

Abstract
Background
Gene Regulatory Networks (GRNs) have been previously studied by using Boolean/multi-state logics. While the gene expression values are usually scaled into the range [0, 1], these GRN inference methods apply a threshold to discretize the data, resulting in missing information. Most of studies apply fuzzy logics to infer the logical gene-gene interactions from continuous data. However, all these approaches require an a priori known network structure.
Results
Here, by introducing a new probabilistic logic for continuous data, we propose a novel logic-based approach (called the LogicNet) for the simultaneous reconstruction of the GRN structure and identification of the logics among the regulatory genes, from the continuous gene expression data. In contrast to the previous approaches, the LogicNet does not require an a priori known network structure to infer the logics. The proposed probabilistic logic is superior to the existing fuzzy logics and is more relevant to the biological contexts than the fuzzy logics. The performance of the LogicNet is superior to that of several Mutual Information-based and regression-based tools for reconstructing GRNs.
Conclusions
The LogicNet reconstructs GRNs and logic functions without requiring prior knowledge of the network structure. Moreover, in another application, the LogicNet can be applied for logic function detection from the known regulatory genes-target interactions. We also conclude that computational modeling of the logical interactions among the regulatory genes significantly improves the GRN reconstruction accuracy.
Keywords: Gene regulatory network, Probabilistic logic, Fuzzy logic, Gene expression data, Bayesian information criterion (BIC), Bayes factor (BF)
Background
The reconstruction of the gene regulatory networks (GRNs) is an important problem in molecular biology, which attempts to represent the causality of regulatory processes. The use of high-throughput microarray technologies to generate gene expression data has significantly facilitated network studies. The DREAM (the Dialogue for Reverse Engineering Assessments and Methods) program was initiated to encourage researchers to develop robust computational tools to infer GRNs from gene expression data [1].
The computational tools for the GRN inference can be classified into different categories. Abstract techniques such as the Principle Component Analysis (PCA) and Mutual Information (MI) [2–7] between genes are largely data-driven models in which the correlations among gene expression data are modelled. At the other extreme, differential equation-based models highly rely on prior knowledge about the network structure and the regulatory interactions. However, the temporal and spatial dynamics of each interaction can be captured by these models [8–10].
The knowledge-based models could rely on the prior information, e.g., reference regulatory networks documented in the databases, and then these reference networks are trimmed based on their consistencies with the gene expressions [11–13]. The prior knowledge is useful for the inference due to the noisy data in the -omics technology. A few differential equation-based and Bayesian models are proposed to reconstruct the GRNs from time-series microarrays, but they do not infer the logics among regulatory genes [14–17].
In the middle between the two extremes, there are Bayesian models, and logic-based models [18–24]. Logic-based models apply either a Boolean logic [20, 21, 25] or a multi-state logic [26–28] to study a priori-specified GRNs by using discretized gene expression data. While the normalized gene expression levels vary in the interval [0, 1], it is assumed in the Boolean networks that each gene is either expressed or not. Boolean logics apply a threshold on the interval [0, 1] to discretize the gene expression levels, resulting in the missing information. To overcome this weakness of the Boolean and the multi-state logics, the fuzzy logic models have been proposed to study the networks from the continuous gene expression data [19, 22]. However, the fuzzy and the multi-state logics study only a network with an a priori-specified structure and do not reconstruct it. Here, we introduce a new logic for continuous data, rather than binary data, called the probabilistic continuous (PC) logic, and accordingly, we propose a logic-based algorithm to reconstruct the GRNs from the continuous gene expression data. This new algorithm, called the LogicNet, is superior to the current logic-based models from several perspectives and has the following properties:
The LogicNet relies on a new kind of logic applicable to continuous data, i.e., the PC logic, for modeling the cooperative, competitive and other types of logical interactions among genes. Regarding the reconstruction of the GRNs from the continuous gene expression data, the performance of the PC logic is superior to that of the fuzzy logic;
Contrary to the current logic-based models, which can analyze only the GRNs with an a priori known structure, the LogicNet requires no prior information or hypothesis about the network structure;
Using the continuous gene expression data in the interval [0, 1], the LogicNet reconstructs the GRN with directed and signed edges. Indeed, the LogicNet infers the underlying biochemical causalities of the regulatory interactions;
The LogicNet infers the underlying logical relationships, e.g., the cooperative (AND, OR), competitive (XOR), and any other types of relationships, among the regulatory genes of a target gene.
Altogether, the main feature of the LogicNet is to improve the current models with the logic detection and not to defeat them in terms of accuracies. To study the regulatory effect of other genes on a target gene, the LogicNet computes the likelihood function for each possible set of regulatory genes with a specified logical interaction. In the LogicNet, the expression levels of the target gene belonging to the interval [0, 1] are intuitively supposed to follow a beta distribution. The parameters of this distribution depend on the type of the logical interaction of the regulatory genes. To prevent the model from over-fitting, the LogicNet applies the Bayesian Information Criterion (BIC) to force a balance between the quality of the fitting and the complexity of the interactions. The significance of the causal interactions is consequently modeled by using the Bayes Factor (BF).
Results
The LogicNet performance is evaluated by using the simulated data from Escherichia coli (E. coli) and also data from the yeast GRNs of DREAM3 [1]. Also, the LogicNet performance is compared to several state-of-the-art tools, i.e., PCA-CMI [3], ARACNe [5], Genie3 [29], Narromi [4], CN [30], and GRNTE [31]. The performance is evaluated by using the true positive rate (TPR), false positive rate (FPR), positive predictive value (PPV), accuracy (ACC) and Matthews’s coefficient constant (MCC) defined as follows:
where TP, FP, TN and FN are the numbers of the true positive, false positive, true negative, and false negative predictions, respectively. The LogicNet has no parameter by which we could calculate the receiver operating characteristic (ROC) curves and the area under the ROC curve (AUC). Therefore, the F-measure, which is the harmonic mean of the TPR and PPV, is used to compare the overall performance of the LogicNet with that of other tools. Although the LogicNet is compared to other tools for detecting undirected/directed network edges, it is also capable of detecting the underlying logic of the regulatory interactions. This capability is one advantage of the LogicNet for reconstructing the GRNs, and no other tool is currently capable of simultaneously detecting the directed network edges and the logic functions.
To evaluate the integrative performance of the LogicNet for the simultaneous detection of the directed edges and the logic functions, we apply a new measure in which we consider a TP if the regulatory genes and the active partition in the Venn diagram are both correctly predicted. In addition, we consider an FP if either the regulatory genes or the active partitions in the Venn diagram are predicted falsely. All other predictions are considered as FN (See the Methods section for more details).
E. coli network with simulated logic functions
Figure 1 A shows the GRN of E. coli from the DREAM3 dataset in which the activatory and the inhibitory interactions are shown by the black and red edges, respectively. Since the logic functions among the regulatory genes are unknown, the E. coli logic functions are simulated with randomly assigned logics of types AND, OR, and XOR. Figure 1. B shows a possible logical network with simulated logics among the regulatory genes, constructed based on the E. coli network in Fig. 1. A. The gene expression samples are then simulated from this logical network, and the LogicNet is applied to predict the directed network and the logic functions. Table 1 shows the LogicNet performance separately for 10 and 50 gene expression samples and 100 repeats of the whole simulation study
Fig. 1.

E. coli GRN and the simulated logic functions among the regulatory genes. a E. coli GRN from DREAM3 is shown. Activatory and inhibitory interactions are shown by the black and red edges, respectively. b E. coli GRN with simulated logic functions among the regulatory genes is shown
Table 1.
The LogicNet performance in predicting the GRNs and the logic functions, for 100 logic function simulations. The performance is evaluated at three levels, i.e., for undirected/directed networks and for directed logical networks in which the integrative detection of the directed edges and logic functions is evaluated
| Sample size | GRN | TPR | FPR | PPV | ACC | MCC | F-measure | |
|---|---|---|---|---|---|---|---|---|
| PC-LogicNet | 10 | Undirected | 0.48 | 0.05 | 0.82 | 0.79 | 0.51 | 0.61 |
| Directed | 0.42 | 0.08 | 0.51 | 0.84 | 0.37 | 0.46 | ||
| Directed Logical | 0.42 | – | 0.52 | – | – | 0.46 | ||
| 50 | Undirected | 0.50 | 0.05 | 0.84 | 0.80 | 0.53 | 0.63 | |
| Directed | 0.44 | 0.08 | 0.53 | 0.84 | 0.39 | 0.48 | ||
| Directed Logical | 0.43 | – | 0.53 | – | – | 0.47 | ||
| Fuzzy-LogicNet | 10 | Undirected | 0.43 | 0.05 | 0.81 | 0.77 | 0.46 | 0.56 |
| Directed | 0.36 | 0.05 | 0.57 | 0.85 | 0.37 | 0.44 | ||
| Directed Logical | 0.09 | – | 0.13 | – | – | 0.10 | ||
| 50 | Undirected | 0.50 | 0.10 | 0.72 | 0.77 | 0.45 | 0.59 | |
| Directed | 0.43 | 0.07 | 0.55 | 0.85 | 0.40 | 0.48 | ||
| Directed Logical | 0.08 | – | 0.10 | – | – | 0.09 |
As indicated in Table 1 for 10 samples, in detecting the undirected and directed GRN of E. coli, the PC-LogicNet reaches the F-measures of 0.61 and 0.46, respectively, which are superior to the performance of PCA-CMI [3], ARACNe [5], Genie3 [29], Narromi [4], CN [30], and GRNTE [31] (see Table 2 for comparisons).
Table 2.
The LogicNet in comparison with PCA-CMI, ARACNe, Genie3, Narromi, CN, and GRNTE in reconstructing the undirected/directed E. coli network, using 10 gene expression samples and 100 repeats of the whole simulation study. Two types of logics, i.e., PC and fuzzy logics, are used separately for reconstructing the GRNs and logic functions in the LogicNet algorithm. Also, the value of c = α + β is set to 1000. The highest accuracies are indicated in boldface. Reported values for the TP, FP, TN, FN are the total of the corresponding values over 100 repeats of the whole simulation study
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC | F-measure |
|---|---|---|---|---|---|---|---|---|---|---|
| Undirected E. coli Network (the edge direction is not taken into account in calculating the performance) | ||||||||||
| PC-LogicNet | 724 | 157 | 2843 | 776 | 0.48 | 0.05 | 0.82 | 0.79 | 0.51 | 0.61 |
| Fuzzy-LogicNet | 640 | 155 | 2845 | 860 | 0.43 | 0.05 | 0.81 | 0.77 | 0.46 | 0.56 |
| PCA-CMI-0.1 | 824 | 1974 | 1026 | 676 | 0.55 | 0.66 | 0.29 | 0.41 | −0.11 | 0.38 |
| PCA-CMI-0.05 | 940 | 2214 | 786 | 560 | 0.63 | 0.74 | 0.30 | 0.38 | −0.11 | 0.40 |
| ARACNe | 160 | 140 | 2860 | 1340 | 0.11 | 0.05 | 0.53 | 0.67 | 0.11 | 0.18 |
| GENIE3-FR-sqrt | 213 | 228 | 2772 | 1287 | 0.14 | 0.08 | 0.48 | 0.66 | 0.10 | 0.22 |
| GENIE3-FR-all | 192 | 235 | 2765 | 1308 | 0.13 | 0.08 | 0.45 | 0.66 | 0.08 | 0.20 |
| Narromi | 490 | 829 | 2171 | 1010 | 0.33 | 0.28 | 0.37 | 0.59 | 0.05 | 0.35 |
| CN | 976 | 2297 | 703 | 524 | 0.65 | 0.77 | 0.30 | 0.37 | −0.12 | 0.41 |
| GRNTE | 420 | 750 | 2241 | 1089 | 0.16 | 0.41 | 0.36 | 0.34 | − 0.28 | 0.22 |
| Directed E. coli Network | ||||||||||
| PC-LogicNet | 624 | 588 | 6912 | 876 | 0.42 | 0.08 | 0.51 | 0.84 | 0.37 | 0.46 |
| Fuzzy-LogicNet | 540 | 405 | 7095 | 960 | 0.36 | 0.05 | 0.57 | 0.85 | 0.37 | 0.44 |
| ARACNe | 120 | 180 | 7320 | 1380 | 0.08 | 0.02 | 0.40 | 0.83 | 0.12 | 0.13 |
| GENIE3-FR-sqrt | 155 | 445 | 7055 | 1345 | 0.10 | 0.06 | 0.26 | 0.80 | 0.07 | 0.15 |
| GENIE3-FR-all | 156 | 444 | 7056 | 1344 | 0.10 | 0.06 | 0.26 | 0.80 | 0.07 | 0.15 |
| Narromi | 275 | 1513 | 5987 | 1225 | 0.18 | 0.20 | 0.15 | 0.70 | −0.02 | 0.17 |
| CN | 616 | 1369 | 6131 | 884 | 0.41 | 0.18 | 0.31 | 0.75 | 0.21 | 0.35 |
| GRNTE | 232 | 1210 | 3030 | 1128 | 0.04 | 0.59 | 0.13 | 0.15 | −0.63 | 0.08 |
Table 1 also shows the integrative performance of the LogicNet in detecting both directed network and logic functions in E. coli. With this integrative measure, the PC- LogicNet reaches an F-measure of 0.46, which is significantly higher than its performance when using the fuzzy logic, i.e., 0.10. It should be noted that in achieving these results, the parameter c, i.e., c = α + β, is set to 1000 (See Methods). In Table 3, the sensitivity of the results is tested for other values of c, i.e., c = 500, 750, 1000 and 1250. As this table indicates, the results are not sensitive to the c values.
Table 3.
The PC-LogicNet performance is evaluated for different values of c = α + β, i.e. c = 500, 750, 1000 and 1250. The PC-LogicNet is applied to reconstruct the directed network and logic functions among the regulatory genes in the E. coli, by using 10 gene expression samples and 100 repeats of the whole simulation study. Reported values for the TP, FP, TN, FN are the total of the corresponding values over 100 repeats of the whole simulation study
| Graph | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC | F-measure | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c = 500 | Undirected a | 716 | 165 | 2835 | 784 | 0.48 | 0.06 | 0.81 | 0.79 | 0.50 | 0.60 |
| Directed b | 616 | 592 | 6908 | 884 | 0.41 | 0.08 | 0.51 | 0.84 | 0.36 | 0.45 | |
| Directed logical c | 614 | 592 | – | 885 | 0.41 | – | 0.51 | – | – | 0.45 | |
| c = 750 | Undirected | 720 | 155 | 2845 | 780 | 0.48 | 0.05 | 0.82 | 0.79 | 0.51 | 0.61 |
| Directed | 620 | 590 | 6910 | 880 | 0.41 | 0.08 | 0.51 | 0.84 | 0.37 | 0.46 | |
| Directed logical | 605 | 590 | – | 899 | 0.40 | – | 0.51 | – | – | 0.45 | |
| c = 1000 | Undirected | 724 | 157 | 2843 | 776 | 0.48 | 0.05 | 0.82 | 0.79 | 0.51 | 0.61 |
| Directed | 624 | 588 | 6912 | 876 | 0.42 | 0.08 | 0.51 | 0.84 | 0.37 | 0.46 | |
| Directed logical | 625 | 588 | – | 868 | 0.42 | – | 0.52 | – | – | 0.46 | |
| c = 1250 | Undirected | 758 | 147 | 2853 | 742 | 0.51 | 0.05 | 0.84 | 0.80 | 0.54 | 0.63 |
| Directed | 658 | 571 | 6929 | 842 | 0.44 | 0.08 | 0.54 | 0.84 | 0.39 | 0.48 | |
| Directed logical | 632 | 571 | – | 845 | 0.43 | – | 0.53 | – | – | 0.47 |
a The edge direction is not taken into account in calculating the performance
b The edge direction is taken into account in calculating the performance
c The integrative performance of the LogicNet in reconstructing both the edge direction and logic function among regulatory genes is evaluated
Yeast network real data
Figure 2 shows two yeast GRNs, i.e., Y2 and Y3, in which activatory and inhibitory interactions are respectively shown by the black and red edges. The microarray gene expression data of these networks are downloaded from the DREAM3 dataset, and the LogicNet is applied for reconstructing the networks. See Table 4 for the predicted edges and logics.
Fig. 2.
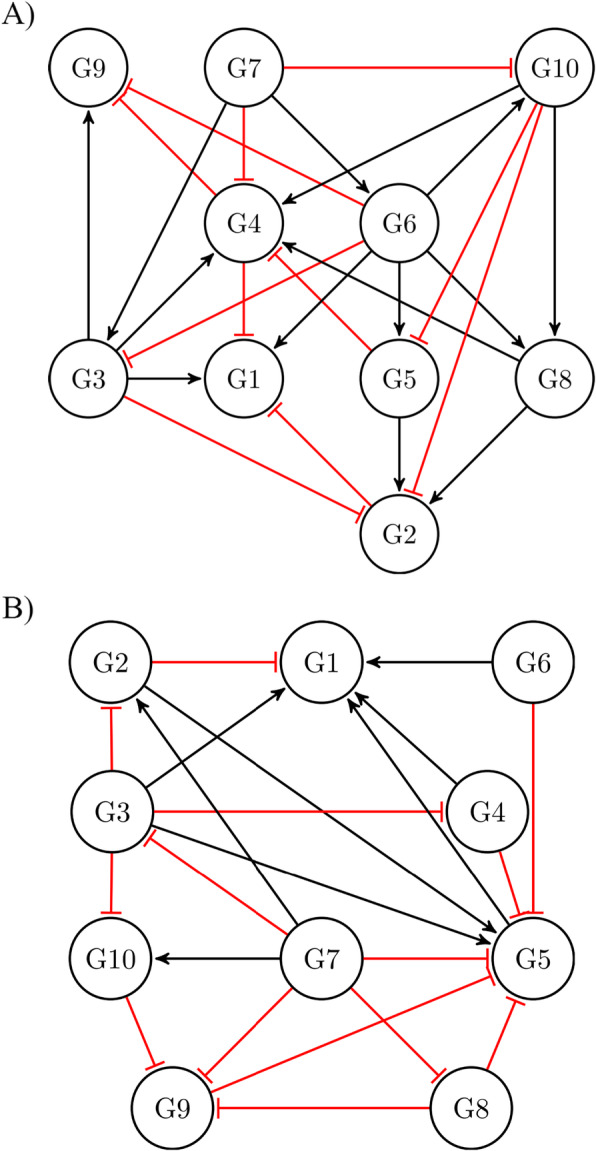
Yeast GRNs. a Yeast network Y2 with 10 nodes and 25 edges, b Yeast network Y3 with 10 nodes and 22 edges, as parts of the DREAM3 dataset
Table 4.
The predicted regulators and logic functions among these regulatory genes in Y2 and Y3 networks, with LogicNet
| Gene | Predicted Regulator/Logic Function |
|---|---|
| Y2 network | |
| G1 | |
| G2 | |
| G3 | |
| G4 | |
| G5 | |
| G6 | |
| G7 | |
| G8 | |
| G9 | |
| G10 | |
| Y3 network | |
| G1 | |
| G2 | |
| G3 | |
| G4 | |
| G5 | |
| G6 | |
| G7 | |
| G8 | – |
| G9 | |
| G10 | |
In Table 5, the performance of the LogicNet in reconstructing the undirected yeast networks is compared with that of other tools, (see Table 6 for the results of predicting the directed networks). As Table 5 illustrates, the LogicNet outperforms the other tools in reconstructing the undirected networks of Y2 and Y3, with an F-measure of 0.60 and 0.74, respectively. Moreover, as shown in Tables 5 and 6, the performance of the PC logic is superior to that of the fuzzy logic, in the majority of cases. These results indicate that the PC logic is more effective and relevant to the biological processes in logic function modeling than the fuzzy logic.
Table 5.
The LogicNet in comparison with PCA-CMI, ARACNe, Genie3, Narromi, CN, and GRNTE in reconstructing the undirected yeast networks (the edge direction is not taken into account in calculating the performance). Yeast networks Y2 and Y3 are reconstructed by using 10 gene expression samples from the DREAM3 dataset. Two types of logics, i.e., the PC and the fuzzy logics, are used separately for reconstructing the GRNs and detecting the logic functions in the LogicNet algorithm. The value of c = α + β is set to 1000. The highest accuracies are indicated in boldface
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC | F-measure |
|---|---|---|---|---|---|---|---|---|---|---|
| Yeast Network Y2 | ||||||||||
| PC-LogicNet | 14 | 10 | 10 | 11 | 0.56 | 0.50 | 0.58 | 0.53 | 0.06 | 0.57 |
| Fuzzy-LogicNet | 14 | 8 | 12 | 11 | 0.56 | 0.40 | 0.64 | 0.58 | 0.16 | 0.60 |
| PCA-CMI-0.1 | 5 | 1 | 19 | 20 | 0.20 | 0.05 | 0.83 | 0.53 | 0.22 | 0.32 |
| PCA-CMI-0.05 | 5 | 2 | 18 | 20 | 0.20 | 0.10 | 0.71 | 0.51 | 0.14 | 0.31 |
| ARACNe | 1 | 0 | 20 | 24 | 0.04 | 0.00 | 1.00 | 0.47 | 0.13 | 0.08 |
| GENIE3-FR-sqrt | 5 | 1 | 19 | 20 | 0.20 | 0.05 | 0.83 | 0.53 | 0.22 | 0.32 |
| GENIE3-FR-all | 3 | 3 | 17 | 22 | 0.12 | 0.15 | 0.50 | 0.44 | −0.04 | 0.19 |
| Narromi | 8 | 2 | 18 | 17 | 0.32 | 0.10 | 0.80 | 0.58 | 0.26 | 0.46 |
| CN | 8 | 5 | 15 | 17 | 0.32 | 0.25 | 0.62 | 0.51 | 0.08 | 0.42 |
| GRNTE | 14 | 9 | 11 | 11 | 0.56 | 0.45 | 0.61 | 0.56 | 0.11 | 0.58 |
| Yeast Network Y3 | ||||||||||
| PC-LogicNet | 17 | 7 | 16 | 5 | 0.77 | 0.30 | 0.71 | 0.73 | 0.47 | 0.74 |
| Fuzzy-LogicNet | 14 | 8 | 15 | 8 | 0.64 | 0.35 | 0.64 | 0.64 | 0.29 | 0.64 |
| PCA-CMI-0.1 | 14 | 2 | 21 | 8 | 0.64 | 0.09 | 0.88 | 0.78 | 0.57 | 0.74 |
| PCA-CMI-0.05 | 15 | 6 | 17 | 7 | 0.68 | 0.26 | 0.71 | 0.71 | 0.42 | 0.70 |
| ARACNe | 3 | 0 | 23 | 19 | 0.14 | 0.00 | 1.00 | 0.58 | 0.27 | 0.24 |
| GENIE3-FR-sqrt | 3 | 1 | 22 | 19 | 0.14 | 0.04 | 0.75 | 0.56 | 0.16 | 0.23 |
| GENIE3-FR-all | 3 | 2 | 21 | 19 | 0.14 | 0.09 | 0.60 | 0.53 | 0.08 | 0.22 |
| Narromi | 6 | 5 | 18 | 16 | 0.27 | 0.22 | 0.55 | 0.53 | 0.06 | 0.36 |
| CN | 17 | 7 | 16 | 5 | 0.77 | 0.30 | 0.71 | 0.73 | 0.47 | 0.74 |
| GRNTE | 10 | 7 | 16 | 12 | 0.45 | 0.30 | 0.59 | 0.58 | 0.15 | 0.51 |
Table 6.
The LogicNet in comparison with ARACNe, Genie3, Narromi, CN, and GRNTE in reconstructing the directed yeast networks. Two Yeast networks, i.e., Y2 and Y3 with 10 genes and 25 edges (Y2)/22 edges (Y3), are reconstructed by the LogicNet by using 10 gene expression samples from the DREAM3 dataset
| Method | TP | FP | TN | FN | TPR | FPR | PPV | ACC | MCC | F-measure |
|---|---|---|---|---|---|---|---|---|---|---|
| Yeast Network Y2 | ||||||||||
| PC-LogicNet | 10 | 20 | 45 | 15 | 0.40 | 0.31 | 0.33 | 0.61 | 0.09 | 0.36 |
| Fuzzy-LogicNet | 8 | 18 | 47 | 17 | 0.32 | 0.28 | 0.31 | 0.61 | 0.04 | 0.31 |
| ARACNe | 0 | 1 | 64 | 25 | 0.00 | 0.02 | 0.00 | 0.71 | −0.07 | – |
| GENIE3-FR-sqrt | 1 | 5 | 60 | 24 | 0.04 | 0.08 | 0.17 | 0.68 | −0.07 | 0.06 |
| GENIE3-FR-all | 1 | 5 | 60 | 24 | 0.04 | 0.08 | 0.17 | 0.68 | −0.07 | 0.06 |
| Narromi | 6 | 5 | 60 | 19 | 0.24 | 0.08 | 0.55 | 0.73 | 0.22 | 0.33 |
| CN | 1 | 5 | 60 | 24 | 0.04 | 0.08 | 0.17 | 0.68 | −0.07 | 0.06 |
| GRNTE | 12 | 17 | 48 | 13 | 0.48 | 0.26 | 0.41 | 0.67 | 0.21 | 0.44 |
| Yeast Network Y3 | ||||||||||
| PC-LogicNet | 11 | 16 | 52 | 11 | 0.50 | 0.24 | 0.41 | 0.70 | 0.25 | 0.45 |
| Fuzzy-LogicNet | 8 | 19 | 49 | 14 | 0.36 | 0.28 | 0.30 | 0.63 | 0.08 | 0.33 |
| ARACNe | 1 | 2 | 66 | 21 | 0.05 | 0.03 | 0.33 | 0.74 | 0.04 | 0.08 |
| GENIE3-FR-sqrt | 2 | 2 | 66 | 20 | 0.09 | 0.03 | 0.50 | 0.76 | 0.13 | 0.15 |
| GENIE3-FR-all | 1 | 5 | 63 | 21 | 0.05 | 0.07 | 0.17 | 0.71 | −0.05 | 0.07 |
| Narromi | 5 | 7 | 61 | 17 | 0.23 | 0.10 | 0.42 | 0.73 | 0.16 | 0.29 |
| CN | 6 | 11 | 57 | 16 | 0.27 | 0.16 | 0.35 | 0.70 | 0.12 | 0.31 |
| GRNTE | 7 | 15 | 53 | 15 | 0.32 | 0.22 | 0.32 | 0.67 | 0.10 | 0.32 |
It should be emphasized that PCA-CMI [3], ARACNe [5], Genie3 [29], Narromi [4], CMI2NI [2], and CN [30] are threshold dependent. These thresholds, e.g., on mutual information between two genes, determine the significance of the regulatory interactions. As these thresholds are user-dependent, and there is no a priori information to determine them, many of the current tools are limited by their dependency on a threshold. However, in the LogicNet, due to the large difference in the likelihoods of the target’s gene expression level under a biologically significant logic and a random logic, we can always decisively infer the significant logic functions with a BF > 100.
Application to the logic function detection
The LogicNet can also be applied to infer the logic functions among the regulatory genes, in the networks with a known structure. For this purpose, we used the previously identified gene regulation in the yeast with 176 Regulatory Factors (RFs) and their target genes [32, 33]. The number of target genes with 1, 2 and 3 RFs are, respectively, 1472, 1013 and 653. To infer the logic function among these regulatory genes, the LogicNet is fed with three well-studied yeast cell-cycle datasets [34, 35]: 1) the alpha-factor time course with 16 time points (0, 7′, …, 119′); 2) cdc15 time course with 25 time points (10′, 30′, …, 290′); and 3) cdc28 time course with 17 time points (0, 10′, …, 160′) for the gene expression samples. After combining all three datasets (5581 genes and 58 time points), the gene expressions for each time point are converted into the interval [0, 1].
For target genes with one RF, we used the LogicNet to characterize the RF1-target logics during the yeast cell cycle. As depicted in Fig. 3. A, we found 1364 RF-target logics of type Target = RF1 and 75 logics of type . The other 33 RF-target logics were of type Target = 1. See Supplementary File 1 for the gene names with RF-target interaction and the corresponding logic function.
Fig. 3.

The number of PC logic functions which are inferred by the LogicNet in the yeast. a The target genes are regulated by one RF. b The target genes are regulated by two RFs
For the target genes with two RFs, we used the LogicNet to characterize the RF1-RF2-target logics by computing the likelihood values for the 16 possible logic functions among two RFs, as shown in Table 7. As depicted in Fig. 3. B, logic functions “Target = RF1VRF2” (i.e., OR logic function), “Target = RF2” and “Target = RF1” are more frequent than the other logic functions for characterizing RF1-RF2-target logics. The OR logic for the RF1-RF2-target interaction indicates that either RF1 or RF2 is enough to activate the expression of their target genes. Also, the non-cooperative logic functions such as “Target = RF2” and “Target = RF1” indicate that only one RF (the dominant RF) controls the target regulation. See Supplementary File 1 for the gene names with RF1-RF2-target interaction and the corresponding logic function. We also used the LogicNet to characterize the RF1-RF2-RF3-target logics by computing the likelihood values for the 256 possible logic functions among three RFs (see Supplementary File 1 for the result).
Table 7.
16 possible PC logic functions between two genes G1 and G2, which regulate the target. The ∪ sign stands for the union of the sets, and ∨, ∧ , ⊕ , and ⨀ stand for the OR, AND, XOR, and XNOR PC logics between G1 and G2
| i | i3 | i2 | i1 | i0 | fi(G1, G2) | Output |
|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 1 | ||
| 2 | 0 | 0 | 1 | 0 | ||
| 3 | 0 | 0 | 1 | 1 | ||
| 4 | 0 | 1 | 0 | 0 | ||
| 5 | 0 | 1 | 0 | 1 | ||
| 6 | 0 | 1 | 1 | 0 | ||
| 7 | 0 | 1 | 1 | 1 | ||
| 8 | 1 | 0 | 0 | 0 | G1G2 = G1 ∧ G2 | |
| 9 | 1 | 0 | 0 | 1 | ||
| 10 | 1 | 0 | 1 | 0 | ||
| 11 | 1 | 0 | 1 | 1 | ||
| 12 | 1 | 1 | 0 | 0 | ||
| 13 | 1 | 1 | 0 | 1 | ||
| 14 | 1 | 1 | 1 | 0 | ||
| 15 | 1 | 1 | 1 | 1 | 1 |
As in previous studies [36], we used RF knockout experiments in the yeast to validate the logic functions which are inferred by the LogicNet. These RF knockout experiments measure the gene expression fold changes, after deleting each RF [37, 38]. If the target is cooperatively regulated by two RFs, e.g., in “Target = RF1VRF2” (OR logic), then it is most likely that the knockout of either RF decreases the target gene expressions. In 412 logic functions “Target = RF1VRF2”, which are inferred by the LogicNet, deleting either RF1 or RF2 decreases the target gene expression by a factor of − 0.016 and − 0.157 in the logarithm scale. For the non-cooperative logic functions, e.g., “Target = RF2”, we found that deleting the dominant RF, i.e., RF2 downregulates the target gene expression more than the removal of RF1. Indeed, in logic function “Target = RF2”, deleting RF1 or RF2 decreases the target gene expression on average by a factor of − 0.022 and − 0.086, respectively, with a standard deviation of 0.37 and 0.34.
Application to RNA-Seq data
LogicNet is also applied to infer GRNs in the early embryonic development data (oocyte to E4.25 blastocyst stages) [39], from single-cell transcriptome sequencing of 48 genes. As described in the original study [39], raw Ct data are first subtracted by the detection limit of 28 and further normalized on a cell-wise basis by subtracting the mean expression of housekeeping genes Actb and Gapdh.
GRNs are then reconstructed for two overlapping subsets of data from 46 genes, i.e., excluding the housekeeping genes which are used for data normalization. The early subset of data includes the cells from oocyte up to 32-cell E3.5 blastocyst stages and the late subset includes the cells from 16-cell morula to 64-cell E4.25 blastocyst stages.
Inferred GRNs using LogicNet are depicted in panels A and B of Fig. 4, respectively for the early and late subsets of cells. As shown in this Figure, GRN for cells from 16-cell morula to 64-cell E4.25 blastocyst stages is more complex than GRN for the early subset of cells. However, in both networks, Grhl2 has an important role as a hub.
Fig. 4.

a For cells from oocyte up to 32-cell E3.5 blastocyst stages. b For cells from 16-cell morula to 64-cell E4.25 blastocyst stages
The LogicNet complexity
To calculate the time complexity of the LogicNet, consider N genes in the network and a sample of n gene expression vectors. For each gene as a target and logic functions including up to k regulatory nodes, we have possible logic functions in the model. Then, having N genes, each considered as a target at a time and a sample size of n, we reach a complexity of for the number of calculations in the model.
Discussion
The PC-LogicNet achieves a considerably higher F-measure than the Fuzzy-LogicNet. This result indicates that the PC logic is more relevant and effective in modeling regulatory gene interactions. Therefore, future studies can benefit from this PC logic in reconstructing the GRNs and detecting the logic functions. Moreover, compared to the previous logic-based models, the LogicNet does not rely on a priori known network structure to infer the logic functions. However, as described in the results section, the LogicNet can be applied for the logic function detection from the known regulatory genes-target interactions.
Moreover, since the parameters of the beta distribution are estimated separately for each sample, the LogicNet can model the gene expression data that follow a multi-modal distribution. This capability is a major advantage of the LogicNet over many existing tools, which have difficulties in modeling the multi-modal gene expression data.
R package of the LogicNet is available at https://github.com/CompBioIPM/LogicNet. Yeast and E.coli data sets, which were used in this study, are also available on this webpage. Parallel programming of the LogicNet algorithm reduces its running time considerably. For a GRN of 10 nodes and 10 gene expression samples, it takes 275 s to run the LogicNet on a 64-bit operating system with an Intel(R) Core (TM) i7-4710HQ CPU @ 3.50 GHz processor and 16 GB RAM.
Conclusion
The LogicNet performance is superior to that of the MI-based and regression-based tools. The low performance of these tools is, to some extent, associated with ignoring the logic function among the regulatory genes. Indeed, compared to the other tools, logic-based models are more accurate for reconstructing the GRNs and more useful for detecting the logic functions, two important problems in biology.
Methods
The LogicNet was developed to infer the existing regulatory interactions of a target gene T and to determine the corresponding logic behind these interactions. The values of the expression level of each gene are normalized into the interval [0, 1]. In the LogicNet, these expression levels are supposed to be the samples of a beta distribution. In this context, the expression level expresses the probability of being an active gene. In other words, an expression level value close to zero indicates a high probability of being off. Accordingly, a regulatory gene with a higher level of activity is more probable to influence other genes. Furthermore, it is assumed that the expression levels of T are the outputs of a continuous logic function whose inputs are the gene expression level of the regulatory genes of T. Hence, each logic function provides an estimate of the expression level of T, or, similarly, an estimate of the probability of the activity of T. We call this function a probabilistic continuous (PC) logic function.
PC logic function
Consider k genes G1, G2, …, Gk regulating the target gene T. Each gene can have an activatory or inhibitory effect on T, denoted by Gi and , respectively. Hence, there are 2k different combinations of the activatory and inhibitory effects of all regulators, e.g.; for k = 1 we have G1 and and for k = 2 we have 4 different combinations of G1G2, , , and . These activatory/inhibitory combinations can be associated with partitions in the Venn diagram of the set of k regulatory genes (Fig. 5). Now for k = 1, 2, and 3, and for the regulatory genes G1, G2, and G3, we use Venn diagram partitions and define PC logic functions as follows:
Fig. 5.

Venn diagram partitions representing different interactions among the regulatory genes influencing the target T. Each partition either exists or does not exist in the corresponding fi(G1, G2, …, Gk) of the logic function. aG1 regulates T. Each partition, i.e., G1 and of the Venn diagram, is possibly on or off in fi(G1). b Both genes G1 and G2 regulate T. The Venn diagram is partitioned into 4 disjoint regions; each is potentially on or off in fi(G1, G2). c Genes G1, G2 and G3 regulate T. The Venn diagram is partitioned into 8 disjoint regions; each is potentially on or off in fi(G1, G2, G3)
| 1 |
| 2 |
| 3 |
where ∪ stands for the union of two sets, and denotes the binary representation of the PC logic function index i. Indeed, the coefficient of each partition in fi(G1, G2, …, Gk) could be 0 or 1, indicating the presence of the corresponding activatory/inhibitory combination in fi(G1, G2, …, Gk), for more details on notations see [40]. Moreover, binary variables in the PC logic function provide a systematic way to generate different logics and these random variables have to be estimated in our maximum likelihood approach, in the next subsections. In general, according to the , there are different PC logic functions fi(G1, G2, …, Gk) for k regulatory genes, where .
The occurrence of each partition in the PC logic function results in the expression of the target gene T. Each partition represents the AND logic between the genes, e.g., f8(G1, G2) = G1G2 = G1 ∧ G2 (Table 7). The union operation between the partitions expresses the logical operation OR, denoted by ∨, e.g. . Figure 6 depicts fi(G1, G2) for i = 3, 6, 8, and 14, corresponding to the PC logics , XOR(G1, G2), AND(G1, G2), and OR(G1, G2), respectively. Note that there is a fundamental difference between the PC and the Boolean logics. The PC logic performs the logical operation on the continuous data, and its output is not restricted to the Boolean values of 0 and 1, but, in contrast, the output is a continuous value in the interval [0, 1].
Fig. 6.

Participating activatory/inhibitory partitions in the Venn diagram for logic functions f3, f6, f8, and f14. The indexes i0, i1, i2 and i3 indicate if the corresponding partition exists in fi(G1, G2), between genes G1 and G2
Probabilistic and fuzzy logics
To define the logical operators for the continuous gene expression data, previous studies usually utilize the fuzzy logic [19, 22], as given in Table 8. However, we propose an alternative logic, i.e., the PC logic, which is based on the probabilistic rules. All Boolean functions can be described by the combination of three basic logical operators: AND, OR, and NOT [40]. The definitions of these basic logical operations for the case of having two regulatory genes G1 and G2 and with the expression levels and are compared in Table 8 for the PC and the fuzzy logics.
Table 8.
The PC logic and the fuzzy logic for the regulatory effects of genes G1 and G2 on the target, utilizing continuous gene expression data. expA and expB denote the expression levels of genes G1 and G2, respectively
| Logic | Probabilistic Logic Def. | Fuzzy Logic Def. |
|---|---|---|
| AND(G1, G2) = G1 ⋀ G2 | ||
| OR(G1, G2) = G1 ⋁ G2 |
In the case of k = 1, only gene G1 is in the causal set of the target gene T. Accordingly, Eq. (1) results in f0(G1) = 0, , , and f3(G1) = 1, where f1(G1) and f2(G1) indicate the inhibitory and activatory effects of gene G1 on T, respectively (see Fig. 6a). By applying probabilistic logics, the output of 16 possible PC logic functions for k = 2 are represented in Table 7. The PC logic function fi(G1, G2, …, Gk) is just an estimator of the probability of the activation of T, i.e., expT.
Likelihood function
Each PC logic function fi(G1, G2, …, Gk) provides an estimate of the expression level of the target gene T. However, there are different PC logic functions for k regulatory genes influencing the target. Therefore, we need to evaluate the likelihood that these PC logic functions will predict the expression level of T. To achieve this goal, we suppose that the expression level of T follows a beta distribution with parameters α and β:
| 4 |
where, 0 ≤ T ≤ 1. We know that in this beta distribution, the expected value of the expression level is . Assuming fi(.) as an unbiased estimator of the target’s expression level, we obtain
| 5 |
In addition, considering α + β = c, where c is a constant, the model parameters are estimated as follows:
| 6 |
To avoid getting zero parameters when fi(.) is either 0 or 1, a small value is added to the estimated α and β in Eq. (6). Then, for n gene expression samples, the logarithm of the likelihood function is
| 7 |
in which, Ts indicates the expression level of the s-th sample of the target gene, and is the PC logic function computed for the corresponding sample.
The c value calibrates the variance of the target gene expression (T) given its regulators, in the beta distribution. As T values are modelled separately for each sample, i.e., T is expected to be close to fi(.), we consider a large value for c to assure a low deviation from fi(.).
Equation 7 is maximized w.r.t the binary variables , representing the on/off state of the 2k partitions in the venn diagram of the k regulatory genes. For this purpose, the current version of the LogicNet evaluates the likelihood under all possible values of these binary variables, i.e., the exact solution.
For the microarray data, the min-max feature scaling is applied to normalize the expressions into the [0, 1] interval, e.g., for a gene A:
The LogicNet is originally proposed to reconstruct the logic based GRNs, from microarrays. However, the count distribution in the RNA-seq data can also be transformed to a distribution close to the Gaussian distribution, using the voom transformation [41]. Then, the min-max feature scaling is applied [41].
Bayesian information criterion (BIC)
The LogicNet computes the likelihood for the expression level of the target gene by considering k regulatory genes. However, increasing the number of regulatory genes may potentially result in model over-fitting. Here, we use the Bayesian Information Criterion (BIC) [42] to strike the right balance between improving the model fitting (likelihood) and making the model more complex. BIC is defined as follows [42]:
| 8 |
In the case of having k regulatory genes, we consider 2k parameters in the model that are associated with 2k partitions of the Venn diagram, where each partition either exists or does not exist in the fi(G1, G2, …, Gk). To this end, the PC logic function with a minimum BIC is considered for each target gene.
Bayes factor (BF)
The PC logic corresponding to the minimum BIC is not necessarily biologically significant and meaningful. To distinguish between random and biologically meaningful logics, the LogicNet applies the Bayes Factor (BF) [43] to test the likelihood significance of the PC logic function with a minimum BIC. The BF is the ratio of the likelihood probabilities for two competing hypotheses as follows:
| 9 |
where M1 is the PC logic function with the minimum BIC and indicates the causal relationships between the regulatory and target genes. M0 is a random logic without a biological significance. Based on the Bayesian literature, a value of BF > 100 means that compared to M0, M1 is decisively supported by data.
The overall workflow of the LogicNet is depicted in Fig. 7. From genes GA, GB, …, and GZ, one gene at a time is considered as the target. Considering gene GA as the target and k = 1, 2, 3, … genes as its regulators, the PC logic functions fi(.) are constructed for different subsets of genes GB, …, and GZ. Then, the likelihood of the expression level of the target gene (i.e., gene GA) is calculated under each PC logic function. BIC is applied to strike the right balance between the likelihood and model complexity, i.e., the number of the regulatory genes. The likelihood significance in the PC logic function with the lowest BIC is consequently evaluated by using the BF. This process is repeated for each gene as the target. The maximum of k in this study is 4.
Fig. 7.

Workflow diagram of the LogicNet to reconstruct the Gene Regulatory Network. Assume n samples are drawn from the expression level of genes GA, GB, …, and GZ. Each time, one gene is considered as the target, and the regulatory effect of other genes on the target is investigated. Here, gene GA is considered as the target. Logic functions consisting of k regulatory genes are constructed for k = 1, 2, 3, and the target gene expression likelihoods are evaluated under different logic functions. To calculate the likelihood, the target’s gene expression is modeled by using a beta distribution whose parameters are identified based on the logic function between the regulatory genes. Then, the Bayesian Information Criterion (BIC) is applied to strike the right balance between the likelihood and model complexity (the number of the regulatory genes). For each target gene, the likelihood significance in the logic function with the lowest BIC is further evaluated by using the Bayes Factor (BF). Only logic functions which are decisively supported by the target gene expression data (with BF > 100) are considered to be significant
The LogicNet integrative performance for directed edges and logic functions
To evaluate the integrative performance of the LogicNet for the simultaneous detection of the directed edges and the logic functions, we apply a new measure in which we consider a TP if the regulatory genes and the active partition in the Venn diagram are both correctly predicted. In addition, we consider an FP if either the regulatory genes or the active partitions in the Venn diagram are predicted falsely. All other predictions are considered as FN. For example, in the case of f14(G1, G2) = G1 ∪ G2 in Fig. 6, three partitions G1G2, and of the Venn diagram are active, and therefore, we consider a TP for the correct prediction of each partition and a FP if either gene G1 or gene G2 or the corresponding active partitions are falsely predicted.
Data and LogicNet availability Project name: LogicNet. Project Home Page:https://github.com/CompBioIPM/LogicNet.
Operating System: Windows and Linux (× 86 and × 64 versions).
Programming Language: Designed in R.
License: Freely available under R-3.0.0 or higher versions.
Any restrictions to use by non-academics: none.
Supplementary information
Additional file 1: Supplementary Table 1. provides more details about the logic function calls that are made by the LogicNet in the regulatory factor-target gene interactions, in the yeast database.
Acknowledgments
We would like to thank Dr. Rosa Aghdam and Dr. Soheil Jahangiri-Tazehkand for their helpful comments and suggestions.
Abbreviations
- ACC
Accuracy
- BF
Bayes Factor
- BIC
Bayesian Information Criterion
- FN
False Negative
- FP
False Positive
- FPR
False Positive Rate
- GRNs
Gene Regulatory Networks
- MCC
Matthews’s Coefficient Constant
- MI
Mutual Information
- PCA
Principle Component Analysis
- PC-Logic
Probabilistic Continuous Logic
- PPV
Positive Predictive Value
- RFs
Regulatory Factors
- ROC
Receiver Operating Characteristic
- T
Target gene
- TN
True Negative
- TP
True Positive
- TPR
True Positive Rate
Authors’ contributions
MS conceived the study and guided its method development and data analysis steps. SAM and ARA developed the method and wrote the manuscript. All three authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the https://github.com/CompBioIPM/LogicNet repository.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-020-03651-x.
References
- 1.Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G. Revealing strengths and weaknesses of methods for gene network inference. Proc Natl Acad Sci. 2010;107(14):6286–6291. doi: 10.1073/pnas.0913357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X, Zhao J, Hao JK, Zhao XM, Chen L. Conditional mutual inclusive information enables accurate quantification of associations in gene regulatory networks. Nucleic Acids Res. 2015;43(5):e31. doi: 10.1093/nar/gku1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Zhao XM, He K, Lu L, Cao Y, Liu J, Hao JK, Liu ZP, Chen L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics (Oxford, Engl) 2012;28(1):98–104. doi: 10.1093/bioinformatics/btr626. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Liu K, Liu ZP, Duval B, Richer JM, Zhao XM, Hao JK, Chen L. NARROMI: a noise and redundancy reduction technique improves accuracy of gene regulatory network inference. Bioinformatics (Oxford, Engl) 2013;29(1):106–113. doi: 10.1093/bioinformatics/bts619. [DOI] [PubMed] [Google Scholar]
- 5.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006;7(1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer PE, Kontos K, Lafitte F, Bontempi G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J Bioinform Syst Biol. 2007;2007(1):79879–9. [DOI] [PMC free article] [PubMed]
- 8.Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modelling of cell signalling pathways. Nat Cell Biol. 2006;8(11):1195–1203. doi: 10.1038/ncb1497. [DOI] [PubMed] [Google Scholar]
- 9.Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W: Rules for Modeling Signal-Transduction Systems. Science's STKE; 2006;2006(344):re6. [DOI] [PubMed]
- 10.Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci. 2000;97(11):5818. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu ZP, Zhang W, Horimoto K, Chen L. Gaussian graphical model for identifying significantly responsive regulatory networks from time course high-throughput data. IET Syst Biol. 2013;7(5):143–152. doi: 10.1049/iet-syb.2012.0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z-P, Wu H, Zhu J, Miao H. Systematic identification of transcriptional and post-transcriptional regulations in human respiratory epithelial cells during influenza a virus infection. BMC Bioinform. 2014;15(1):336. doi: 10.1186/1471-2105-15-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z-P. Towards precise reconstruction of gene regulatory networks by data integration. Quant Biol. 2018;6(2):113–128. [Google Scholar]
- 14.Qian L, Wang H, Dougherty ER. Inference of Noisy nonlinear differential equation models for gene regulatory networks using genetic programming and Kalman filtering. IEEE Trans Signal Process. 2008;56(7):3327–3339. [Google Scholar]
- 15.Li Y, Chen H, Zheng J, Ngom A. The max-min high-order dynamic Bayesian network for learning gene regulatory networks with time-delayed regulations. IEEE/ACM Transact Comput Biol Bioinform. 2016;13(4):792–803. doi: 10.1109/TCBB.2015.2474409. [DOI] [PubMed] [Google Scholar]
- 16.Yang B, Liu S, Zhang W. Reverse engineering of gene regulatory network using restricted gene expression programming. J Bioinforma Comput Biol. 2016;14(5):1650021. doi: 10.1142/S0219720016500219. [DOI] [PubMed] [Google Scholar]
- 17.Yang B, Bao W. RNDEtree: regulatory network with differential equation based on flexible neural tree with novel criterion function. IEEE Access. 2019;7:58255–58263. [Google Scholar]
- 18.Kim HD, Shay T, O'Shea EK, Regev A. Transcriptional regulatory circuits: predicting numbers from alphabets. Science (New York, NY) 2009;325(5939):429–432. doi: 10.1126/science.1171347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aldridge BB, Saez-Rodriguez J, Muhlich JL, Sorger PK, Lauffenburger DA. Fuzzy logic analysis of kinase pathway crosstalk in TNF/EGF/insulin-induced Signaling. PLoS Comput Biol. 2009;5(4):e1000340. doi: 10.1371/journal.pcbi.1000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saez-Rodriguez J, Alexopoulos LG, Epperlein J, Samaga R, Lauffenburger DA, Klamt S, Sorger PK. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol Syst Biol. 2009;5:331. doi: 10.1038/msb.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saez-Rodriguez J, Alexopoulos LG, Zhang M, Morris MK, Lauffenburger DA, Sorger PK. Comparing signaling networks between normal and transformed hepatocytes using discrete logical models. Cancer Res. 2011;71(16):5400–5411. doi: 10.1158/0008-5472.CAN-10-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Z, Hahn J: Fuzzy Modeling of Signal Transduction Networks, vol. 64; 2009.
- 23.Zielinski R, Przytycki PF, Zheng J, Zhang D, Przytycka TM, Capala J. The crosstalk between EGF, IGF, and insulin cell signaling pathways--computational and experimental analysis. BMC Syst Biol. 2009;3:88. doi: 10.1186/1752-0509-3-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA. Logic-based models for the analysis of cell Signaling networks. Biochemistry. 2010;49(15):3216–3224. doi: 10.1021/bi902202q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alizad-Rahvar AR, Sadeghi M. Ambiguity in logic-based models of gene regulatory networks: an integrative multi-perturbation analysis. PLoS One. 2018;13(11):e0206976. doi: 10.1371/journal.pone.0206976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mai Z, Liu H. Boolean network-based analysis of the apoptosis network: irreversible apoptosis and stable surviving. J Theor Biol. 2009;259(4):760–769. doi: 10.1016/j.jtbi.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 27.Wu M, Yang X, Chan C. A dynamic analysis of IRS-PKR Signaling in liver cells: a discrete Modeling approach. PLoS One. 2009;4(12):e8040. doi: 10.1371/journal.pone.0008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlatter R, Schmich K, Avalos Vizcarra I, Scheurich P, Sauter T, Borner C, Ederer M, Merfort I, Sawodny O. ON/OFF and beyond--a boolean model of apoptosis. PLoS Comput Biol. 2009;5(12):e1000595. doi: 10.1371/journal.pcbi.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5(9):e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aghdam R, Ganjali M, Zhang X, Eslahchi C. CN: a consensus algorithm for inferring gene regulatory networks using the SORDER algorithm and conditional mutual information test. Mol BioSyst. 2015;11(3):942–949. doi: 10.1039/c4mb00413b. [DOI] [PubMed] [Google Scholar]
- 31.Castro JC, Valdés I, Gonzalez-García LN, Danies G, Cañas S, Winck FV, Ñústez CE, Restrepo S, Riaño-Pachón DM. Gene regulatory networks on transfer entropy (GRNTE): a novel approach to reconstruct gene regulatory interactions applied to a case study for the plant pathogen Phytophthora infestans. Theor Biol Med Model. 2019;16(1):7. doi: 10.1186/s12976-019-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jothi R, Balaji S, Wuster A, Grochow JA, Gsponer J, Przytycka TM, Aravind L, Babu MM. Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol Syst Biol. 2009;5(1):294. doi: 10.1038/msb.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne J-B, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2(1):65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 35.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9(12):3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D, Yan K-K, Sisu C, Cheng C, Rozowsky J, Meyerson W, Gerstein MB. Loregic: a method to characterize the cooperative logic of regulatory Factors. PLoS Comput Biol. 2015;11(4):e1004132. doi: 10.1371/journal.pcbi.1004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reimand J, Vaquerizas JM, Todd AE, Vilo J, Luscombe NM. Comprehensive reanalysis of transcription factor knockout expression data in Saccharomyces cerevisiae reveals many new targets. Nucleic Acids Res. 2010;38(14):4768–4777. doi: 10.1093/nar/gkq232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Z, Killion PJ, Iyer VR. Genetic reconstruction of a functional transcriptional regulatory network. Nat Genet. 2007;39(5):683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- 39.Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell. 2010;18(4):675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Nelson VP, Nagle HT, Carroll BD, Irwin JD: Digital logic circuit analysis and design: prentice-hall, Inc.; 1995.
- 41.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29–9. [DOI] [PMC free article] [PubMed]
- 42.Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6(2):461–464. [Google Scholar]
- 43.Berger J, Pericchi L. Bayes Factors. Wiley StatsRef: Statistics Reference Online. 2015;1-14. 10.1002/9781118445112.stat00224.pub2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Table 1. provides more details about the logic function calls that are made by the LogicNet in the regulatory factor-target gene interactions, in the yeast database.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the https://github.com/CompBioIPM/LogicNet repository.