aromatic VOCs (e.g., C7H6N2O6 in toluene/NOX SOA). Therefore, in future ambient analysis or source apportionment studies, the identified NACs with high mass contributions (e.g, C10H7NO3 for naphthalene) to total SOA in this study could be used as source markers for secondary BrC derived from specific aromatic VOCs in urban areas, when and where biomass burning is less prominent; the monitoring data of aromatic VOCs can serve as supporting evidence, benefiting the identification of the dominant VOC precursor. Those NACs might also be used to estimate the SOA contributions of aromatic VOCs to ambient organic aerosol with the organic tracer-based method by Kleindienst et al.(51) However, the associated uncertainties due to the variability in mass contribution of specific NACs in different SOA samples and the influence from biomass burning and photobleaching need further study.
Contributions of NACs to Absλ
In general, the NACs in aromatic SOA samples had higher contributions to Abs365 than their mass contributions (Figures 2 and 3), suggesting that most of the identified NACs are strong BrC chromophores. Similar to their mass compositions, the C7H7NO4 and C7H7NO5 compounds had the largest contributions to Abs365 (40.5 ± 15.2% and 8.25 ± 1.79%) in m-cresol/NOX SOA; C10H7NO3 and C10H7NO4 were the characteristic compounds in Abs365 contribution (NOX 13.2 ± 5.62% and 4.52 ± 1.56%; H2O2 8.09 ± 8.84% and 1.49 ± 1.61%) for naphthalene SOA; the C6H5NO4 and C6H5NO5 compounds lead the contributions to Abs365 (17.5 ± 9.28% and 10.1 ± 4.22%) in benzene/NOX SOA; the C7H7NO4 and C7H6N2O6 compounds are the two major Abs365 contributors (6.68 ± 3.09% and 2.43 ± 1.08%) in toluene/NOX SOA. In this work, 4-nitrocatechol, 2-methyl-4-nitroresorcinol, and 2-nitrophloroglucinol were used as the reference compounds for C6H5NO4, C7H7NO4, C6H5NO5, and C7H7NO5 in estimating their Abs365 contributions. All three compounds have peak absorption at around 350 nm (Figure S11), explaining the shoulders observed in the UV/vis spectra for benzene/NOX and m-cresol/NOX samples in Figure 1i,k. In ethylbenzene/NOX samples, the C8H7NO4 and C7H5NO5 compounds had comparable mass contributions to C6H5NO4 and C8H9NO4 (Figure 2), but the contributions to Abs365 were dominated by the latter two compounds (Figure 3). This is because the Abs365 contribution from C8H7NO4 and C7H5NO5 was estimated using two carboxylic acid compounds (3,5-dimethyl-4-nitrobenzoic acid and 2-methyl-5-nitrobenzoic acid) with very low absorption at wavelengths above 350 nm (Figure S11).
At 400 nm, the light absorption of 2-nitro-1-naphthol and 2-nitrophloroglucinol, two surrogate compounds used to estimate the light absorption of C10H7NO3, C10H7NO4, C6H5NO5, and C7H7NO5, respectively, almost reaches the maximum in their UV/vis spectra (Figure S11). While the light absorption of the reference compounds (4-nitrocatechol and 2-methyl-4-nitroresorcinol) for C6H5NO4 and C7H7NO4 is much lower at 400 nm than at 365 nm (Figure S11). As a result, the NACs generated from naphthalene/NOX experiments had the highest average contribution (47.4 ± 20.0%) to Abs400, followed by m-cresol/NOX (39.3 ± 2.94%) and benzene/NOX (36.5 ± 9.56%) samples (Figure S13). Since all the reference compounds have their maximum absorption below 450 nm, the Abs450 contributed by NACs (<20%) in different experiments was lower than their contributions to Abs400 by more than 50%. At 500 and 550 nm, very few identified compounds (e.g., C6H5NO4, C6H4N2O6) had contributions to the light absorption of sample extracts, and the sum of their contributions was no more than 3% of the total SOA (Tables S10 and S11). Zhang et al.(32) and Teich et al.(52) measured the light absorption from NACs in ambient particles, which accounted for no more than 10% (0.02–9.86%) of the aqueous extract light absorption at 365–370 nm, much lower than the total NACs contribution to Abs365 of benzene/NOX, naphthalene/NOX, and m-cresol/NOX SOA in this work. This might be due to the fact that the ambient SOA is derived from the photochemical reaction of a complex mixture of VOCs, and most of them (e.g., biogenic VOC, toluene, and trimethylbenzene) have weak potential in generating NACs. Additionally, atmospheric processes (e.g., aging and photobleaching) can remove BrC chromophores in the atmosphere.
To understand if the difference in bulk MAEλ observed in Figure 1 between NOX and H2O2 conditions is caused by the difference in NACs production, the average contributions of NACs to MAE at 365, 400, and 450 nm for the different experiments were calculated using eq 1. The difference in bulk MAE and MAE contributions from NACs between aromatic/NOX and aromatic/H2O2 samples at 365, 400, and 450 nm is provided in Table S12. The difference in MAE contributions from NACs explained the highest fraction (59.0%) of the bulk MAE difference between m-cresol/NOX and m-cresol/H2O2 samples at 365 nm. However, for other reactions, no more than 50% of the bulk MAE differences between NOX and H2O2 conditions could be explained by the difference in MAE contributions from NACs, especially at long wavelengths (∼450 nm). For 1,3,5-trimethylbenzene, 1,2,4-trimethylbenzene, and m-xylene, the explainable fractions could be even lower (<10%). However, these three aromatic VOCs show no significant differences (Table S5) in bulk MAE365 between NOX and H2O2 conditions. These results suggested that besides the identified NACs in this work, other light-absorbing products could be generated in reactions with NOX and contributing to the differences in bulk MAE between NOX and H2O2 experiments. Lin et al.(26, 41) investigated the molecular characterization of BrC in biomass burning aerosols and toluene/NOX SOA. They identified a list of nitrogen-containing compounds with carbon number up to 48; Di Lorenzo et al.(53) found that most BrC absorption was due to high molecular weight (HMW > 500) molecules in both fresh and aged biomass burning aerosols. As such, the unexplained differences in bulk MAE between NOX and H2O2 experiments might be ascribed to unidentified nitrogen-containing compounds and HMW components with extremely low volatility.
Atmospheric Implications
The stronger light absorption of aromatic SOA compared to biogenic SOA reveals the potential significance of aromatic VOCs as important precursors for secondary BrC; the comparisons of bulk MAE spectra derived from SOA extracts of NOX versus H2O2 conditions imply that the secondary BrC should have more impact on aerosol absorption in urban atmospheres with abundant aromatic VOCs and NOX. Fifteen NAC chemical formulas were derived from aromatic SOA samples. The average total amount of characteristic compounds in each sample can explain up to 10% of SOA mass, which is greater than other anthropogenic tracers (e.g., phthalic acid, 2,3-dihydroxy-4-oxopentanoic acid) in aromatic SOA reported by Kleindienst et al.(51, 54) Some of the identified NACs are predominantly associated with specific aromatic VOCs (e.g., C6H5NO5 for benzene, C10H7NO3 for naphthalene, and C7H7NO5 for m-cresol), suggesting potential application of these compounds in ambient BrC or PM source apportionment studies for the estimation of aromatic SOA contribution. However, the uncertainties associated with mass contribution, as induced by the application of surrogate compounds and the difference between reactions (residence time) in chamber and that in the atmosphere, need to be addressed in future work; the influences from biomass burning and atmospheric processing also need to be evaluated. Up to 50% of bulk Absλ and MAEλ could be attributed to the characteristic NACs at 365 and 400 nm for m-cresol/NOX, benzene/NOX, and naphthalene/NOX SOA, which had the strongest light absorption among all types of SOA, indicating that NACs could be important BrC chromophores in the atmosphere. Little light absorption of SOA extracts could be explained by identified NACs at wavelengths longer than 450 nm, and further work is needed to elucidate the structures and quantity of HMW BrC components in aromatic SOA with strong chromophores in the visible region.
The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
Supplementary Material
Acknowledgment
The U.S. Environmental Protection Agency, through its Office of Research and Development, funded and collaborated in the research described here under Contract EP-C-15-008 to Jacobs Technology, Inc.
Supporting research data used in the writing of this article may be obtained upon request to Amara Holder (holder.amara@epa.gov).
Footnotes
The authors declare no competing financial interest.
References
- 1.IPCC. Assessment Record: Climate Change 2007; Cambridge University Press: New York, 2007. [Google Scholar]
- 2.Andreae MO; Ramanathan V Climate’s Dark Forcings Science 2013, 340 (6130) 280–281DOI: 10.1126/science.1235731 [DOI] [PubMed] [Google Scholar]
- 3.Sokolik IN; Winker DM; Bergametti G; Gillette DA; Carmichael G; Kaufman YJ; Gomes L; Schuetz L; Penner JE Introduction to special section: Outstanding problems in quantifying the radiative impacts of mineral dust J. Geophys. Res. 2001, 106 (D16) 18015–18027DOI: 10.1029/2000JD900498 [DOI] [Google Scholar]
- 4.Bond TC; Bergstrom RW Light absorption by carbonaceous particles: An investigative review Aerosol Sci. Technol. 2006, 40 (1) 27–67DOI: 10.1080/02786820500421521 [DOI] [Google Scholar]
- 5.Bond TC; Doherty SJ; Fahey DW; Forster PM; Berntsen T; DeAngelo BJ; Flanner MG; Ghan S; Kärcher B; Koch D; Kinne S; Kondo Y; Quinn PK; Sarofim MC; Schultz MG; Schulz M; Venkataraman C; Zhang H; Zhang S; Bellouin N; Guttikunda SK; Hopke PK; Jacobson MZ; Kaiser JW; Klimont Z; Lohmann U; Schwarz JP; Shindell D; Storelvmo T; Warren SG; Zender CS Bounding the role of black carbon in the climate system: A scientific assessment J. Geophys. Res. 2013, 118 (11) 5380–5552DOI: 10.1002/jgrd.50171 [DOI] [Google Scholar]
- 6.Lafon S; Sokolik IN; Rajot JL; Caquineau S; Gaudichet A. Characterization of iron oxides in mineral dust aerosols: Implications for light absorption J. Geophys. Res. 2006, 111 (D21) D21207DOI: 10.1029/2005JD007016 [DOI] [Google Scholar]
- 7.Chung SH; Seinfeld JH Global distribution and climate forcing of carbonaceous aerosols J. Geophys. Res. 2002, 107 (D19) AAC 14–1– AAC 14–33DOI: 10.1029/2001JD001397 [DOI] [Google Scholar]
- 8.Myhre G; Samset BH; Schulz M; Balkanski Y; Bauer S; Berntsen TK; Bian H; Bellouin N; Chin M; Diehl T; Easter RC; Feichter J; Ghan SJ; Hauglustaine D; Iversen T; Kinne S; Kirkevåg A; Lamarque JF; Lin G; Liu X; Lund MT; Luo G; Ma X; van Noije T; Penner JE; Rasch PJ; Ruiz A; Seland Ø; Skeie RB; Stier P; Takemura T; Tsigaridis K; Wang P; Wang Z; Xu L; Yu H; Yu F; Yoon JH; Zhang K; Zhang H; Zhou C. Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations Atmos. Chem. Phys. 2013, 13 (4) 1853–1877DOI: 10.5194/acp-13-1853-2013 [DOI] [Google Scholar]
- 9.Saleh R; Hennigan CJ; McMeeking GR; Chuang WK; Robinson ES; Coe H; Donahue NM; Robinson AL Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions Atmos. Chem. Phys. 2013, 13 (15) 7683–7693DOI: 10.5194/acp-13-7683-2013 [DOI] [Google Scholar]
- 10.Saleh R; Robinson ES; Tkacik DS; Ahern AT; Liu S; Aiken AC; Sullivan RC; Presto AA; Dubey MK; Yokelson RJ; Donahue NM; Robinson AL Brownness of organics in aerosols from biomass burning linked to their black carbon content Nat. Geosci. 2014, 7 (9) 647–650DOI: 10.1038/ngeo2220 [DOI] [Google Scholar]
- 11.Washenfelder RA; Attwood AR; Brock CA; Guo H; Xu L; Weber RJ; Ng NL; Allen HM; Ayres BR; Baumann K; Cohen RC; Draper DC; Duffey KC; Edgerton E; Fry JL; Hu WW; Jimenez JL; Palm BB; Romer P; Stone EA; Wooldridge PJ; Brown SS Biomass burning dominates brown carbon absorption in the rural southeastern United States Geophys. Res. Lett. 2015, 42 (2) 653–664DOI: 10.1002/2014GL062444 [DOI] [Google Scholar]
- 12.Zhang X; Kim H; Parworth CL; Young DE; Zhang Q; Metcalf AR; Cappa CD Optical properties of wintertime aerosols from residential wood burning in Fresno, CA: Results from DISCOVER-AQ 2013 Environ. Sci. Technol. 2016, 50 (4) 1681–1690DOI: 10.1021/acs.est.5b04134 [DOI] [PubMed] [Google Scholar]
- 13.Yang M; Howell SG; Zhuang J; Huebert BJ Attribution of aerosol light absorption to black carbon, brown carbon, and dust in China – interpretations of atmospheric measurements during EAST-AIRE Atmos. Chem. Phys. 2009, 9 (6) 2035–2050DOI: 10.5194/acp-9-2035-2009 [DOI] [Google Scholar]
- 14.Chung CE; Ramanathan V; Decremer D. Observationally constrained estimates of carbonaceous aerosol radiative forcing Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (29) 11624–11629DOI: 10.1073/pnas.1203707109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Y; Ramanathan V; Kotamarthi VR Brown carbon: a significant atmospheric absorber of solar radiation? Atmos. Chem. Phys. 2013, 13 (17) 8607–8621DOI: 10.5194/acp-13-8607-2013 [DOI] [Google Scholar]
- 16.Wang X; Heald CL; Ridley DA; Schwarz JP; Spackman JR; Perring AE; Coe H; Liu D; Clarke AD Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon Atmos. Chem. Phys. 2014, 14 (20) 10989–11010DOI: 10.5194/acp-14-10989-2014 [DOI] [Google Scholar]
- 17.Nakayama T; Matsumi Y; Sato K; Imamura T; Yamazaki A; Uchiyama A. Laboratory studies on optical properties of secondary organic aerosols generated during the photooxidation of toluene and the ozonolysis of α-pinene J. Geophys. Res. 2010, 115 (D24) D24204DOI: 10.1029/2010JD014387 [DOI] [Google Scholar]
- 18.Lambe AT; Cappa CD; Massoli P; Onasch TB; Forestieri SD; Martin AT; Cummings MJ; Croasdale DR; Brune WH; Worsnop DR; Davidovits P. Relationship between oxidation level and optical properties of secondary organic aerosol Environ. Sci. Technol. 2013, 47 (12) 6349–6357DOI: 10.1021/es401043j [DOI] [PubMed] [Google Scholar]
- 19.Liu PF; Abdelmalki N; Hung HM; Wang Y; Brune WH; Martin ST Ultraviolet and visible complex refractive indices of secondary organic material produced by photooxidation of the aromatic compounds toluene and m-xylene Atmos. Chem. Phys. 2015, 15 (3) 1435–1446DOI: 10.5194/acp-15-1435-2015 [DOI] [Google Scholar]
- 20.Liu J; Lin P; Laskin A; Laskin J; Kathmann SM; Wise M; Caylor R; Imholt F; Selimovic V; Shilling JE Optical properties and aging of light-absorbing secondary organic aerosol Atmos. Chem. Phys. 2016, 16 (19) 12815–12827DOI: 10.5194/acp-16-12815-2016 [DOI] [Google Scholar]
- 21.Nguyen TB; Lee PB; Updyke KM; Bones DL; Laskin J; Laskin A; Nizkorodov SA Formation of nitrogen- and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols J. Geophys. Res. 2012, 117 (D1) D01207DOI: 10.1029/2011JD016944 [DOI] [Google Scholar]
- 22.Lee AKY; Zhao R; Li R; Liggio J; Li S-M; Abbatt JPD Formation of light absorbing organo-nitrogen species from evaporation of droplets containing glyoxal and ammonium sulfate Environ. Sci. Technol. 2013, 47 (22) 12819–12826DOI: 10.1021/es402687w [DOI] [PubMed] [Google Scholar]
- 23.Lin P; Laskin J; Nizkorodov SA; Laskin A. Revealing brown carbon chromophores produced in reactions of methylglyoxal with ammonium sulfate Environ. Sci. Technol. 2015, 49 (24) 14257–14266DOI: 10.1021/acs.est.5b03608 [DOI] [PubMed] [Google Scholar]
- 24.Updyke KM; Nguyen TB; Nizkorodov SA Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors Atmos. Environ. 2012, 63, 22–31DOI: 10.1016/j.atmosenv.2012.09.012 [DOI] [Google Scholar]
- 25.Lin Y-H; Budisulistiorini SH; Chu K; Siejack RA; Zhang H; Riva M; Zhang Z; Gold A; Kautzman KE; Surratt JD Light-absorbing oligomer formation in secondary organic aerosol from reactive uptake of isoprene epoxydiols Environ. Sci. Technol. 2014, 48 (20) 12012–12021DOI: 10.1021/es503142b [DOI] [PubMed] [Google Scholar]
- 26.Lin P; Liu JM; Shilling JE; Kathmann SM; Laskin J; Laskin A. Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene Phys. Chem. Chem. Phys. 2015, 17 (36) 23312–23325DOI: 10.1039/C5CP02563J [DOI] [PubMed] [Google Scholar]
- 27.Edney EO; Kleindienst TE; Jaoui M; Lewandowski M; Offenberg JH; Wang W; Claeys M. Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOX/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States Atmos. Environ. 2005, 39 (29) 5281–5289DOI: 10.1016/j.atmosenv.2005.05.031 [DOI] [Google Scholar]
- 28.Kleindienst TE; Conver TS; McIver CD; Edney EO Determination of secondary organic aerosol products from the photooxidation of toluene and their implications in ambient PM2.5 J. Atmos. Chem. 2004, 47 (1) 79–100DOI: 10.1023/B:JOCH.0000012305.94498.28 [DOI] [Google Scholar]
- 29.Kleindienst TE; Edney EO; Lewandowski M; Offenberg JH; Jaoui M. Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2 Environ. Sci. Technol. 2006, 40 (12) 3807–3812DOI: 10.1021/es052446r [DOI] [PubMed] [Google Scholar]
- 30.Hecobian A; Zhang X; Zheng M; Frank N; Edgerton ES; Weber RJ Water-soluble organic aerosol material and the light-absorption characteristics of aqueous extracts measured over the Southeastern United States Atmos. Chem. Phys. 2010, 10 (13) 5965–5977DOI: 10.5194/acp-10-5965-2010 [DOI] [Google Scholar]
- 31.Chen Y; Bond TC Light absorption by organic carbon from wood combustion Atmos. Chem. Phys. 2010, 10 (4) 1773–1787DOI: 10.5194/acp-10-1773-2010 [DOI] [Google Scholar]
- 32.Zhang X; Lin Y-H; Surratt JD; Weber RJ Sources, Composition and Absorption Ångström Exponent of Light-absorbing Organic Components in Aerosol Extracts from the Los Angeles Basin Environ. Sci. Technol. 2013, 47 (8) 3685–3693DOI: 10.1021/es305047b [DOI] [PubMed] [Google Scholar]
- 33.Metcalf AR; Loza CL; Coggon MM; Craven JS; Jonsson HH; Flagan RC; Seinfeld JH Secondary organic aerosol coating formation and evaporation: Chamber studies using black carbon seed aerosol and the single-particle soot photometer Aerosol Sci. Technol. 2013, 47 (3) 326–347DOI: 10.1080/02786826.2012.750712 [DOI] [Google Scholar]
- 34.Song C; Gyawali M; Zaveri RA; Shilling JE; Arnott WP Light absorption by secondary organic aerosol from α-pinene: Effects of oxidants, seed aerosol acidity, and relative humidity J. Geophys. Res. 2013, 118 (20) 11741–11749DOI: 10.1002/jgrd.50767 [DOI] [Google Scholar]
- 35.Desyaterik Y; Sun Y; Shen X; Lee T; Wang X; Wang T; Collett JL Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China J. Geophys. Res. 2013, 118 (13) 7389–7399DOI: 10.1002/jgrd.50561 [DOI] [Google Scholar]
- 36.Gratien A; Johnson SN; Ezell MJ; Dawson ML; Bennett R; Finlayson-Pitts BJ Surprising formation of p-cymene in the oxidation of alpha-pinene in air by the atmospheric oxidants OH, O-3, and NO3 Environ. Sci. Technol. 2011, 45 (7) 2755–2760DOI: 10.1021/es103632b [DOI] [PubMed] [Google Scholar]
- 37.Iinuma Y; Böge O; Gräfe R; Herrmann H. Methyl-nitrocatechols: Atmospheric tracer compounds for biomass burning secondary organic aerosols Environ. Sci. Technol. 2010, 44 (22) 8453–8459DOI: 10.1021/es102938a [DOI] [PubMed] [Google Scholar]
- 38.Sato K; Hatakeyama S; Imamura T. Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition J. Phys. Chem. A 2007, 111 (39) 9796–9808DOI: 10.1021/jp071419f [DOI] [PubMed] [Google Scholar]
- 39.Sato K; Takami A; Kato Y; Seta T; Fujitani Y; Hikida T; Shimono A; Imamura T. AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: effects of chemical structure on SOA aging Atmos. Chem. Phys. 2012, 12 (10) 4667–4682DOI: 10.5194/acp-12-4667-2012 [DOI] [Google Scholar]
- 40.Claeys M; Vermeylen R; Yasmeen F; Gómez-González Y; Chi X; Maenhaut W; Mészáros T; Salma I. Chemical characterisation of humic-like substances from urban, rural and tropical biomass burning environments using liquid chromatography with UV/vis photodiode array detection and electrospray ionisation mass spectrometry Environ. Chem. 2012, 9 (3) 273–284DOI: 10.1071/EN11163 [DOI] [Google Scholar]
- 41.Lin P; Aiona PK; Li Y; Shiraiwa M; Laskin J; Nizkorodov SA; Laskin A. Molecular characterization of brown carbon in biomass burning aerosol particles Environ. Sci. Technol. 2016, 50 (21) 11815–11824DOI: 10.1021/acs.est.6b03024 [DOI] [PubMed] [Google Scholar]
- 42.Martins LD; Andrade MF; Freitas ED; Pretto A; Gatti LV; Albuquerque ÉL; Tomaz E; Guardani ML; Martins MHRB; Junior OMA Emission factors for gas-powered vehicles traveling through road tunnels in São Paulo, Brazil Environ. Sci. Technol. 2006, 40 (21) 6722–6729DOI: 10.1021/es052441u [DOI] [PubMed] [Google Scholar]
- 43.Nelson PF; Tibbett AR; Day SJ Effects of vehicle type and fuel quality on real world toxic emissions from diesel vehicles Atmos. Environ. 2008, 42 (21) 5291–5303DOI: 10.1016/j.atmosenv.2008.02.049 [DOI] [Google Scholar]
- 44.Lewis AC; Evans MJ; Hopkins JR; Punjabi S; Read KA; Purvis RM; Andrews SJ; Moller SJ; Carpenter LJ; Lee JD; Rickard AR; Palmer PI; Parrington M. The influence of biomass burning on the global distribution of selected non-methane organic compounds Atmos. Chem. Phys. 2013, 13 (2) 851–867DOI: 10.5194/acp-13-851-2013 [DOI] [Google Scholar]
- 45.George IJ; Hays MD; Herrington JS; Preston W; Snow R; Faircloth J; George BJ; Long T; Baldauf RW Effects of cold temperature and ethanol content on VOC emissions from light-duty gasoline vehicles Environ. Sci. Technol. 2015, 49 (21) 13067–13074DOI: 10.1021/acs.est.5b04102 [DOI] [PubMed] [Google Scholar]
- 46.George IJ; Hays MD; Snow R; Faircloth J; George BJ; Long T; Baldauf RW Cold temperature and biodiesel fuel effects on speciated emissions of volatile organic compounds from diesel trucks Environ. Sci. Technol. 2014, 48 (24) 14782–14789DOI: 10.1021/es502949a [DOI] [PubMed] [Google Scholar]
- 47.Gilman JB; Lerner BM; Kuster WC; Goldan PD; Warneke C; Veres PR; Roberts JM; de Gouw JA; Burling IR; Yokelson RJ Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US Atmos. Chem. Phys. 2015, 15 (24) 13915–13938DOI: 10.5194/acp-15-13915-2015 [DOI] [Google Scholar]
- 48.Forrister H; Liu J; Scheuer E; Dibb J; Ziemba L; Thornhill KL; Anderson B; Diskin G; Perring AE; Schwarz JP; Campuzano-Jost P; Day DA; Palm BB; Jimenez JL; Nenes A; Weber RJ Evolution of brown carbon in wildfire plumes Geophys. Res. Lett. 2015, 42 (11) 4623–4630DOI: 10.1002/2015GL063897 [DOI] [Google Scholar]
- 49.Lee HJ; Aiona PK; Laskin A; Laskin J; Nizkorodov SA Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon Environ. Sci. Technol. 2014, 48 (17) 10217–10226DOI: 10.1021/es502515r [DOI] [PubMed] [Google Scholar]
- 50.Zhao R; Lee AKY; Huang L; Li X; Yang F; Abbatt JPD Photochemical processing of aqueous atmospheric brown carbon Atmos. Chem. Phys. 2015, 15 (11) 6087–6100DOI: 10.5194/acp-15-6087-2015 [DOI] [Google Scholar]
- 51.Kleindienst TE; Jaoui M; Lewandowski M; Offenberg JH; Lewis CW; Bhave PV; Edney EO Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location Atmos. Environ. 2007, 41 (37) 8288–8300DOI: 10.1016/j.atmosenv.2007.06.045 [DOI] [Google Scholar]
- 52.Teich M; van Pinxteren D; Wang M; Kecorius S; Wang Z; Müller T; Močnik G; Herrmann H. Contributions of nitrated aromatic compounds to the light absorption of water-soluble and particulate brown carbon in different atmospheric environments in Germany and China Atmos. Chem. Phys. 2017, 17 (3) 1653–1672DOI: 10.5194/acp-17-1653-2017 [DOI] [Google Scholar]
- 53.Di Lorenzo RA; Washenfelder RA; Attwood AR; Guo H; Xu L; Ng NL; Weber RJ; Baumann K; Edgerton E; Young CJ Molecular-size-separated brown carbon absorption for biomass-burning aerosol at multiple field sites Environ. Sci. Technol. 2017, 51 (6) 3128–3137DOI: 10.1021/acs.est.6b06160 [DOI] [PubMed] [Google Scholar]
- 54.Kleindienst TE; Jaoui M; Lewandowski M; Offenberg JH; Docherty KS The formation of SOA and chemical tracer compounds from the photooxidation of naphthalene and its methyl analogs in the presence and absence of nitrogen oxides Atmos. Chem. Phys. 2012, 12 (18) 8711–8726DOI: 10.5194/acp-12-8711-2012 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.