

SUMMARY
Stresses associated with disease may pathologically remodel the proteome by both increasing interaction strength and altering interaction partners, resulting in proteome-wide connectivity dysfunctions. Chaperones play an important role in these alterations, but how these changes are executed remains largely unknown. Our study unveils a specific N-glycosylation pattern used by a chaperone, Glucose-regulated protein 94 (GRP94), to alter its conformational fitness and stabilize a state most permissive for stable interactions with proteins at the plasma membrane. This “protein assembly mutation’ remodels protein networks and properties of the cell. We show in cells, human specimens, and mouse xenografts that proteome connectivity is restorable by inhibition of the N-glycosylated GRP94 variant. In summary, we provide biochemical evidence for stressor-induced chaperone-mediated protein mis-assemblies and demonstrate how these alterations are actionable in disease.
Graphical Abstract
In Brief
Yan et al. show how N-glycosylation transforms a chaperone from a folding to a scaffolding protein that remodels protein connectivity, with the end result of proteome-wide dysfunction. This specific modification, exploited by cancer cells for enhanced fitness, is an actionable target in disease.
INTRODUCTION
Changes in the fitness of the endoplasmic reticulum (ER) and the function of the proteins within it are associated with various diseases, including cancer, autoimmune disorders, neurodegeneration, and cardiac dysfunction (Giampietri et al., 2015; Luoma, 2013; Mekahli et al., 2011; Ozcan and Tabas, 2012; Yadav et al., 2014). Understanding the nature of such dysfunctions is central to how we treat these diseases.
An important component and regulator of ER function is Glucose-regulated protein 94 (GRP94), also called gp96 or endoplasmin. GRP94 is one of four HSP90 paralogs and shares 50% amino acid homology with the cytosolic chaperone HSP90 (Marzec et al., 2012; McCaffrey and Braakman, 2016; Zhu and Lee, 2015). In normal cells, GRP94 functions in ER quality control, buffers Ca2+ levels, and is a key chaperone in the folding of “client” proteins. These clients include the transforming growth factor b (TGF-β)-associated protein GARP, insulin-like growth factors, Toll-like receptors (TLRs), and integrins (Ansa-Addo et al., 2016; Eletto et al., 2010).
Although primarily localized to the ER, GRP94 is also found in the cytosol, at the cell surface, and extracellularly (Ansa-Addo et al., 2016; Lee, 2014; Wiersma et al., 2015). This phenomenon is often associated with and enhanced under conditions of chronic stress, such as being associated with disease (Altmeyer et al., 1996; Booth and Koch, 1989; Lee, 2014; Wiersma et al., 2015; Wu et al., 2016; Zheng et al., 2001). For example, in dendritic cells, forced overexpression of cell-surface GRP94 results in spontaneous autoimmune disease due to the GRP94-mediated enhancement of TLR function and its subsequent downstream signaling through MyD88 (Liu et al., 2003). Similarly, pathogens use surface GRP94 to infect host cells (Martins et al., 2012), and extracellular GRP94 complexed with immunoglobulin Gs (IgGs) plays a pathologic role in type 1 diabetes (Pagetta et al., 2014). Finally, in breast cancer (BC) cells, chronic proteome stress induced by overexpression of HER2 kinase leads to enhanced translocation of GRP94 to the plasma membrane (PM) (Chavany et al., 1996; Li et al., 2015; Patel et al., 2013). There, GRP94 maintains the stability of HER2 and its enhanced downstream signaling (Patel et al., 2013). Although the function of these disease-associated GRP94 pools is becoming better understood, little is known about how stress structurally modifies the ER chaperone to alter its function.
Stress also appears to modulate post-translational modifications (PTMs) on GRP94 (Cloutier and Coulombe, 2013). GRP94 isolated from normal tissues is primarily monoglycosylated (Wearsch and Nicchitta, 1996), whereas the heterologous overexpression of murine GRP94 in COS cells or of canine GRP94 in Sf21 insect cells results in an altered GRP94 glycosylation pattern, with several hyperglycosylated forms observed (Qu et al., 1994). Similar species were also noted in preparations of endogenous GRP94 isolated from mouse sarcomas (Feldweg and Srivastava, 1995). However, the functional significance of such modifications is poorly understood. Earlier studies showed that both nonglycosylated and glycosylated GRP94 species associate with various client proteins, including Ig light chain, a1-antitrypsin, and plasma IgGs (Marzec et al., 2012; Pagetta et al., 2014). A hyperglycosylated GRP94 was also reported, but it is a non-functional form targeted for degradation in an OS-9-mediated, ER-associated protein degradation (ERAD)-independent, lysosomal-like mechanism (Cherepanova et al., 2019; Dersh et al., 2014).
Although stress is a common hallmark of disease, it is mostly studied as a damager of proteins and of their function (Solimini et al., 2007). In this context, chaperones, such as GRP94, are important in stress regulation, as they may correct and influence such damage through folding or dis-aggregation and degradation (Brehme and Voisine, 2016). Accordingly, changes in chaperone expression have been extensively studied in disease, and GRP94 overexpression has been implicated in cancer (Buc Calderon et al., 2018; Lee, 2014).
Stress, however, also alters how proteins interact (Harper and Bennett, 2016), a feature also influenceable by chaperones (Ellis, 2013). Accordingly, structurally modified chaperone pools, termed epichaperomes, may form under stress and act as scaffolds to pathologically remodel cellular processes by mediating aberrant protein-protein interactions and, in turn, creating a state of proteome-wide connectivity dysfunction (Dart, 2016; Joshi et al., 2018). Although it was first reported in cancer (Pillarsetty et al., 2019; Rodina et al., 2016), recent evidence in Parkinson and Alzheimer disease (Inda et al., 2020; Kishinevsky et al., 2018) proposes that chaperone-mediated protein mis-assembly, and in turn protein connectivity dysfunction, has broad disease relevance. Yet, a biochemical mechanism for this phenomenon has been difficult to pinpoint, and evidence for how a chaperone may switch from a folding protein into a maladaptive scaffolder remains to be provided.
Based on these previous reports, we propose that disease-associated stresses may greatly modify the proteome, creating intracellular pools of structurally and functionally heterogeneous proteins and protein assemblies. We hypothesize these protein pools are chaperone-mediated assemblies that portend disease-associated activity by remodeling proteome-wide connectivity and, thus, function. To address our hypothesis, we combine chemical biology tools and complementary biochemical and functional approaches, with a specific interest on functions and modifications induced by proteome stress associated with malignant transformation and mediated by GRP94 modifications. We use established cancer cell lines, fresh patient bio-specimens, and cell and patient-derived xenografts in mice and ex vivo as disease models.
We identify a biochemical mechanism whereby aberrant N-glycosylation of a fraction of the cellular pool of the chaperone GRP94 remodels its location and conformation and, in turn, its interaction strength and interaction partners (i.e., connectivity). The outcomes are aberrantly remodeled protein pathways and, in turn, a pathologic cellular phenotype. We, therefore, provide a missing link in chaperone-mediated protein connectivity dysfunction by demonstrating how stress hijacks the customary role of a protein, turning it from a folder into a remodeler of protein connectivity.
RESULTS
GRP94 Inhibition Is Lethal in a Subset of Tumor Cells
To better understand how oncogenic stress alters the proteome through chaperone-mediated modifications, we first investigated the vulnerability for apoptosis (i.e., sensitivity) of a panel of cancer cell lines (n = 64) encompassing 11 distinct tumor types (Figure 1A) to GRP94 inhibition by PU-WS13. PU-WS13 is a small molecule whose selectivity arises from its ability to bind to an allosteric pocket of GRP94 that only partly overlaps with the ATP-binding pocket and is not accessible in the closely related paralog HSP90 (Gewirth, 2016; Patel et al., 2013, 2015; Shrestha et al., 2016; Stothert et al., 2017). It shows >100-fold selectivity over HSP90 and no interaction with kinases when tested at 10 mM in a 98-kinase panel (Patel et al., 2015). As a control, we used PU29F, a selective inhibitor of cytosolic HSP90 (Patel et al., 2013).
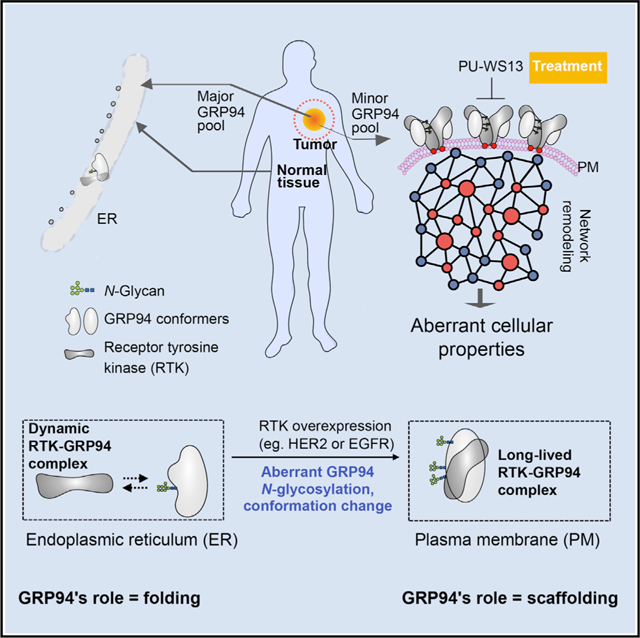
Figure 1. Sensitivity of Cancer Cells and Primary Specimens to GRP94 Inhibition by PU-WS13.
(A) Viability of cancer cell lines (n = 64) treated for 72 h with PU-WS13 (10 μM) or PU-29F (20 μM). Mean values of triplicate experiments are graphed. Negative values depict killing of the initial cell population.
(B) Correlative analysis between RTK (i.e., HER2 and EGFR) levels and cell viability for BC cells in (A). Pearson’s r, two-tailed, n = 12.
(C) Ex vivo sensitivity of primary BC explants treated for 48 h with PU-WS13 (10 μM), PU-29F (20 μM), or taxol (1 μM) evaluated as indicated in the associated schematic. Mean values from 2 slices per condition are shown.
(D) Western blot analysis of MDA-MB-468 cells treated for 24 h with PU-WS13 (0, 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 μM). PU-WS13, WS13. Graph, mean of 3 independent experiments; error bars, SEM.
(E) Same as in (C) for an EGFR+ BC specimen treated with PU-WS13 (20 μM) or vehicle for 24 h. Scale bar, 200 μm.
See also Figure S1.
We found that only a subset of these cell lines were vulnerable to PU-WS13, as measured by ATP levels and annexin V staining (Figures 1A and S1A). To understand the basis of the differential response, we focused further on BC in which vulnerability correlated with receptor tyrosine kinase (RTK) status (i.e., HER2 or EGFR [epidermal growth factor receptor]) and most sensitive samples exhibited greater RTK levels than the insensitive ones (n = 12 cell lines; Figure 1B and S1E). Sensitivity to PU-WS13 was retained in RTK-overexpressing (RTK+) primary breast tumors (n = 5 fresh BC tissue slices; Figure 1C) and esophagogastric tumors (n = 2 patient-derived xenografted tumors; Figure S1B). These effects were GRP94 specific, as there was no overlap with sensitivity to inhibition of cytosolic HSP90 or to taxol, a standard-of-care chemotherapy in BC (Figures 1A and 1C). PU-WS13 treatment of these cell lines and primary specimens was sufficient to reduce the steady-state level of the RTKs and impair downstream signaling through these receptors (Figures 1D, S1B, and S1C; see p-ERK and p-STAT3). This is consistent with previous findings for GRP94 knockdown or inhibition in HER2+ BC cells (Li et al., 2015; Patel et al., 2013) and with GRP94 knockdown in EGFR+ BC cell lines (Figure S1D).
We found that suppressing GRP94 function by PU-WS13, or analogs HJP-149 and SO-33, was more toxic to BC cell lines overexpressing HER2 and EGFR than was direct inhibition of the RTK by either a kinase inhibitor or an anti-RTK antibody, as judged using PARP cleavage as a marker of cell death (Figure S1C). Conversely, non-transformed human mammary epithelial cells remained unaffected by treatment with PU-WS13. In the fresh BC tissue explants (Corben et al., 2014; Rodina et al., 2016), we found that normal cells adjacent to the cancer cells remained unaltered at concentrations of PU-WS13 that induced apoptosis in >70% of the tumor cells. This was seen in the benign lobules with associated acini and ducts (white arrows, Figure 1E) that remained unaltered in the same treated section in which treated tumor cells showed pyknotic nuclei and apoptotic debris, nuclear morphological changes that are indicative of apoptosis (black arrows, Figure 1E).
GRP94 Is Heterogeneous in Cancer
Total GRP94 levels were comparable between the different cancer cell lines assessed for sensitivity to GRP94 inhibition (Figure S1E), suggesting that chaperone concentration alone was not responsible for the different responses to inhibition. To understand the cause for heightened sensitivity to GRP94 inhibition in some cell lines, we analyzed the GRP94 isolated from sensitive and resistant cell lines for residence in stable protein complexes, cellular localization, conformation, and PTM.
We ran cell homogenates from both inhibitor-sensitive and -resistant cancer cells on native gels in buffers near the physiological pH (Figure 2A). In addition to the ~242-kDa dimer that is characteristic of non-transformed cells (Wearsch and Nicchitta, 1996), we also observed a number of distinct and indistinct high-molecular-weight (HMW) GRP94 species above the main 242-kDa band when blotting with the 9G10 antibody, which recognizes the charged linker region (residues 290–350) and is sensitive to the conformation of GRP94 (Edwards et al., 1984; Vogen et al., 2002). These species were significantly enriched in the PU-WS13-sensitive cell lines (Figure 2A), indicating an enrichment of the 9G10-recognized conformation in the inhibitor-sensitive cell lines, even though the total amount of GRP94 in all cell lines was comparable. This difference was not due to protein unfolding or degradation under the experimental conditions (Figures S2A and S2B). In native PAGE carried out at near-physiological pH (to avoid denaturation), the signal may reflect both binding of other proteins and the protein’s own conformation. Such complexity influences the subsequent immunoblotting step. The signal observed in native PAGE may, therefore, reflect both complexation and retained native conformation of the proteins, which is recognized by the antibody. When cells were treated with PU-WS13 prior to native PAGE analysis, we observed a complete disappearance of the HMW GRP94 species but no change in total GRP94, as detected under denaturing conditions (Figure 2B).
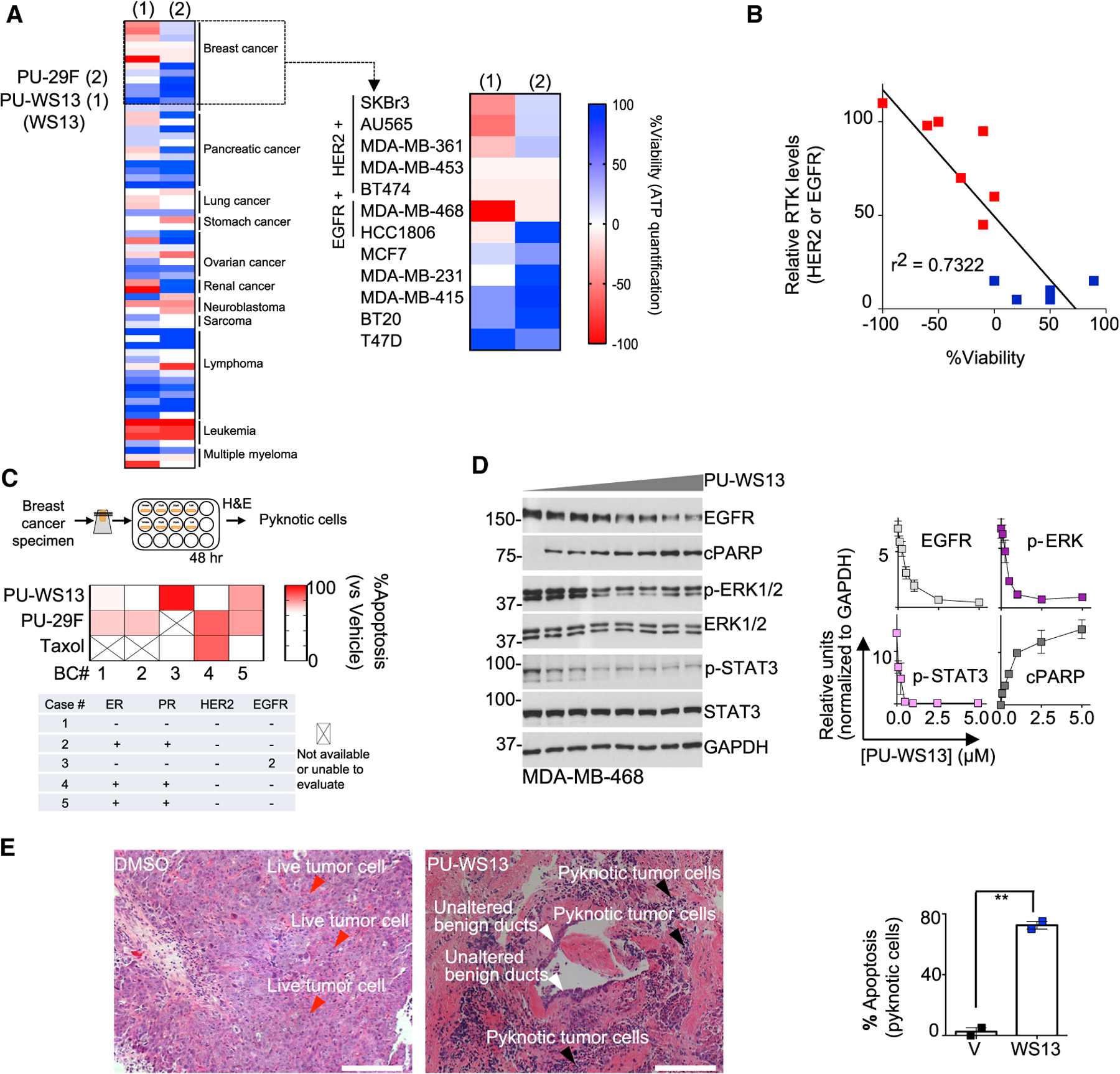
Figure 2. GRP94 Is Biochemically and Functionally Heterogeneous in Cancer Cells.
(A and B) Native-PAGE (top) and SDS-PAGE (bottom) separation followed by immunoblot with the 9G10 anti-GRP94 antibody in un-treated cell lines (A) or in those treated for 4 h with PU-WS13 (10 mM) (B). Each data point is an individual cell line; lanes 1–11 are BT474, MDA-MB-468, SKBr3, AU565, MDA-MB-361, HCC1806,MCF7, MDA-MB-231, T47D, BT20, and HMEC. Graph, mean. Error bar, SEM; unpaired t test, **p < 0.01, ***p < 0.001.
(C) Biochemical signature of GRP94 in distinct cellular compartments of a PU-WS13-sensitive (SKBr3) and a -resistant (MCF7) cell line. Cells were treated as in (B) prior to fractionation. WCL, whole cell lysate; C, cytosol; TM, total membrane; F1, cytosol and ER/Golgi; F2, plasma membrane. HSP70, cellular fractionation and loading control; LE and HE, low and high exposure, respectively. Red arrow, lower motility GRP94 specific to F2; blue arrow, higher motility GRP94 specific to F1. Graph, mean; n = 4. Error bar, SEM; unpaired t test, ***p < 0.001.
(D) Representative immunofluorescence (IF) staining of SKBr3 cells (n = 50) treated as in (B). Calnexin, ER marker; 4′,6-diamidino-2-phenylindole (DAPI), blue, nuclear stain; PU-WS13, WS13. Scale bar, 10 mm. Graph, mean. Error bar, SEM; unpaired t test, ****p < 0.0001.
(E) Same as in (C) for HER2 normalized to vehicle-treated cells. Graph, mean; n = 9 for native-PAGE and n = 5 for SDS-PAGE. Error bar, SEM; unpaired t test, ***p < 0.001.
(F) IF staining of SKBr3 cells (n = 20) treated for 4 h with vehicle (DMSO), PU-WS13 (WS13, 10 μM), or PU-29F (29F, 20 μM). Mean Pearson’s correlation coefficient (PCC) R value is shown corresponding to the colocalization of HER2 and EEA1. Scale bar, 10 μm. Graph, mean. Error bar, SEM; one-way ANOVA with Dunnett’s post hoc, ***p < 0.001, ****p < 0.0001.
(G) Western blot analysis of the protein cargo isolated by streptavidin-immobilized WS13-B (a biotinylated version of PU-WS13) from SKBr3 TM extracts. See also Figures S2 and S3.
We next profiled the subcellular localization of GRP94 (Figure S2C) in SKBr3 cells, a PU-WS13-sensitive cell line, and in MCF7 cells, an insensitive cell line. The total (whole-cell lysate [WCL]) GRP94 levels in these two cell lines were comparable, as judged by western blotting of SDS-PAGE separations, whereas substantially more GRP94 was located in the PM fraction (F2) in SKBr3 cells than in MCF7 cells (Figure 2C, WCL versus F2/F1 where F1 is the GRP94 pool found in the ER, Golgi, and cytosol).
In SKBr3 cells, we also observed that F2 was enriched in a ~100-kDa GRP94 species compared to the same fraction in MCF7 cells. This species ran slower than the normal 94-kDa species on SDS gels and may reflect additional PTMs (Figure 2C, red versus blue arrows), as we demonstrate further. The distinctive mobility of this species was also apparent in the F2 fraction of SKBr3 cells under native PAGE separation and in experiments in which surface proteins were isolated by biotinylation (Figure S3A). Finally, PU-WS13 treatment resulted in a decrease in the HMW species noted in F2 but had no effect on the GRP94 pool in F1 (Figure 2C, native separation).
In summary, the viability data presented above indicate that GRP94 is essential in only some cancer cells. Essentiality correlates with RTK overexpression and potentially, with an increase in the translocation of GRP94 to the PM, the formation of GRP94-containing HMW electrophoretic complexes, a preference for particular GRP94 conformations, and increased GRP94 PTM. We, therefore, proceeded to investigate the contribution of each factor to the observed HMW species: complexation, conformation, and PTM.
GRP94 Complexation Contributes to Its Heterogeneity
Because HER2, an abundant RTK in SKBr3 cells, co-localizes with GRP94 at the PM in these cells (Figure 2D; Li et al., 2015; Patel et al., 2013), we probed the cellular fractions of this cell line for HER2. HER2 detected by western blotting on native gels was observed as a HMW species in the WCL, total membrane (TM), and PM fractions (Figure 2E). These HMW HER2 species, absent in MCF7 cells, correspond to the same fractions that contained the HMW GRP94 species. Short-term treatment of SKBr3 cells with PU-WS13 reduced the amount of HMW HER2 in the F2 pool (Figure 2D, immunofluorescence; Figure 2E, native PAGE) without majorly changing the overall levels of HER2 (Figure 2E, WCL by SDS-PAGE), paralleling the observations seen with GRP94.
We observed that HER2 at the PM, but not intracellular HER2, co-localized with early endosomes following short-term treatment with PU-WS13 (Figure 2F, EEA1 staining), indicating that GRP94 inhibition initiates a clearance of PM-resident HER2, which involves internalization and endosomal sorting to lysosomes for degradation (Cortese et al., 2013; Raja et al., 2008). Conversely, PU-29F affected intracellular HER2 pools.
Finally, we introduced a biotinylated PU-WS13 reagent, PU-WS13-B, which we found to preferentially isolate the GRP94 found in the F2, and not in the ER, Golgi, or cytosolic fractions (F1) (Figure S3B), and to enrich for the 100-kDa GRP94 species (Figure 2G, red arrow). This probe pulled down HER2 along GRP94 (Figure 2G). Similar results were seen in EGFR+ BC cells, in which EGFR, another RTK, was GRP94 bound (Figure S3C). We found PU-WS13-B preferentially isolated GRP94 bound to HER2 over free GRP94 (Figure S3D). Conversely, the G4420 antibody, which recognizes amino acids 733–750 in the C-terminal domain of GRP94, was able to capture both the HER2 bound and the free GRP94 (Figure S3D).
Conformation Is Key in HMW GRP94 Formation
We next determined if GRP94’s conformational state contributes to formation of the HMW protein pool. We probed the HMW pool with two anti-GRP94 antibodies, the conformation-specific 9G10 antibody and the G4420 antibody, which is not known to discriminate between different conformational states of the chaperone. A dose-dependent immunocapture of GRP94 and HER2 by the G4420 antibody was associated with a dose-dependent decrease in the HMW GRP94 and HER2 pools noted on native PAGE and a decrease in both GRP94 and HER2 levels in the supernatant noted on western blot (Figure 3A). Conversely, the antibody 9G10 captured GRP94 but not HER2; nonetheless, both the GRP94 and HER2 HMW pools were diminished on native PAGE, but only GRP94 was reduced in the supernatant following 9G10 immunocapture, consistent with a conformational change induced by this antibody, which is associated with the release of the bound cargo.
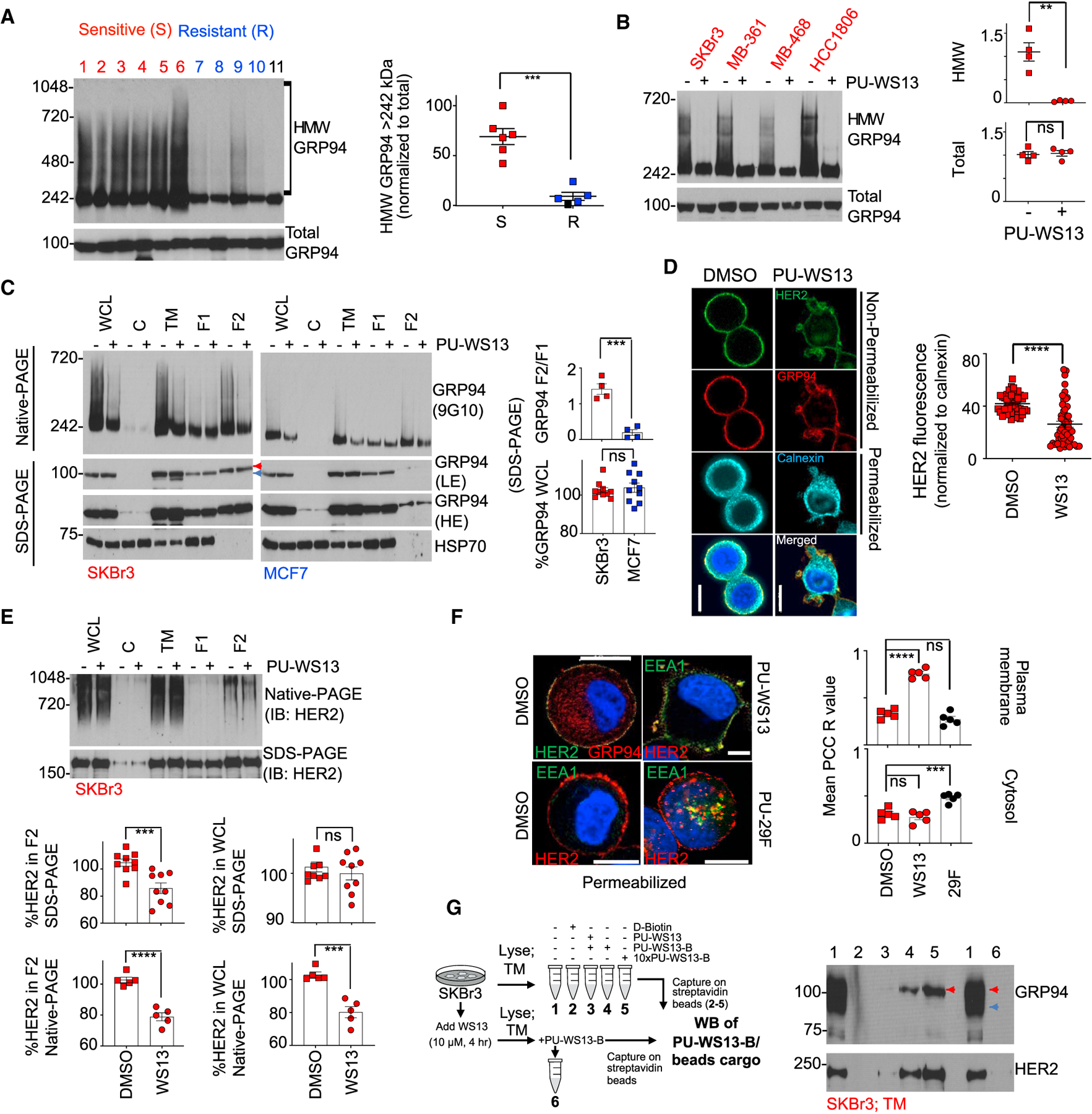
Figure 3. A Specific GRP94 Conformation Enables the Formation of Stable HMW GRP94 and HER2 Pools.
(A) Anti-GRP94 antibody captured cargo from SKBr3 cell extracts, and the remaining supernatant was separated under native and denaturing conditions and immunoblotted with the indicated antibodies.+, 1 times antibody amount; ++, 2 times antibody amount. IgG, control; red arrow, unspecific signal.
(B) Immuno-capture as in (A) of SKBr3 cells treated with PU-WS13 in a dose and time-dependent manner. Graph, mean of 3 independent experiments; error bars, SEM.
(C) Summary of the findings.
As noted above, PU-WS13-B preferentially captured the PM-localized GRP94 that was bound to HER2. To see if this corresponds to a specific conformation of the chaperone, we treated cells with PU-WS13 prior to immunocapture with the two GRP94 antibodies (Figure 3B). Increasing amounts of PU-WS13 or increased duration of PU-WS13 exposure dramatically reduced the amount of GRP94 captured by the 9G10 antibody, indicating thattheinhibitor changesGRP94to a conformation thatis no longer recognized by the antibody. Conversely, PU-WS13 treatment increased the amount of both GRP94 captured by the G4420 antibody and HER2 pulled down by the immunocapture of PU-WS13-bound GRP94, indicating that the ligand promotes the chaperone conformation that associates with surface-resident HER2.
Although these findings show that PU-WS13 induces a conformational change in GRP94, it also indicates that a specific conformation of GRP94 exists that enables the formation of the GRP94 and HER2 stable HMW protein pools (Figure 3C). By adopting this conformation, GRP94 stabilizes the PM-bound HER2, an essential mechanism for the oncogenic function of RTKs.
Hyperglycosylation Favors HMW GRP94 Formation
GRP94 is post-translationally modified by phosphorylation and glycosylation (Cala, 2000), and we asked whether these PTMs contribute to the formation or stability of the HMW GRP94 complexes. Treatment with Endoglycosidase H (Endo H) or Peptide:N-glycosidase F (PNGase F), but not with lambda-protein phosphatase, led to a change in the electrophoretic mobility of GRP94 (Figure S4A). The mobility shift due to glycosylation differed between cellular fractions. The F2 fraction exhibited a greater mobility shift than the ER F1 fraction. This indicates that both species are N-glycosylated but that more residues are glycosylated on the GRP94 protein localized at the PM than GRP94 in other locations, such as the ER. Because an N-linked carbohydrate chain has a mass of 1.5–3 kDa, the slower migrating GRP94 species we detected in the F2 likely contains two or more N-glycan-modified Asn residues. We term this F2-specific species as hyper-N-glycosylated GRP94 (hgGRP94).
Because hgGRP94 was the predominant species in the F2 fraction, we investigated the effect of removing N-glycosylation on the stability of the HMW GRP94 and HER2 complexes (Figure 4A). Following deglycosylation under native conditions, we probed for GRP94 and HER2 HMW complexes on native gels or captured the GRP94 complexes with immobilized PU-WS13-B or with the two GRP94 antibodies G4420 or 9G10. Glycan removal significantly reduced the amounts of HMW GRP94 and HER2 species seen on native gels, indicating that glycosylation is important for the stability of these complexes (Figure 4B). The amount of the GRP94 and HER2 (or EGFR) cargo captured by PU-WS13-B and G4420 pulldown was also decreased by deglycosylation treatment (Figures 4C and S4B). The effect was more dramatic when the sugars were completely stripped from Asn (such as by PNGase F), with near-complete disappearance of the HMW GRP94 species on native PAGE (Figure S4C), similar to what we observed upon PU-WS13 treatment. Complete deglycosylation also abolished GRP94’s binding to PU-WS13-B (Figure S4C).
Figure 4. The N-Glycan Content of GRP94 Regulates the Stability and Function of the HMW GRP94-HER2 PM-Associated Pool.
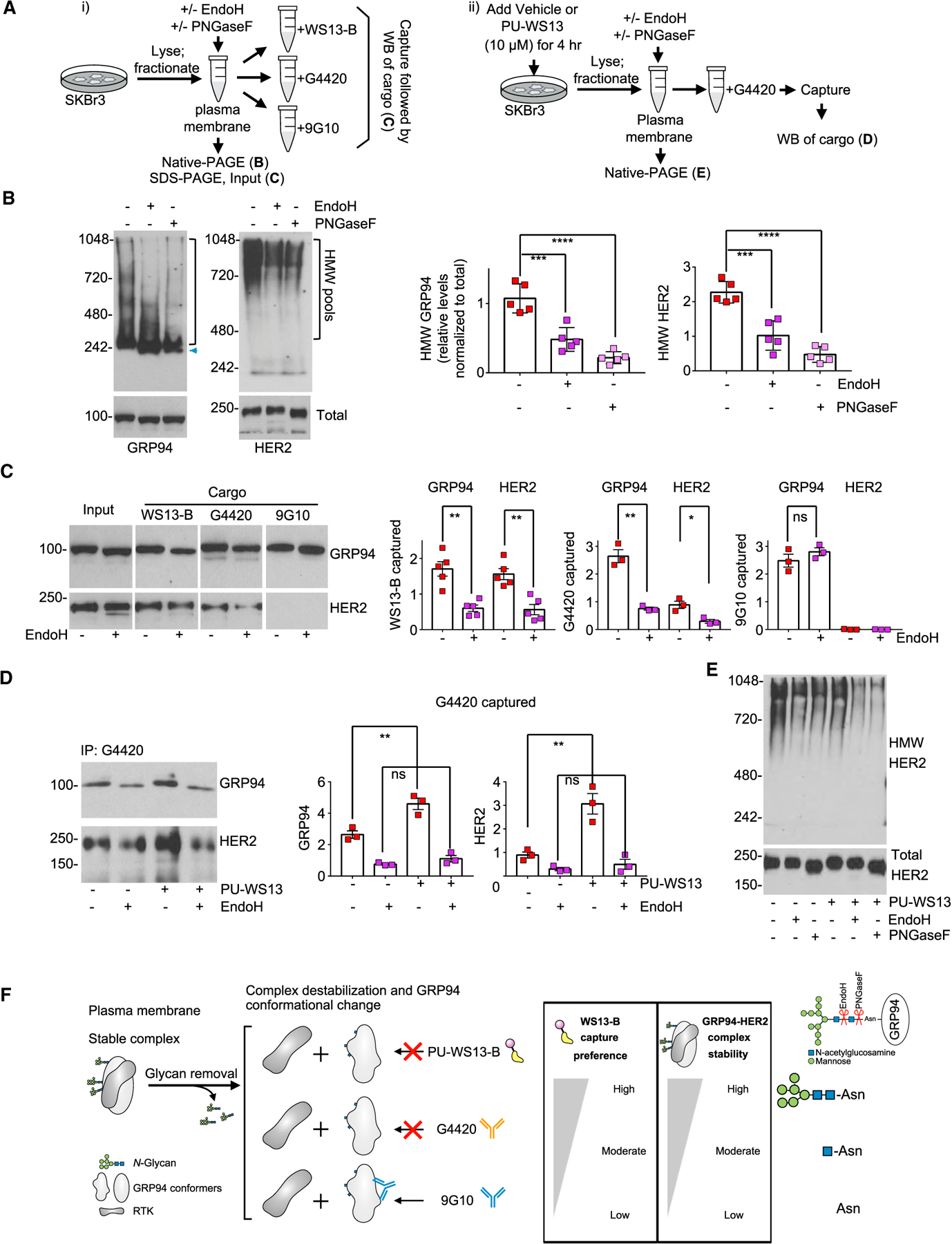
(A) Schematic of the experimental design to investigate the role of N-glycans on the conformation, stability, and function of the HMW GRP94 pools. WS13-B, biotinylated PU-WS13.
(B) PM pool of GRP94 and HER2 in SKBr3 extracts treated under native conditions as in (A) with the indicated enzymes. Graph, mean; error bars, SEM; n = 5; unpaired t test, ***p < 0.001, ****p < 0.0001.
(C) Western blot analysis of GRP94 and HER2 isolated from extracts treated as in (A) and (B). Graph, mean; error bars, SEM; n = 3 to 5; unpaired t test, **p < 0.01, *p < 0.05.
(D) Western blot analysis of cells treated for 4 h with PU-WS13 (10 μM) prior to N-glycan removal and immuno-capture with the G4420 anti-GRP94 antibody as in (A). Graph, mean; error bars, SEM; n = 3; unpaired t test, **p < 0.01.
(E) Biochemical signature of HER2 in extracts as in (C) and (D).
(F) Summary of the findings.
See also Figure S4.
As shown above, G4420 favors the PU-WS13-bound GRP94-HER2 complex, and indeed, we observed significantly more GRP94 and HER2 captured by the antibody in cells pre-treated with PU-WS13 (Figure 4D). N-Glycan removal, however, equally diminished the amount of GRP94 and HER2 captured by G4420 (Figure 4D) and the amount of HMW HER2 observed on native gels (Figure 4E), substantiating further that the N-glycans are key for maintaining the HMW GRP94 and HER2 pools (Figures 4F and S4C).
GRP94 contains six potential N-glycan acceptor sites, and under normal conditions, the protein is predominantly monoglycosylated at N217 (Cloutier and Coulombe, 2013; Schwarz and Aebi, 2011). We performed glycosylation site mapping by mass spectrometry and identified N62, N217, and N502 as putative glycosylated Asn sites on the hgGRP94 variant. Through knock out and mutagenesis of endogenous GRP94 by CRISPR-Cas9, we generated, isolated, and expanded four homozygous clones—N62Q, N217A, N62Q/N217A, and knockout (KO) (Figure 5A). Using these clones, we confirmed that more than one Asn was glycosylated on hgGRP94 (see Endo H treatment, Figure 5B). We found that N62 was a key residue needed for the formation of the HMW GRP94 pool, as evidenced by native PAGE (Figures 5C and S5A), insensitivity to PU-WS13 (see GRP94 pools on native PAGE, Figure 5C; see p-ERK and EGFR on SDS-PAGE, Figures 5D and S5B), a decrease in RTK signaling activity (see ERK downstream signaling, Figures 5E and S5B), diminished interaction with the G4220 antibody (Figure 5F), and a significant decrease in GRP94 and RTK localized at the PM (Figures 5G and S5C) in the N62Q-containing clones when compared to the wild type (WT). GRP94 KO mimicked the effects observed with the N62Q-containing mutants (i.e., a limited EGFR pool translocated to the PM, no hyperactive ERK as noted by p-ERK levels, and insensitivity of basal p-ERK levels to PU-WS13). Conversely, N217 mutagenesis had little effect on these GRP94 and RTK functions.
Figure 5. Glycosylation at N62 of GRP94 Is a Key Regulator in the Formation of the HMW GRP94 Variant and Is Important for Its Oncogenic Activity.
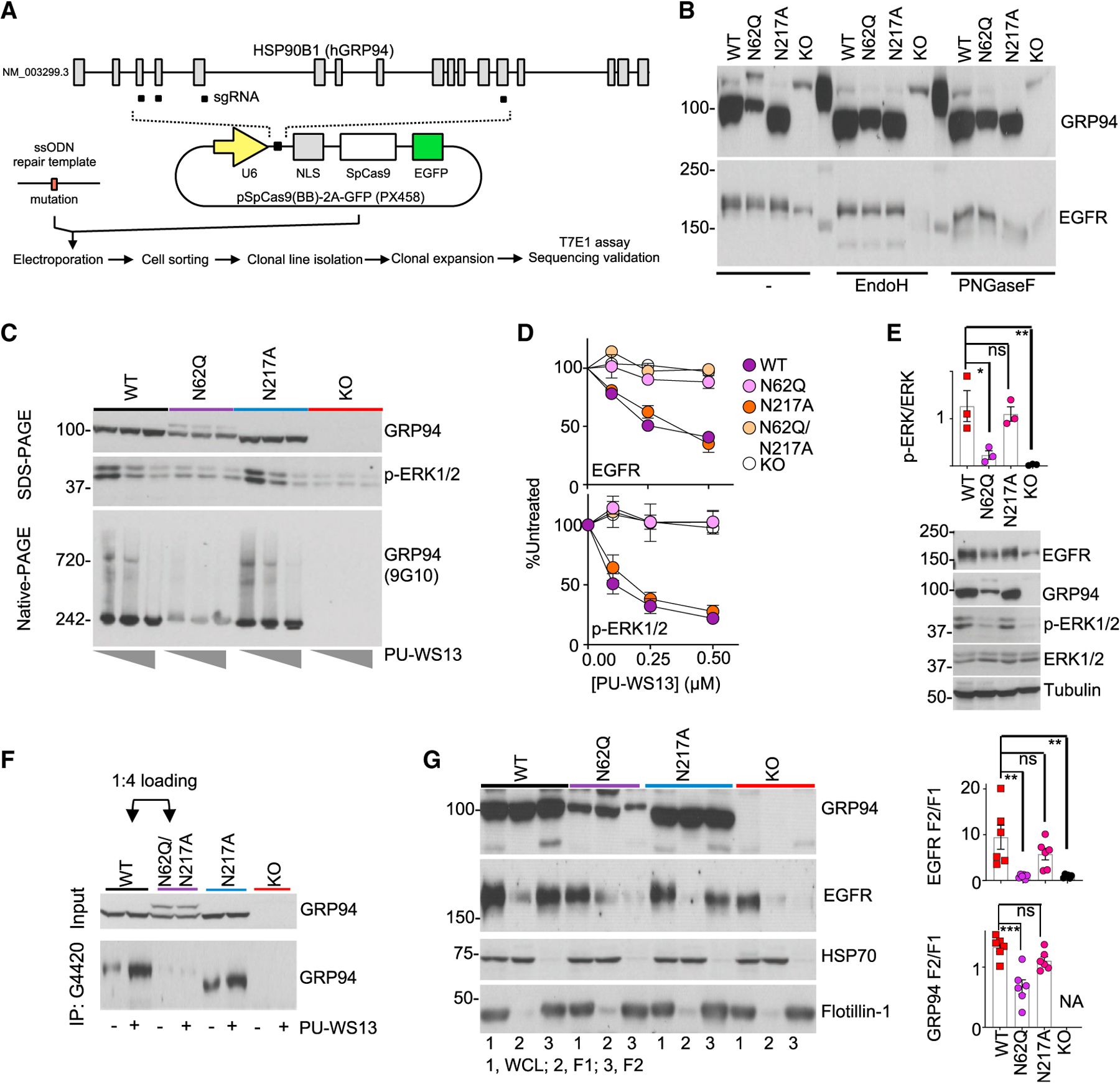
(A) Schematic for clone generation and validation. MDA-MB-468 cells (WT) and clones containing the indicated GRP94 mutants were then used for the analyses below.
(B) The effect of GRP94 N-glycan mutagenesis or KO evaluated on glycosylation load.
(C) GRP94 HMW formation (measured by blotting for GRP94 HMW species on native-PAGE and ERK activity on SDS-PAGE) and its sensitivity to PU-WS13, in cells treated for 24 h with 0, 0.25, or 1 μM PU-WS13.
(D) Steady-state levels of EGFR and activity of its downstream signaling (measured by p-ERK levels) in cells treated for 24 h with PU-WS13. Graph, mean ± SEM; quantification of 3 western blot analyses.
(E) Baseline activity of EGFR-downstream signaling measured by western blot analysis of p-ERK and ERK. Graph, mean (n = 3). Error bar, SEM; one-way ANOVA with Dunnett’s post hoc, *p < 0.05, **p < 0.01.
(F) GRP94 immunocapture in clones pre-treated for 4 h with vehicle (−) or PU-WS13 (2.5 μM). Four times more lysate from the N62Q-containing clone was loaded to normalize for the GRP94 input.
(G) Cellular localization of EGFR and GRP94. 1, WCL; 2, F1; 3, F2 fractions. Tubulin, HSP70, and Flotillin-1 are controls for equal loading and for cellular fractionation purity; NA, not available. Graph, mean ± SEM; one-way ANOVA with Dunnett’s post hoc; n = 6 individual experiments. ***p < 0.001, **p < 0.01.
See also Figure S5.
Collectively, these data indicate that GRP94’s ability to form long-lived, stable complexes with RTKs at the PM (as opposed to the dynamic interactions needed for RTK folding by GRP94 in the ER) (Eletto et al., 2010) is dependent on a specific hyperglycosylation pattern, with N62 being a key residue for the observed switch of GRP94 from a folding, ER chaperone to an oncogenic protein that stabilizes and activates RTKs at the PM.
HMW GRP94: An Oncogenic Gain of Function
Because HMW GRP94 is observed in cells that have high PM expression and oncogenic downstream signaling of RTKs, we wondered if the accumulation of GRP94 at the PM was sufficient to initiate such oncogenic signaling. To address this question, we used a construct that directs myc-tagged GRP94 to the PM by deletion of the Lys-Asp-Glu-Leu (KDEL) sequence and incorporation of a transmembrane domain from platelet-derived growth factor receptor (Zheng et al., 2001). We used a Meth A fibrosarcoma cell line that was stably transfected with this construct, TM96, and we compared the properties of this cell line to those of WT Meth A cells.
Fractionation of cell extracts expressing TM96 showed that the TM96 GRP94 construct was found only in the F2 fraction, but not the ER (F1) or cytoplasmic (C) fractions (Figure 6A). TM96-expressed GRP94 participated in the formation of stable HMW GRP94 complexes, as evidenced by the characteristic electrophoretic migration pattern on native gels (Figure 6A), its glycosylation status suggestive of hyperglycosylation (Figure S6A), and its sensitivity to PU-WS13 (Figure S6B). Its introduction was sufficient to increase the PM expression of proteins that require GRP94 for cell surface presentation, such as LRP6 (Liu et al., 2013; Figure 6B), and to increase the neoplastic nature of these cells (Figures 6A and 6B, see p-p65 and p-ERK; Figure 6C, see anchorage-independent growth). TM96-expressed GRP94 also augmented the formation of intracellular stable HMW complexes incorporating HSP90 (Figure 6A, HSP90 native PAGE), also referred to as HSP90-incorporating epichaperomes, which act as molecular scaffolding platforms that augment the activity of cytosolic protein pathways, including signaling pathways (Joshi et al., 2018; Kourtis et al., 2018; Rodina et al., 2016). PU-WS13 treatment was sufficient to reverse these effects, as evidenced by inhibition of the activated but not baseline signaling (Figure 6B) and the loss of the HMW GRP94 pool located at the PM (Figure S5B). Importantly, the increase in the HMW GRP94 species upon TM96 transfection significantly increased the vulnerability of cells to PU-WS13 treatment (Figure 6D).
Figure 6. Accumulation of hgGRP94 at the PM Is Sufficient to Augment HMW GRP94 Levels, Stabilize Receptors at the PM, and RewireProtein Networks in the Cytosol.
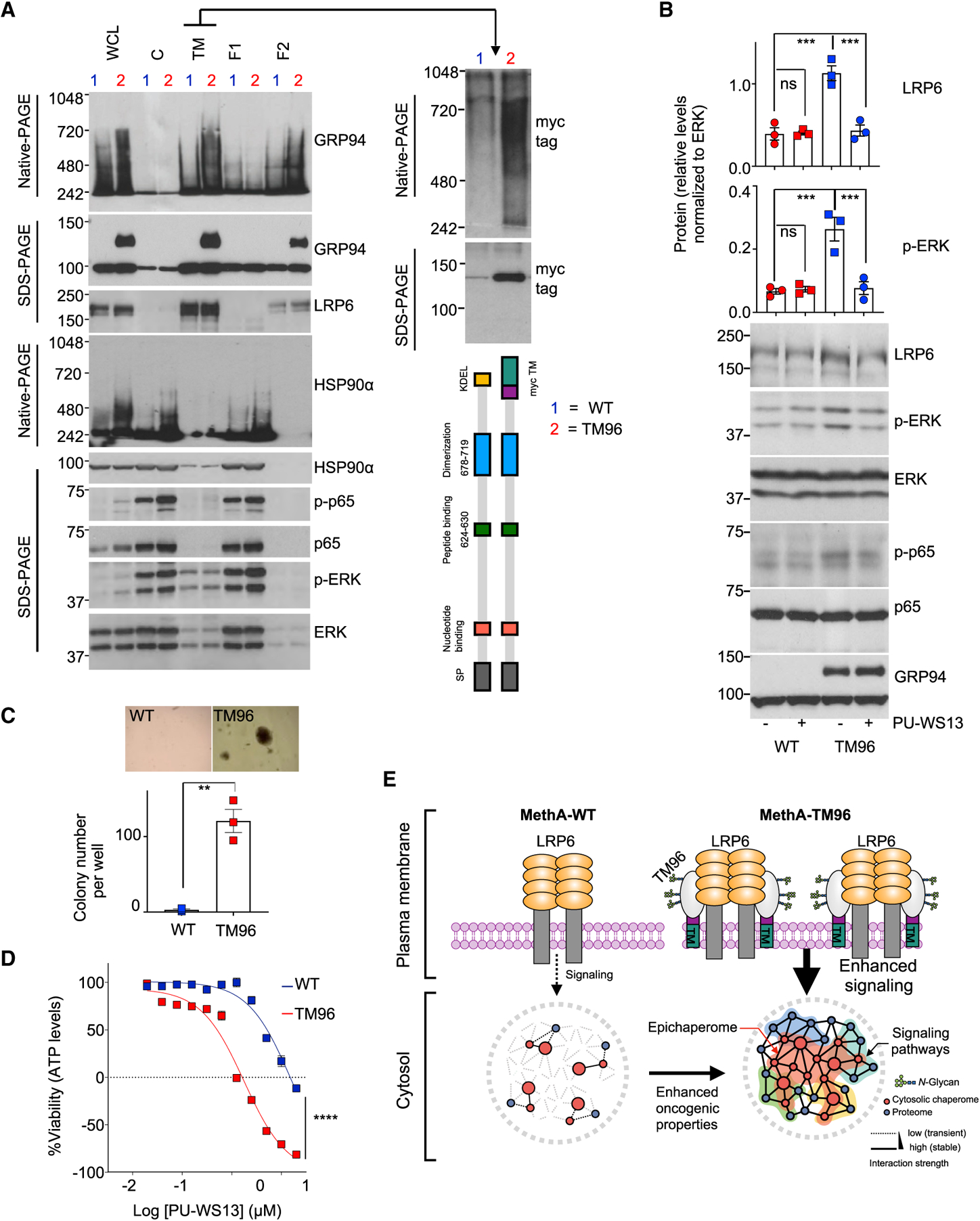
(A) Biochemical profile of GRP94 in the indicated cell fractions obtained from cells containing the WT (1) or the TM96 (2) GRP94 construct. LRP6, control for PM proteins; p-p65, p-ERK control for signaling activity and HSP90-incorporating epichaperomes for oncogenic activation of cytosolic chaperomes; myc, control for the presence of TM96. Gels are representative of three individual experiments.
(B) Western blot of indicated cells treated for 24 h with PU-WS13 (10 μM). Graphs, mean ± SEM; n = 3; one-way ANOVA with Dunnett’s post hoc, ***p < 0.001.
(C) Anchorage-independent growth in soft agar examined for MethA WT and TM96 cells at day 21. Graph, mean ± SEM; n = 3 experiments; unpaired t test, **p < 0.01.
(D) Viability of cells treated with PU-WS13 for 72 h. Graph, mean ± SEM; n = 4; two-way ANOVA, ****p < 0.0001. Negative y axis values depict killing of the initial cell population.
(E) Schematic of the findings.
See also Figure S6.
Together with the above findings in BC, these results indicate that an increase in HMWGRP94 at the PM is sufficient to alter the properties of PM proteins, resulting downstream in the rewiring of the cytosolic protein networks for increased signaling output (Figure 6E). Importantly, this gain of function brought about through the formation of HMW GRP94 species also increased the cells’ dependence on this oncogenic mechanism, as indicated by increased vulnerability to its inhibition.
HMW GRP94 Is an Actionable Target in Cancer
Because PU-WS13 exhibits a preference for the GRP94 pool incorporated into stable HMW complexes located at the PM of cancer cells, we can use this to address the targetability and safety of inhibiting this unusual GRP94 variant in cancer. Treatment is a balance between target engagement and therapeutic index, and we evaluated whether target suppression can be safely achieved by PU-WS13 in vivo. To understand target engagement during the study, we measured tumor and tissue pharmacokinetics (PK) and pharmacodynamics (PD) after either a single dose of PU-WS13 administered intraperitoneally (i.p.) or at the end of a long-term treatment (see Figure S7A for study design). Both AU565 HER2+ and MDA-MB-468 EGFR+ breast tumors were established orthotopically.
We observed that a dose of 75 mg/kg PU-WS13 significantly engaged the target, as indicated by significant RTK downregulation (Figures 7A and 7B) and supported by the PU-WS13 concentration recorded in these tumors (Figures 7C). HSP70 levels, which are a marker of HSP90 inhibition (Yuno et al., 2018), remained unchanged at this dose, indicating that inhibition of this close paralog during treatment by PU-WS13 did not occur (Figures S7B and S7C). We, therefore, investigated the efficacy of 75 mg/kg and 125 mg/kg PU-WS13 given three times weekly (Monday, Wednesday, and Friday), every other day (qod) or daily (qd), with weekends off (Figure 7D). We observed significant and dose and schedule-dependent effects of PU-WS13 (Figures 7E–7G), with complete tumor growth suppression observed under the daily treatment paradigm. Similar results were also noted when tumors were established subcutaneously (Figures 7H and 7I).
Figure 7. The HMW GRP94 Tumor Variant Is an Actionable Target.
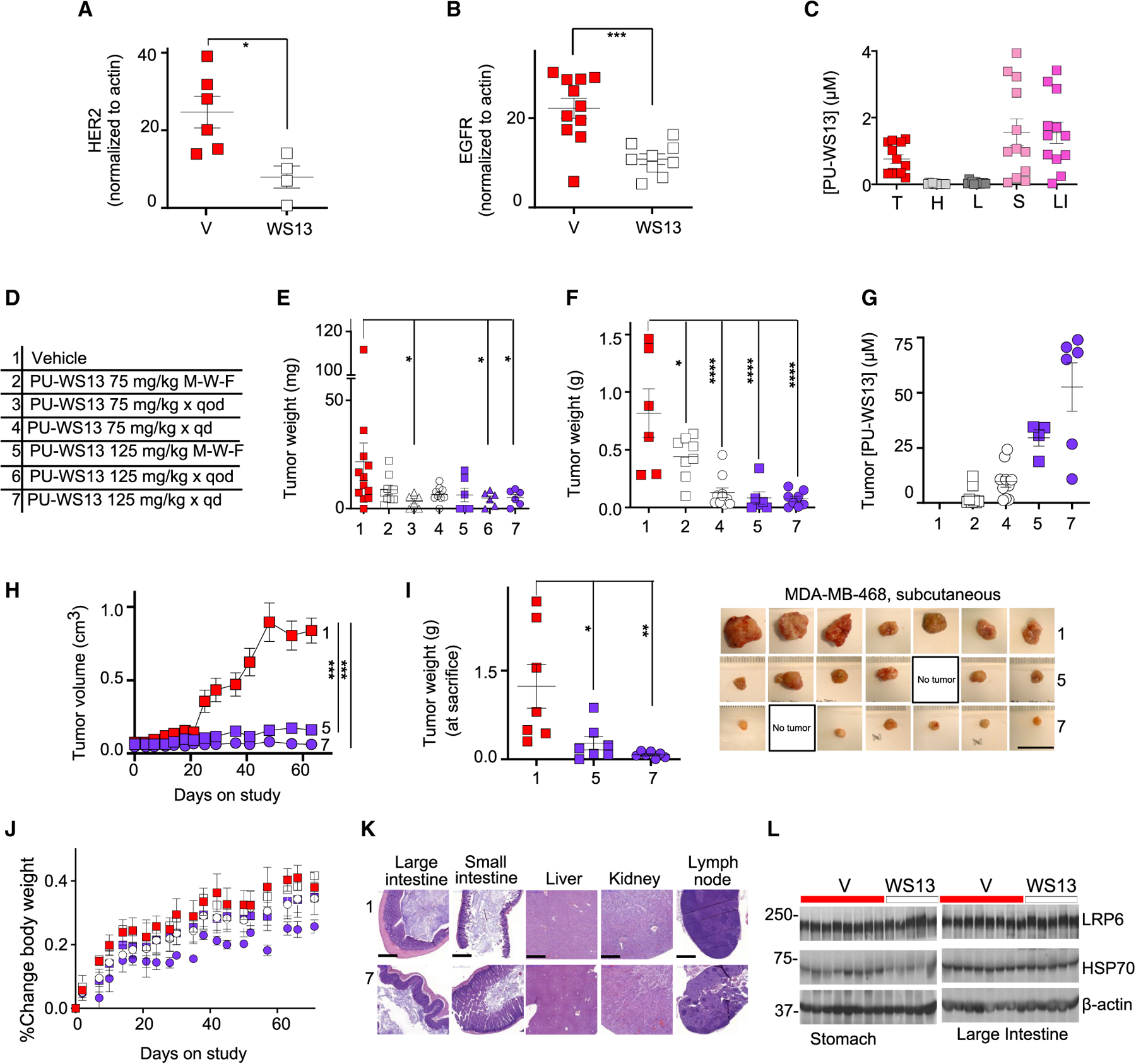
(A and B) PD analyses at 24 h after one dose of PU-WS13 (75 mg/kg, i.p.) administered to mice bearing AU565 (A) or MDA-MB-468(B) orthotopic tumors. Graphs, mean ± SEM; n = 6 for vehicle (V) and n = 4 for PU-WS13 (WS13) for AU565; n = 11 for V and n = 9 for WS13, for MDA-MB-468; unpaired two tailed t test, ***p <0.001, *p < 0.05. See Figure S6 for representative western blot analyses.
(C) Molar concentration of PU-WS13 in individual tumors and tissues for mice (n = 12) as in (B). Graphs, mean ± SEM. T, tumor; H, heart; L, lung; S, stomach; LI, large intestine.
(D) Dose and schedule paradigms used in testing the efficacy and safety of PU-WS13. 75MWF, 75 mg/kg given Monday, Wednesday, and Friday; qod, every other day; qd, every day, Monday through Friday.
(E) Tumor weight measurements of mice as in (A) after 64 days of treatment as indicated in (D). 1, n = 12; 2, n = 9; 3, n = 6; 4, n = 9; 5, n = 6; 6, n = 6; 7, n = 6, pooled data from two experiments. Graph, mean ± SEM; two-tailed Mann Whitney test, *p < 0.05.
(F) Tumor weight measurements of mice as in (B) after 87 days of treatment as indicated in (D). 1, n = 6; 2, n = 8; 4, n = 10; 5, n = 6; 7, n = 8, pooled data from two experiments. Graph, mean ± SEM; one-way ANOVA, Dunnett’s multiple comparisons, *p < 0.05, ****p < 0.0001.
(G) Concentration of PU-WS13 in individual tumors as in (F).
(H) Tumor volume monitoring during treatment as in (D) of mice bearing subcutaneous MDA-MB-468 tumors. Graph, mean ± SEM; n = 7 per cohort; two-way ANOVA, ****p < 0.0001.
(I) Tumor weight measurements of mice as in (H). Graph, mean ± SEM; one-way ANOVA, Dunnett’s multiple comparisons, *p < 0.05, **p < 0.01. Picture of individual tumors is also shown. No tumor, no macroscopic tumor evident at sacrifice. Scale bar, 2.5 cm.
(J) Body weight monitoring of mice as in (F). Graph, mean ± SD.
(K) H&E-stained tissues and organs from cohorts 1 and 7 as in (F). Scale bar, 500 μm.
(L) Western blot of indicated tissues from individual mice treated as in (B). LRP6, control for HMW GRP94 selectivity; HSP70, control for GRP94 over HSP90 selectivity.
See also Figure S7.
PU-WS13 was well tolerated. Even for the long-term treatment regimens that delivered 37 to 62 doses of PU-WS13 to mice over 87 days, we observed no treatment-related toxicities: mice retained a normal weight throughout treatment (Figure 7J). We conducted complete necropsies and analyzed hematology and serum chemistry panels on vehicle-treated mice and on mice receiving the 125-mg/kg dose five times per week for 87 days (Figure 7K; Table S1). All hematological and clinical chemistry findings were within normal parameters, and histopathology conducted on major organs showed no toxic changes induced by PU-WS13.
We also evaluated gastrointestinal (GI) tract LRP6 levels after PU-WS13 administration (Figure 7L). Housekeeping GRP94 is essential for folding and regulating physiologic functions of the Wnt receptor LRP6 (Rachidi et al., 2015), and it is expected that compounds, such as PU-WS13, selectively targeting the tumor-specific HMW GRP94 variant will act on tumor functions while leaving housekeeping GRP94 functions unaltered at similar or higher concentrations as those seen in the tumor. Because most small molecules, including PU-WS13, are largely cleared by the GI tract, it is a body site most exposed to such agents over the time they spend in the body. Indeed, the concentration of PU-WS13 detected in the stomach and large intestine and the exposure to agent over 24 h were much higher in these organs than in the tumor (Figure 7C); nonetheless, no decrease in LRP6 levels were detected by western blotting.
DISCUSSION
Our study identifies a GRP94 variant in cancer, whereby by altering N-glycosylation, a new protein conformationally, dynamically, and functionally distinct from the GRP94 of normal cells is created. In place of a chaperone that is confined to the ER and makes transient interactions with and folds client proteins, a specific increase in N-glycosylation promotes a conformational state that allows for stable interactions with oncoproteins at the PM. In this context, hyperglycosylation is a modality used by GRP94 to alter its conformational fitness and stabilize a state most permissive for stable interactions. Through this stabilization, the functions of these proteins are enhanced, and cellular protein pathways are aberrantly remodeled—N-glycosylation, thus, transforms a chaperone, GRP94, from a folding to a scaffolding protein that remodels protein connectivity, with an end result of proteome-wide dysfunction. Therefore, the N-glycosylation pattern of GRP94 we identify is a specific modification exploited by cancer cells to alter the customary role of a chaperone.
The aberrantly N-glycosylated GRP94 variant is present only in some tumors, is independent of total GRP94 levels, and is absent or scarce in non-transformed cells. We found that the functions of one class of oncoproteins, RTKs, are modified by this GRP94 variant and only in cancer cells driven by RTK overexpression. RTK overexpression is a form of proteome stress, and under these conditions, GRP94 N-glycosylation at specific sites is key both to enhance the presence of these proteins at the PM by forming stable complexes with the RTKs and to maintain RTKs in a state that enables aberrant downstream signaling and a rewiring of cytosolic protein pathways.
N-linked glycosylation is among the most ubiquitous protein modifications in eukaryotes. It is implicated in a myriad of housekeeping functions, including modification of a protein’s folding capacity, stability, and oligomerization and aggregation status; ER quality control and protein trafficking; host cell-surface interactions; and modulation of enzyme activity (Lee et al., 2015). Changes in glycosylation are observed in cancer where they affect the interaction and, subsequently, the activation capacity of RTKs (Mereiter et al., 2019). Conversely, to our knowledge, there is no report of N-glycosylation increasing the oncogenic properties of a protein indirectly by modulating its complexation.
Presumably, N-glycosylation does not induce significant changes in a protein’s structure but decreases protein conformational dynamics, likely leading to an increase in protein stability (Lee et al., 2015). In this view, N-glycans act like molecular glues, holding together residues around the glycosylation sites through favorable interactions made with nearby protein residues, thus resulting in the stabilization of a specific protein conformation or disfavoring others (Solá and Griebenow, 2006). Stabilization of a specific conformation favoring the formation of complexes of enhanced stability was recently shown for HSP90, the cytosolic paralog of GRP94 (Inda et al., 2020; Kishinevsky et al., 2018; Kourtis et al., 2018; Rodina et al., 2016). There, an oncogenic stress, such as MYC hyperactivation, or a neuronal damaging stress, such as tau overexpression, redistributes the cytosolic pool of molecular chaperones and helpers into complexes of enhanced stability. These stable assemblies, termed epichaperomes, function as multi-component scaffolds to provide a framework on which the cell’s complement of proteins can work more efficiently or differently than they would without chaperone participation. A similar role for the hgGRP94 variant in the context of disease is suggested by our data. The heterogeneous HMW GRP94 species observed on native gels likely consists of numerous other proteins besides the RTKs we have thus far identified. The identity of these other components, as well as their role in working with GRP94 to regulate the oncogenic HER2 and EGFR functions, is not yet known.
Because both HSP90 and GRP94 are stable as dimers even at high concentrations, a conformational change is necessary to drive stress-induced complexation alteration. This may lead to the unmasking of a site that is absent in the dimer, giving rise to a new protein-protein interaction platform and a new quaternary structure that reverts the chaperone from a folding to a scaffolding protein. Although for HSP90 the identity of such a conformational stabilizer remains to be elucidated, for GRP94, our study suggests it to be, at least in part, regulated by N-glycosylation. Apparently, in most cases, glycosylation does not induce permanent secondary structural formations but rather prompts local conformational changes close to the glycosylation site. Often, they result in a b turn, infrequently a b strand, and rarely an a helix (Mitra et al., 2006). Whether such a conformational switch changes the direct interaction of GRP94 with HER2 or whether it mediates the creation of a stable multimeric platform with co-chaperones and other factors that mediate HER2 stabilization, similarly to that seen for HSP90, remains to be seen.
We show that inhibition of the hgGRP94 variant with compounds such as PU-WS13 is feasible in cancer cells, human primary tumor specimens, and xenografted tumors in mice. Although GRP94 is abundant in most cells of the mammalian body, it is clear that the housekeeping variant and the N-glycosylated variant targeted in cancers are different, rendering compounds such as PU-WS13 selective for the cancer form. By introducing these PU-type chemical probes, we also demonstrate that the N-glycosylated GRP94 variant and the specific aberrant proteins and cellular processes enabled by this variant are targetable in disease. Thus, inhibitors of the N-glycosylated GRP94 variant are an example of a “targeted protein degradation-based therapeutic” that acts specifically on dysfunctions and protein networks enabled by this variant, thus sparing the normal folding functions of GRP94.
Although we mainly evaluated the expression and significance of the hgGRP94 variant in BC, several lines of evidence suggest that it is implicated in other cancers as well. RTK overexpression (e.g., EGFR, HER2, and MET) is observed in a variety of cancer cells and in cells of a tumor-supportive microenvironment (Butti et al., 2018; Contessa et al., 2008, 2010; Siddals et al., 2011; Tan et al., 2018; Turrini et al., 2017). Overall, several tumors are characterized by an aberrant expression of RTKs (FGFR1 in lung and BC; Dutt et al., 2011; Reis-Filho et al., 2006), FGFR3 in breast and bladder cancer (Fischbach et al., 2015; Helsten et al., 2016), ERBB4 in breast and gastric cancer (Kim et al., 2016; Shi et al., 2012), FLT3 in colon cancer (Moreira et al., 2015), KIT in melanoma and gastrointestinal stromal tumor (GIST) (Carvajal et al., 2011; Tabone et al., 2005), and PDGFRA in glioblastoma multiforme (GBM) (Nobusawa et al., 2011). RTK amplifications also allow tumor cells to escape therapeutic treatment (MET and HER2 amplification can be detected in EGFR mutant lung cancers that become resistant to EGFR TKI therapy; Yu et al., 2013). Because EGFR overexpression is often a side effect of radiation therapy (Cuneo et al., 2015), targeting the GRP94 variant with PU-WS13 may also radiosensitize tumors.
It is noteworthy that inhibition of the GRP94 variant is more toxic to EGFR+ tumors than the direct inhibition of EGFR by kinase inhibitors or anti-EGFR antibodies. Approximately half of all triple-negative BCs (TNBCs) and inflammatory BCs overex-press EGFR. Nonetheless, clinical trials of EGFR inhibitors have reported little or no benefit (Masuda et al., 2012). It is believed that the ineffectiveness of these treatments is due to crosstalk between EGFR and c-Met or other RTKs because strategies that knocked down EGFR, either by small interfering RNA (siRNA) or by mixtures of antibodies that induced robust degradation of EGFR, led to reduced viability of TNBC cells (Ferraro et al., 2013; Mueller et al., 2012). As per our findings, GRP94 inhibition by PU-WS13 also induces robust EGFR degradation and apoptosis in TNBC cells, and this effect may provide a therapeutic advantage over tyrosine kinase inhibitors.
In conclusion, our study reports that increasing the interaction strength between GRP94 and RTKs and other receptors at the PM, which we found to be regulated by a specific N-glycosylation pattern, is a mechanism used by a chaperone to enhance the stabilization and interaction of certain proteins. Our findings identify a biochemical mechanism whereby stress remodels a chaperone from a folding to a scaffolding protein, creating a state of chaperone-mediated protein connectivity dysfunction. Here, aberrant N-glycosylation of a fraction of the cellular pool of the chaperone GRP94 remodels GRP94 location and conformation and, in turn, its interaction strength and interaction partners, with the outcome being aberrantly remodeled protein pathways and a pathologic cellular phenotype. We, thus, propose that the HMW form of GRP94 is an example of a “protein assembly mutation” (Nussinov et al., 2019), a proteome malfunction defined by defective protein-protein interaction that portends pathologic activity. This variant is a target for cancers and other diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Gabriela Chiosis (chiosisg@mskcc.org).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
LC-MS data that support the findings of this study have been deposited in MassIVE with the accession number MSV000085459, [ftp://massive.ucsd.edu/MSV000083484/].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
De-identified pathology discarded specimens were obtained in accordance with the guidelines and approval of the Institutional Review Board #09–121 (breast cancer, PI: Dr. Modi) and #10–018 (esophagogastric cancer, PI: Dr. Janjigian) at Memorial Sloan Kettering Cancer Center (New York, NY). The patients provided signed informed consent prior to participation.
Animals
All animal studies were conducted in compliance with MSKCC’s Institutional Animal Care and Use Committee (IACUC) guidelines under an approved protocol. All mice were housed at MSKCC animal facility and Research Animal Resource Center (RARC) provided husbandry and clinical care. Athymic nude mice (Hsd:Athymic Nude-Foxn1nu, female, 20–25 g, 8 weeks old; RRID:MGI:5652489) were obtained from Envigo and NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, female, 20–25 g, 8 weeks old, IMSR Cat# JAX:005557, RRID:IMSR_JAX:005557) were obtained from the Jackson Laboratory.
Human Cell Lines
Cell lines were obtained from laboratories at MSKCC, or purchased from the American Type Culture Collection (ATCC) or Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ). Cells were cultured as per the providers’ recommended culture conditions. Cells were authenticated using short tandem repeat profiling and tested for mycoplasma before and after use. The breast cancer cell lines (MDA-MB-468 (HTB-132), HCC1806 (CRL-2335), MDA-MB-231 (HTB-26), MDA-MB-415 (HTB-128), MCF-7 (HTB-22), BT474 (HTB-20), BT20 (HTB-19), MDA-MB-361 (HTB-27), SKBr3 (HTB-30), MDA-MB-453 (HTB-131), T47D (HTB-133), AU565 (CRL-2351) and the non-transformed cell line HMEC (human primary mammary epithelial cells, PCS-600–010) were obtained from the American Type Culture Collection (ATCC). The pancreatic cancer cell lines include: MiaPaCa2 (CRL-1420), Panc-1 (CRL-1469), BxPc-3 (CRL-1687), Capan-1 (HTB-79), SU.86.86 (CRL-1837), HPAF2 (CRL-1997), ASPC-1 (CRL-1682), PL45 (CRL-2558), CFPAC (CRL-1918), Capan-2 (HTB-80) were purchased from ATCC; 931102 and 931019 are patient derived cell lines provided by Dr. Y. Janjigian, MSKCC. The lung cancer cell lines NCI-H3122, NCI-H299 were kindly provided by Dr. M. Moore, MSKCC; NCI-H1373 and NCI-H525 were obtained from Dr. N. Lecomte, MSKCC. The stomach cancer cell lines SNU-1 (CRL-5971) and NCI-N87 (CRL-5822) were obtained from ATCC. The ovarian cancer cell lines PEO-1, PEO-4, OVCAR4, OV1847, A2780, IGROV-1 and OVCAR5 were kindly provided by Dr. D. Solit, MSKCC. The renal cancer lines SKRC38 and SKRC52 were provided by Dr. S. Larson, MSKCC. Neuroblastoma cells SY5Y (CRL-2266) was purchased from ATCC; LAN5 and SMS-KCNR were obtained from the Children’s Oncology Group (COG). Ewing’s sarcoma cells TC71 and A673 were kindly provided by Dr. S. Ambati, MSKCC. Lymphoma cell lines include: SU-DHL-6 (CRL-2959), Toledo (CRL-2631), Farage (CRL-2630) and BC3 (CRL-2277) were purchased from ATCC; HBL-1, MD901 and U2932 were kindly provided by J. Angel Martinez-Climent, Centre for Applied Medical Research, Pamplona, Spain; Karpas422 (ACC-32), RCK8 (ACC-561) and SU-DHL-4 (ACC-495) were obtained from the DSMZ; OCI-LY1, OCI-LY3 and OCI-LY7 were obtained from the Ontario Cancer Institute; TMD8 was kindly provided by L. M. Staudt, NIH. Leukemia cell lines KASUMI-1 (CRL-2724), K562 (CCL-243) were purchased from ATCC; MOLM-13 (ACC-554) was obtained from DSMZ. Multiple Myeloma cell lines U266, PCNY1 and MM.1R were kindly provided from Dr. Z. Li, OSU.
Murine Cell Lines
Wild-type and TM96 expressing MethA fibrosarcoma cells were kindly provided by Dr. Z. Li, OSU. The cells were established as previously reported (Zheng et al., 2001) and cultured in RPMI medium with 10% heat-inactivated FBS (VWR) and 1% Penicillin/Streptomycin.
METHOD DETAILS
Reagents
PU-WS13, PU-WS13-biotin, inactive-WS13-biotin, PU29F, PU-H71, HJP-149 and SO-33 were synthesized using a previously reported protocol (Patel et al., 2013, 2015; Rodina et al., 2016). Briefly, PU-WS13 was synthesized via CuI-catalyzed coupling of 8-mercaptoadenine with 3,5-dichloroiodobenzene at 110°C resulting in 8-(3,5-dichloro-phenylsulfanyl)adenine in 72% yield, which was heated with 3-(tertbutoxycarbonyl-isopropyl-amino)-propyl tosylate in DMF at 80°C under nitrogen protection for 30 min. The Boc-deprotection of resulting tert-butyl(3-(6-amino-8-((3,5-dichlorophenyl)thio)-9H-purin-9-yl)propyl)(isopropyl)carbamate from the previous step with TFA gave crude material which upon purification by preparatory TLC with CH2Cl2:MeOH-NH3 (7N) at 20:1 yielded PU-WS13 (45% yield). PU-WS13-biotin was synthesized through alkylation of 8-(3,5-dichloro-phenyl sulfanyl)adenine at position N9 with N-(8-bromooctyl)phthalimide in the presence of Cs2CO3 in DMF at room temperature to obtain 2-(8-(6-amino-8-((3,5-dichlorophenyl)thio)-9H-purin-9-yl)octyl)isoindoline-1,3-dione in 21% yield. The resulting product from the previous step was subjected to phathalimide-deprotection with hydrazine hydrate in a mixture of CH2Cl2 and CH3OH to afford 9-(3-aminohexyl)-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine in 50% yield. In the final step, the free amine was reacted with NHS-active ester of biotin in DMF to result in a crude residue of PU-WS13-biotin. This residue was purified by preparatory TLC (CH2Cl2-MeOH-NH3 (7N), 10:1)to give 34% yield of PU-WS13-biotin. The synthetic route to inactive-WS13-biotin comprises S-alkylation of 8-mercaptoadenine with 1-iodo-2-methoxyethane in aqueous KOH solution providing 87% yield of 8-((2-methoxyethyl)thio)-9H-purin-6-amine. Further reaction with N-(8-bromooctyl)phthalimide, followed by phathalimide-deprotection and coupling with NHS-active ester of biotin in DMF gave crude inactive-WS13-biotin. This resulting residue was purified by preparatory TLC (CH2Cl2:MeOH-NH3 (7N), 10:1) to give in 72% yield the inactive-WS13-biotin. For the synthesis of HJP-149, C-S coupling was achieved by reacting 8-mercaptoadenine with 1-chloro-3,5-diiodobenzene in the presence CuI and neocuproine in DMF at 110°C. The resulting product was then N9-alkylated with 1,3-dibromopropane affording 9-(3-bromopropyl)-8-((3-chloro-5-iodophenyl)thio)-9H-purin-6-amine in 25% yield. In the final step, the reaction of bromo derivative with excess of isopropylamine, followed by purification using preparative TLC (CH2Cl2:MeOH-NH3 (7N), 20:1 or 15:1) afforded HJP-149 [8-((3-chloro-5-iodophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine] in 83% yield. Synthesis of SO-33 includes cyclocondensation of 2,4,5,6-tetraaminopyrimidine sulfate with CS2 in ethanol to obtain 2,6-diamino-9H-purine-8-thiol in quantitative yields. Next, copper-catalyzed coupling of 2,6-diamino-9H-purine-8-thiol with 3,5-dichloroiodobenzene resulted in 65% yield of 8-((3,5-dichlorophenyl)thio)-9H-purine-2,6-diamine. Transformation of C2-amino group of 8-((3,5-dichlorophenyl)thio)-9H-purine-2,6-diamine to Cl was achieved with SbCl3 and t-BuONO in 10:1 mixture of DCE and DMSO at 80°C resulting in 2-chloro-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine. The sequential reaction of 2-chloro-8-((3,5-dichlorophenyl)thio)-9H-purin-6-amine with 1,3-dibromopropane, followed by treating the bromo derivative from the previous step with excess isopropylamine resulted in crude SO-33. The crude residue was purified by preparatory TLC (CH2Cl2-MeOH-NH3 (7N), 20:1) to give 60% yield of SO-33 [2-chloro-8-((3,5-dichlorophenyl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine]. Synthesis of PU29F commenced with the coupling of 2,4,5,6-tetraaminopyrimidine with the acid fluoride of the 3,4,5-trimethoxyphenylacetic acid resulting in N-(2,4,6-triaminopyrimidin-5-yl)-2-(3,4,5-trimethoxyphenyl)acetamide. The acid fluoride was generated by treating phenylacetic acid derivative with cyanuric fluoride and pyridine in CH2Cl2. The acetamide derivative was cyclized to 8-(3,4,5-trimethoxybenzyl)-9H-purine-2,6-diamine by heating it in alcoholic NaOMe. Further transformation of the C2-amino group to fluorine was conducted by a modified Schiemann diazotization-fluorodediazoniation reaction in HF/pyridine and NaNO2 to yield 2-fluoro-8-(3,4,5-trimethoxybenzyl)-9H-purin-6-amine. The fluoro derivative was then N9-alkylated using the Mitsunobu reaction with 2-isopropoxyethan-1-ol in the presence of PPh3 and di-tert-butylazodi-carboxylate to give PU29F [2-Fluoro-9-(2-isopropoxy-ethyl)-8-(3,4,5-trimethoxy-ben-zyl)-9H-purin-6-ylamin] in 86% yield. For the synthesis of PU-H71, the cyclocondensation of 4,5,6-triaminopyrimidine sulfate with CS2 gave 8-mercaptoadenine in quantitative yield which was used directly without additional purification. CuI-catalyzed coupling of 8-mercaptoadenine with 5-iodobenzo[d][1,3]dioxole at 110°C resulted in 8-(benzo[1,3]dioxol-5-ylsulfanyl)adenine in 58% yield. Next, iodination of 8-(benzo[1,3]dioxol-5-ylsulfanyl)adenine with NIS/TFA afforded 8-(6-iodo-benzo[1,3]dioxol-5-ylsulfanyl)adenine which was heated with 3-(tertbutoxycarbonyl-isopropyl-amino)-propyl tosylate and Cs2CO3 in DMF at 80°C under nitrogen atmosphere. The resulting tertbutyl(3-(6-amino-8-((6-iodobenzo[d][1,3]dioxol-5-yl)thio)-9H-purin-9-yl)propyl)(isopropyl)carbamate underwent Boc-deprotection with TFA at room temperature to give crude product which was further purified by preparatory thin-layer chromatography on silica gel (CHCl3/MeOH/NH4OH at 10:1:0.5) to afford PU-H71 in 98% yield. All final compounds were characterized using 1H-NMR, 13C-NMR and HRMS and their purity determined by HPLC to be higher than 98%. Taxol (S1150), Erlotinib (S1023) and Lapatinib (S2111) were purchased from Selleckchem. Cetuximab was received as leftover from the MSKCC Clinical Pharmacy. Lambda protein Phosphatase (Lambda PPase, P0753S), Endo H (P0703S) and PNGase F (P0709S) were purchased from NEB Inc. High capacity Streptavidin Agarose (20361) was purchased from ThermoFisher Scientific.
Cell Fractionation and Immunoblotting
Cells were either treated with DMSO (vehicle) or indicated compounds and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% sodium deoxycholate and 0.5% NP40) supplemented with cocktail protease inhibitors (Roche) to produce whole-cell lysates. Lysates for cytosol and total membrane fractions were harvested and processed using ProteoExtract Subcellular Proteome Extraction Kit (Millipore Sigma) following the manufacturer’s instructions. Plasma membrane proteins were prepared using the Minute Kit (Invent Biotechnologies Inc.) according to manufacturer’s instructions. Protein concentrations were determined using the BCA kit (Pierce). Ten to fifty micrograms of total protein were examined by immunoblotting with indicated antibodies. The following primary antibodies were used: HSP90 (SMC-107) from Stressmarq; HER2 (28–0004), myc (R950–25) from Invitrogen; Calnexin (610523) from BD Biosciences; HSP70 (SPA-810), GRP94 (SPA-850) from Enzo; GAPDH (ab8245), GRP78 (ab21685), HSP90α (ab2928) from Abcam; cleaved PARP (G7341) from Promega; EGFR (4267), LRP6 (2560), p-AKT (S473) (9271), AKT (4691), Caspase 7 (9494), p-ERK1/2 (T202/Y204) (4377), ERK1/2 (4695), p-STAT3 (9145), STAT3 (12640), p-p65 (S536) (3033), p65 (8242), Flotillin-1 (3253) from Cell Signaling Technology; GRP94 (G4420), a-Tubulin (T5168), b-actin (A1978) and Concanavalin A-HRP (L6397) from Sigma-Aldrich. The blots were washed with TBS/0.1% Tween 20 and incubated with appropriate HRP-conjugated secondary antibodies. Chemiluminescent signal was detected with Enhanced Chemiluminescence Detection System (GE Healthcare) according to the manufacturer’s instructions.
Native Gel Electrophoresis
Cells were lysed in the RIPA buffers (WCL), or fractionated (C, TM, F1, F2) and diluted with Felts buffer (20 mM HEPES pH 7.3, 50 mM KCl, 2 mM DTT, 5 mM MgCl2, 20 mM Na2MoO4 and 0.01% NP40 with 0.1 mg/mL BGG). Twenty-five to one hundred mg of protein was loaded onto 5% native gel and resolved at 4°C. After running, gels were soaked in Tris-Glycine-SDS running buffer for 15 min prior to gel transfer for 75 min at 100 v, then immunoblotted as described above.
Native Gel Electrophoresis under Denaturing Condition
Cell lysates were mixed with 10 M urea (dissolved in Felts buffer) to reach the final concentrations of 2M, 4M, 6M and 8M. The mixtures were incubated for 15 min at room temperature and the volumes were adjusted by using Felts buffer. Immediately after the reaction, the samples were mixed with native loading buffer, and loaded onto native gel and resolved at 4°C. The GRP94 and HER2 signals were then detected by immunoblotting as described above.
Chemical bait Precipitation and Coimmunoprecipitation
Protein extracts were prepared in the indicated buffers and diluted in Felts buffer. Samples were incubated with D-biotin (Control), Inactive-WS13-biotin (Control), PU-WS13-biotin (GRP94 bait) or GRP94 antibodies for 3 hr at 4°C, followed by incubation with High Capacity Streptavidin agarose beads (ThermoFisher Scientific) or Protein A/G agarose beads (Roche) for another 2 hr at 4°C. The beads were washed with cold Felts buffer three times and subjected to immunoblotting.
Sequential Capture
PU-WS13-biotin beads were prepared by incubating 20 mM PU-WS13-biotin (chemical bait) with High Capacity Streptavidin agarose beads (ThermoFisher Scientific) for 3 hr at 4°C followed by washing with Felts buffer for three times. Antibody beads were prepared by incubating 9G10 or G4420 anti-GRP94 antibodies with protein A/G agarose beads (Roche) for 2 hr at 4°C followed by washing with Felts buffer for three times. The pre-formed chemical bait or antibody bait was then added into the cell lysate and the mixture was incubated on a rotator for 3 hr at 4°C. After separating the beads by centrifugation, the supernatant was collected and incubated with new pre-formed chemical bait or antibody bait. The sequential capture experiment was carried out by repeating the chemical precipitation (CP)/immunoprecipitation (IP) three times before the final IP with the indicated antibody bait. Captured cargos at each step were washed with Felts buffer three times before loading onto SDS-PAGE and subjecting to immunoblotting.
siRNA knock-down of GRP94
Transient transfections were carried out using Lipofectamine RNAiMax reagent (ThermoFisher) according to the manufacturer’s instructions. siGRP94 (Gene HSP90B1) and scramble siRNA were purchased from QIAGEN. Cells were transfected with 5 nM or 20 nM siRNA. The knockdown efficiency and other cellular markers were evaluated at 72 hr post transfection by immunoblotting.
Immunofluorescence
Cells were seeded and grown onto Lab-Tek II chamber slides for 24 hr before the experiment. After washing two times with cold PBS, cells were fixed by treating at RT for 20 min with 2% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS containing 10% FBS for 10 min and blocked with 2% BSA for 1 hr. After washing four times with PBS, primary antibodies were added into the chambers, and incubated overnight at 4°C. The cells were washed again with PBS, followed by incubation with the secondary antibody for 1 hr at RT. The cells were finally washed again with PBS for four times and mounted with ProLong Diamond Anti-fade Mountant with DAPI (Molecular Probes). Slides were cured for 2 hr at RT and stored at 4°C overnight before imaging under microscope (× 63 oil lens, Leica Upright Confocal SP5). Protein co-association was quantified using Coloc2 plugin in Fiji software to determine the mean Pearson’s Correlation Coefficient R value (PCC R value). Background was subtracted by using a Rolling-Ball Background Substraction, threshold regression was set as costes and the costes randomizations were set at 10. The following primary antibodies were used for the immunofluorescence: HER2 (Zymed; 28004; 1:50), HER2-FITC (BD; 340553; 1:200), GRP94 (Sigma; G4420; 1:100), EEA1 (Abcam; ab70521; 1:100) and Calnexin (Abcam; 22595; 1:100).
Enzymatic Deglycosylation and Dephosphorylation
Cell lysates were treated with Endo H or PNGase F according to the manufacturer’s instructions. After reacted at 37°C for 1h, the samples were mixed with protein loading buffer and subjected to immunoblotting. For the deglycosylation reactions under native condition (used for further pulldown assays), the lysates were first diluted in Felts buffer and then incubated with the enzymes without protein denaturing on ice overnight. The deglycosylated samples were further used for detatured or native gel electrophoresis, chemical or immuno precipitation experiments. To compare GRP94 protein’s sensitivity to different enzymes (i.e., Figure S4A), same amount of cell lysate was diluted into 1 × enzyme reaction buffer (1 × Protein MetalloPhosphatase (PMP) buffer for lambda PPase, 1 × glyco buffer × for Endo H or 1 × glyco buffer 2 for PNGase F. The control sample was diluted into PMP buffer. Enzymes were added into the reaction tubes and incubated for 1hr at 30°C for PPase, at 37 C for Endo H and PNGase F. Immediately after the reaction was finished, the samples were mixed with the protein loading buffer, heated at 95°C for 5 min, stored on ice before loading into the gels.
Assessment of Cell-Surface Proteins
Cell surface protein isolation kit (Pierce) was used to purify the cell surface proteins according to the manufacturer’s instructions. Briefly, cell surface proteins were biotinylated by incubating the live cells with Sulfo-NHS-SS-biotin for 30 min at 4°C. The reaction was quenched and cells were lysed. The biotinylated surface proteins were affinity-purified by NeutrAvidin Agarose beads, and then eluted with protein loading buffer and subjected to immunoblotting. The flow-through was used as intracellular protein fraction.
CRISPR/Cas9 mediated Knock-Out and targeted Mutagenesis of endogenous GRP94
Design and construction of CRISPR sgRNAs: GRP94 CRISPR sgRNAs were designed using the online tools - CRISPOR (http://crispor.tefor.net/) and CHOPCHOP (https://chopchop.cbu.uib.no/). All sgRNA sequences were further validated using the online Nucleotide BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch) to target unique genomic sites. sgRNAs against hGRP94 used in this study are: sg1948, GAAGAAGCTATTCAGTTGGA; sg3859, CAACGATACCCAGCACATCT. The single-stranded oligos were synthesized by Intergrated DNA Technologies, cloned into PX458 (pSpCas9(BB)-2A-GFP, Addgene plasmid #48138; Ran et al., 2013) via BsaI, and the positive clones were validated by plasmid sequencing (Genewiz). Cell transfection and GFP sorting: ssODN repair template were synthesized by Genewiz and the sequences used for mutagenesis are: N62Q, gaactatgc catgcaatatttgcttacctaactgatttccttagagaggaggaggctattcagttggatggattaCAGgcatcacaaataagagaacttaga-gagaagtcggaaaagtttgccttcca; N217A, ctattccgccttccttgtagcagataaggttattgtcacttc aaaacacaacGCTgatacccagcacatctgggagtct-gactccaatgaattttctgtaattgctgacccaagaggaaacac. Breast cancer cells, MDA-MB-468, were transfected using Neon nucleofection system (ThermoFisher) according to the manufacturer’s instructions. Briefly, 1 × 106 cells were mixed with 2 μg PX458-sgRNA plasmid, 4 μL ssODN repair template (10 μM) in 100 μL Electroporation tip. Parameters were set at 1100 pulse voltage, 30 ms pulse width, 2 pulses. 48 hr post-transfection, GFP positive cells were sorted and collected via Flow. The positive cells were re-seeded into the dishes and left to recover for additional 24 hr. Isolation of clonal cell lines by dilution: Cells were dissociated from the plates and then passed through a 35 mm cell strainer to prevent cell clumping. After counting the cell number, the cells were diluted to a final concentration of 0.5 cells per 100 μL and plated into 96 well plates. The plates were closely inspected every other day one week after the plating. Single cell clones were expanded and transferred into 12-well or 6-well plates for further validation. T7 endonuclease I assay: Genomic regions flanking the CRISPR sgRNA target sites were PCR amplified with Fusion Flash High-Fidelity PCR Master Mix (F548S, ThermoFIsher Scientifics) using gene-specific primers. PCR products were purified with MinElute PCR Purification Kit (-QIAGEN) and hybridized in PCR buffer (95°C, 5 min; 95–85°C at −2°C/s; 85–25°C at −0.1°C/s; hold at 4°C). After treatment with T7 endonuclease I (2.5 U, NEB) at 37°C for 1 hr, the fragments were subjected to electrophoresis in a 2.5% agarose gel and visualized by staining with SyBR Safe DNA Gel stain (Invitrogen). Clone cell validations: When the single clone expanded to grow to a significant cell number, the genomic DNAs were extracted via the Phenol:Chloroform method and subjected for PCR and sequencing. The sequencing primers used in this study are: N62-F, ccattttaacccccaagaca; N62-R, atcaggccgtgaacctattt; N217-F, cactttcagaaaaggccataaaa; N217-R, caggaaaattaaggcccaga. Whole cell lysates were also validated by immunoblotting with GRP94 antibodies.
Soft Agar Colony Formation Assay
6-well plates were coated with a bottom layer of 2 mL 1% low-melting-point agarose (Invitrogen) dissolved in the complete culture medium. Cell suspension in culture medium containing 0.4% low-melting-point agarose was then added on the top of the layer. Colony growth was quantified and images were taken after staining with 0.05% crystal violet.
Cell Viability Assessment
Annexin V staining.Cells were labeled with Annexin V-FITC and 7AAD after PU-WS13 treatment for 48 hr, as previously reported (Rodina et al., 2016). The samples were analyzed by flow cytometry (LSR-II, BD Biosciences). FlowJo software was used for data analysis (FlowJo LLC)). The necrotic cells were defined as Annexin V+/7AAD+, and the early apoptotic cells were defined as Annexin V+/7AAD–. ATP based cell viability assessment. Cell viability was assessed using CellTiter-Glo luminescent Cell Viability Assay (Promega) after a 72 hr PU-WS13 treatment. The method determines the number of viable cells in culture based on quantification of ATP amount, which signals the presence of metabolically active cells, and was performed as previously reported (Rodina et al., 2007).
Ex vivo Studies
The fresh tissue slicing method maintains tissue integrity and architecture within an intact tumor-microenvironment-macroenvironment context throughout treatment, providing a more clinically-relevant means to assess the inhibitors’ effects. This is important because interactions among tumor and stromal cells are known to play a major role in cancer growth and progression and in the anti-tumor efficacy of agents. De-identified pathology discarded specimens were obtained in accordance with the guidelines and approval of the Institutional Review Board# 09–121 (PI: Dr. Modi). The primary breast cancer specimens or fresh esophagogastric PDX samples were processed as reported before (Corben et al., 2014). Briefly, the sample was delivered in a fresh state, harvested in a sterile environment under 30 minutes from the surgical procedure. Tumor tissue was chosen from the periphery of the index lesion to avoid potential frank central necrosis (cell death). The necrotic tissue may be grossly recognizable by any of the following criteria: loss of color or paleness of the tissue; loss of strength in which necrotic tissue is soft and friable; a distinct demarcation between the necrotic and viable tissue. Immediate after sampling is received, the sample was placed in wet ice and transported to the laboratory for ex vivo fresh tissue sectioning. Samples were then embedded into 5% Agarose gel and cut into 200 mm thick sections on a Leica VT 1000S vibratome. The live sections were transferred into 24-well tissue culture plates and treated for 24 hr or 48 hr with the indicated concentration of PU-WS13. Sections were then fixed in 4% formalin for 1hr at room temperature, and transferred into 70% ethanol. Following paraffin embedding, sectioning and mounting, the sections were stained with hematoxylin and eosin (H&E) and evaluated by the pathologists. Apoptosis and necrosis of the tumor cells was assessed by reviewing all the H&E slides of the case (controls and treated ones) blindly. The effect to any precursor lesions (if present) and any off-target effects to benign surrounding tissue was also evaluated.
In vivo Studies in Mice
For the breast cancer model: Athymic nude mice (Hsd:Athymic Nude-Foxn1nu, female, 20–25 g, 6 weeks old; RRID:MGI:5652489) were obtained from Envigo and were allowed to acclimatize at the MSKCC vivarium for 1 week prior to implanting tumors. Tumor xenografts were established subcutaneously into the dorsal flank or orthotopically into the 4th mammary fat pad. Tumors were initiated by subcutaneous injection of 5 × 106 cells for MDA-MB-468 or orthotopic injection of 5 × 106 cells for AU565 in a 200 μL cell suspension of a 1:1 v/v mixture of PBS with reconstituted basement membrane (BD matrigel, Collaborative Biomedical Products Inc.). Tumor Volume was determined by measurement with Vernier calipers, calculated using the formula-length × width2 × 0.5 and analyzed on indicated days as the median tumor volume ± SD. Mice were randomized prior to treatments, and euthanized after similar PU-WS13 treatment periods and at a time before tumor reached a size that resulted in discomfort or difficulty in physiological functions in the individual treatment group, in accordance with the IUCAC protocol. For the esophagogastric cancer model: Esophagogastric PDX model was generated as previously described (Mattar et al., 2018). Briefly, patient specimens (~0.5 g) collected under the approved IRB protocol (10–018, PI: Dr. Janjigian) were minced, mixed with Matrigel (1:1 v/v) and implanted subcutaneously in 8 weeks old female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, the Jackson Laboratory, IMSR Cat# JAX:005557, RRID:IMSR_ JAX:005557). When tumors reached 1–1.5 cm3, they were excised and transplanted in recipient mice, and models were considered successfully established following 3 rounds of transplantation.
Pharmacokinetic (PK) and Pharmacodynamic (PD) Studies
PK study. Frozen tumors or tissues were dried and weighed prior to homogenization in acetonitrile/H2O (3:7). PU-WS13 was extracted in methylene chloride, and the organic layer was separated and dried under vacuum. Samples were reconstituted in mobile phase. Concentrations of PU-WS13 in tissue or plasma were determined by high-performance LC-MS/MS. PU-H71 was added as the internal standard. Compound analysis was performed on the 6410 LC-MS/MS system (Agilent Technologies) in multiple reaction monitoring mode using positive-ion electrospray ionization. For tissue samples, a Zorbax Eclipse XDB-C18 column (2.1 × 50 mm, 3.5 μm) was used for the LC separation, and the analyte was eluted under an isocratic condition (80% H2O + 0.1% HCOOH: 20% CH3CN) for 3 min at a flow rate of 0.4 mL/min. For plasma samples, a Zorbax Eclipse XDB-C18 column (4.6 × 50 mm, 5 μm) was used for the LC separation, and the analyte was eluted under a gradient condition (H2O+0.1% HCOOH:CH3CN, 95:5 to 70:30) at a flow rate of 0.35 mL/min. PD study. Tumors or tissues were homogenized in tissue lysis buffer (50 mM Tris-HCl pH 7.5, 50 mM KCl, 150 mM NaCl, 2 mM EDTA, 0.1% Sodium deoxycholate, 0.5% NP40, 0.5% Triton X-100, 0.5% SDS) using Bullet Blender Tissue Homogenizer (Next Advance Inc.). Protein concentrations were determined using the BCA kit (Pierce) according to the manufacturer’s instructions. Protein lysates (20–100 μg) were electrophoretically resolved by SDS/PAGE, transferred to nitrocellulose membrane, and probed with the indicated antibodies.
Efficacy and Toxicology Studies
Efficacy studies. Mice bearing MDA-MB-468 or AU565 xenograft tumors reaching a volume of 100–150 mm3 were treated i.p. with PU-WS13 (75mg/kg or 125mg/kg, dissolved in 60mM citrate buffer (pH 4.0) with 30% Captisol) or vehicle, on a 3-times or 5 times per week schedule, as indicated. Tumor volume (in mm3) was determined by measurement with Vernier calipers, and was calculated as the product of its length × width2 × 0.5. Mice were sacrificed after similar PU-WS13 treatment periods, and at a time before tumors reached a size that resulted in a discomfort or difficulty in physiological functions of mice in the individual treatment group, in accordance with our IUCAC protocol. All animals were observed daily for mortality from the time of animal receipt through the end of the study. Body weights for all animals were recorded no more than three times, but no fewer than once per week during the administration of the test article. All mice were observed for clinical symptoms at the time the animals were received and on all days in which the test article was administered. Toxicology studies. The study assessed the safety and relevant toxicities of PU-WS13 administered by i.p. injection (125 mg/kg, five injections a week) over a chronic administration period (87 days). On the final day of the efficacy study, mice were anesthetized with isoflurane and approximately 100 μL of whole blood was collected from the orbital plexus of each mouse into a labeled tube containing EDTA anticoagulant. Within 2 hr of blood collection, complete necropsies hematology and clinical chemistry were analyzed. A necropsy was performed on each animal. Gross examinations of each animal including internal organs were performed by a pathologist and any macroscopic lesions or other abnormal findings were recorded using standard terminology. For histopathology, tissues were collected and preserved in formalin. After at least 24 hr in fixative, the tissues were embedded in paraffin, sectioned and stained with hematoxylin and eosin or subjected to Immunostaining. All tissues slides were examined by a pathologist. Lesions were recorded using morphologic diagnoses following standardized nomenclature.
Identification of N-Glycosylation sites using nano LC-Mass Spectrometry (LC-MS/MS)
Samples were treated with EndoH as described. Initially, non-treated samples, as well as samples treated with EndoH were utilized to develop the mass spectrometric workflow used. Protein samples were separated by SDS-PAGE and processed using standard published protocols (Rodina et al., 2016) with the following modifications: Gel regions containing endoplasmin were generously excised and subjected to in-gel tryptic digestion with 200–300ng Trypsin (Trypsin Gold, Mass Spectrometry Grade, Promega) overnight, and after acidification with 10% formic acid (final concentration of 0.5%–1% formic acid) resulting peptides were desalted using hand packed reversed phase Empore C18 Extraction Disks (3M, St. Paul, MN, USA) using a method described before (Rappsilber et al., 2007). Desalted peptides were concentrated to a very small droplet by vacuum centrifugation and reconstituted in 10 mL 0.1% formic acid in water. Approximately 90% of the peptide material was used for liquid chromatography followed by tandem mass spectrometry (LC-MS/MS). A Q Exactive HF mass spectrometer was coupled directly to an EASY-nLC 1000 (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a self-packed 75 μm × 20-cm reverse phase column (360 mm OD, 75 mm ID, 10 mm ID tip Picotip emitter, New Objective, Woburn MA column packed with ReproSil-Pur C18, 3 μM beads, Dr. Maisch GmbH, Germany) for peptide separation. Peptides were eluted with a 3%–40% acetonitrile gradient over 110 min at a flow rate of 250 nL/min. The mass spectrometer was operated in DDA mode with survey scans acquired at a resolution of 120,000 (at m/z 200) over a scan range of 300–1750 m/z. Up to 15 most abundant precursors from the survey scan were selected with an isolation window of 1.6 Th for fragmentation by higher-energy collisional dissociation with normalized collision energy (NCE) of 27. The maximum injection time for the survey and MS/MS scans was 60 ms and the ion target value (AGC) for both scan modes was set to 3e6.
Mass Spectrometry Data Processing
All mass spectra were first converted to mgf peak list format using Proteome Discoverer 1.4 (Thermo Fisher Scientific, Waltham, MA, USA) and the resulting mgf files searched against a human UniProt protein database using Mascot (Matrix Science, London,UK; version 2.5.0; www.matrixscience.com). Decoy protein sequences with reversed sequence were added to the database to allow for the calculation of false discovery rates (FDR). The search parameters were as follows: (i) up to two missed tryptic cleavages were allowed; (ii) precursor ion mass tolerance = 10 ppm; (iii) fragment ion mass tolerance = 0.3 Da; and (iv) variable protein modifications were allowed for N-linked glycosylation [N-Acetylhexosamine (HexNAc)], methionine oxidation, deamidation of asparagine and glutamines, and protein N-terminal acetylation. MudPit scoring was typically applied using significance threshold score p < 0.01. Decoy database search was always activated and, in general, for merged LS-MS/MS analysis of a gel lane with p < 0.01, false discovery rate averaged around 1%. Generated .RAW files further were analyzed with Thermo Scientific Xcalibur (version 4.1.31.9) software 4 (Thermo Fisher Scientific, Waltham, MA, USA). Layouts containing target precursors (modified by HexNAc) were created with mass accuracy set to 5ppm. The intensity of each specific precursor was extracted and noted. We also noted the presence of the oxonium ion at 204.087 and GlcNAc fragment ions at m/z 126.055, 138.055, 168.065, and 186.075 in each MS/MS spectrum of modified peptides (Hägglund et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were collected and statistical analyses performed using GraphPad Prism (version 6; GraphPad Software) or R statistical package. In each group of data, estimate variation was taken into account and is indicated in each figure as SD or SEM. If a single panel is presented, data are representative of 2 or 3 biological or technical replicates, as indicated. P values for unpaired comparisons between two groups with comparable variance were calculated by two-tailed Student’s t test. Pearson’s tests were used to identify correlations among variables. Significance for all statistical tests was shown in figures for not significant (NS), p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) and p < 0.0001 (****). No samples or animals were excluded from analysis, and sample size estimates were not used. Animals were randomly assigned to groups. Studies were not conducted blinded, with the exception of all patient specimen histological analyses.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HSP90 | StressMarq Biosciences | Cat# SMC-107, RRID:AB_854214 |
| Mouse monoclonal anti-Calnexin | BD Biosciences | Cat# 610523, RRID:AB_397883 |
| Rabbit polyclonal anti-HER2 | Invitrogen | Cat# 28–0004 |
| Mouse monoclonal anti-HSP70 | Enzo Life Sciences | Cat# ADI-SPA-810, RRID:AB_10615203 |
| Rat monoclonal anti-GRP94 | Enzo Life Sciences | Cat# ADI-SPA-850, RRID:AB_11179746 |
| Mouse monoclonal anti-GAPDH | Abcam | Cat# ab8245, RRID:AB_2107448 |
| Rabbit polyclonal anti-GRP78 | Abcam | Cat# ab21685, RRID:AB_2119834 |
| Rabbit polyclonal anti-HSP90α | Abcam | Cat# ab2928, RRID:AB_303423 |
| Mouse monoclonal anti-EEA1 | Abcam | Cat# ab70521, RRID:AB_1603734 |
| Rabbit polyclonal anti-Calnexin | Abcam | Cat# ab22595, RRID:AB_2069006 |
| Rabbit polyclonal anti-Cleaved PARP | Promega | Cat# G7341, RRID:AB_430876 |
| Rabbit monoclonal anti-EGFR | Cell Signaling Technology | Cat# 4267, RRID:AB_2246311 |
| Rabbit monoclonal anti-LRP6 | Cell Signaling Technology | Cat# 2560, RRID:AB_2139329 |
| Rabbit polyclonal anit-p-AKT (S473) | Cell Signaling Technology | Cat# 9271, RRID:AB_329825 |
| Rabbit monoclonal anti-AKT | Cell Signaling Technology | Cat# 4691, RRID:AB_915783 |
| Mouse monoclonal anti-Caspase 7 | Cell Signaling Technology | Cat# 9494, RRID:AB_2068141 |
| Rabbit monoclonal anti-p-ERK1/2 (T202/Y204) | Cell Signaling Technology | Cat# 4377, RRID:AB_331775 |
| Rabbit monoclonal anti-ERK1/2 | Cell Signaling Technology | Cat# 4695, RRID:AB_390779 |
| Rabbit monoclonal anti-p-STAT3 (Y705) | Cell Signaling Technology | Cat# 9145, RRID:AB_2491009 |
| Rabbit monoclonal anti-STAT3 | Cell Signaling Technology | Cat# 12640, RRID:AB_2629499 |
| Rabbit polyclonal anti-Flotillin-1 | Cell Signaling Technology | Cat# 3253, RRID:AB_2106734 |
| Rabbit monoclonal anti-p-p65 (S536) | Cell Signaling Technology | Cat# 3033, RRID:AB_331284 |
| Rabbit monoclonal anti-p65 | Cell Signaling Technology | Cat# 8242, RRID:AB_10859369 |
| Rabbit polyclonal anti-GRP94 | Sigma-Aldrich | Cat# G4420, RRID:AB_477017 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat# T5168, RRID:AB_477579 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat# A1978, RRID:AB_476692 |
| Concanavalin A-HRP | Sigma-Aldrich | Cat# L6397 |
| anti-HER2-FITC | BD Biosciences | Cat# 340553, RRID:AB_2099395 |
| Annexin V-FITC | BD Biosciences | Cat# 556419, RRID:AB_2665412 |
| Goat anti-rat IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2006, RRID:AB_1125219 |
| Goat anti-Rabbit Ig-HRP | SouthernBiotech | Cat# 4010–05, RRID:AB_2632593 |
| Sheep Anti-Mouse IgG-HRP | GE Healthcare | Cat# NXA931, RRID:AB_772209 |
| Mouse monoclonal anti-Myc | Thermo Fisher Scientific | Cat# R950–25, RRID:AB_2556560 |
| Goat anti-Rabbit IgG-Cy3 | Thermo Fisher Scientific | Cat# A10520, RRID:AB_2534029 |
| Goat anti-Rabbit IgG-Cy5 | Thermo Fisher Scientific | Cat# A10523, RRID:AB_2534032 |
| Goat anti-Mouse IgG-Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A28175, RRID:AB_2536161 |
| Bacterial | ||
| Stbl3 Chemically Competent E. coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PU-WS13 | Chiosis Lab | N/A |
| PU-29F | Chiosis Lab | N/A |
| PU-H71 | Chiosis Lab | N/A |
| HJP-149 | Chiosis Lab | N/A |
| SO-33 | Chiosis Lab | N/A |
| PU-WS13-biotin | Chiosis Lab | N/A |
| Inactive-WS13-biotin | Chiosis Lab | N/A |
| Paclitaxel | Selleck Chemicals | Cat#S1150 |
| Erlotinib | Selleck Chemicals | Cat# S1023 |
| Lapatinib | Selleck Chemicals | Cat# S2111 |
| Cetuximab | Leftover from the MSKCC pharmacies | N/A |
| Critical Commercial Assays | ||
| Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit | Invent Biotechnologies | Cat# SM005 |
| ProteoExtract® Subcellular Proteome Extraction Kit | Millipore Sigma | Cat# 539790 |
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | Cat# G9242 |
| T7E1 assay | NEB | Cat# M0302 |
| De-glycosylation by Endo H | NEB | Cat# P0703 |
| De-glycosylation by PNGase F | NEB | Cat# P0709 |
| Lambda Protein Phosphatase | NEB | Cat# P0753 |
| Phusion Flash High-Fidelity PCR Master Mix | Thermo Fisher Scientific | Cat # F548 |
| Neon Transfection System 100 μL Kit | Thermo Fisher Scientific | Cat# MPK10025 |
| Pierce Cell Surface Protein Isolation Kit | Thermo Fisher Scientific | Cat# 89881 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat# 13778075 |
| Deposited Data | ||
| Proteomic Data | This study | MassIVE (MSV000085459); ftp://massive.ucsd.edu/MSV000085459/ |
| Experimental Models: Cell Lines | ||
| Human: HMEC | ATCC | Cat# PCS-600–010 |
| Human: SKBr3 | ATCC | Cat# HTB-30, RRID:CVCL_0033 |
| Human: MDA-MB-468 | ATCC | Cat# HTB-132, RRID:CVCL_0419 |
| Human: HCC1806 | ATCC | Cat# CRL-2335, RRID:CVCL_1258 |
| Human: MDA-MB-231 | ATCC | Cat# HTB-26, RRID:CVCL_0062 |
| Human: MDA-MB-415 | ATCC | Cat# HTB-128, RRID:CVCL_0621 |
| Human: MCF-7 | ATCC | Cat# HTB-22, RRID:CVCL_0031 |
| Human: BT-474 | ATCC | Cat# HTB-20, RRID:CVCL_0179 |
| Human: BT-20 | ATCC | Cat# HTB-19, RRID:CVCL_0178 |
| Human: MDA-MB-361 | ATCC | Cat# HTB-27, RRID:CVCL_0620 |
| Human: MDA-MB-453 | ATCC | Cat# HTB-131, RRID:CVCL_0418 |
| Human: T47D | ATCC | Cat# HTB-133, RRID:CVCL_0553 |
| Human: AU565 | ATCC | Cat# CRL-2351, RRID:CVCL_1074 |
| Mouse: MethA WT | Dr. Z. Li, OSU | N/A |
| Mouse: MethA TM96 | Dr. Z. Li, OSU | N/A |
| Human: MiaPaCa2 | ATCC | Cat# CRL-1420, RRID:CVCL_0428 |
| Human: Panc1 | ATCC | Cat# CRL-1469, RRID:CVCL_0480 |
| Human: BxPC3 | ATCC | Cat# CRL-1687, RRID:CVCL_0186 |
| Human: CAPAN1 | ATCC | Cat# HTB-79, RRID:CVCL_0237 |
| Human: SU86.86 | ATCC | Cat# CRL-1837, RRID:CVCL_3881 |
| Human: HPAF2 | ATCC | Cat# CRL-1997, RRID:CVCL_0313 |
| Human: 931019 | Dr. Y. Janjigian, MSKCC | N/A |
| Human: 931102 | Dr. Y. Janjigian, MSKCC | N/A |
| Human: ASPC1 | ATCC | Cat# CRL-1682, RRID:CVCL_0152 |
| Human: CAPAN2 | ATCC | Cat# HTB-80, RRID:CVCL_0026 |
| Human: PL45 | ATCC | Cat# CRL-2558, RRID:CVCL_3567 |
| Human: CFPAC | ATCC | Cat# CRL-1918, RRID:CVCL_1119 |
| Human: NCI-H3122 | Dr. M. Moore, MSKCC | RRID:CVCL_5160 |
| Human: NCI-H2228 | Dr. M. Moore, MSKCC | RRID:CVCL_1543 |
| Human: NCI-H1373 | Dr. N. Lecomte, MSKCC | RRID:CVCL_1465 |
| Human: NCI-H525 | Dr. N. Lecomte, MSKCC | N/A |
| Human: NCI-N87 | ATCC | Cat# CRL-5822, RRID:CVCL_1603 |
| Human: SNU-1 | ATCC | Cat# CRL-5971, RRID:CVCL_0099 |
| Human: PEO-1 | Dr. D. Solit, MSKCC | RRID:CVCL_Y032 |
| Human: PEO-4 | Dr. D. Solit, MSKCC | RRID:CVCL_2690 |
| Human: OVCAR4 | Dr. D. Solit. MSKCC | RRID:CVCL_1627 |
| Human: OV1847 | Dr. D. Solit. MSKCC | RRID:CVCL_D703 |
| Human: A2780 | Dr. D. Solit, MSKCC | RRID:CVCL_0134 |
| Human: IGROV-1 | Dr. D. Solit, MSKCC | RRID:CVCL_1304 |
| Human: OVCAR5 | Dr. D. Solit, MSKCC | RRID:CVCL_1628 |
| Human: SKRC38 | Dr. S. Larson, MSKCC | RRID:CVCL_6189 |
| Human: SKRC52 | Dr. S. Larson, MSKCC | RRID:CVCL_6198 |
| Human: LAN5 | Children’s Oncology Group (COG) | RRID:CVCL_0389 |
| Human: SMS-KCNR | Children’s Oncology Group (COG) | RRID:CVCL_7134 |
| Human: SY5Y | ATCC | Cat# CRL-2266, RRID:CVCL_0019 |
| Human: TC71 | Dr. S Ambati, MSKCC | RRID:CVCL_2213 |
| Human: A673 | Dr. S Ambati, MSKCC | RRID:CVCL_0080 |
| Human: SU-DHL4 | DSMZ | Cat# ACC-495, RRID:CVCL_0539 |
| Human: SU-DHL6 | ATCC | Cat# CRL-2959, RRID:CVCL_2206 |
| Human: Toledo | ATCC | Cat# CRL-2631, RRID:CVCL_3611 |
| Human: Karpas422 | DSMZ | Cat# ACC-32, RRID:CVCL_1325 |
| Human: OCI-Ly1 | Ontario Cancer Institute | RRID:CVCL_1879 |
| Human: OCI-Ly7 | Ontario Cancer Institute | RRID:CVCL_1881 |
| Human: Farage | ATCC | Cat# CRL-2630, RRID:CVCL_3302 |
| Human: RCK8 | DSMZ | Cat# ACC-561, RRID:CVCL_1883 |
| Human: HBL1 | Dr. J. Angel Martinez-Climent | RRID:CVCL_4213 |
| Human: OCI-Ly3 | Ontario Cancer Institute | RRID:CVCL_8800 |
| Human: U2932 | Dr. J. Angel Martinez-Climent | RRID:CVCL_1896 |
| Human: MD901 | Dr. J. Angel Martinez-Climent | RRID:CVCL_D709 |
| Human: TMD8 | Dr. L. M. Staudt, NIH | RRID:CVCL_A442 |
| Human: BC3 | ATCC | Cat# CRL-2277, RRID:CVCL_1080 |
| Human: Kasumi-1 | ATCC | Cat# CRL-2724, RRID:CVCL_0589 |
| Human: MOLM13 | DSMZ | Cat# ACC-554, RRID:CVCL_2119 |
| Human: K562 | ATCC | Cat# CCL-243, RRID:CVCL_0004 |
| Human: U266 | Dr. Z. Li, OSU | RRID:CVCL_0566 |
| Human: PCNY1 | Dr. Z. Li, OSU | N/A |
| Human: MM.1R | Dr. Z. Li, OSU | Cat# CRL-2975, RRID:CVCL_8794 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Hsd:Athymic Nude-Foxn1nu | ENVIGO RMS, INC. | Outbred, Stock #: 069, RRID:MGI:5652489 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | IMSR Cat# JAX:005557, RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| List of oligos See | STAR Methods | N/A |
| Recombinant DNA | ||
| PX458 | Addgene | RRID:Addgene_48138 |
| Software and Algorithms | ||
| Prism V6 | GraphPad software | https://www.graphpad.com |
| FloJo | FlowJo LLC | https://www.flowjo.com |
| LAS AF LITE | Leica | https://leica-las-af-lite.software.informer.com/ |
| FIJI | IMAGEJ | https://imagej.net/Fiji |
| Proteome Discoverer 1.4 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30795 |
| Thermo Scientific Xcalibur | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30487 |
| Mascot | Matrix Science | http://www.matrixscience.com/ |
| Other | ||
| Protein A Agarose | Roche | Cat# 11134515001 |
| Protein G Agarose | Roche | Cat# 11243233001 |
| Pierce High Capacity Streptavidin Agarose | Thermo Fisher Scientific | Cat# 20361 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32109 |
| UltraPure LMP Agarose | Thermo Fisher Scientific | Cat# 16520–100 |
| ProLong Gold Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36935 |
| Lab-Tek II Chamber Slide System | Thermo Fisher Scientific | Cat# 154461 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28908 |
| RPMI with 1% Penicillin/Streptomycin | MSKCC Media Core | N/A |
| DMEM with 1% Penicillin/Streptomycin | MSKCC Media Core | N/A |
| FBS | VWR | Cat# 97068–085 |
| 7-AAD Viability Staining Solution | Biolegend | Cat# 420404 |
| Corning® Matrigel® Basement Membrane Matrix, Phenol Red-free | Corning | Cat# 356237 |
Highlights.
N-glycosylation transforms a chaperone, GRP94, from a folder into a scaffolding protein
These changes are pathologic in nature as they remodel proteome-wide connectivity
The N-glycosylated GRP94 variant is a small and distinct fraction of the GRP94 pool
Proteome dysfunctions mediated by the N-glycosylated GRP94 variant are actionable
ACKNOWLEDGMENTS
This work is supported in part by the US National Institutes of Health (NIH) (R01 CA172546, R56 AG061869, R01 CA155226, P01 CA186866, P30 CA08748, and P50 CA192937), the Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of the Memorial Sloan Kettering Cancer Center; T.W. was supported by the Lymphoma Research Foundation; G.C. was supported in part by the Steven A. Greenberg charitable trust and the Solomon Program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107840.
DECLARATION OF INTERESTS
G.C., P.Y., H.J.P., W.S., C.Y., and T.T. are inventors on patents covering PU-WS13 and associated composition of matter. G.C. is a founder of Samus Therapeutics and a member of its board of directors.
REFERENCES
- Altmeyer A, Maki RG, Feldweg AM, Heike M, Protopopov VP, Masur SK, and Srivastava PK (1996). Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int. J. Cancer 69, 340–349. [DOI] [PubMed] [Google Scholar]
- Ansa-Addo EA, Thaxton J, Hong F, Wu BX, Zhang Y, Fugle CW, Metelli A, Riesenberg B, Williams K, Gewirth DT, et al. (2016). Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Curr. Top. Med. Chem 16, 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, and Koch GL (1989). Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell 59, 729–737. [DOI] [PubMed] [Google Scholar]
- Brehme M, and Voisine C (2016). Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity. Dis. Model. Mech 9, 823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buc Calderon P, Sennesael AL, and Glorieux C (2018). Glucose-regulated protein of 94 kDa contributes to the development of an aggressive phenotype in breast cancer cells. Biomed. Pharmacother 105, 115–120. [DOI] [PubMed] [Google Scholar]
- Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, and Kundu GC (2018). Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol. Cancer 17, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cala SE (2000). GRP94 hyperglycosylation and phosphorylation in Sf21 cells. Biochim. Biophys. Acta 1496, 296–310. [DOI] [PubMed] [Google Scholar]
- Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, et al. (2011). KIT as a therapeutic target in metastatic melanoma. JAMA 305, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavany C, Mimnaugh E, Miller P, Bitton R, Nguyen P, Trepel J, White-sell L, Schnur R, Moyer J, and Neckers L (1996). p185erbB2 binds to GRP94 in vivo. Dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J. Biol. Chem 271, 4974–4977. [DOI] [PubMed] [Google Scholar]
- Cherepanova NA, Venev SV, Leszyk JD, Shaffer SA, and Gilmore R (2019). Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J. Cell Biol 218, 2782–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier P, and Coulombe B (2013). Regulation of molecular chaperones through post-translational modifications: decrypting the chaperone code. Biochim. Biophys. Acta 1829, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, and Lawrence TS (2008). Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 68, 3803–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, and Lawrence TS (2010). Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res 16, 3205–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corben AD, Uddin MM, Crawford B, Farooq M, Modi S, Gerecitano J, Chiosis G, and Alpaugh ML (2014). Ex vivo treatment response of primary tumors and/or associated metastases for preclinical and clinical development of therapeutics. J. Vis. Exp, e52157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese K, Howes MT, Lundmark R, Tagliatti E, Bagnato P, Petrelli A, Bono M, McMahon HT, Parton RG, and Tacchetti C (2013). The HSP90 inhibitor geldanamycin perturbs endosomal structure and drives recycling ErbB2 and transferrin to modified MVBs/lysosomal compartments. Mol. Biol. Cell 24, 129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuneo KC, Nyati MK, Ray D, and Lawrence TS (2015). EGFR targeted therapies and radiation: Optimizing efficacy by appropriate drug scheduling and patient selection. Pharmacol. Ther 154, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart A (2016). Tumorigenesis: Networking: a survival guide. Nat. Rev. Cancer 16, 752. [DOI] [PubMed] [Google Scholar]
- Dersh D, Jones SM, Eletto D, Christianson JC, and Argon Y (2014). OS-9 facilitates turnover of nonnative GRP94 marked by hyperglycosylation. Mol. Biol. Cell 25, 2220–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, Chande A, Tanaka KE, Stransky N, Greulich H, et al. (2011). Inhibitor sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One 6, e20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DP, Weigel NL, Schrader WT, O’Malley BW, and McGuire WL (1984). Structural analysis of chicken oviduct progesterone receptor using monoclonal antibodies to the subunit B protein. Biochemistry 23, 4427–4435. [DOI] [PubMed] [Google Scholar]
- Eletto D, Dersh D, and Argon Y (2010). GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol 21, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ (2013). Assembly chaperones: a perspective. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldweg AM, and Srivastava PK (1995). Molecular heterogeneity of tumor rejection antigen/heat shock protein GP96. Int. J. Cancer 63, 310–314. [DOI] [PubMed] [Google Scholar]
- Ferraro DA, Gaborit N, Maron R, Cohen-Dvashi H, Porat Z, Pareja F, Lavi S, Lindzen M, Ben-Chetrit N, Sela M, and Yarden Y (2013). Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA 110, 1815–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach A, Rogler A, Erber R, Stoehr R, Poulsom R, Heidenreich A, Schneevoigt BS, Hauke S, Hartmann A, Knuechel R, et al. (2015). Fibroblast growth factor receptor (FGFR) gene amplifications are rare events in bladder cancer. Histopathology 66, 639–649. [DOI] [PubMed] [Google Scholar]
- Gewirth DT (2016). Paralog Specific Hsp90 Inhibitors—A Brief History and a Bright Future. Curr. Top. Med. Chem 16, 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampietri C, Petrungaro S, Conti S, Facchiano A, Filippini A, and Ziparo E (2015). Cancer Microenvironment and Endoplasmic Reticulum Stress Response. Mediators Inflamm. 2015, 417281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hägglund P, Bunkenborg J, Elortza F, Jensen ON, and Roepstorff P (2004). A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res 3, 556–566. [DOI] [PubMed] [Google Scholar]
- Harper JW, and Bennett EJ (2016). Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, and Kurzrock R (2016). The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next Generation Sequencing. Clin. Cancer Res 22, 259–267. [DOI] [PubMed] [Google Scholar]
- Inda MC, Joshi S, Wang T, Bolaender A, Gandu S, Koren J, Che AY, Taldone T, Yan P, Sun W, et al. (2020). The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun 11, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, and Chiosis G (2018). Adapting to stress—chaperome networks in cancer. Nat. Rev. Cancer 18, 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Jung HH, Do IG, Bae S, Lee SK, Kim SW, Lee JE, Nam SJ, Ahn JS, Park YH, and Im YH (2016). Prognostic value of ERBB4 expression in patients with triple negative breast cancer. BMC Cancer 16, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishinevsky S, Wang T, Rodina A, Chung SY, Xu C, Philip J, Taldone T, Joshi S, Alpaugh ML, Bolaender A, et al. (2018). HSP90-incorporating chaperome networks as biosensor for disease-related pathways in patient-specific midbrain dopamine neurons. Nat. Commun 9, 4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Lazaris C, Hockemeyer K, Balandrán JC, Jimenez AR, Mullenders J, Gong Y, Trimarchi T, Bhatt K, Hu H, et al. (2018). Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat. Med 24, 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS (2014). Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Qi Y, and Im W (2015). Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep 5, 8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sun L, Hou J, Gui M, Ying J, Zhao H, Lv N, and Meng S (2015). Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. Int. J. Cancer 137, 512–524. [DOI] [PubMed] [Google Scholar]
- Liu B, Dai J, Zheng H, Stoilova D, Sun S, and Li Z (2003). Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. USA 100, 15824–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Staron M, Hong F, Wu BX, Sun S, Morales C, Crosson CE, Tomlinson S, Kim I, Wu D, and Li Z (2013). Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc. Natl. Acad. Sci. USA 110, 6877–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma PV (2013). Elimination of endoplasmic reticulum stress and cardiovascular, type 2 diabetic, and other metabolic diseases. Ann. Med 45, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins M, Custódio R, Camejo A, Almeida MT, Cabanes D, and Sousa S (2012). Listeria monocytogenes triggers the cell surface expression of Gp96 protein and interacts with its N terminus to support cellular infection. J. Biol. Chem 287, 43083–43093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec M, Eletto D, and Argon Y (2012). GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, and Ueno NT (2012). Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat 136, 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar M, McCarthy CR, Kulick AR, Qeriqi B, Guzman S, and de Stanchina E (2018). Establishing and Maintaining an Extensive Library of Patient-Derived Xenograft Models. Front. Oncol 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey K, and Braakman I (2016). Protein quality control at the endoplasmic reticulum. Essays Biochem. 60, 227–235. [DOI] [PubMed] [Google Scholar]
- Mekahli D, Bultynck G, Parys JB, De Smedt H, and Missiaen L (2011). Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Per-spect. Biol 3, a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereiter S, Balmaña M, Campos D, Gomes J, and Reis CA (2019). Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 36, 6–16. [DOI] [PubMed] [Google Scholar]
- Mitra N, Sinha S, Ramya TN, and Surolia A (2006). N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem. Sci 31, 156–163. [DOI] [PubMed] [Google Scholar]
- Moreira RB, Peixoto RD, and de Sousa Cruz MR (2015). Clinical Response to Sorafenib in a Patient with Metastatic Colorectal Cancer and FLT3 Amplification. Case Rep. Oncol 8, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K, and Boerner JL (2012). Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 14, R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobusawa S, Stawski R, Kim YH, Nakazato Y, and Ohgaki H (2011). Amplification of the PDGFRA, KIT and KDR genes in glioblastoma: a population-based study. Neuropathology 31, 583–588. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Jang H (2019). Protein ensembles link genotype to phenotype. PLoS Comput. Biol 15, e1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, and Tabas I (2012). Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med 63, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagetta A, Tramentozzi E, Tibaldi E, Cendron L, Zanotti G, Brunati AM, Vitadello M, Gorza L, and Finotti P (2014). Structural insights into complexes of glucose-regulated Protein94 (Grp94) with human immunoglobulin G. relevance for Grp94-IgG complexes that form in vivo in pathological conditions. PLoS One 9, e86198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, et al. (2013). Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, et al. (2015). Structure-activity relationship in a purine-scaffold compound series with selectivity for the endo plasmic reticulum Hsp90 paralog Grp94. J. Med. Chem 58, 3922–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillarsetty N, Jhaveri K, Taldone T, Caldas-Lopes E, Punzalan B, Joshi S, Bolaender A, Uddin MM, Rodina A, Yan P, et al. (2019). Paradigms for Precision Medicine in Epichaperome Cancer Therapy. Cancer Cell 36, 559–573.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D, Mazzarella RA, and Green M (1994). Analysis of the structure and synthesis of GRP94, an abundant stress protein of the endoplasmic reticulum. DNA Cell Biol. 13, 117–124. [DOI] [PubMed] [Google Scholar]
- Rachidi S, Sun S, Wu BX, Jones E, Drake RR, Ogretmen B, Cowart LA, Clarke CJ, Hannun YA, Chiosis G, et al. (2015). Endoplasmic reticulum heat shock protein gp96 maintains liver homeostasis and promotes hepatocellular carcinogenesis. J. Hepatol 62, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SM, Clubb RJ, Bhattacharyya M, Dimri M, Cheng H, Pan W, Ortega-Cava C, Lakku-Reddi A, Naramura M, Band V, and Band H (2008). A combination of Trastuzumab and 17-AAG induces enhanced ubiquitinylation and lysosomal pathway-dependent ErbB2 degradation and cytotoxicity in ErbB2-overexpressing breast cancer cells. Cancer Biol. Ther 7, 1630–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, et al. (2006). FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res 12, 6652–6662. [DOI] [PubMed] [Google Scholar]
- Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y, et al. (2007). Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol 3, 498–507. [DOI] [PubMed] [Google Scholar]
- Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz F, and Aebi M (2011). Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol 21, 576–582. [DOI] [PubMed] [Google Scholar]
- Shi J, Yao D, Liu W, Wang N, Lv H, He N, Shi B, Hou P, and Ji M (2012). Frequent gene amplification predicts poor prognosis in gastric cancer. Int. J. Mol. Sci 13, 4714–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha L, Patel HJ, and Chiosis G (2016). Chemical Tools to Investigate Mechanisms Associated with HSP90 and HSP70 in Disease. Cell Chem. Biol 23, 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddals KW, Allen J, Sinha S, Canfield AE, Kalra PA, and Gibson JM (2011). Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J. Biol. Chem 286, 16623–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solá RJ, and Griebenow K (2006). Chemical glycosylation: new insights on the interrelation between protein structural mobility, thermodynamic stability, and catalysis. FEBS Lett. 580, 1685–1690. [DOI] [PubMed] [Google Scholar]
- Solimini NL, Luo J, and Elledge SJ (2007). Non-oncogene addiction and the stress phenotype of cancer cells. Cell 130, 986–988. [DOI] [PubMed] [Google Scholar]
- Stothert AR, Suntharalingam A, Tang X, Crowley VM, Mishra SJ, Webster JM, Nordhues BA, Huard DJE, Passaglia CL, Lieberman RL, et al. (2017). Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep 7, 17951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabone S, Théou N, Wozniak A, Saffroy R, Deville L, Julié C, Callard P, Lavergne-Slove A, Debiec-Rychter M, Lemoine A, and Emile JF (2005). KIT overexpression and amplification in gastrointestinal stromal tumors (GISTs). Biochim. Biophys. Acta 1741, 165–172. [DOI] [PubMed] [Google Scholar]
- Tan HY, Wang N, Lam W, Guo W, Feng Y, and Cheng YC (2018). Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 17, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrini R, Pabois A, Xenarios I, Coukos G, Delaloye JF, and Doucey MA (2017). TIE-2 expressing monocytes in human cancers. OncoImmunology 6, e1303585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogen S, Gidalevitz T, Biswas C, Simen BB, Stein E, Gulmen F, and Argon Y (2002). Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J. Biol. Chem 277, 40742–40750. [DOI] [PubMed] [Google Scholar]
- Wearsch PA, and Nicchitta CV (1996). Purification and partial molecular characterization of GRP94, an ER resident chaperone. Protein Expr. Purif 7, 114–121. [DOI] [PubMed] [Google Scholar]
- Wiersma VR, Michalak M, Abdullah TM, Bremer E, and Eggleton P (2015). Mechanisms of Translocation of ER Chaperones to the Cell Surface and Immunomodulatory Roles in Cancer and Autoimmunity. Front. Oncol 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BX, Hong F, Zhang Y, Ansa-Addo E, and Li Z (2016). GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv. Cancer Res 129, 165–190. [DOI] [PubMed] [Google Scholar]
- Yadav RK, Chae SW, Kim HR, and Chae HJ (2014). Endoplasmic reticulum stress and cancer. J. Cancer Prev 19, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, and Riely GJ (2013). Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res 19, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuno A, Lee MJ, Lee S, Tomita Y, Rekhtman D, Moore B, and Trepel JB (2018). Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol 1709, 423–441. [DOI] [PubMed] [Google Scholar]
- Zheng H, Dai J, Stoilova D, and Li Z (2001). Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J. Immunol 167, 6731–6735. [DOI] [PubMed] [Google Scholar]
- Zhu G, and Lee AS (2015). Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol 230, 1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
LC-MS data that support the findings of this study have been deposited in MassIVE with the accession number MSV000085459, [ftp://massive.ucsd.edu/MSV000083484/].