

The treatment of acute myelogenous leukemia (AML) relies heavily on cytarabine (Ara-C) containing chemotherapy. Even newly approved multi-kinase inhibitors improved survival only when combined with standard Ara-C containing chemotherapy1. When given at intermediate doses, Ara-C induces remission in 50–80% younger patients with AML, whereas patients older than 60 years fare better with low dose Ara-C. However, resistance eventually develops and both groups have a poor prognosis after relapse2,3. Sapacitabine (CNDAC) is a newer nucleoside analog that provides similar clinical benefit when compared with low dose Ara-C in elderly patients4. CNDAC and Ara-C are nucleoside analogs that become incorporated during DNA replication. Mechanistically, CNDAC induces single strand nicks that become converted to DNA double strand (ds) breaks, which are repaired by the homologous recombination repair (HR) pathway,5 and efficient DNA repair via HR represents a potential resistance mechanism that limits the damage caused by CNDAC5. In contrast, Ara-C stalls DNA replication, which is repaired by the nucleotide excision pathway6. Since nucleoside analogs remain the best therapeutic strategy against AML, we evaluated whether targeting DNA repair could synergistically improve their efficacy in AML.
HSP90 is a molecular chaperone that is critical for the folding and proper function of multiple proteins that regulate DNA repair pathways7. In this study, we focused on consequence of targeting the HSP90 client proteins, Chk1 and Rad51. Chk1 becomes recruited to DNA double strand breaks and in turn activates signaling cascades that result in cell cycle arrest and DNA repair8. Of these, γ-H2AX acts a sensor of DNA damage and repair by forming foci at the sites of damaged DNA that get resolved upon successful repair9. Similarly, Rad51 plays a key role in HR repair and the presence of Rad51 foci can be used as a marker of successful ongoing HR10. Inhibition of HSP90 causes the degradation of client proteins via the ubiquitin-proteasome pathway and can be used to target DNA repair7. When the AML cell lines OCI-AML3 and MV4–11 were exposed to the HSP90 inhibitor, onalespib, there was a time dependent decrease in the levels of the HSP90 clients, Chk1 and Rad51 (Fig 1A). Similarly, exposure of primary AML blasts to onalespib for either 24 or 36 hours resulted in depletion of Chk1 and Rad51 (Fig 1B).
Figure 1. HSP90 inhibition depletes levels of DNA repair proteins in AML and to compromise double strand break repair.
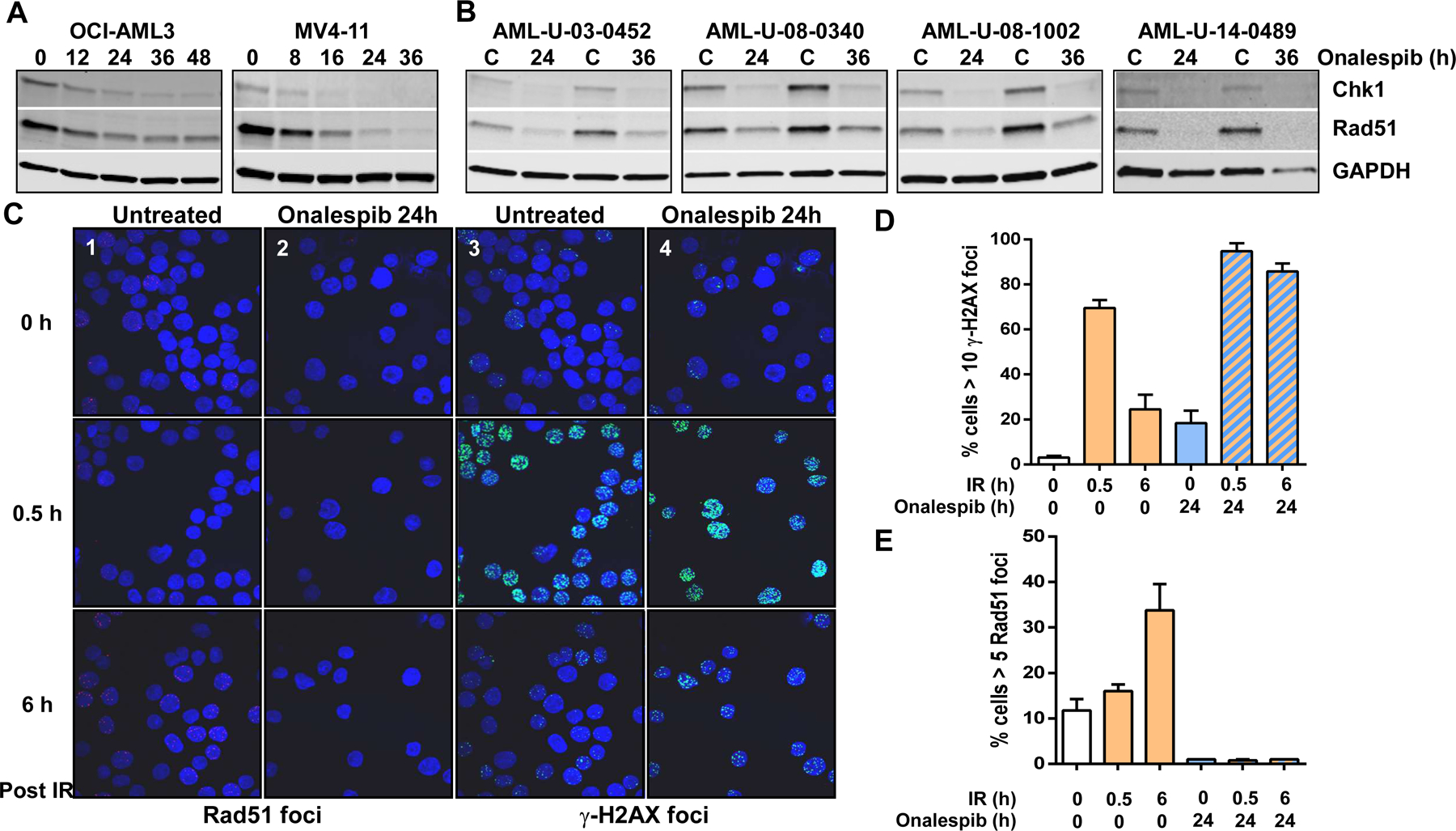
(A) Immunoblot analysis of Chk1, Rad51 and GAPDH (loading control) in OCI-AML3 and MV4-11 cells after exposure to a 0.25 µM onalespib for various times. (B) Immunoblot analysis of Chk1, Rad51 and GAPDH (loading control) in AML blasts cells after exposure to a 0.25 µM onalespib for 24 or 36 h. (C) OCI-AML3 cells were left untreated (control) or exposed to 0.25 µM onalespib for 24 h before undergoing DNA damge with 2 Gy IR. Cells were harvested 0.5 or 6 h post radiation and attached to microscope slides by centrifugation for 4 min at 600 rpm in a cytospin centrifuge. Slides were fixed in cold methanol, washed in PBS and incubated for 10 min with 5% bovine serum albumin (BSA), 0.1% Tween-20 and 0.1% Saponin in PBS. All subsequent steps were performed with 0.1% Saponin in the media. Cells were then incubated overnight at 4°C with primary antibodies, mouse anti-phospho-histone H2AX (γ-H2AX-S139; 1:1000; Millipore) and rabbit anti-Rad51 (1:1000; Cell signaling). The cells were then washed and incubated with secondary antibodies goat anti-mouse IgG Alexa Fluor® 488 (1:1000, Life technologies) and donkey anti-rabbit IgG IgG Alexa Fluor®555 (1:1000, Life technologies). DNA was stained with 100 nM 4,6 diamidino-2-phenylindole for 15 min. A total of 200 cells/slide were counted and cells with 10 foci of phospho-histone H2AX and 5 or more Rad51 foci were scored as positive. (D) Quantitation of cells postive for Rad51 or (E) γ-H2AX foci.
To elucidate the consequence of HSP90 inhibition on HR repair we used ionizing radiation (IR) as an established model for DNA double strand damage-induced signaling and repair8. OCI-AML3 exposed to IR, had a significant increase in the number of Rad51 foci at 6 h post IR (Fig 1C, panel-1 and Fig 1D) indicating active HR repair. Correspondingly, the large number of γ-H2AX foci found at 0.5 h after IR decreased by 6 h post-IR indicating efficient repair of DNA damage (Fig 1C, panel-3 and Fig 1E). In contrast, pre-exposure to onalespib resulted in a complete lack of formation of Rad51 foci at 6 h post IR (Fig 1C, panel-2 and Fig 1D) as well as a persistence of γ-H2AX in most cells at 6 h post IR suggesting unrepaired double strand DNA breaks (Fig 1C, panel-4 and Figure 1E).
We then tested whether HSP90 inhibition would sensitize AML cells to sapacitabine (CNDAC), a clinically active double strand break-inducing agent as measured by inhibition of growth in colony forming assays. Exposure to onalespib (0.1 µM) alone for 24 and 48 h caused a modest decrease in viability of OCI-AML3 cells (Fig 2A) and a more pronounced loss of viability of MV4-11 cells (Fig 2B) as measured by colony forming assays; likely because MV4-11 cells express FLT3-ITD, a HSP90 client protein and key survival factor in AML. Exposure to CNDAC (0.1 µM) alone for 24 h also resulted in a modest loss of viability in OCI-AML3 cells and in MV4-11 cells (Fig 2A and 2B). However, pretreatment with onalespib for 24 h significantly sensitized both OCI-AML3 and MV4-11 cells to CNDAC (Fig 2A and 2B). Although colony formation assays remain the gold standard in measuring loss of replicative potential, they do not distinguish whether the decreases in colony number arise due to prolonged cell cycle arrest or due to apoptotic cell death. To determine this, OCI-AML3 cells were exposed to onalespib and CNDAC at the same concentrations and time conditions as used in the colony forming assay before being harvested and assayed for cell cycle arrest using propidium iodide staining and induction of apoptotic cell death via Annexin V assays. We found that when cells were exposed to low concentrations of CNDAC (0.1 µM) for 24h, they did not affect cell cycle transit. However, exposure to onalespib for 24 or 48 h modestly increased the fraction of cells arrested in S (14%) and G2 (20%) phase which were unchanged in cells exposed to onalespib prior to exposure to CNDAC (Fig 1C). Parallel measurements of annexin V positivity indicated that OCI-AML3 cells did not significantly increase the percentage of apoptotic cells upon exposure to either CNDAC or onalespib alone for 24 h. The percentage of apoptotic cells increased by 10% in response to onalespib for 48h which did not change when cells were exposed to onalespib prior to CNDAC (Supp. Fig 1A). These results indicate that the onalespib-mediated loss of Chk1 likely induces cell cycle arrest, which does not increase after the addition of CNDAC. However, cells pre-exposed to onalespib show a depletion in Rad51 and inhibition in HR repair, which in turn impairs the ability to repair CNDAC-induced lesions leading to a synergistic decrease in viability.
Figure 2. Effect of the HSP90 inhibitor onalespib alone or incombination with cytarabine and sapacitabine on AML cell lines, and effects of onalespib on AML blasts and a MOLM-13 derived AML mouse model.
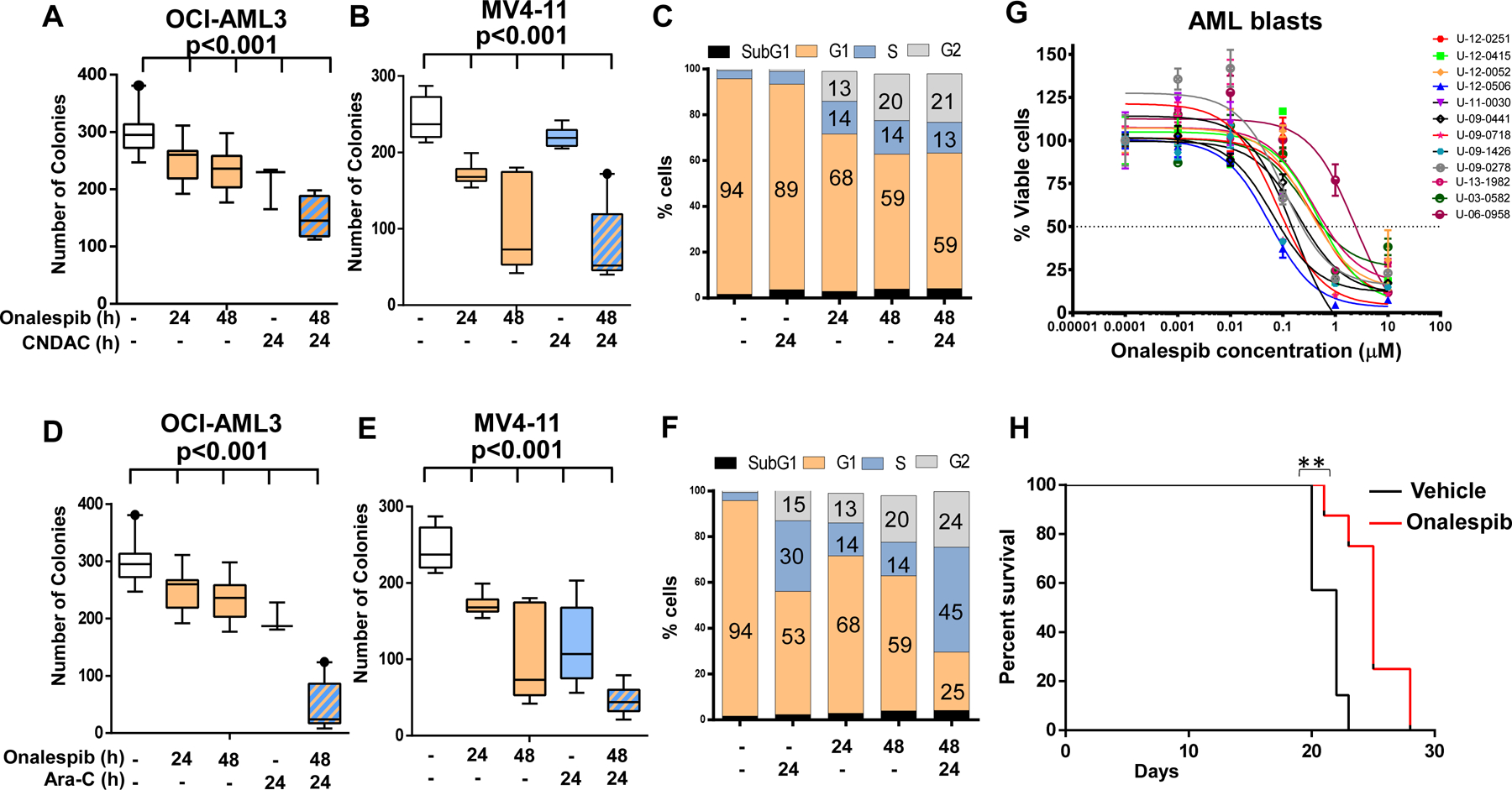
(A and B) Onalespib exposure sensitizes OCI-AML3 and MV4-11 cells to sapacitabine (CNDAC). Cells were treated with onalespib for 24 and 48h (0.1 µM for OCI-AML3 and 0.05 µM for MV4-11), CNDAC, 0.1 µM for 24 h or onalespib for 24 h before being exposed to CNDAC for an additional 24 h before being washed into fresh media and plated in MethoCult™ for 7–14 days and assayed for colony formation using a GelCount scanner (Oxford Optronix). (C) OCI-AML3 cells were treated with onalespib, 0.1 µM for 24 and 48h, CNDAC, 0.1 µM for 24 h or onalespib for 24 h before being exposed to CNDAC for an additional 24 h, fixed in 70% ethanol, stained with propidium iodide (PI) and analyzed by flow cytometery for cell cycle fractions. (D and E) Onalespib expsure sensitizes OCI-AML3 and MV4-11 cells to cytarabine (Ara-C). Cells were treated with onalespib for 24and 48h (0.1 µM for OCI-AML3 and 0.05 µM for MV4-11), Ara-C 2.5 µM for 24 h or onalespib for 24 h before being exposed to Ara-C for an additional 24 h before being washed into fresh media and plated in MethoCult™ for 7–14 days and assayed for colony formation using a GelCount scanner (Oxford Optronix). Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Values are the mean ± SEM from at least three independent experiments. ***p<0.001, unpaired t-test, two-tailed. (F) OCI-AML3 cells were treated with onalespib, 0.1 µM for 24 and 48h, Ara-C, 2.5 µM for 24 h or onalespib for 24 h before being exposed to Ara-C for an additional 24 h, fixed in 70% ethanol, stained with propidium iodide (PI) and analyzed by flow cytometery for cell cycle fractions. (G) Effect of onalespib on AML blasts. AML blasts were plated on HS5 Stromal cells to maintain survival in culture. Cells were exposed to varying doses of onalspib for 96 h before evaluating viability with the MTT assay. (H). Twenty female NOD SCID gamma (NSG) mice (6-wk old, Jackson Laboratory, Bar Harbor, MA) were intravenously injected with luciferase-labeled MOLM-13 cells (0.7 x 106 cells/100 μL) and randomly divided into two groups. One week post injection, the mice were treated with vehicle (15% Captosil) or onalespib (45 mg/kg body weight) twice weekly by intrperitoneal injection and followed for survival.** p=0.0019, Mantel-Cox test.
In addition to its role in HR, Chk1 is critical in mediating cell recovery following the replicative stress caused by nucleoside analogues such as cytarabine (Ara-C); high levels of Chk1 are linked to a poor outcome whereas pharmacological inhibition of Chk1 sensitizes cells to Ara-C11. Since HSP90 inhibition depletes Chk1 levels, we determined whether it would sensitize AML cells to Ara-C. Low doses of Ara-C alone modestly decreased colony growth in both OCI-AML3 and MV4-11 cells (Fig 2C and 2D) whereas pre-treatment with onalespib for 24 h sensitized AML cells to Ara-C (Fig 2C and 2D). When we determined whether these treatments caused cell cycle arrest or apoptosis, we found that cells exposed to Ara-C (2.5 µM) for 24 h became arrested in S phase (30%). In parallel cells exposed to onalespib for 24 to 48 h modestly increased S (14%) and G2 fractions (20%). However, cells pretreated with onalespib before exposure to Ara-C showed a further increase in the fraction of cells arrested in S-Phase (45%). Parallel evaluation of apoptotic cell death showed that there was no significant increase in the percentage of apoptotic cells over control in any condition except that of Ara-C alone at 24 h (30%). These results show that Ara-C stalls replication forks to induce S-phase arrest. In addition, exposure to onalespib results in the loss of Chk1, inducing modest cell cycle arrest. However, cells exposed to the combination of onalespib and Ara-C strongly induce S-phase arrest which may explain the mechanism by which onalespib sensitizes OCI-AML3 cells to Ara-C.
We next evaluated the action of onalespib on AML blasts in vitro and in an MOLM13 driven mouse model of AML in vivo. AML blasts from twelve patients were plated on HS5-stromal cells to keep them viable in vitro and were exposed to varying doses of onalespib. Our results showed a dose-dependent decrease in cell viability (Fig 2E). Additionally, in an in vivo model of AML generated by luciferase expressing MOLM13 cells in NSG mice, treatment with 45 mg/kg of onalespib resulted in statistically significant (p=0.0019, Log-rank Mantel-Cox test) increase in survival compared to treatment with placebo (Fig 2E).
As single agents, HSP90 inhibitors have shown efficacy in a small subset of patients whose tumors are driven by oncogenes that are sensitive HSP90 client proteins. Although HSP90 inhibitors effectively inhibit DNA repair processes in AML blasts and represent an attractive strategy to broadly target these cells by increasing their sensitivity to nucleoside analogs such as cytarabine and sapacitabine their clinical utility has been hampered by toxicities and insufficient length of target inhibition12. Therefore, it may be practical to identify druggable targets within DNA repair pathways using HSP90 inhibitors but to utilize targeted inhibitors of DNA repair proteins such as ATM or Chk1 that were identified as client proteins of HSP90 for use in clinical trials with the aim of sensitizing AML cells to nucleoside analogs while minimizing toxicity.
Supplementary Material
Supplementary Fig 1 OCI-AML3 cells were treated with onalespib, 0.1 µM for 24 and 48h, CNDAC, 0.1 µM for 24 h or onalespib for 24 h before being exposed to CNDAC for an additional 24 h. Cells were harvested and incubated with annexin V–fluorescein isothiocyanate (BD Biosciences) and propidium iodide (PI) for 15 minutes. Fluorescence of at least 10000 cells was determined on a BD Biosciences FACSCalibur flow cytometer to determine the percentage of apoptotic cells within the population.
Acknowledgements
This work was supported in part by grants from the National Cancer Institute R01CA223165 and P30CA016058. We thank the Leukemia Tissue Bank at The Ohio State University Comprehensive Cancer Center, Columbus, OH, for sample processing and storage services.
Footnotes
The authors have no conflict of interest.
References
- 1.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377(5):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowenberg B, Pabst T, Vellenga E, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med 2011;364(11):1027–1036. [DOI] [PubMed] [Google Scholar]
- 3.Wu D, Duan C, Chen L, Chen S. Efficacy and safety of different doses of cytarabine in consolidation therapy for adult acute myeloid leukemia patients: a network meta-analysis. Scientific Reports. 2017;7(1):9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burnett AK, Russell N, Hills RK, et al. A randomised comparison of the novel nucleoside analogue sapacitabine with low-dose cytarabine in older patients with acute myeloid leukaemia. Leukemia. 2015;29(6):1312–1319. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Wang Y, Benaissa S, et al. Homologous recombination as a resistance mechanism to replication-induced double-strand breaks caused by the antileukemia agent CNDAC. Blood. 2010;116(10):1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strom SS, Estey E, Outschoorn UM, Garcia-Manero G. Acute myeloid leukemia outcome: role of nucleotide excision repair polymorphisms in intermediate risk patients. Leuk Lymphoma. 2010;51(4):598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of Hsp90 Compromises the DNA Damage Response to Radiation. Cancer Research. 2006;66(18):9211–9220. [DOI] [PubMed] [Google Scholar]
- 8.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends in Cell Biology. 2016;26(1):52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012;920:613–626. [DOI] [PubMed] [Google Scholar]
- 10.Gachechiladze M, Skarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. 2017;141(7):1286–1294. [DOI] [PubMed] [Google Scholar]
- 11.David L, Fernandez-Vidal A, Bertoli S, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal 2016;9(445):ra90. [DOI] [PubMed] [Google Scholar]
- 12.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 2012;18(1):64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1 OCI-AML3 cells were treated with onalespib, 0.1 µM for 24 and 48h, CNDAC, 0.1 µM for 24 h or onalespib for 24 h before being exposed to CNDAC for an additional 24 h. Cells were harvested and incubated with annexin V–fluorescein isothiocyanate (BD Biosciences) and propidium iodide (PI) for 15 minutes. Fluorescence of at least 10000 cells was determined on a BD Biosciences FACSCalibur flow cytometer to determine the percentage of apoptotic cells within the population.