

Abstract
Essential genes are those whose loss of function compromises organism viability or results in profound loss of fitness. Recent gene-editing technologies have provided new opportunities to characterize essential genes. Here, we present an integrated analysis that comprehensively and systematically elucidates the genetic and regulatory characteristics of human essential genes. First, we found that essential genes act as ‘hubs’ in protein–protein interaction networks, chromatin structure and epigenetic modification. Second, essential genes represent conserved biological processes across species, although gene essentiality changes differently among species. Third, essential genes are important for cell development due to their discriminate transcription activity in embryo development and oncogenesis. In addition, we developed an interactive web server, the Human Essential Genes Interactive Analysis Platform (http://sysomics.com/HEGIAP/), which integrates abundant analytical tools to enable global, multidimensional interpretation of gene essentiality. Our study provides new insights that improve the understanding of human essential genes.
Keywords: human essential genes, multi-omic, integrated analysis, cell development, web server
Introduction
Essential genes are indispensable for organism survival and the maintenance of basic cell and tissue functions [1–3]. The systematic identification of essential genes in different organisms [4] has provided critical insights into the molecular bases of many biological processes [5]. Such information may be useful for applications in areas such as synthetic biology [6] and drug target identification [7, 8]. The identification of human essential genes is a particularly attractive area of research because of the potential for medical applications [9, 10]. Utilizing gene-editing technologies based on CRISPR-Cas9 and retroviral gene-trap screens, three independent genome-wide studies [11–13] identified essential genes that are indispensable for human cell viability. The results agreed very well among the three studies, confirming the robustness of the evaluation approaches. All of these studies [11–13] showed that ∼10% of the ∼20 000 genes in human cells are essential for cell survival, highlighting the intrinsic buffering mechanisms of eukaryotic genomes against genetic and environmental insults [14].
In a recent review, Pavelka et al. [15] stated that gene essentiality is not a fixed property but instead depends strongly on environmental and genetic contexts and can be altered during short- or long-term evolution. However, this paradigm leaves some questions unresolved, as we do not know how essential genes are interconnected within cells, why these genes are essential or what their underlying mechanisms may be. Furthermore, we do not know whether these genes are associated with disease (e.g. cancer) or have the potential to be exploited as targets for therapeutic strategies. With the recent and rapid development of next-generation sequencing and other experimental technologies, we now have access to myriad data on genomic sequences, epigenetic modifications, structures and disease-related information. These data will enable researchers to examine human essential genes from multiple perspectives.
In light of this background, we performed a comprehensive study of human essential genes, including their genomic, epigenetic, proteomic, evolutionary and embryonic patterning characteristics. Genetic and regulatory characteristics were studied to understand what makes these genes essential for cell survival. We analyzed the evolutionary status of human essential genes and their profiles during embryonic development. Our findings suggest that human essential genes are important for lineage segregation. Essential genes have important implications for drug discovery, which may inform the next generation of cancer therapeutics (Figure 1). Finally, we developed a new web server, the Human Essential Genes Interactive Analysis Platform (HEGIAP), to facilitate the global research community’s comprehensive exploration of human essential genes.
Figure 1.
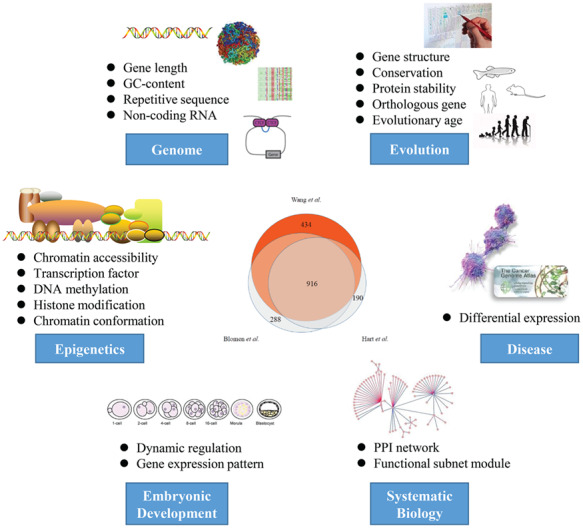
Comprehensive overview of the integrated analysis of human essential genes. In this study, we performed a systemically analysis of human essential. By integrating multi-omics (genome, epigenome and proteome) data, we characterized the evolutionary nature of essential genes and provided new insights into embryonic development and tumorigenesis.
Results
Multi-level essentiality of human essential genes
Essential genes define the key biological functions that are required for cell growth, proliferation and survival. To characterize human essential genes, we first compared three essential gene sets generated by different experimental methods [11–13]; more than 60% of essential genes were cataloged in at least two data sets (Venn diagram in Figure 1). Here, we focused on the essential genes detected by Wang et al. [11], who defined a CRISPR score (CS) for the assessment of gene essentiality. Briefly, low CSs indicate high degrees of essentiality and vice versa. Here, we used data from the KBM7 cell line for further analysis. We first evaluated the cell-line specificity of CSs to ensure the robustness of our analysis in terms of reflecting the general properties of gene essentiality in different cell types. First, according to Wang et al. [11], a predominant part (98.99%) of essential genes in the KBM7 cell line were not cell-line specific; they identified only 19 such genes. Essential genes in the KBM7 cell line also represent more than two-thirds of the essential genes identified in the other two studies. Using these data, we calculated Pearson coefficients of CS correlations between cell lines for all screened genes to assess the similarity of essentiality between cell lines. The essentialities of all screened genes in multiple cell lines showed high degrees of correlation (Pearson coefficient: maximum, 0.83; minimum, 0.79). In addition, the results of our analyses using CSs for different cell lines and using multi-omics data from different cell types were robust, demonstrating the reliability of the data sets used.
We divided all protein-coding genes into 10 groups (CS0–CS9) according to ascending CS values, where group CS0 was composed of essential genes. This detailed classification provided a good representation of the various features in subsequent analyses.
Protein essentiality: high transcription activity and stability, `hub’ of the PPI network
We first analyzed the expression levels of human genes in 2916 individuals from the Genotype-Tissue Expression Program [16]. Essential genes were highly expressed compared with nonessential genes (P < 1 × 10−50, Welch’s t-test; Cohen’s d = 8.76), and the expression level decreased as gene essentiality decreased (the CS value increased; r2 = 0.42, P = 0.04; Figure 2A). Furthermore, an analysis using publicly available human protein stability data [17] showed that proteins encoded by essential genes showed more stability than did other proteins (P = 1.10 × 10−18, Welch’s t-test; Supplementary Figure S1A). This observation was consistent with the findings of a recent study [18], which showed that highly expressed proteins are stable because they are designed to tolerate translational errors that would lead to the accumulation of toxic misfolded species.
Figure 2.
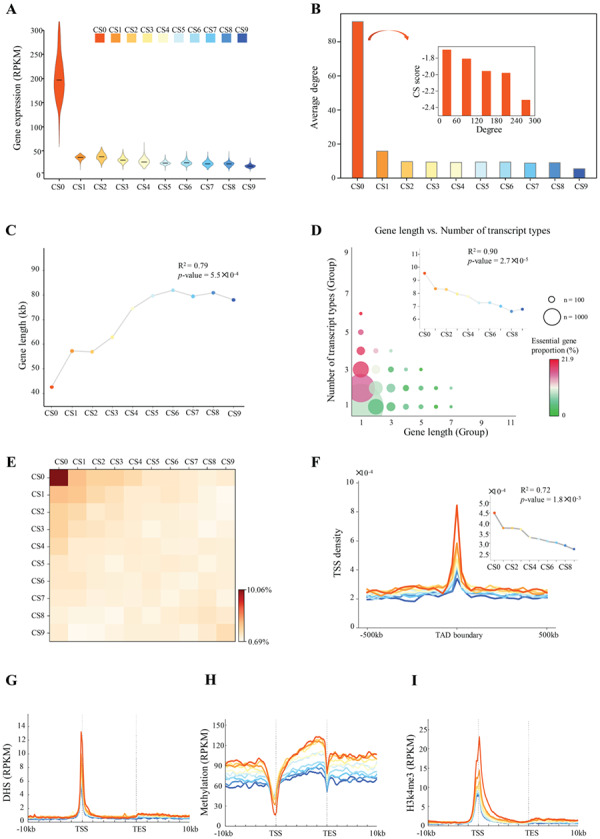
General properties of human essential genes. (A) Violin plot showing gene expression for 2916 individuals from the GTEx program. The mean expression level was calculated for each group of genes (CS0–CS9). (B) Degree of connectivity for each gene group. Inset, relationship between the degree of connectivity and CS value in group CS0 (human essential genes). (C) Relationship between gene essentiality and gene length. R2 and P-values from linear regression are shown. (D) Relationship between gene length and number of transcript types. R2 and P-values from linear regression are shown. (E) Heatmap showing the colocalization of gene TSSs. (F) TSS density surrounding TAD boundary (IMR90 cell line). Inset, average TSS densities within regions (50 kb upstream, TAD boundary and 50 kb downstream). (G–I) Profiles showing mean signals for chromatin accessibility (G), methylation level (H) and H3k4me3 density (I).
In many model organisms, essential genes tend to encode abundant proteins that engage extensively in protein–protein interactions (PPIs) [19]. We constructed a PPI network for each CS group (Supplementary Figure S1B). Essential genes showed significantly more connectivity than did other genes (P = 2.73 × 10−266, Welch’s t-test; Figure 2B), and the degree of connectivity was correlated negatively with the CS in the essential gene set (r = −0.29, P = 1.22 × 10−37; insets in Figure 2B). We then calculated the distribution of genes over the range of connectivity. We found that with higher degrees of connectivity, there were fewer genes (Supplementary Figure S2A) but greater proportions of essential genes (Supplementary Figure S2B). As a novel study showed that long noncoding RNAs (lncRNAs) are important players in regulatory networks [20], we investigated genes whose expression levels were altered significantly following CRISPRi knockout of lncRNAs, identified in a previous study [21]. We found significantly a greater proportion of essential genes among all differentially expressed genes (DEGs) than among genes that showed no significant alteration in expression (Supplementary Figure S2C). This result suggests that essential genes are more likely to be regulated by lncRNAs. Finally, we performed a gene ontology (GO) analysis for each group. Essential genes were enriched in fundamental biological processes, such as rRNA processing, translational initiation, mRNA splicing and DNA replication, and nonessential genes were less significantly enriched in other processes (Supplementary Figure S3).
In summary, essential genes are highly expressed and associated with important biological processes. Proteins encoded by essential genes are stable and located at connection hubs in PPI networks. Taken together, these results show the essentiality of essential genes at the protein level.
Structural essentiality: high density in the genome and 3D structure lead to a `hub’ of chromatin organization
In general, gene length affects the stability of the kinetics of genetic switches and thus the dynamics of gene expression [22]. We found that human essential genes were much shorter than nonessential genes (P = 1.38 × 10−53, Welch’s t-test; Figure 2C), consistent with the results of a previous study of Escherichia coli [22]. Generally, long genes were likely to contain more diverse transcripts in the human genome (r = 0.34, P = 1.0 × 10−100, Pearson correlation; Supplementary Figure S4A). Therefore, fewer types of transcripts were expected in essential genes, as they were short. However, a greater variety of transcripts was found in essential genes compared with nonessential genes (P = 7.02 × 10−42, Welch’s t-test; Figure 2D), and the number of transcript types decreased as essentiality decreased (r2 = 0.90, P = 2.70 × 10−5, Pearson correlation; insets in Figure 2D), indicating that mRNAs transcribed by essential genes were highly variable. GC content is associated with DNA stability, and variations in GC content within the genome result in variations in staining intensity in chromosomes [19]. We thus examined the distribution of GC content. Our result showed that genes with moderate to high degrees of essentiality (CS0–CS4) tended to have a slightly higher GC content than did other genes (CS5–CS9) in both the promoter regions and gene bodies (Supplementary Figure S4B). Furthermore, as Alu repetitive elements in the genome have been found to be regulators of gene expression [23–26] and as the Alu repetitive component may contribute to prevention of DNA damage [27], we investigated repetitive elements within essential genes. DNA sequences for Alu elements were first identified and masked using RepeatMasker [28]. We found that Alu repetitive elements were significantly enriched in essential genes compared with those in nonessential genes (P < 1 × 10−50, Welch’s t-test; Supplementary Figure S4C). Together, these results indicate that essential genes are formed with high GC content and colocalize with Alu repetitive elements, suggesting high DNA stability of essential genes.
We next examined the genomic distribution of essential genes and found that the transcription start sites (TSSs) of these genes tended to cluster (Figure 2E; Supplementary Figure S5A). We then investigated the three-dimensional (3D) structural organization of essential genes. Previous studies have shown that topologically associated domains (TADs) are highly conserved between cell types and species and that proximity to the TAD boundary likely contributes to the stabilization of gene expression [29–33]. We detected TADs using high-throughput/high-resolution chromosome conformation capture (Hi-C) data from Jin et al. [34] and Rao et al. [35]. We observed a significantly greater density of essential than nonessential genes within TAD boundaries (P < 1 × 10−16, Welch’s t-test; Figure 2F; Supplementary Figure S5B–E). As chromatin may be associated with proteins’ affinity for each other, resulting in chromatin loops [36], we calculated intra-TAD local (<100 kb) Hi-C contacts for each gene set and examined the distribution of gene TSSs in chromatin loop anchors. Essential genes contained more local contacts (P < 1 × 10−43, Welch’s t-test; Supplementary Figure S6A and B), and TSS density of essential genes that located in chromatin loop anchors was greater than that of nonessential gene groups (Supplementary Figure S6C). As the formation of architectural loops depends strongly on the protein CTCF [37], we examined the distribution of CTCF signals detected by ChIP-seq [38]. As expected, CTCF was more likely to bind near essential genes (Supplementary Figure S6D). We used the recently introduced SPRITE experimental protocol [39] to identify active hubs of inter-chromosomal interactions that arranged around nuclear speckles. We found that essential genes were enriched on these experimentally verified structural hubs (Supplementary Figure S6E).
In summary, essential genes are structurally essential due to their high GC content, highly enriched Alu repetitive elements and central location in the chromosomal scaffold.
Epigenetic essentiality: the enrichment of epigenetic marks leads to an epigenetic regulatory network `hub’
The epigenetic modification of chromatin provides the necessary plasticity for cells to respond to environmental and positional cues and enables the maintenance of acquired information without changing the DNA sequence [40]. To study the epigenetic information on essential genes, we took advantage of recent high-throughput genomic assays [41, 42]. We first examined the chromatin accessibility of essential genes. Strong DNase I hypersensitivity (DHS) signals were observed in the promoters of essential genes, and this enrichment decreased as gene essentiality decreased (Figure 2G). Moreover, more transcription factor binding sites were detected in the promoters of essential genes (Supplementary Figure S7). We then examined the DNA methylation pattern of essential genes. By analyzing data obtained with two sequencing-based methods, the DNA immunoprecipitation (MeDIP-seq) and methylation-sensitive restriction enzyme (MRE-seq) methods [43], we found that the methylation levels of gene promoters increased as gene essentiality decreased (MRE-seq: r2 = 0.93, P = 8.6 × 10−6; MeDIP-seq: r2 = 0.93, P = 5.8 × 10−6, Welch’s t-test; Figure 2H; Supplementary Figure S8A). Methylation levels were higher in the gene body regions of essential genes than in other genes (MRE-seq, P = 1.5 × 10−5; MeDIP-seq, P = 9.6 × 10−20), supporting the highly transcribed nature of essential genes [44]. Next, we examined two histone modifications—trimethylation of H3 lysine 4 (H3K4me3) and trimethylation of H3 lysine 27 (H3K27me3)—associated with transcription activation and gene repression [45–47], respectively. Similar to chromatin accessibility, H3K4me3 signals were strongly enriched in the promoters of essential genes, and H3K4me3 density in gene promoters increased while gene essentiality increased (Figure 2I). In contrast, H3K27me3 signals were weaker in the promoters of essential genes compared with nonessential genes (Supplementary Figure S8B). Finally, we studied the abundance of noncoding RNAs (ncRNAs), which play a key role in regulating gene expression [48, 49]. The density of ncRNAs in gene promoters decreased as essentiality decreased (r2 = 0.40, P = 0.049; Supplementary Figure S8C).
These observations of both histone modification marks show that essential genes tend to be located in the most active chromatin regions. Therefore, essential genes are hubs of epigenetic modification. As these modifications have important regulatory functions, these epigenetic hubs may, in turn, contribute to gene essentiality. For instance, inactive genes with low expression levels in the essential gene group seem to be `outliers’ because most essential genes are highly expressed. We suppose that a fraction of these inactive genes are essential, not because they are highly expressed and are direct modulators of critical cellular processes, but because they are located at epigenetic hubs. CRISPR-based screening may negatively affect the integrity of these epigenetic hubs, hampering cell proliferation. Based on the hypothesis that epigenetic hub location contributes to a gene’s high degree of essentiality, we believe that some inactive genes located near epigenetic hubs, for instance in highly accessible chromatin regions, are more likely to be essential, even if they are not actively transcribed like typical essential genes; hereafter, we refer to these genes as inactive epigenetic hub genes. A high degree of essentiality of an inactive epigenetic hub gene may be due predominantly to its location near an epigenetic hub. Therefore, such genes may be more enriched in the CS0 group than among genes with lower degrees of essentiality (CS1–CS9). In the analysis (Materials and methods) performed to test this hypothesis, we found that inactive epigenetic hub genes were more likely to be in the essential gene group than in the nonessential gene groups (Supplementary Figure S8D). Thus, disruption of these genes may hamper cell proliferation not by their seized expression, but by the disruption of epigenetic hub integrity. This result further supports the role of essential genes as epigenetic hubs.
In summary, these results provide epigenetic evidence for the role of essential genes as hubs of active epigenetic modification.
Evolutionary nature of human essential genes
Highly and widely expressed genes have been found to have originated early and to be conserved across species [50]. We next investigated the universal distribution of evolutionary rates of essential genes. Using gene age categories defined in a study [51] based on inferred gene origination times, we showed that, as expected, essential genes on average were older (Figure 3A) and significantly more conserved (P = 9.40 × 10−9, Welch’s t-test; Supplementary Figure S9) than were nonessential genes. However, a small subset of essential genes was notably young (Figure 3B); we found that the proportion of essential genes among human-specific genes (gene age group 13) was significantly larger than that of other genes (gene age groups 1–11; Figure 3C), indicating that human-specific genes are more likely to be essential in humans. Similar results were obtained for the other three cell types (Supplementary Figure S10). GO analysis of essential genes in the two youngest groups (human and chimpanzee) showed significant enrichment in the regulation of GTPase activity (Supplementary Figure S11A). Enrichment of these young essential genes in immune-related functions may indicate that these young and essential genes compress a few of cell-type-specific essential genes in hematopoietic lineages and that these genes take up a considerable proportion due to their very small amount of these young and essential genes. We also found that these youngest essential genes were shorter, less conserved and not as actively expressed as other essential genes (Supplementary Figure S11B). In addition, `old’ genes have been reported to be longer than `young’ genes [50, 52, 53]; however, most essential genes in this study were `old,’ but they were shorter than other genes on average (Figure 3D).
Figure 3.
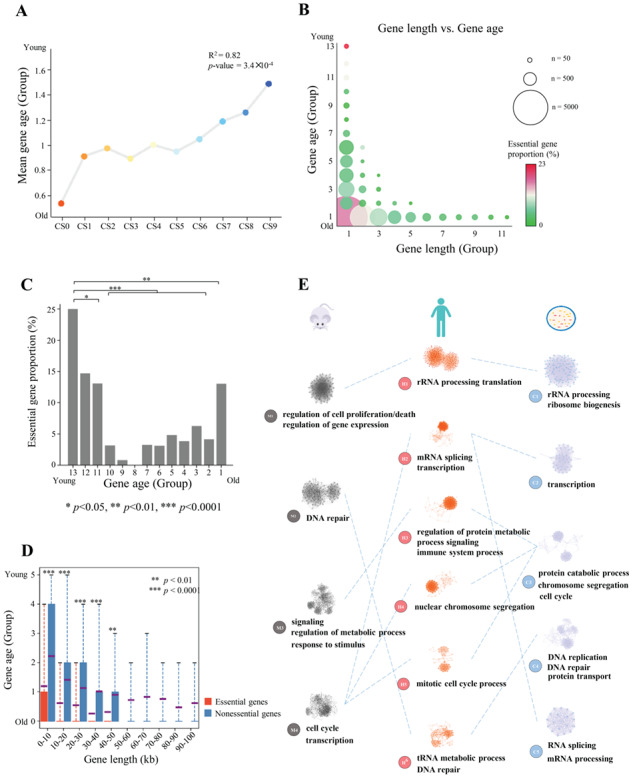
Evolutionary nature of human essential genes. (A) Relationship between gene essentiality and gene age. R2 and P-values from linear regression are shown. (B) Scatterplot of gene length and evolutionary age. Circle size indicates the number of genes, and color represents the essential gene proportion. (C) Essential gene proportions in all 13 gene age groups. (D) Analysis of age differences between essential and nonessential genes grouped by gene length. Red boxes, age data for essential genes. Blue boxes, age data for nonessential genes. Violet line, mean value. Gene groups containing <100 genes are not shown. (E) Essential gene-associated specific functional modules. The network of specific functional interactions among the 1878 human essential genes was clustered using a graph theory clustering algorithm to elucidate gene modules. Six clusters that containing ≥40 genes (H1–H6) were tested for functional enrichment by using genes annotated with GO biological process terms. Representative processes and pathways enriched within each cluster are presented alongside the cluster label. Enriched functions provide a landscape of the potential effects of cellular functions for essential genes. Similar functional processes were shared by essential genes in mouse (four subnet modules) and S. cerevisiae (five subnet modules).
Evolutionary age is defined based on the presence of a homolog in a wide range of species from single-celled organisms to primates [54]. However, the essentiality of a gene can change during the course of evolution [15]. We investigated the essentiality of homologous genes in humans and four other species (mouse, Danio rerio, Drosophila melanogaster and Saccharomyces cerevisiae). We observed 329, 66, 15 and 143 essential genes in humans that were also essential in the four other species, respectively, but only 150, 9, 2 and 2 shared genes in random controls, indicating that essential genes were significantly more conserved than were other genes (P < 1 × 10−5, permutation test), consistent with previous findings [55]. Interestingly, more than half of the genes found to be essential in humans were nonessential in other species and vice versa (Supplementary Figure S12), also consistent with the findings of Hart et al. [55]. Gene essentiality may change in different species because genes or functions could arise separately or be lost or replaced by others during evolution; in this way, the biological network could become more robust [15]. To test this hypothesis, we compared PPI networks among species using essential genes annotated in various species retrieved from DEG database [4]. We first constructed a PPI network with essential genes for each species using a previously described network topology method [56] and detected subnet modules (densely connected regions that can represent molecular complexes) using the MCODE algorithm [57]. We then performed gene set enrichment analysis [58] for each subnet module. Interestingly, similar biological processes were observed between human and other species, although the essential genes were quite different (Figure 3E); for instance, rRNA processing was enriched in human, mouse and S. cerevisiae, but less than 18% of human essential genes were essential in these species (Supplementary Figure S12). Protein localization analysis using existing annotation data [59] also showed that a larger fraction of human essential genes encode proteins located in cytoplasm than those encoded by human nonessential genes, whereas percentages of proteins located in membrane and extracellular for human essential genes are lower than those for human nonessential genes (Supplementary Figure S13). These observations showed similar protein localization propensity in human and in prokaryotes [60] for essential genes in comparison with nonessential genes and may also support similar functions of essential genes between species. In addition, the percentage of proteins located in nucleus is higher for human essential genes than for human nonessential genes (Supplementary Figure S13).
Our observations suggest that essentialomes are enriched in genes required for essential processes.
Transcription activation of essential genes during cell development
Dynamic expression of essential genes in early embryo development
Cell fate decisions contribute fundamentally to the development and homeostasis of complex tissue structures in multicellular organisms. The key to understand the different fates of apparently identical cells lies in the emergence of transcriptional programs [61]. We next characterized essential genes in mammalian embryonic development. Due to the lack of experimental data from human embryos, we used data from mouse preimplantation embryos. To examine whether genetic and epigenetic features were consistent in human and mouse, we calculated the distributions of gene expression and epigenetic information in human and mouse embryonic development. We observed strong correlations (Supplementary Figure S14), which supported the use of mouse data to study human essential genes. During embryo development, the expression levels of essential genes were progressively increased, and two significant increasing were observed in two-cell embryos and the inner cell mass (ICM), which corresponding to zygotic genome activation [62, 63] and the 1st cell fate decision, respectively. In contrast, the expression levels of nonessential genes were significantly lower than those of essential genes during the entire preimplantation period (P < 1 × 10−100, Welch’s t-test), and genes in groups CS7–CS9, which labeled as the least essentiality, were silent after the two-cell embryo stage, similar to the maternal mRNA degradation process (Figure 4A). To further understand the dynamic changes in the transcription activity of essential genes during embryo development, we investigated chromatin state dynamics. Accessible chromatin and active histone modifications were highly enriched in the promoters of essential genes compared with those of nonessential genes (Figures 2G and I and 4B and C). In addition, chromatin was progressively accessible and the H3K4me3 density progressively increased (Figure 4B and C). However, essential genes were least methylated during embryo preimplantation (Figure 4E). These observations suggest that the expression of essential genes is required for embryo development and that both chromatin accessibility and epigenetic modifications contribute to the formation of transcriptional programs in essential genes.
Figure 4.
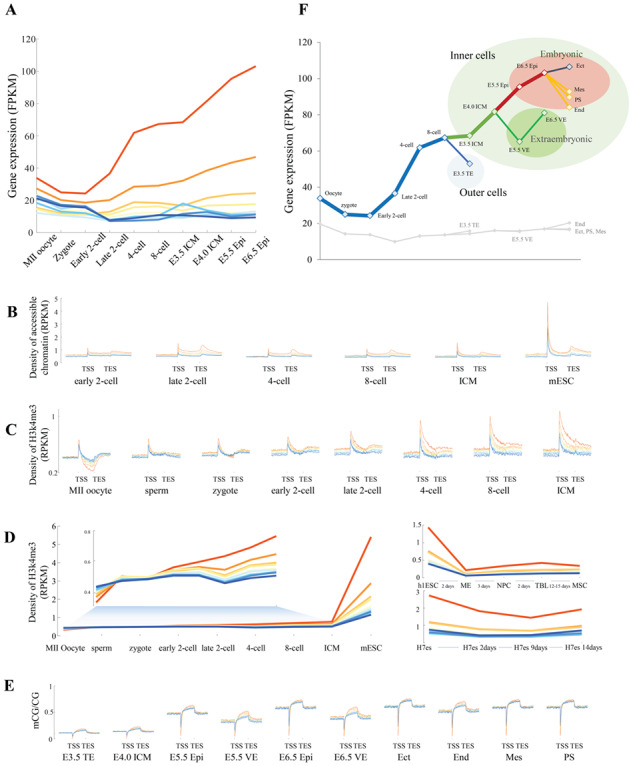
Essential genes in mouse embryo development. (A) Expression levels of essential genes (red lines) and other gene groups at each developmental stage. (B) Density of accessible chromatin surrounding TSSs and TESs of genes at each developmental stage. (C) Density of active H3k4me3 modifications surrounding TSSs and TESs of genes at each developmental stage. (D) Dynamics of H3k4me3 density in gene bodies in preimplantation mouse embryos (left) and postimplantation human embryos (right). (E) Profiles of CG methylation surrounding TSSs of genes at each developmental stage. (F) Dynamics of gene expression for essential and other genes at each developmental stage. TE means trophectoderm; ICM, inner cell mass; VE, visceral endoderm; Epi, epiblast; Ect, ectoderm; End, endoderm; Mes, mesoderm; PS, primitive streak.
To gain insight into the potential function of essential genes during embryo preimplantation, we examined the gene expression pattern at each developmental stage. Interestingly, differential patterns of transcription were observed during early lineage specification (Figure 4F). Essential genes were highly expressed in the ICM, which gives rise to the entire fetus, but much less expressed in the trophectoderm (TE), the outer layer of the blastocyst-stage embryo. During the subsequent formation of the primitive endoderm (PE) and epiblast (Epi), essential genes were also more highly expressed in embryonic tissues than in extraembryonic tissues. Finally, during the formation of the three germ layers, essential genes were highly expressed in the ectoderm, which was derived from the anterior epiblast by embryonic day 6.5 (E6.5), but weakly expressed in the primitive streak (PS) and PS-derived mesoderm and endoderm [61]. Compared with essential genes, nonessential genes were weakly expressed and showed no apparent pattern during embryo development. Thus, the transcription of essential genes is required for lineage segregation, especially for the development of the fetal-origin part of the placenta.
Essential genes are differentially expressed in cancer and normal tissues
Given the fundamental role played by essential genes, it is unsurprising that they represent current and potential novel targets of many antimicrobial and anticancer compounds [64–66]. To further investigate the therapeutic implications of essential genes, we examined the relationship between essential genes and cancer genes. The identification of cancer genes has varied markedly among studies [67–71]; for instance, more than two-thirds of cancer genes identified in one study were not identified as such in another study (Supplementary Figure S15A). Thus, we compared genes from these studies separately and found that essential genes were significantly enriched among cancer genes relative to all protein-coding genes (Figure 5A). For instance, five well-known oncogenes—BRCA1, BRCA2, MYC, EZH2 and SMARCB1—were essential in terms of human cell survival and were associated with chromatin stability, remodeling and modification.
Figure 5.
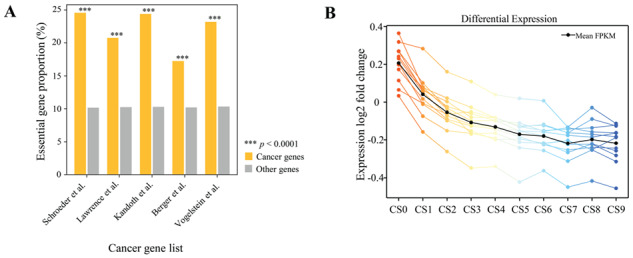
Relationships between human essential genes and cancer. (A) Proportions of human essential genes in five cancer gene lists and in all human genes. ***P < 0.0001, Fisher exact test. (B) Differential expression of genes in normal and tumor tissues from the TCGA database. Each colored line represents a mean differential expression fold change (among all donors, from normal to cancerous for each donor) by tumor type.
As reported above, essential genes were more strongly expressed than nonessential genes in cancer and normal cells (Supplementary Figure S15B). We next determined whether essential genes were differentially expressed between cancer and normal cells. Twenty-three cancer types were examined using gene expression data from the TCGA project. Interestingly, the expression levels of essential genes were significantly higher in cancer cells than in normal cells (P = 8.5 × 10−6, paired-sample t-test; Figure 5B), whereas nonessential genes exhibited similar transcription activity in both cancer and normal cells. These results suggest that essential genes are more sensitive to tumorigenesis and may be superior targets for further drug screening and development.
To further understand the potential function of essential genes in drug screening, we identified 297 significantly DEGs using TCGA data (Materials and methods and Supplementary Table S1). Using the DrugBank database [72], we then identified 135 candidate drugs for these 297 DEGs (Supplementary Table S2). Some of these candidate drugs (e.g. the antineoplastic agents pemetrexed, decitabine, doxorubicin, mitoxantrone and capecitabine) have already been matured in drug-targeting strategies for oncology programs. Other candidate drugs (e.g. the anti-infective agents trifluridine and fleroxacin and the antibacterial agents enoxacin, pefloxacin and ciprofloxacin) may also play roles in cancer treatment, and more research and clinical experiments are required.
HEGIAP: an interactive web server for the study of essential genes
We developed the interactive web server HEGIAP (http://sysomics.com/HEGIAP/), which integrates abundant analytical and visual tools to provide multi-level interpretation of the essentiality of single genes. HEGIAP provides an overall gene property graph, which shows gene length, number of transcript types and the distributions of exons and introns of each transcript. Boxplots are provided that describe properties of the 10 gene groups (CS0–CS9), including but not limited to gene length, protein length, exon count and counts of repetitive elements near promoter regions. Graphs show the corresponding value and group number for each selected gene. For a selected gene, the web server provides histone modification, methylation and chromatin accessibility profiles and a Hi-C contact map of chromatin structure, all of which have been shown to be correlated significantly with the CS value. Multigene analysis is available for the comprehensive examination of groups of genes (Figure 6).
Figure 6.
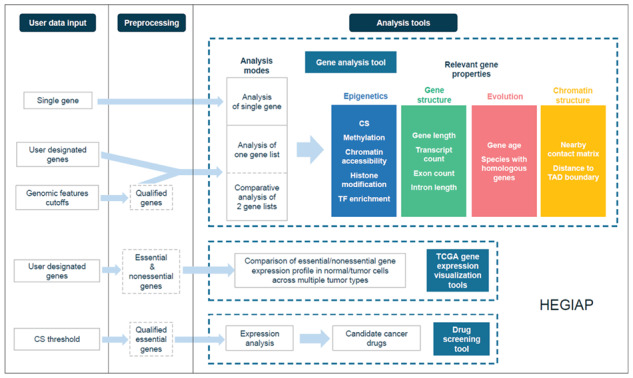
Integrative analysis of individual genes using HEGIAP. HEGIAP provides different analysis modules to enable a multi-view exploration of gene essentiality measured by the CS.
HEGIAP supports both feature- and gene-oriented analyses. In feature-oriented analysis, users can obtain all of the genes that meet chosen screening thresholds for multiple features. They can examine the CS distribution or any other property, enabling exploration of possible correlations between the CS and other genomic features. In classic gene-oriented analysis, users specify their chosen genes and are provided with a comprehensive view of their essentiality and genomic features. A comparative analysis of two different gene lists is provided to facilitate free exploration of the variation in genomic features between genes of interest or between genes screened by degree of essentiality or any other property. Tools are provided to identify genes with aberrant epigenetic modification levels or genomic features based on their essentiality.
To facilitate examination of the difference in essential gene expression level between cancer cells and normal cells, HEGIAP provides a tool for the direct visualization of expression profiles across multiple TCGA tumor types for any group of genes uploaded. Genes are also grouped into essential and nonessential subgroups whose expression profiles are shown for further comparison.
HEGIAP identifies genes that are differentially expressed in tumor and normal tissues. It has a drug-screening tool that is based on the assumption that essential genes have predictive power for the identification of candidate drugs for cancer. Users can set a CS threshold and acquire a list of drugs that significantly suppress the expression of cancer-specific highly expressed genes filtered by this threshold. The user-friendly interface of HEGIAP was constructed using R Shiny, and it requires no plug-in installation for users running any popular web browser.
Discussion
Three types of `hubs’ location of essential genes
Essential genes have been identified to be important content in multiple life-science research domains including those of genetic networks [20, 73, 74], developmental phenotypes [75], evolution [76], cancer therapy [77] and drug discovery [78]. Our work extends previous findings by revealing three `hub’ locations of essential genes. First, essential genes are `hubs’ of PPI networks. As described in the `centrality-lethality’ rule, genes and proteins with high degrees of connectivity tend to be essential because their inactivation is more likely to disrupt overall network architecture [15]. Our statistical analysis confirmed that gene essentiality is correlated significantly with the degree of connectivity and that proteins encoded by essential genes are very stable, tolerating translational errors. Second, our work revealed a structural ‘hub’ of essential genes. Not only are essential genes clustered densely in the genome; these clusters have a 3D structural organization. Third, essential genes are sensitive `hubs’ of epigenetic modification, which contributes to their high expression levels. As essential genes are centers of epigenetic modification and chromatin structure, their high transcription activity levels may further promote the expression of surrounding genes; that is, essential genes may act as the ‘seeds’ of a transcription factory, where endogenous genes are replicated, transcribed and repaired [79–81]. Furthermore, this three-‘hub’ model for essential genes indicates that gene knockdown in CRISPR experiments has the following effects on the transcriptional regulatory system in a cell: the gene will be down-regulated; the expression of other genes in the same PPI network will change; and the chromatin structure and epigenetic signal surrounding the CRISPR site may also change, which may affect the regulation of many other genes.
No gene is absolutely essential; only functions can be so
Consistent with previous findings [50], we confirmed that most essential genes are old. However, we also found that an unexpectedly high proportion of the youngest, human-specific genes are essential and play a role in the regulation of GTPase activity, although we cannot rule out the possibility that this may reflect enrichment of the very few cell-type-specific essential genes in hematopoietic cells, which could easily comprise a considerable fraction of all young and essential genes. Although essential genes are highly expressed and genes with high expression levels tend to be conserved across species, we noticed great variation in essential genes among species. By further examining the PPI networks constructed by essential genes, we found that although gene essentiality changes across species, the biological processes were conserved. This observation provides new insight supporting the idea that no gene is absolutely essential; only functions can be so [15].
Implications for gene editing and synthetic biology
Major innovations in our ability to edit genome sequences have enabled cost-effective and straightforward genome editing in yeasts, plants and animals [82, 83]. The identification of three ‘hub’ locations of essential genes suggests that the effects of gene editing (or gene therapy) on cells should be examined with consideration not only of the target gene and its signaling pathways but also of the associated epigenetic environment and the context of chromatin structure. Furthermore, essential genes can be used as a preferred gene set or important reference for gene interactions in synthetic biology. Additionally, in cancer research, they could facilitate drug discovery, offer promise as markers and may be useful for the identification of clinical therapeutic applications.
In summary, our work provides very valuable information that improves our understanding of human essential genes. Due to the limitations of experimental approaches, further work is required not only to understand the evolutionary plasticity of essential genes across various species but also to gain more evidence on the three `hub’ locations of essential genes. These studies will facilitate our understanding of the design principles of transcription regulatory networks, higher-level organization of vital processes and principles underlying drug resistance.
Materials and methods
Data set
Data description for each evaluation and figure, software or package generating the figures and other related details are provided in Supplementary Table S3.
For human essential genes
Data on DHS, DNA methylation, histone modifications, CTCF and evolutionary conservation were downloaded from the ENCODE project and RoadMap database. Hi-C data were obtained from GSE43070 [34] and GSE63525 [35]. TAD boundaries were detected according to a previously described protocol [84]. Position-specific weight matrices of transcription factors were downloaded from the TRANSFAC and JASPAR databases. Data on ncRNAs were downloaded from the NONCODE database [85] (http://www.noncode.org). Essential gene data for different species were obtained from the DEG database [86] (http://www.essentialgene.org/). Cancer data were downloaded from the TCGA project. Drug data were obtained from the DrugBank database. Human gene annotations were obtained from the GENCODE database (V21).
For essential genes during mouse embryo preimplantation
The transposase-accessible chromatin followed by sequencing assay (ATAC-seq) was obtained from GSE66390 [87]. Histone modification H3K4me3 data for the early two-cell, four-cell and eight-cell stages of mouse embryos and ICMs were obtained from GSE71434 [88] and the ENCODE project. Histone modification H3K4me3 data for H1hESC and H1hESC-derived cells were obtained from a previous study [89]. Histone modification H3K4me3 data for H7es and H7es-derived cells were obtained from the ENCODE project. Mouse gene annotations were obtained from the Mouse Genome Informatics database [90].
Division of genes into groups by CS value
Given the high consistency of essential genes in different cell lines, we used essential genes in the KBM7 cell line for this study. CS value of KBM7 cell line is provided by Wang et al. [11]. Genes were sorted by ascending CS value in KBM7 cell line and divided into 10 groups (CS0–CS9). Specifically, group CS0 is composed of 1878 essential genes as reported by Wang et al. The 1st nine gene groups (CS0–CS8) contains the same number of genes. The rest of genes with highest CS are assigned to group CS9. Corresponding CS threshold of each group is shown in Supplementary Table S4.
Hi-C data processing
For H1hES cell, Hi-C contact matrices were constructed and then normalized using HOMER (http://homer.ucsd.edu/homer/). For the GM12878, IMR90 and K562 cell lines, Hi-C contact matrices and loops were obtained from a previous study [35] and normalized using the SQRTVC method.
Calculating relative abundances of inactive epigenetic hub genes
The observed proportion of genes in each essentiality group with expression levels lower than a specific threshold and whose promoter regions were highly accessible (defined by a specific cutoff of DNase-seq tag density in a 2 kb region upstream of the TSS) was calculated. Then, for each group, the expected probability that a gene was an inactive epigenetic hub gene (defined as the product of the proportion of genes with low expression levels and that of genes located at highly accessible chromatin sites, using the same cutoffs) was calculated. The relative possibility that a gene was an inactive epigenetic hub gene was defined using the observed/expected ratio. To ensure that the analysis was not biased toward the selection of particular cutoff values, we applied widely ranging cutoff selection parameters [expression cutoff: minimum = 2, maximum = 10 (FPKM), step = 0.5; chromatin accessibility cutoff: minimum = top 50% of all genes, maximum = top 95% of all genes, step = 1%]. All 170 cutoff combinations yielded similar and significant results.
PPI network construction
Protein–protein associations were obtained from the STRING database (version: 10.5) [91]. Based on these associations, PPI network was constructed using Cytoscape [92]. Using CentiScaPe [56], we computed specific centrality parameters to describe the network topology and then calculated the degree of connectivity for each node in the PPI network. Densely connected regions in large PPI networks were detected using the molecular complex detection method [57]. A GO analysis was performed using DAVID [93].
Profiling of epigenetic information
For each gene, the gene body and 10 kb upstream and downstream segments were each broken into 50 bins. The ChIP-seq density (RPKM) in these regions was calculated and combined to obtain 150 bins spanning 10 kb upstream, the gene body and 10 kb downstream. The average combined profiles for genes are shown.
Screening of pan-cancer candidate genes
Twelve tumor types (COAD, KICH, BLCA, KIRC, CHOL, UCEC, PRAD, KIRP, LIHC, CESC, LUAD and BRCA) from the TCGA project were used to screen pan-cancer candidate genes. DEGs were first quantified in each cancer–normal tissue pair. The 5000 top-scoring DEGs among all genes and the 1000 top-scoring DEGs among essential genes were further combined, and genes in both of these sets were used as candidate genes for each cancer type. Pan-cancer candidate genes were defined as those that were candidates in at least eight cancer types.
Key Points
We performed a very detailed classification of human protein-coding genes (including essential and nonessential genes) and found general correlations between gene essentiality level and major features.
We extended previous work by integrating multi-omics (genome, epigenome and proteome) data into a new model of the three types of `hub’ location of essential genes in PPI networks, chromatin structure and epigenetic regulation, which enables multidimensional understanding of essential genes.
We conducted, to our knowledge, the 1st systematic analysis of essential genes from the view of 3D chromatin structure, dissecting chromatin loop anchors, TAD boundaries and intra-TAD local contacts, and revealed the ‘hub’ location of essential genes in spatial chromatin conformation.
Our work extended knowledge on two features of essential genes. First, although previous studies indicated that `old’ genes are generally longer than `young’ genes, we found that essential genes are old but unexpectedly short. Second, although long genes are generally more likely to contain more diverse transcripts than short genes, the short essential genes contained a larger variety of transcripts. These two characters of essential genes may be important for the stability of cell functions.
We developed HEGIAP, a web server that provides multiple tools for further visualization and analysis of essential genes.
Supplementary Material
Hebing Chen, Zhuo Zhang and Hao Li work at the Beijing Institute of Radiation Medicine.
Ruijiang Li, Wanying Li and Chenghui Zhao are Master’s students at the Beijing Institute of Radiation Medicine.
Shuai Jiang, Hao Hong and Xin Huang are PhD students at the Beijing Institute of Radiation Medicine.
Xiaochen Bo is a Professor at the Beijing Institute of Radiation Medicine.
Funding
Major Research Plan of the National Natural Science Foundation of China (U1435222); National Natural Science Foundation of China (31801112 and 61873276).
References
- 1. Koonin EV. How many genes can make a cell: the minimal-gene-set concept. Annu Rev Genomics Hum Genet 2000;1:99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol 2003;1:127–36. [DOI] [PubMed] [Google Scholar]
- 3. Gerdes S, Edwards R, Kubal M, et al. Essential genes on metabolic maps. Curr Opin Biotechnol 2006;17:448–56. [DOI] [PubMed] [Google Scholar]
- 4. Luo H, Lin Y, Gao F, et al. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res 2014;42:574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giaever G, Chu AM, Ni L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002;418:387–91. [DOI] [PubMed] [Google Scholar]
- 6. Lartigue C, Glass JI, Alperovich N, et al. Genome transplantation in bacteria: changing one species to another. Science 2007;317:632–8. [DOI] [PubMed] [Google Scholar]
- 7. Galperin MY, Koonin EV. Searching for drug targets in microbial genomes. Curr Opin Biotechnol 1999;10:571–8. [DOI] [PubMed] [Google Scholar]
- 8. Hu W, Sillaots S, Lemieux S, et al. Essential gene identification and drug target prioritization in Aspergillus fumigatus. PLoS Pathog 2007;3:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liao BY, Zhang JZ. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A 2008;105:6987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen WH, Minguez P, Lercher MJ, et al. OGEE: an online gene essentiality database. Nucleic Acids Res 2012;40:D901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang T, Birsoy K, Hughes NW, et al. Identification and characterization of essential genes in the human genome. Science 2015;350:1096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blomen VA, Majek P, Jae LT, et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015;350:1092–6. [DOI] [PubMed] [Google Scholar]
- 13. Hart T, Chandrashekhar M, Aregger M, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015;163(6):1515–26. [DOI] [PubMed] [Google Scholar]
- 14. Hartman JLIV, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science 2001;291:1001–4. [DOI] [PubMed] [Google Scholar]
- 15. Rancati G, Moffat J, Typas A, et al. Emerging and evolving concepts in gene essentiality. Nat Rev Genet 2017;19(1):34–49. [DOI] [PubMed] [Google Scholar]
- 16. GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yen HS, Xu Q, Chou DM, et al. Global protein stability profiling in mammalian cells. Science 2008;322:918–23. [DOI] [PubMed] [Google Scholar]
- 18. Leuenberger P, Ganscha S, Kahraman A, et al . Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 2017;355(6327):1–13. [DOI] [PubMed] [Google Scholar]
- 19. Furey TS, Haussler D. Integration of the cytogenetic map with the draft human genome sequence. Hum Mol Genet 2003;12:1037–44. [DOI] [PubMed] [Google Scholar]
- 20. Wang T, Yu H, Hughes NW, et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017;168:890–903e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu SJ, Horlbeck MA, Cho SW, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017;355(6320). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ribeiro AS, Häkkinen A, Lloyd-Price J. Effects of gene length on the dynamics of gene expression. Comput Biol Chem 2012;41:1–9. [DOI] [PubMed] [Google Scholar]
- 23. Su M, Han D, Boydkirkup J, et al. Evolution of Alu elements toward enhancers. Cell Rep 2014;7:376–85. [DOI] [PubMed] [Google Scholar]
- 24. Hasler J, Strub K. Alu elements as regulators of gene expression. Nucleic Acids Res 2006;34:5491–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu Z, Jin K, Crabbe MJC, et al. Enrichment analysis of Alu elements with different spatial chromatin proximity in the human genome. Protein Cell 2016;7:250–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shapiro JA, Sternberg R. Why repetitive DNA is essential to genome function. Biol Rev Camb Philos Soc 2005;80:227–50. [DOI] [PubMed] [Google Scholar]
- 27. Patchsung M, Settayanon S, Pongpanich M, et al. Alu siRNA to increase Alu element methylation and prevent DNA damage. Epigenomics 2018;10:175–85. [DOI] [PubMed] [Google Scholar]
- 28. Smit A, Hubley R, Green P. RepeatMasker Open-4.0. 2013–2015 http://repeatmasker.org (10 April 2015, date last accessed).
- 29. Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015;518:331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagano T, Lubling Y, Stevens TJ, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013;502:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sexton T, Yaffe E, Kenigsberg E, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012;148:458–72. [DOI] [PubMed] [Google Scholar]
- 33. Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012;485:381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013;503:290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krijger PH, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol 2016;17:771–82. [DOI] [PubMed] [Google Scholar]
- 37. Handoko L, Xu H, Li G, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 2011;43:630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. MB G. Architecture of the human regulatory network derived from ENCODE data. Nature 2012;489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quinodoz S, Ollikainen N, Tabak B, et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 2018;174(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 2017;18. [DOI] [PubMed] [Google Scholar]
- 41. Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010;11:191–203. [DOI] [PubMed] [Google Scholar]
- 42. Zhu J, Adli M, Zou JY, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 2013;152:642–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 2010;28:1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zemach A, Mcdaniel IE, Silva P, et al. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010;328:916–9. [DOI] [PubMed] [Google Scholar]
- 45. Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298:1039–43. [DOI] [PubMed] [Google Scholar]
- 46. Krogan NJ, Dover J, Wood A, et al. The Paf1 complex is required for histone h3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 2003;11:721–9. [DOI] [PubMed] [Google Scholar]
- 47. Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005;120:169–81. [DOI] [PubMed] [Google Scholar]
- 48. Mattick JS. The genetic signatures of noncoding RNAs. PLoS Genet 2009;5:e1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qu Z, Adelson DL. Evolutionary conservation and functional roles of ncRNA. Front Genet 2012;3:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen WH, Trachana K, Lercher MJ, et al. Younger genes are less likely to be essential than older genes, and duplicates are less likely to be essential than singletons of the same age. Mol Biol Evol 2012;29:1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010;11:208–19. [DOI] [PubMed] [Google Scholar]
- 52. Alba MM, Castresana J. Inverse relationship between evolutionary rate and age of mammalian genes. Mol Biol Evol 2005;22:598–606. [DOI] [PubMed] [Google Scholar]
- 53. Wolf YI, Novichkov PS, Karev GP, et al. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci USA 2009;106:7273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yin H, Ma L, Wang G, et al. Old genes experience stronger translational selection than young genes. Gene 2016;590:29–34. [DOI] [PubMed] [Google Scholar]
- 55. Hart T, Chandrashekhar M, Aregger M, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015;163:1515–26. [DOI] [PubMed] [Google Scholar]
- 56. Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009;25:2857–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pierleoni A, Martelli PL, Fariselli P, et al. eSLDB: eukaryotic subcellular localization database. Nucleic Acids Res 2007;35:208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peng C, Gao F. Protein localization analysis of essential genes in prokaryotes. Sci Rep 2015;4:6001–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zernicka-Goetz M, Morris SA, Bruce AW. Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat Rev Genet 2009;10:467. [DOI] [PubMed] [Google Scholar]
- 62. Schultz RM. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum Reprod Update 2002;8:323–31. [DOI] [PubMed] [Google Scholar]
- 63. Schier AF. The maternal-zygotic transition: death and birth of RNAs. Science 2007;316:406–7. [DOI] [PubMed] [Google Scholar]
- 64. Lu Y, Deng JY, Rhodes JC, et al. Predicting essential genes for identifying potential drug targets in Aspergillus fumigatus. Comput Biol Chem 2014;50:29–40. [DOI] [PubMed] [Google Scholar]
- 65. Roemer T, Jiang B, Davison J, et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 2003;50:167–81. [DOI] [PubMed] [Google Scholar]
- 66. Paul MLS, Kaur A, Geete A, et al. Essential gene identification and drug target prioritization in Leishmania species. Mol Biosyst 2014;10:1184–95. [DOI] [PubMed] [Google Scholar]
- 67. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schroeder MP, Rubio-Perez C, Tamborero D, et al. OncodriveROLE classifies cancer driver genes in loss of function and activating mode of action. Bioinformatics 2014;30:I549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 2014;42:D1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Costanzo M, VanderSluis B, Koch EN, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016;353(6306):1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huttlin EL, Bruckner RJ, Paulo JA, et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017;545:505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dickinson ME, Flenniken AM, Ji X, et al. High-throughput discovery of novel developmental phenotypes. Nature 2016;537:508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Albalat R, Canestro C. Evolution by gene loss. Nat Rev Genet 2016;17:379–91. [DOI] [PubMed] [Google Scholar]
- 77. Patel SJ, Sanjana NE, Kishton RJ, et al. Identification of essential genes for cancer immunotherapy. Nature 2017;548:537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017;16:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Papantonis A, Cook PR. Transcription factories: genome organization and gene regulation. Chem Rev 2013;113:8683–705. [DOI] [PubMed] [Google Scholar]
- 80. Zhang Y, Wong CH, Birnbaum RY, et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013;504:306–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012;148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014;157:1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014;32:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schmitt AD, Hu M, Jung I, et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep 2016;17:2042–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhao Y, Li H, Fang SS, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 2016;44:D203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Luo H, Lin Y, Gao F, et al. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res 2014;42:D574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wu J, Huang B, Chen H, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016;534:652–7. [DOI] [PubMed] [Google Scholar]
- 88. Zhang B, Zheng H, Huang B, et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 2016;537:553–7. [DOI] [PubMed] [Google Scholar]
- 89. Xie W, Schultz MD, Lister R, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013;153:1134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blake JA, Eppig JT, Kadin JA, et al. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res 2017;45:D723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.