

Abstract
Research interest in the development of histone deacetylase 8 (HDAC8) activators has substantially increased since loss-of-function HDAC8 mutations were found in patients with Cornelia de Lange syndrome (CdLS). A series of N-acetylthioureas (e.g., TM-2–51) have been identified as HDAC8-selective activators, among others; however, their activation mechanisms remain elusive. Herein, we performed molecular dynamics (MD) simulations and fragment-centric topographical mapping (FCTM) to investigate the mechanism of HDAC8 activation. Our results revealed that improper binding of the coumarin group of fluorescent substrates leads to the “flipping out” of catalytic residue Y306, which reduces the enzymatic activity of HDAC8 towards fluorescent substrates. A pocket between the coumarin group of the substrate and thed catalytic residue Y306 was filled with the activator TM-2–51, which not only enhanced binding between HDAC8 and the fluorescent substrate complex but also stabilized Y306 in a catalytically active conformation. Based on this newly proposed substrate-dependent activation mechanism, we performed structure-based virtual screening and successfully identified low-molecular-weight scaffolds as new HDAC8 activators.
Keywords: HDAC8, activator, molecular dynamics simulation, virtual screening
1. Introduction
As a member of the histone deacetylase (HDAC) protein family, histone deacetylase 8 (HDAC8) catalyzes the deacetylation of acetyl-lysine residues within histone and nonhistone proteins[1, 2]. HDAC8 also serves as fatty acid deacylase and catalyzes the hydrolysis of long chain fatty acyl lysine[3]. HDAC8 plays important roles in diverse processes, including energy homeostasis, microtubule integrity, muscle contraction and sister chromatid separation[4]. Consequently, HDAC8 exhibits various functions in human pathophysiology and may serves as a therapeutic target for many diseases, such as cancer[5, 6], X-linked intellectual disability[7, 8], and infections[9, 10].
Cornelia de Lange syndrome (CdLS) is an inherited congenital anomaly resulting from mutations in the cohesin-loading protein NIPBL or core components of the cohesin complex (SMC1A, SMC3, and RAD21)[11–13]. HDAC8 was identified as the lysine deacetylase that catalyzes human SMC3 deacetylation and shown to be essential for cohesin recycling in the cell cycle. To date, 16 different missense mutations in HDAC8 have been identified in children diagnosed with CdLS[8, 14]. Importantly, in both cellular and clinical studies, a loss of HDAC8 activity resulted in increased SMC3 acetylation as well as decreased cohesin at localized sites and caused features of CdLS. Therefore, interest in developing HDAC8 activators in the search for new therapeutic strategies for CdLS has gained substantial interest[15].
Despite the numerous studies on HDAC8 inhibitors, only a limited number of HDAC8 activators have been reported[16, 17]. For example, among a series of N-acylthiourea derivatives identified as HDAC8 activators, compound TM-2–51 exhibited the highest potency and isoform selectivity (Fig. 1)[18]. The activation feature of TM-2–51 manifests as a decrease in the Km value of the enzyme for a fluorescent substrate and an increase in the catalytic turnover rate of the enzyme[17]. More recently, TM-2–51 was reported to rescue the catalytic activity of CdLS-related HDAC8 mutants[15], further highlighting the therapeutic benefits of HDAC8-selective activators. However, the molecular details regarding the mechanism of HDAC8 activation remain unclear. Subsequent experiments found that the observed activation effect is dependent on the use of a fluorescent substrate in the enzyme activity assay and that TM-2–51 may not promote HDAC8 activity against native substrates[16, 19]. In 2017, Mukhtar et al. reported a new series of HDAC8 activators, which show activation effect against both AMC peptide substrate and AMC-less peptide substrate (Fig. 1)15.
Fig. 1.
To elucidate the mechanism of HDAC8 activation, we performed molecular dynamics (MD) simulations as well as fragment-centric topographical mapping (FCTM) of HDAC-substrate complexes. Based on our computational results, we have explained previous experimental observations. In addition, we suggest a “sandwich-like” binding mode for the known activator TM-2–51 and propose a substrate-dependent mechanism for HDAC8 activation. Similar to our previously proposed mechanism of SIRT1 activation by resveratrol, TM-2–51 may restore tight binding between HDAC8 and specific “loosely bound” substrates, such as fluorescent substrates, by binding a pocket formed by HDAC8-substrate complex formation. Structure-based virtual screening against the unoccupied pocket within the protein-substrate interface successfully identified new HDAC8 activators, with the most potent fragment, HA-16, shown to possess an EC50 of 11.1 μM. This newly proposed substrate-dependent mechanism further improves our understanding of HDAC8 activation, and the newly identified agonists provide more chemotypes for the design of novel substrate-dependent HDAC8 activators.
2. Results and Discussions
2.1. Molecular dynamics simulations of HDAC-substrate complexes with or without the activator TM-2–51
Crystal structures of HDAC8 with a fluorescent substrate (AMC substrate) have been determined[20] [21], and both of these structures indicate a dimeric arrangement in which the coumarin group of the substrates exhibit head-to-head packing (Fig. 2). Besides, similar π-π stacking interactions formed by AMC groups have also been observed in the crystal structure of SIRT5, implying its potential role in crystal packing[22]. We analyzed the crystal structure of an HDAC8 (Y306F)-fluorescent substrate complex (PDB: 2V5W) and identified a pocket (Fig. 2A) formed by the coumarin group of the substrate and the F306 residue in HDAC using AlphaSpace[23]. This pocket was clearly occupied by the coumarin group of the substrate in the other monomer in the dimer structure but remained unoccupied in the monomer structure (Fig. 2). Previous study already indicated that HDAC8 is catalytically active as a monomer in vitro and has been proposed to function as a monomer in vivo[24].
Fig. 2.
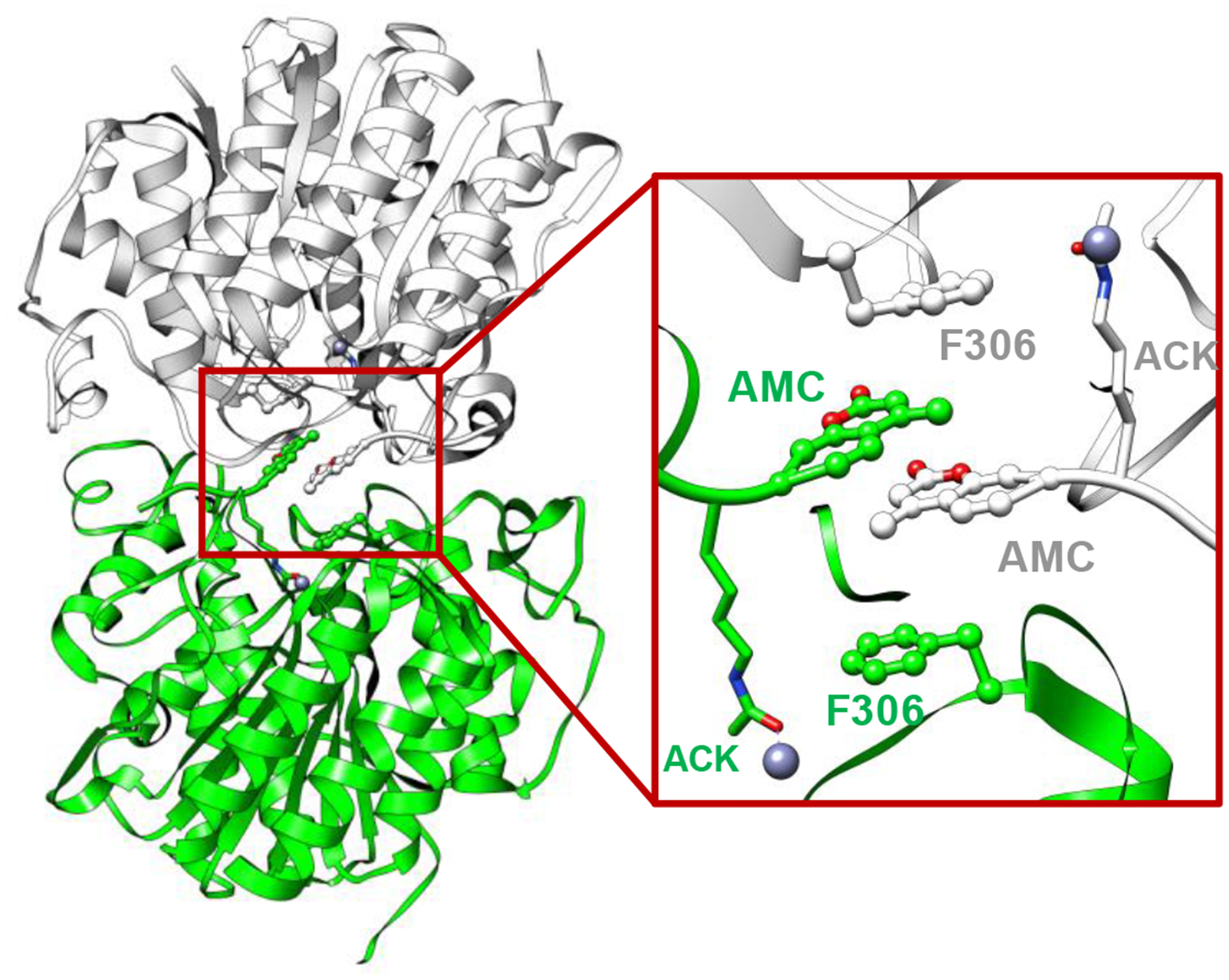
Crystal structure of the HDAC8-substrate complex (PDB: 2V5W). Two monomers are illustrated as white and green ribbons. The Fluor de Lys substrates are highlighted with their coumarin groups presented as a ball-and-stick model, and acetyl-lysine residues are presented as a stick model.
To gain further insights into the mechanism of HDAC8 activation, we performed MD simulations using HDAC8 and an AMC substrate (Table 1). For comparison, we also constructed another HDAC8-substrate complex using a native substrate[25]. Consistent with experimental observations, the binding of HDAC8 with a native substrate was more favorable for catalysis than its binding with a fluorescent substrate[25, 26]. As shown in Fig. 3, a packing defect of the +1 coumarin group led to the “flipping out” of the catalytic residue Y306, while the +1 Phe residue in the native substrate adopted a proper interaction with respect to Y306 (Fig. 3C and 3D). In addition, binding of the AMC group of the fluorescent substrate was less stable than that of the Phe group of the native substrate during MD simulations (Fig. 3B). These computational results suggested that the unoccupied pocket (Fig. 3A) led to the defect in packing between the coumarin group and Y306, which may explain the weak catalytic activity of HDAC8 towards fluorescent substrates[2].
Table 1.
Comparison of proteins, substrate sequences, presence of activator, experimental data, and simulation results of three MD systems.
| MD System | Protein | Substrate | Activator | Experimental enzyme activity | MD simulation results | |
|---|---|---|---|---|---|---|
| Y306 conformer | Substrate binding | |||||
| 1 | HDAC8 | RHK(ac)K(ac)AMC | - | Low | Inactive | Loose |
| 2 | HDAC8 | RHK(ac)K(ac)AMC | TM-2–51 | Recurred | Activate | Tight |
| 3 | HDAC8 | ISK(ac)FDa | - | Normal | Activate | Tight |
| 4 | HDAC1 | RHK(ac)K(ac)AMC | - | Normal | Activate | Tight |
Native substrate of HDAC8 [25]
Fig. 3.
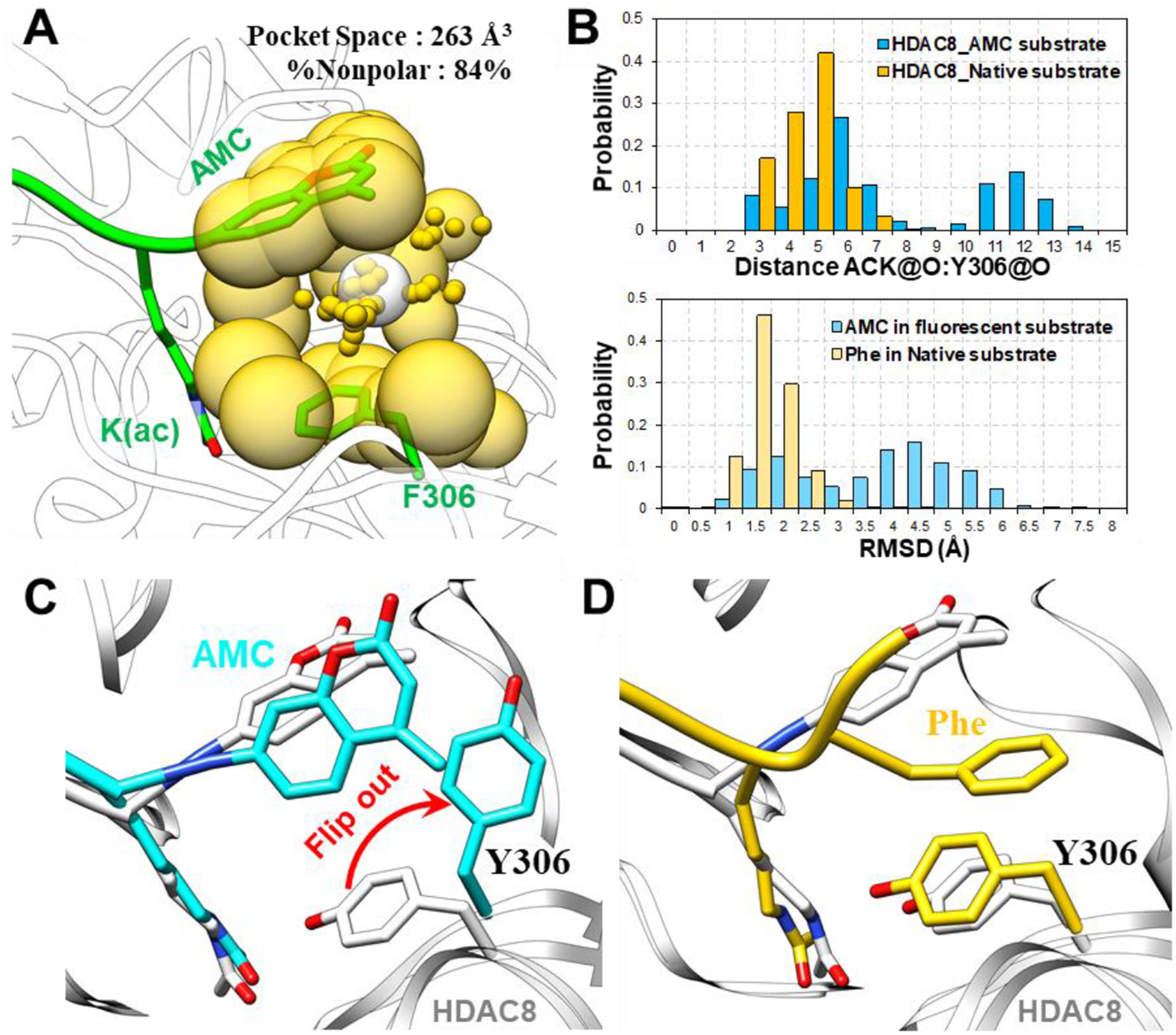
(A) Pocket analysis of the HDAC8 (Y306F)-substrate monomer using AlphaSpace revealed an unoccupied pocket formed by the coumarin group and F306. (B) The distance between ACK@O and Y306@O, which is essential for the catalytic reaction, and the RMSD values of the +1 positions in substrates during MD simulations of HDAC8 with a fluorescent substrate and native substrate are shown. (C) Comparison of a representative MD snapshot of the HDAC8-fluorescent substrate complex (colored cyan) and a crystal structure (PDB: 2V5W, colored white). (D) Comparison of a representative MD snapshot of the HDAC8-native substrate complex (colored yellow) and a crystal structure (PDB: 2V5W, colored white).
Interestingly, the volume of the pocket (263 Å3) was very similar to the ligand volume of the known HDAC8-selective activator TM-2–51 (220 Å3, Fig. S1A in Supporting Information). In addition, molecular docking of TM-2–51 against HDAC8-substrate complex also suggested the preference in binding with the pocket formed by coumarin and Y306 (Fig. S1B in Supporting Information). Thus, we hypothesize that TM-2–51 binds the pocket and enhances the binding of HDAC8 with fluorescent substrates.
Three representative docking poses were selected and subjected to MD simulations (Fig. S2 in Supporting Information). Interestingly, we observed the stable binding of TM-2–51 in the HDAC8-substrate complex starting from docking pose II. As shown in Fig. 4A, TM-2–51 targeted both the pocket and an adjunct pocket and exhibited a “sandwich-like” binding mode with the HDAC8-fluorescent substrate complex. As expected, the binding of TM-2–51 not only stabilized binding of the coumarin group but also locked Y306 in a catalytically active conformation (Fig. 4). Based on these results, we have suggested a potential TM-2–51 binding mode and elucidated the activation mechanism of the HDAC8 activator.
Fig. 4.
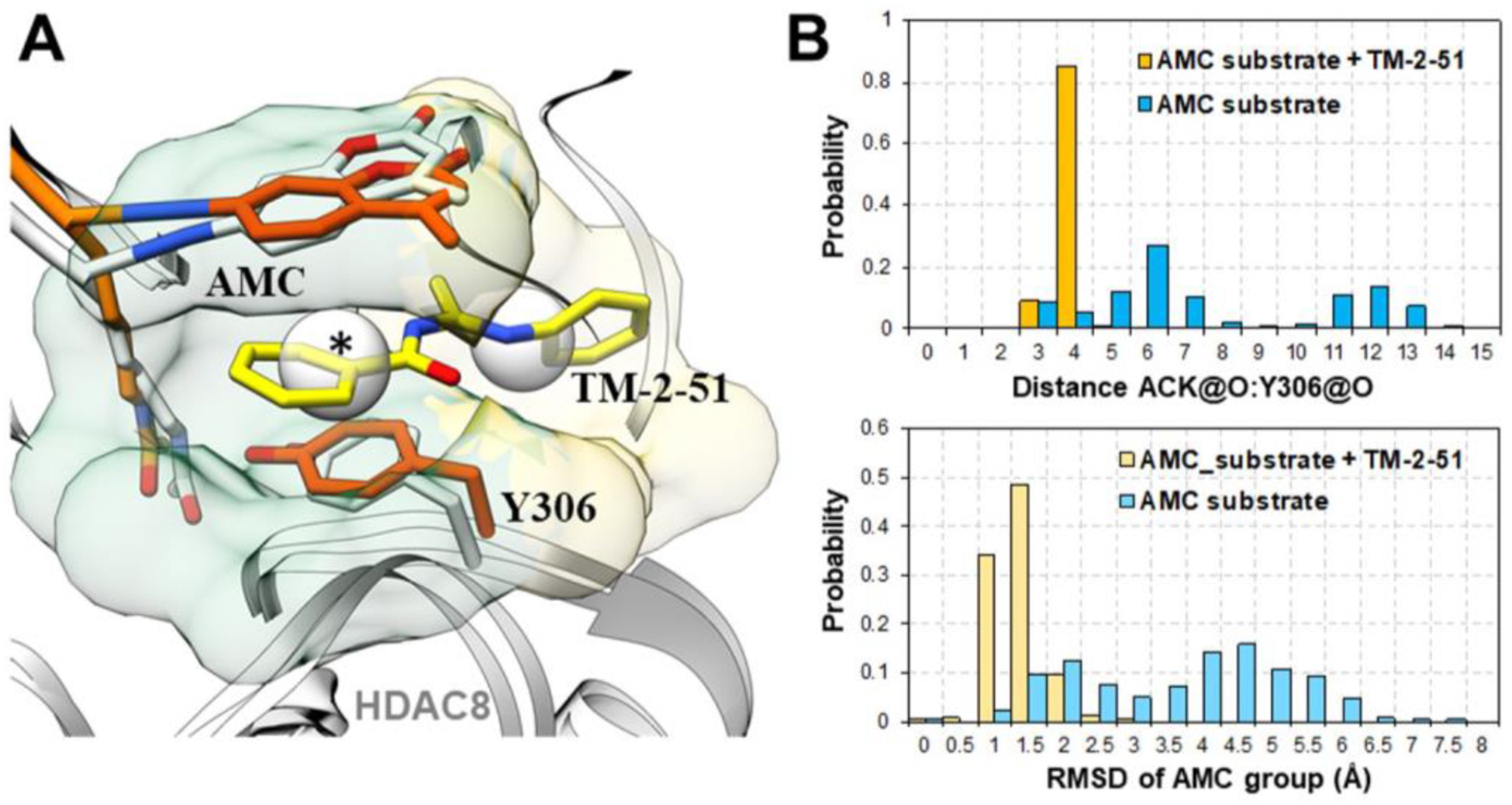
(A) Comparison of representative MD snapshots of an HDAC8-fluorescent substrate complex (colored orange) with bound activator TM-2–51 (colored yellow) and a crystal structure (PDB: 2V5W, colored white). The pocket (transparent green surface, marked with *) and adjunct pocket (transparent yellow surface) occupied by the activator TM-2–51 were detected by AlphaSpace. (B) The distance between ACK@O and Y306@O, which is essential for catalytic reaction, and the RMSD values of the AMC groups in substrates during MD simulations of HDAC8-fluorescent substrate with or without the bound activator are shown.
Notably, TM-2–51 is a selective HDAC8 activator with no effects against other HDAC isoforms, such as HDAC1. To further test our proposed activation mechanism of TM-2–51, we then performed MD simulations using a HDAC1-fluorescent substrate complex. Consistent with the experimental data, the binding of HDAC1 to the fluorescent substrate was more stable than that of HDAC8[26] (Fig. 5). Compared to HDAC8, HDAC1 possesses a larger L1 loop proximal to the active site that directly interacts with both Y306 and the AMC group (Fig. 5). The pocket is occupied by the L1 loop of HDAC1, leaving no room for TM-2–51 to bind, so we did not observe defects in packing between Y306 and the AMC group. This result explained the isoform selectivity of TM-2–51.
Fig. 5.
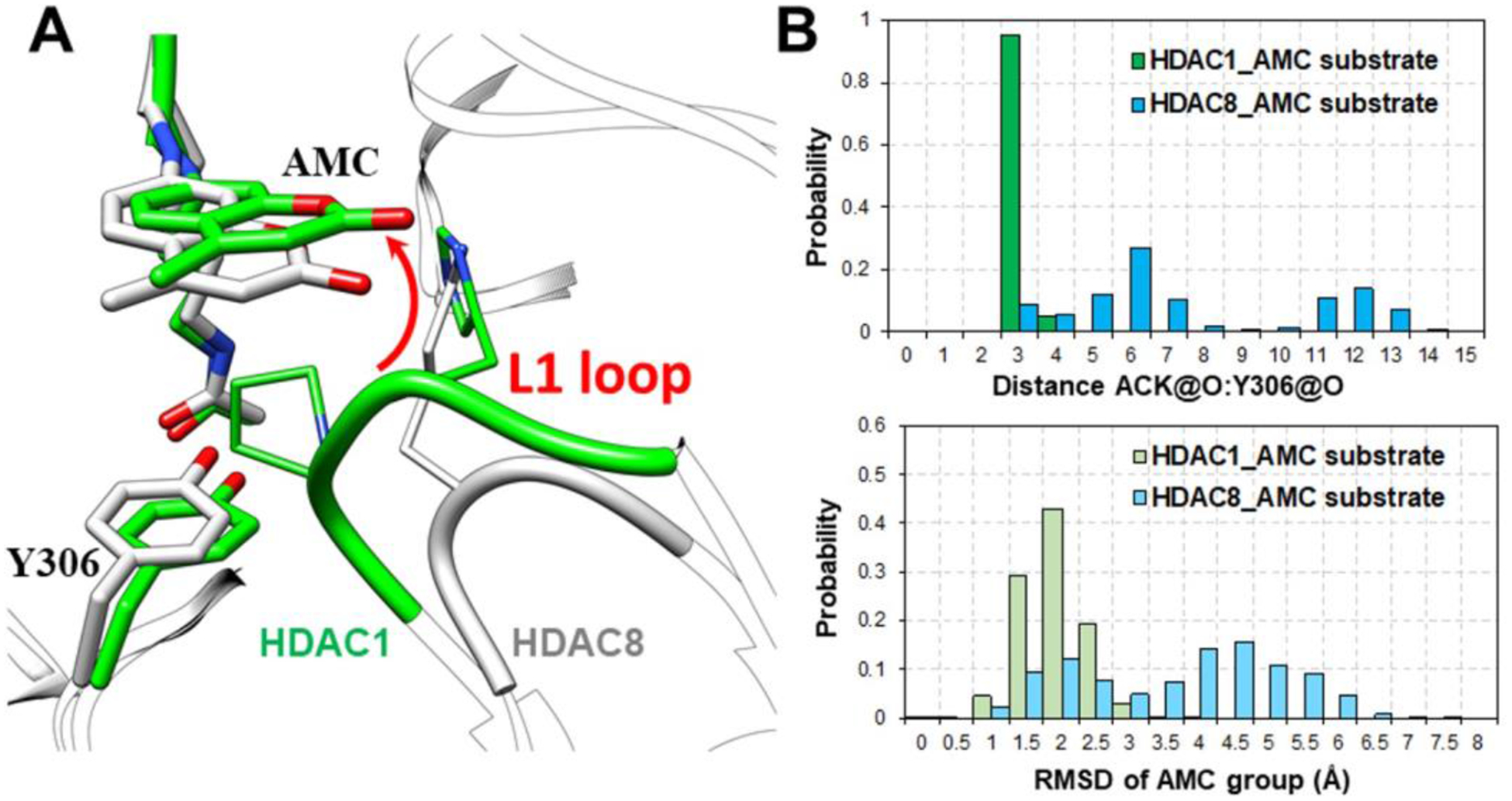
(A) Comparison of HDAC8-fluorescent substrate crystal structure (2V5W, colored in white) and representative MD snapshot of HDAC1-fluorescent substrate complex (colored in green). (B) The distance between ACK@O and Y306@O, which is essential for catalytic reaction, and the RMSD values AMC group in substrates during MD simulations of HDAC1-fluorescent substrate and HDAC8-fluorescent substrate.
2.2. Substrate-dependent activation mechanism
Based on the above results, here, we propose a detailed mechanism of substrate-dependent HDAC8 activation. This mechanism explains the lower catalytic activity of HDAC8 towards fluorescent substrates than the native substrate, suggests a “sandwich-like” TM-2–51 binding mode, and reveals how TM-2–51 selectively promotes the interaction between HDAC8 and fluorescent substrates.
As summarized in Fig. 6, HDAC8 tightly binds the native substrate due to direct interaction between the +1 Phe residue in the substrate and catalytic residue Y306. However, fluorescent substrates fail to form a stable interaction with HDAC8 due to the presence of a pocket between the +1 coumarin group and the Y306 residue; this pocket impairs proper HDAC8-substrate interactions, and defects in packing of the coumarin group may lead to the “flipping out” of catalytic residue Y306. Importantly, the known activator TM-2–51 can bind the complementary pocket, therefore stabilizing the HDAC8-substrate complex in the catalytically required conformation. Moreover, other known HDAC8 activators (compound 4 and 7 in Fig. 1) are also able to bind with this pocket according to molecular docking results (Supporting information Fig. S3). On the other hand, HDAC1 possesses a larger loop than HDAC8. In HDAC1, this loop occupies the pocket and forms stable interactions with the coumarin group. Thus, TM-2–51 cannot further enhance the binding between HDAC1 and its fluorescent substrate.
Fig. 6.
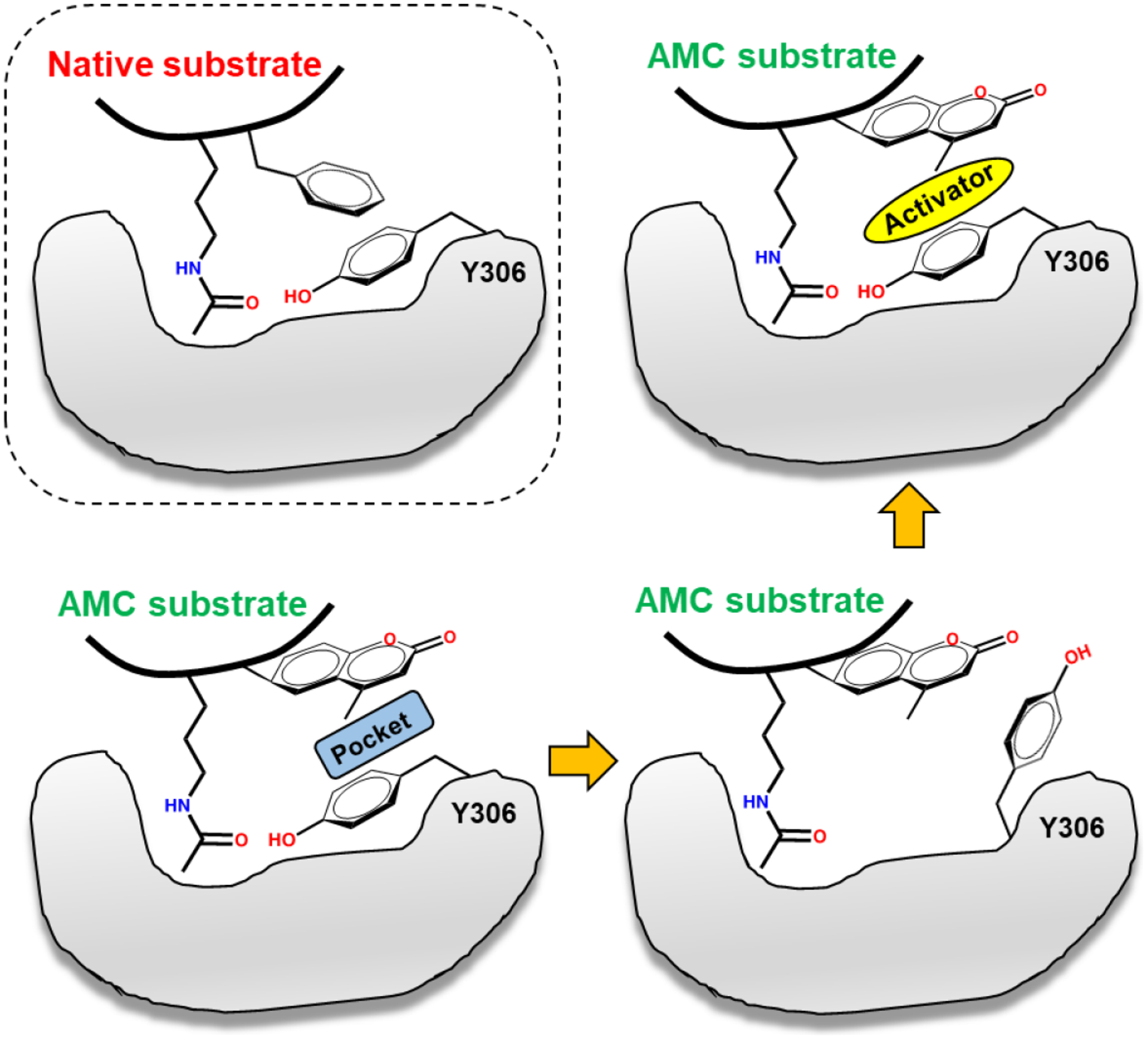
Substrate-dependent activation mechanism of HDAC8.
This new mechanism highlights the importance of aromatic amino acid residues in the +1 position of HDAC8 substrates and suggests that small-molecule activators activate only “loosely bound” substrates (e.g., fluorescent substrates) by restoring proper protein-substrate interactions. In addition, many nuclear proteins have been found to contain aromatic amino acids at the C-terminal ends of their acetylated lysine residues; these proteins serve as targets for various HDAC isozymes[27].
A previously reported crystal structure of HDAC3 in complex with the deacetylase-activating domain (DAD) revealed the important role of the inositol tetraphosphate (IP4) molecule in corepressor assembly and HDAC3 activation[28]. Interestingly, the IP4-binding site in HDAC3 is close to our predicted binding site for the HDAC8 activator (Fig. S4 in Supporting Information). Furthermore, MD simulations also suggested that the IP4 molecule at the HDAC3-DAD interface promotes the inward conformation of Y298 (Y306 in HDAC8)[29], which is similar to our newly proposed mechanism of HDAC8 activation. Thus, our results further suggest that class I HDACs adopt a similar activation mechanism in which small-molecule activators bind a pocket close to the L1 loop and stabilize HDAC in the catalytically required conformation.
Notably, in conjunction with HDAC8 activation, the activation of SIRT1, which belongs to the class III HDAC family, by resveratrol has been controversial[30, 31]. Recently, such controversy has been mitigated by the demonstration that SIRT1 cleaves physiological peptide substrates harboring aromatic amino acids at the +1 position of the acetylated lysine substrate[32]. A similar substrate-dependent mechanism of SIRT1 activation was described in our previous study in which resveratrol serves as a protein-substrate stabilizer[33].Thus, stabilization of protein-substrate interactions may be a general mechanism of substrate-dependent small-molecule activators. Searching for disease-related “loosely bound” substrates and identifying small molecules that are compatible with tight protein-substrate binding are possible strategies for the rational design of enzyme activators.
2.3. Novel low-molecular-weight HDAC8 activators
To further verify this newly proposed activation mechanism as well as discover novel scaffolds for the design of HDAC8 activators, we performed structure-based virtual screening using AutoDock Vina. According to the proposed activation mechanism of TM-2–51, sandwich-like π-π-π stacking interactions between Y306 in HDAC8 and the activator and fluorescent substrate are important. On the other hand, low-molecular-weight fragments not only complement the pocket (volume = 263 Å3), but also serve as valuable scaffolds for future drug design. Therefore, a total of 1,046 low-molecular-weight fragments containing at least one aromatic ring, which selected from commercial database, were subjected to structure-based virtual screening. Based on the calculated binding energies as well as the occupancy of the pocket, twenty fragments were selected and tested for their activation effect on HDAC8 enzymatic activity (Fig. S5 in Supporting Information). As shown in Fig. 7A, six fragments induced 1.3–2-fold activation (20 μM) of a fluorescent substrate, with the most potent fragments, HA-1 and HA-16, possess EC50 values of 11.1 μM and 16.9 μM, respectively (Fig. 7B). The predicted model also suggested a binding mode for these new activators similar to that of TM-2–51 (Fig. 8). These results not only support our proposed mechanism of HDAC8 activation but also provide a series of novel scaffolds for the design of new HDAC8 activators.
Fig. 7.
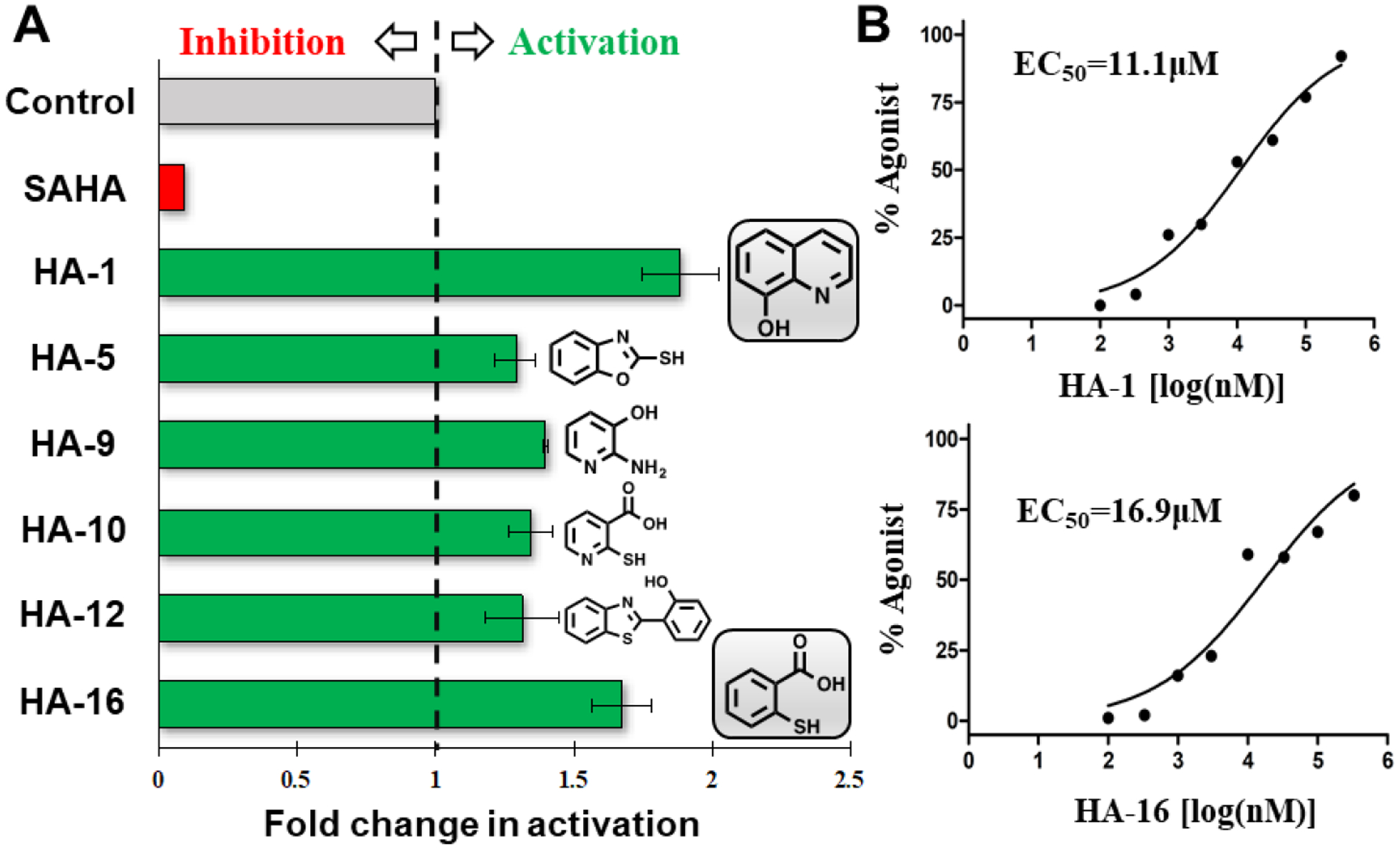
(A) Chemical structures and activation rates of six new HDAC8 activators (20 μM). (B) Dose-dependent activation curves of fragments HA-1 and HA-16.
Fig. 8.
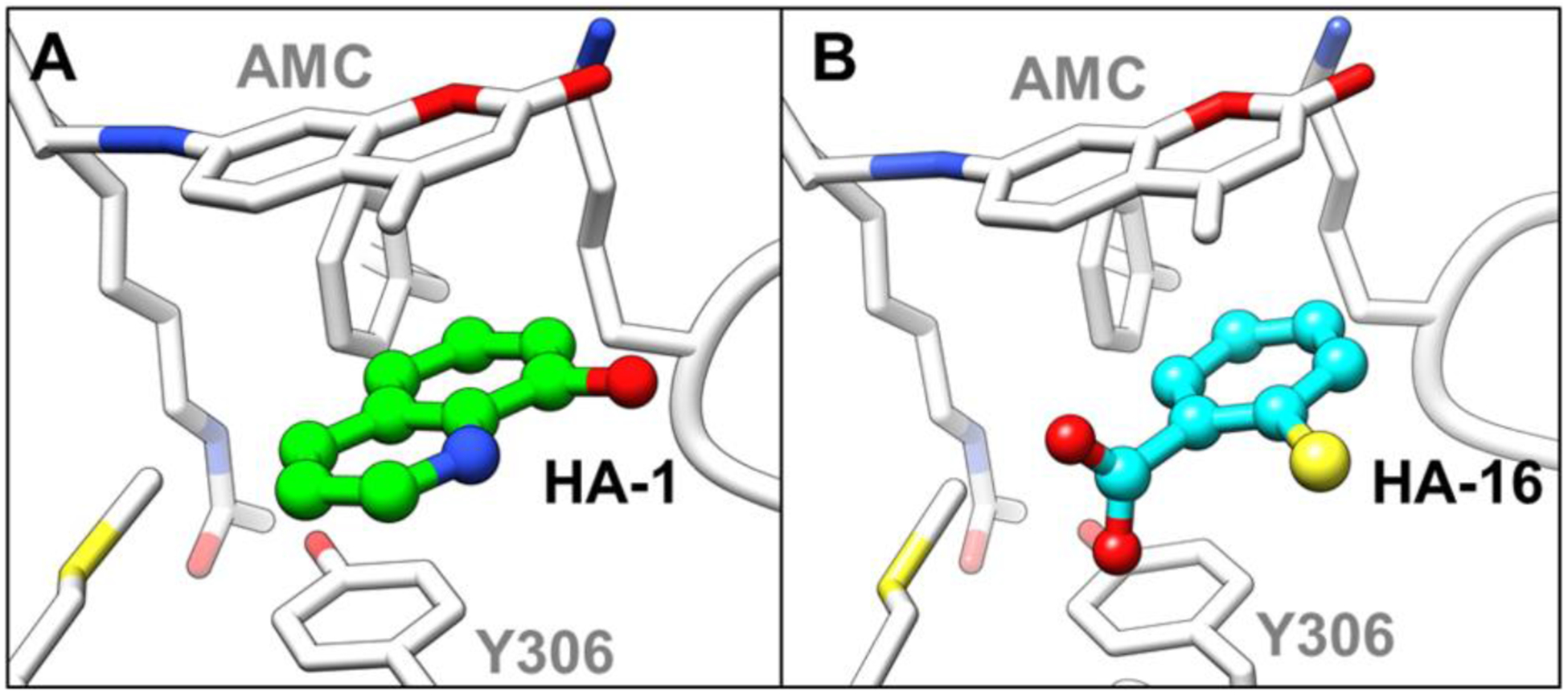
Predicted mode of HA-1 (A) and HA-16 (B) binding in the HDAC8-fluorescent substrate complex.
3. Conclusion
In summary, we propose a new mechanism of HDAC8 activation in which the activation of HDAC8 is dependent on stabilization of the interactions between proteins and specific “loosely bound” substrates (such as fluorescent substrates) that contain specific chemical groups, resulting in catalytically unfavorable conformation changes. Thus, identifying disease-related “loosely bound” substrates will be one of the primary tasks in the future development of HDAC8 activators.
Notably, six new HDAC8 activators were identified on the basis of our proposed activation mechanism. Although these new activators were identified using a fluorescent substrate, these results further support our proposed mechanism of HDAC8 activation and have significant implications for the further rational design of substrate-specific HDAC8 activators.
4. Materials and Methods
4.1. Structure preparation
The crystal structure of an HDAC8-fluorescent substrate complex (PDB: 2V5W[20]) was used as the starting structure for the HDAC8 simulation system. First, the structure of the native peptide was modeled to be a fluorescent substrate using the tleap module in AmberTools 15[34]. The crystal structure of HDAC1 (PDB: 4BKX[35]) was used as the starting structure for the HDAC1 simulation system, and the structure of a fluorescent substrate was adapted from the HDAC8 crystal structure (PDB: 2V5W). The protonation states of charged residues were determined based on pKa calculations via the PDB2PQR server[36]. The protonation states of catalytic residues were determined based on our previous quantum mechanical/molecular mechanical (QM/MM) study[37]. For the system consisting of the HDAC8-fluorescent substrate complex with a bound activator, the known activator TM-2–51 was docked into the pocket formed by the coumarin group and Y306 using AutoDock Vina[38]. Three representative docking poses (I, II and III) of TM-2–51 were selected from the top 10 scored conformers.
4.2. MD simulation
MD simulations were performed using the AMBER14 package[39] with the ff14SB force field[40] for the protein, the TIP3P model for water molecules, and the short-long effective function 2 (SLEF2) force field[41] for zinc ions. Partial charges for the 7-Amino-4-methylcoumarin (AMC) group in the fluorescent substrate and small-molecule activators were fit using the basis sets HF/6–31 G(d) at the Hartree-Fock level of theory (Gaussian 09, revision E.01)[42]. The restrained electrostatic potential (RESP) method[43] in Antechamber was employed to fit the charges to each atomic center, followed by GAFF parameterization to obtain complete topological descriptions of the ligands. Each system was solvated with explicit TIP3P water in a rectangular periodic box and neutralized with Na+ counterions. A series of minimizations, equilibrations, and standard MD simulations were performed following our previous study[33] on GPUs using Particle Mesh Ewald Molecular Dynamics (PMEMD)[44, 45]. For each simulation system, three independent runs were carried out for 100 ns with periodic boundary conditions, and snapshots were saved every 10 ps for analysis. The SHAKE algorithm was applied to constrain all bonds involving hydrogen atoms. The particle mesh Ewald (PME) method with a 10 Å cutoff for the nonbonded interactions was used in energy minimizations and MD production. The Berendsen thermostat method was used to maintain the system temperature at 300 K. All other parameters were default values.
4.3. FCTM
FCTM of protein-substrate/ligand interfaces was performed using AlphaSpace[23, 46], which identifies and represents concave spaces across a protein surface using a geometric model based on Voronoi tessellation. Concave spaces were represented as a set of alpha-atom/alpha-space pairs, which were then clustered into discrete fragment-centric pockets. Pocket occupation was evaluated based on the distance between its associated alpha-atom and the closest atom to the ligand using a 1.6 Å cutoff.
4.4. Structure-based virtual screening
A total of 1,046 fragment-like compounds (Number of rings ≥ 1 and M.W. ≤ 250 Da) selected from commercial library (Sigma-Aldrich and Aladdin) are used in virtual screening. Ligand structures were prepared using the structure preparation module of SYBYL-x; these preparations included: (1) the addition of missing hydrogens, (2) the calculation of atomic Gasteiger-Hückel charges, and (3) structural minimization using the MMFF94 force field. Virtual screening was performed using AutoDock Vina[38]. The search space box used for docking was centered around the pocket formed by the coumarin group and Y306. The pocket occupancy of each ligand was measured using AlphaSpace. According to their Vina scores as well as pocket occupancies, a total of 20 fragments were selected for biological testing. The selected compounds were purchased from Sigma-Aldrich and Aladdin with purity of higher than 95% (confirmed by the supplier).
4.5. HDAC8 activation assay
All of the enzymatic reactions were conducted at 37°C for 30 minutes. The 50 μL reaction mixture contained 25 mM Tris (pH 8.0), 1 mM MgCl2, 0.1 mg/mL BSA, 1.37 mM NaCl, 2.7 mM KCl, HDAC8 and the fluorescent substrate (Fluor de Lys). Compounds were diluted in 10% DMSO, and 5 μL of each diluted compound was added to a 50 μL reaction volume such that the final concentration of DMSO was 1% in all reactions. The pan-HDAC inhibitor SAHA was employed as negative control. The assay was performed by quantitating the fluorescent product in solution following enzyme reaction. Fluorescence was then analyzed with an excitation wavelength of 350–360 nm and an emission wavelength of 450–460 nm using a SpectraMax M5 microtiter plate reader. EC50 values were calculated using nonlinear regression with normalized dose-response fit using GraphPad Prism 8 software.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81874288), Shandong Provincial Natural Science Foundation (ZR2019LZL004), the China-Australia Centre for Health Sciences Research (CACHSR no. 2019GJ07) and Young Scholars Program of Shandong University and Research Funds of Shandong Academy of Medical Science for Young Scholar (Grant No. 2018-32). Y. Z. would acknowledge the support by NIH (R35-GM127040). The authors gratefully acknowledge NYU-ITS and NYUAD for providing computational resources.
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
Appendix A. Supplementary material
Supplementary data to this article can be found online
References
- 1.Hu E, Chen Z, Fredrickson T, et al. , Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor, J Biol Chem, 2000; 275: 15254–15264. [DOI] [PubMed] [Google Scholar]
- 2.Wolfson NA, Pitcairn C, Fierke CA, HDAC8 substrates: Histones and beyond, Biopolymers, 2013; 99: 112–126.23175386 [Google Scholar]
- 3.Aramsangtienchai P, Spiegelman NA, He B, et al. , HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine, ACS Chem Biol, 2016; 11: 2685–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakrabarti A, Oehme I, Witt O, et al. , HDAC8: a multifaceted target for therapeutic interventions, Trends Pharmacol Sci, 2015; 36: 481–492. [DOI] [PubMed] [Google Scholar]
- 5.Oehme I, Deubzer HE, Wegener D, et al. , Histone Deacetylase 8 in Neuroblastoma Tumorigenesis, Clin Cancer Res, 2009; 15: 91–99. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Du CL, Lv Z, et al. , The Up-Regulation of Histone Deacetylase 8 Promotes Proliferation and Inhibits Apoptosis in Hepatocellular Carcinoma, Digest Dis Sci, 2013; 58: 3545–3553. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser FJ, Ansari M, Braunholz D, et al. , Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance, Hum Mol Genet, 2014; 23: 2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deardorff MA, Bando M, Nakato R, et al. , HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle, Nature, 2012; 489: 313–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marek M, Kannan S, Hauser AT, et al. , Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni, Plos Pathog, 2013; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamauchi Y, Boukari H, Banerjee I, et al. , Histone Deacetylase 8 Is Required for Centrosome Cohesion and Influenza A Virus Entry, Plos Pathog, 2011; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pie J, Gil-Rodriguez MC, Ciero M, et al. , Mutations and Variants in the Cohesion Factor Genes NIPBL, SMC1A, and SMC3 in a Cohort of 30 Unrelated Patients With Cornelia de Lange Syndrome, Am J Med Genet A, 2010; 152a: 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Revenkova E, Focarelli ML, Susani L, et al. , Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA, Hum Mol Genet, 2009; 18: 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deardorff MA, Kaur M, Yaeger D, et al. , Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation, Am J Hum Genet, 2007; 80: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decroos C, Christianson NH, Gullett LE, et al. , Biochemical and Structural Characterization of HDAC8 Mutants Associated with Cornelia de Lange Syndrome Spectrum Disorders, Biochemistry, 2015; 54: 6501–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decroos C, Bowman CM, Moser JAS, et al. , Compromised Structure and Function of HDAC8 Mutants Identified in Cornelia de Lange Syndrome Spectrum Disorders, ACS Chem Biol, 2014; 9: 2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhtar YM, Huang YJ, Liu JJ, et al. , Acetanilide and bromoacetyl-lysine derivatives as activators for human histone deacetylase 8, Bioorg Med Chem Lett, 2017; 27: 2319–2323. [DOI] [PubMed] [Google Scholar]
- 17.Singh RK, Mandal T, Balsubramanian N, et al. , Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate, Bioorg Med Chem Lett, 2011; 21: 5920–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh RK, Cho K, Padi SKR, et al. , Mechanism of N-Acylthiourea-mediated Activation of Human Histone Deacetylase 8 (HDAC8) at Molecular and Cellular Levels, J Biol Chem, 2015; 290: 6607–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toro TB, Pingali S, Nguyen TP, et al. , KDAC8 with high basal velocity is not activated by N-acetylthioureas, Plos One, 2016; 11: e0146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vannini A, Volpari C, Gallinari P, et al. , Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex, EMBO Rep, 2007; 8: 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dowling DP, Gantt SL, Gattis SG, et al. , Structural Studies of Human Histone Deacetylase 8 and Its Site-Specific Variants Complexed with Substrate and Inhibitors, Biochemistry, 2008; 47: 13554–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang HL, Liu S, Wu CY, et al. , Interactions between sirtuins and fluorogenic small-molecule substrates offer insights into inhibitor design, RSC Adv, 2017; 7: 36214–36222. [Google Scholar]
- 23.Rooklin D, Wang C, Katigbak J, et al. , AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein-Protein Interaction Interfaces, J Chem Inf Model, 2015; 55: 1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang XJ, Seto E, The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men, Nat Rev Mol Cell Bio, 2008; 9: 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olson DE, Udeshi ND, Wolfson NA, et al. , An Unbiased Approach To Identify Endogenous Substrates of “Histone” Deacetylase 8, ACS Chem Biol, 2014; 9: 2210–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schultz BE, Misialek S, Wu JS, et al. , Kinetics and comparative reactivity of human class I and class IIb histone deacetylases, Biochemistry, 2004; 43: 11083–11091. [DOI] [PubMed] [Google Scholar]
- 27.Riester D, Hildmann C, Grunewald S, et al. , Factors affecting the substrate specificity of histone deacetylases, Biochem Biophys Res Commun, 2007; 357: 439–445. [DOI] [PubMed] [Google Scholar]
- 28.Watson PJ, Fairall L, Santos GM, et al. , Structure of HDAC3 bound to co-repressor and inositol tetraphosphate, Nature, 2012; 481: 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arrar M, Turnham R, Pierce L, et al. , Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3, Protein Sci, 2013; 22: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borra MT, Smith BC, Denu JM, Mechanism of human SIRT1 activation by resveratrol, J Biol Chem, 2005; 280: 17187–17195. [DOI] [PubMed] [Google Scholar]
- 31.Pacholec M, Bleasdale JE, Chrunyk B, et al. , SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1, J Biol Chem, 2010; 285: 8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hubbard BP, Gomes AP, Dai H, et al. , Evidence for a Common Mechanism of SIRT1 Regulation by Allosteric Activators, Science, 2013; 339: 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou XB, Rooklin D, Fang H, et al. , Resveratrol serves as a protein-substrate interaction stabilizer in human SIRT1 activation, Sci Rep, 2016; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salomon-Ferrer R, Case DA, Walker RC, An overview of the Amber biomolecular simulation package, Wires Comput Mol Sci, 2013; 3: 198–210. [Google Scholar]
- 35.Millard CJ, Watson PJ, Celardo I, et al. , Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates, Mol Cell, 2013; 51: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolinsky TJ, Czodrowski P, Li H, et al. , PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations, Nucleic Acids Res, 2007; 35: W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu RB, Wang SL, Zhou NJ, et al. , A Proton-Shuttle Reaction Mechanism for Histone Deacetylase 8 and the Catalytic Role of Metal Ions, J Am Chem Soc, 2010; 132: 9471–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trott O, Olson AJ, Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading, J Comput Chem, 2010; 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Case DA, Babin V, Berryman J, et al. , Amber 14, 2014.
- 40.Maier JA, Martinez C, Kasavajhala K, et al. , ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J Chem Theory Comput, 2015; 11: 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong WJ, Wu RB, Zhang YK, Thiol Versus Hydroxamate as Zinc Binding Group in HDAC Inhibition: An Ab Initio QM/MM Molecular Dynamics Study, J Comput Chem, 2015; 36: 2228–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frisch M, Trucks G, Schlegel H, et al. , GAUSSIAN09. Revision E. 01. Gaussian Inc., Wallingford, CT, USA, 2009. [Google Scholar]
- 43.Cieplak P, Cornell WD, Bayly C, et al. , Application of the Multimolecule and Multiconformational Resp Methodology to Biopolymers - Charge Derivation for DNA, Rna, and Proteins, J Comput Chem, 1995; 16: 1357–1377. [Google Scholar]
- 44.Salomon-Ferrer R, Gotz AW, Poole D, et al. , Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J Chem Theory Comput, 2013; 9: 3878–3888. [DOI] [PubMed] [Google Scholar]
- 45.Gotz AW, Williamson MJ, Xu D, et al. , Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J Chem Theory Comput, 2012; 8: 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rooklin D, Modell AE, Li H, et al. , Targeting Unoccupied Surfaces on Protein-Protein Interfaces, J Am Chem Soc, 2017; 139: 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.