

Abstract
Objective:
We examined the effects of treatment with soluble epoxide hydrolase inhibitor (sEHi) and epoxyeicosatrienoic acids (EETs) analogue (EET-A), given alone or combined, on blood pressure (BP) and ischemia/reperfusion myocardial injury in rats with angiotensin II (ANG II)-dependent hypertension.
Methods:
Ren-2 transgenic rats (TGR) were used as a model of ANG II-dependent hypertension and Hannover Sprague–Dawley rats served as controls. Rats were treated for 14 days with sEHi or EET-A and BP was measured by radiotelemetry. Albuminuria, cardiac hypertrophy and concentrations of ANG II and EETs were determined. Separate groups were subjected to acute myocardial ischemia/reperfusion injury and the infarct size and ventricular arrhythmias were determined.
Results:
Treatment of TGR with sEHi and EET-A, given alone or combined, decreased BP to a similar degree, reduced albuminuria and cardiac hypertrophy to similar extent; only treatment regimens including sEHi increased myocardial and renal tissue concentrations of EETs. sEHi and EET-A, given alone or combined, suppressed kidney ANG II levels in TGR. Remarkably, infarct size did not significantly differ between TGR and Hannover Sprague–Dawley rats, but the incidence of ischemia-induced ventricular fibrillations was higher in TGR. Application of sEHi and EET-A given alone and combined sEHi and EET-A treatment were all equally effective in reducing life-threatening ventricular fibrillation in TGR.
Conclusion:
The findings indicate that chronic treatment with either sEHi or EET-A exerts distinct antihypertensive and antiarrhythmic actions in our ANG II-dependent model of hypertension whereas combined administration of sEHi and EET-A does not provide additive antihypertensive or cardioprotective effects.
Keywords: epoxyeicosatrienoic acid analogue, epoxyeicosatrienoic acid, hypertension, infarct size, ischemic arrhythmia, kidney, renin–angiotensin system, soluble epoxide hydrolase inhibitor
INTRODUCTION
Acute myocardial infarction affects millions of patients worldwide and remains a serious medical problem, even though early coronary reperfusion by primary percutaneous intervention effectively limits the infarct size. It is now common knowledge that while myocardial reperfusion helps rescue the myocardium, it also triggers a second wave phenomena described as myocardial reperfusion injury [1,2]. It is estimated that up to 50% of the final infarct size is caused by reperfusion injury; unfortunately no clinically relevant protective therapies against this injury are available [3–6]. Although the mortality of patients following acute myocardial infarction has decreased, the number of patients who survive and develop heart failure within 1 year is progressively increasing [2,3,7]. As the infarct size is critically important in the process of injurious left ventricle (LV) remodeling and development and progression of heart failure, limiting the myocardial infarct size is imperative: it is estimated that reduction in the myocardial infarct size by barely 5% substantially improves the clinical outcome [3,4,8].
Therefore, new treatment strategies for the protection of the myocardium against ischemia/reperfusion have been explored; however, even though a number of preclinical studies proposed promising cardioprotective strategies, attempts at the therapeutic application proved largely unsatisfactory [2–6]. This translation failure can be ascribed to some limitations of the experimental research process. Indeed, while preclinical studies enriched our knowledge of the pathophysiology of ischemia/reperfusion injury, the major problem was that they were mostly performed in relatively young and healthy animals. Therefore, the results can hardly apply to patients in whom diverse comorbidities substantially influence the severity of myocardial ischemia/reperfusion injury and limit the effects of the potentially cardioprotective therapies [2–6,9]. Hence, a critical need for preclinical studies that would evaluate cardioprotective treatment strategies in animal models that exhibit at least some of the major risk factors for ischemic heart disease [6,9,10]. As a prominent risk factor for cardiovascular disease is hypertension, especially when associated with established LV hypertrophy (LVH) and increased activity of the renin–angiotensin system (RAS) [3,11–14], we aimed to evaluate the myocardial susceptibility to ischemia/reperfusion injury in Ren-2 transgenic hypertensive rats (TGR). Remarkably, the relevant studies in hypertensive rats with established LVH have brought conflicting results [15–27]. TGR strain used here [strain name: TGR(mRen2)27] presents a unique angiotensin II (ANG II)-dependent model in which the development of hypertension is attributed to a single gene alteration and to activation of the RAS. Importantly, TGR display marked LVH [25,28,29] which, together with hypertension and RAS hyperactivity, completes the triad of features which are thought to influence the severity of ischemia/reperfusion injury in patients.
It is now widely recognized that the opening of the mitochondrial permeability transition pore (MPTP) in the first minutes of reperfusion is the critical mediator of reperfusion-induced cardiomyocyte death, and preventing its opening either by pharmacological or genetic means led to substantial reduction in myocardial infarct size [30–32]. Therefore, the optimal cardioprotective therapy should inhibit the opening of MPTP during the reperfusion phase and possibly offset some major risk factors for ischemic heart disease. Considering this knowledge, we focused on the potential relevant role of epoxyeicosatrienoic acids (EETs), cytochrome P-450 (CYP)-derived metabolites of arachidonic acid: growing evidence indicates that increased EETs bioavailability exhibits cardioprotective actions by inhibiting the opening of MPTP [33–35] and lowers blood pressure (BP) [36–39]. Therefore, elevation of EETs levels is considered a promising therapeutic approach with antihypertensive and cardioprotective effects in patients with cardiovascular diseases [31–41]). However, the studies involving increasing EETs levels showed some limitations that have not yet been addressed. First, in the majority of studies, only acute or short-term (a few days’) effects of increased EETs levels/actions were examined. Second, antihypertensive effects of EETs were mostly reported from experiments in which EETs levels were raised in the initial phase of hypertension; when the treatment was applied in established hypertension, the results were divergent [25,26,36–49]. Third, it has to be considered that EETs are biologically unstable and are fast degraded to relatively inactive dihydroxyeicosatrienoic acid (DHETE) by the soluble epoxide hydrolase (sEH). In most studies tissue EETs bioavailability was increased by blocking sEH, however, this approach might prove less successful whenever baseline endogenous EETs formation is compromised (e.g. as a consequence of some inflammatory disorders, treatment with drugs that inhibit CYP activity, etc.) [50–52]. Therefore, an alternative approach to target EETs in the treatment of cardiovascular diseases consists of application of EETs-agonistic analogues that are designed to resist degradation and show good stability. However, the experience with this new approach is limited and the results are inconsistent [26,53–57].
Based on the above knowledge and considerations, we decided, first, to examine if chronic treatment with either an orally active sEH inhibitor or orally active EETs agonistic analogue will exhibit long-term antihypertensive, renoprotective and cardioprotective actions in TGR in the sustained phase of ANG II-dependent hypertension with developed LVH. Second, considering the known and possibly unknown limitations of either pharmacological approach targeting EETs and intended to treat cardiovascular disease, we decided to make the first attempt to combine the two therapeutic variants: sEH blockade and administration of EETs analogue, in hope to achieve additive antihypertensive, renoprotective and cardioprotective effects.
METHODS
Ethical approval, animals and chemicals
The studies were performed in accordance with the guidelines and practices established by the Animal Care and Use Committee of the Institute for Clinical and Experimental Medicine, Prague, which accord with the national law and the European Union policy (EEC Council Directive 86/609, OJL 358–1, December 1987). All animals used in the present study were bred at the Center of Experimental Medicine of this Institute, from stock animals supplied by the Max Delbrück Center for Molecular Medicine, Berlin, Germany, which is accredited by the Czech Association for Accreditation of Laboratory Animal Care. Heterozygous TGR were generated by breeding male homozygous TGR with female homozygous Hannover–Sprague Dawley (HanSD) rats as described in the original study [58]; age-matched HanSD rats served as transgene-negative normotensive controls. The reason for using heterozygous TGR is that they develop hypertension with BP levels comparable with those in hypertensive patients (SBP about 180 mmHg] and therefore they seem to be a suitable model for studying the role of hypertension and endogenous RAS in modifying the severity of myocardial ischemia/reperfusion injury [25,27–29,58]. Dissimilarly, homozygous TGR develop malignant hypertension: already at the age of 5 weeks they display SBP over 220 mmHg, with marked signs of early end-organ damage; without treatment these animals begin to die at the age 8 weeks [59,60]. Therefore, homozygous TGR serve as a model of malignant hypertension and are not appropriate for our present studies. The animals were kept on a 12-h/12-h light/dark cycle. Throughout the experiments they were fed a normal-salt, normal-protein diet (0.45% NaCl, 19–21% protein) and had free access to tap water. Male TGR and HanSD rats at the initial age of 10 weeks were used for experiments: at this age TGR are already in the sustained phase of hypertension with established marked LVH and substantial activation of endogenous RAS, as demonstrated in previous studies, including ours [28,29,49]. cis-4-[4-(3-Adamantan-1-yl-ureido) cyclohexyloxy] benzoic acid was used as the soluble epoxide hydrolase inhibitor (sEHi), and given in drinking water at 3 mg/l. The dose of cis-4-[4-(3-adamantan-1-yl-ureido) cyclohexyloxy] benzoic acid was selected based on our recent studies in which it elicited substantial increases in tissue concentration of EETs without altering plasma and tissue ANG II levels [61]; the aim was to assess antihypertensive and cardioprotective effects of EETs elevation alone. The 14,15-EET analogue [disodium (S)-2-(13-(3-pentyl)ureido)-tridec-8(Z)-enamido)succinate, EET-A] was given in drinking water, and its concentration was adjusted in a way that the daily dose was 10 mg/kg of body weight. Recent studies, including our own, have shown that with this dosage plasma EET-A concentrations were above the range of half maximal inhibitory concentration (IC50) [55–57].
Experimental design
Series 1: effects of soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue given alone and of combined soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue treatment on blood ppressure (radiotelemetry), urinary sodium excretion, albuminuria and cardiac hypertrophy
In accordance with the current recommendations for BP measurement in experimental animals, we employed a radiotelemetry system for direct BP measurements [62]. Rats were anesthetized with a combination of tiletamine, zolazepam (Zoletil; Virbac SA, Carros Cedex, France; 8 mg/kg), and xylazine (Rometar, Spofa, Czech Republic; 4 mg/kg), given intramuscularly, and TA11PA-C40 radiotelemetric probes (Data Sciences International, St. Paul, Minnesota, USA) were implanted for direct BP measurements as described previously [57,61]. The rats were allowed 7 days to recover before basal BP was recorded and only animals with stable BP records at the end of this recovery period were used for experiments. Basal BP was determined for 3 days and then the treatment was initiated and continued for 14 days. In the animals implanted with radiotransmitters, 24-h urine samples were collected before the start of pharmacological treatment (day −1), and then on days 1, 6 and 13 of treatment, to assess daily sodium excretion and albuminuria. At the end of experiments, all animals were killed by an overdose of thiopental sodium (Sandoz, Basel, Switzerland) and the ratio of LV weight (LVW) to tibia length was used to evaluate the degree of LVH [48,49,57,61]. The following groups were examined:
HanSD rats/untreated (n = 6)
HanSD rats/sEHi (n = 6)
HanSD rats/EET-A (n = 6)
HanSD rats/sEHi + EET-A (n = 6)
TGR/untreated (n = 9)
TGR/sEHi (n = 9)
TGR/EET-A (n = 8)
TGR/sEHi + EET-A (n = 9)
Series 2: effects of soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue given alone for 14 days and of combined soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue treatment on kidney tissue epoxyeicosatrienoic acids, dihydroxyeicosatrienoic acid, hydroxyeicosatrienoic acids, plasma renin activity, renal renin activity, plasma and kidney angiotensin II levels and myocardial CYP2C23, CYP4A1 and soluble epoxide hydrolase inhibitor protein expression
The aim was to gain a detailed insight into the role of interaction of EET-based therapies and the RAS in mediating possible antihypertensive, renoprotective and cardioprotective actions of chronic treatment with sEHi and EET-A. Because it is known that plasma and tissue ANG II concentrations in anesthetized animals are higher than those measured in rats decapitated in conscious state, and that normotensive animals exhibit greater increases in renin secretion in response to anesthesia and surgery than ANG II-dependent hypertensive animals, at the end of studies the rats were killed by decapitation and plasma renin activity (PRA), renal renin activity (RRA) and ANG II levels were measured by radioimmunoassay as described previously [28,29]. The concentrations of arachidonic acid metabolites: 5,6-EET, 8,9-EET, 11,12-EET and 14–15-EET and DHETEs were separately measured in the kidney cortex and LV myocardium and then pooled for presentation. The samples were extracted, separated by reverse-phase HPLC and analyzed by negative-mode electrospray ionization and tandem MS as described previously [47–49]. The results are shown as total concentrations of EETs and DHETEs: it is recognized that the former are biologically most active products of the CYP epoxygenase pathway [36–39]. Similarly, 5-HETE (hydroxyeicosatrienoic acid), 8-HETE, 9-HETE, 11-HETE, 12-HETE, 15-HETE, 19-HETE and 20-HETE were separately determined and pooled for presentation: these metabolites are biologically the most active products of the CYP hydroxylase pathway [37,39]. All these parameters were assessed in the same experimental groups as in series 1 (n = 7 in each group). Western blot analysis of protein expression of CYP2C23, the enzyme that is predominantly responsible for the formation of EETs, and of sEH, the enzyme responsible for the conversion of EETs to DHETEs, was performed as described previously [49], with the levels normalized against β-actin. In addition, the protein expression for CYP4A, the enzyme responsible for the formation of HETEs, was analysed in the renal and LV tissue as described previously [63].
Series 3: effects of soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue given alone and of combined soluble epoxide hydrolase inhibitor and epoxyeicosatrienoic acid analogue treatment on myocardial infarct size and the incidence and severity of ventricular arrhythmias induced by ischemia/reperfusion procedure
Separate experimental groups, as in series 1, were employed (n = 11 in each group). Rats were subjected to regional myocardial ischemia/reperfusion as described previously, using an open-chest model [25,26,64,65]. Animals were anesthetized with pentobarbital sodium (60 mg/kg body weighti.p.). A heparinized cannula was placed in the left carotid artery for BP monitoring with a pressure transducer (Gould P23Gb; Gould Instruments Systems, Valley View, Ohio, USA) and the readings were analyzed by our custom-designed software. Rats were ventilated with room air at 68 strokes/min (tidal volume, 1.2 ml/100 g body weight) using a rodent ventilator (Ugo Basile, Italy). Heart rate was derived from the BP curve. The rectal temperature was maintained between 36.5 and 37.5 °C by a servo-controlled heated table throughout the experiment. Left thoracotomy was performed, and a silk suture 5–0 (Chirmax, Czech Republic) was placed loosely around the left anterior descending coronary artery 1–2 mm distal to its origin. After 10-min stabilization, regional ischemia was induced by tightening of the suture threaded through a polyethylene tube. After a 20-min occlusion period, the ligature was released and reperfusion of previously ischemic tissue continued for 3 h.
Determination of infarct size
The infarct size and the area at risk were determined as described previously [25,26,64,65], by staining with potassium permanganate and 2,3,5-triphenyltetrazolium chloride (Sigma Aldrich, Prague, Czech Republic), respectively. Briefly, hearts were excised and washed with saline via aorta. The area at risk was delineated by perfusion of 5% potassium permanganate after coronary artery re-occlusion. Frozen hearts were cut into slices 1 mm thick, stained with 1% 2,3,5-triphenyltetrazolium chloride (pH 7.4, 37 °C) for 30 min, and fixed in formaldehyde solution. The infarct size and area at risk were determined by computerized planimetry and the infarct size was normalized to the area at risk (infarct size/area at risk). The weight of the LV was determined and the area at risk was normalized to the LV (area at risk/LV). This approach has been validated in many experimental studies and is currently accepted as the ‘gold standard’ for measurement of myocardial infarct size in small animals [10].
Analysis of arrhythmias
The incidence and severity of ventricular arrhythmias during the 20-min ischemic insult and during the first 3-min of reperfusion were assessed according to the recommendation of the Lambeth Conventions [66], which was established 25 years ago and is currently accepted as the ‘gold standard’ for the analysis of incidence and severity of arrhythmias. Briefly, premature ventricular complexes (PVCs) occurring as singles, salvos (2 or 3 PVCs) or tachycardia (a run of 4 or more consecutive PVCs) were counted separately. The incidence of ventricular tachycardia and ventricular fibrillation were also evaluated. Ventricular fibrillation lasting more than 2 min was considered as sustained and animals were not treated to reverse theses abnormalities. The hearts from animals exhibiting sustained ventricular fibrillations were, in accordance with generally accepted approach [10,27], excluded from further evaluation for determination of infarct size. Such hearts are exposed to a tremendous ischemic insult that further alters the infarct size. However, the data were used for analysis of incidence and severity of arrhythmias. The severity of arrhythmias in each group was evaluated by an arrhythmia score according to the incidence of the most severe form of arrhythmia that occurred in each individual heart (heart with single PVCs were given a score of 1, salvos – 2, ventricular tachycardia – 3, reversible ventricular fibrillation – 4 and sustained ventricular fibrillation – 5) as described previously in our studies [25,26,64,65]. This standardized approach for determination of infarct size and arrhythmias enables a comparison of effects of various cardioprotective strategies inside one research group throughout the time, but also with other research groups worldwide [9,20–27,33,34,64,65].
Statistical analysis
All values are expressed as mean±SEM. Graph-Pad Prism software (Graph Pad Software, San Diego, California, USA) was used and statistical analysis was performed using Student’s t test, Wilcoxon’s signed-rank test for unpaired data or one-way analysis of variance when appropriate. One-way analysis of variance for repeated measurements, followed by Student–Newman–Keuls test was performed for the analysis within groups (e.g. before and after treatment with either sEHi or EET-A). Differences in the number of PVCs between the groups were compared by the Kruskal–Wallis nonparametric test. The incidence of tachycardia and fibrillation was examined by Fischer’s exact test. Values exceeding the 95% probability limits (P<0.05) were considered statistically significant. The data that were not normally distributed (arrhythmias) were expressed as median±interquartile range.
RESULTS
Effects of treatments on blood pressure, urinary sodium excretion, albuminuria and indices of cardiac hypertrophy
As shown in Fig. 1a, mean SBP in HanSD rats remained within the normotensive range (123–128 mmHg) throughout the study and was not altered by sEHi or EET-A given alone or combined sEHi and EET-A treatment. TGR were markedly hypertensive before the treatments (183–188 mmHg) and sEHi or EET-A given alone or combined elicited significant and similar decreases in SBP, by about 35–40 mmHg.
FIGURE 1.
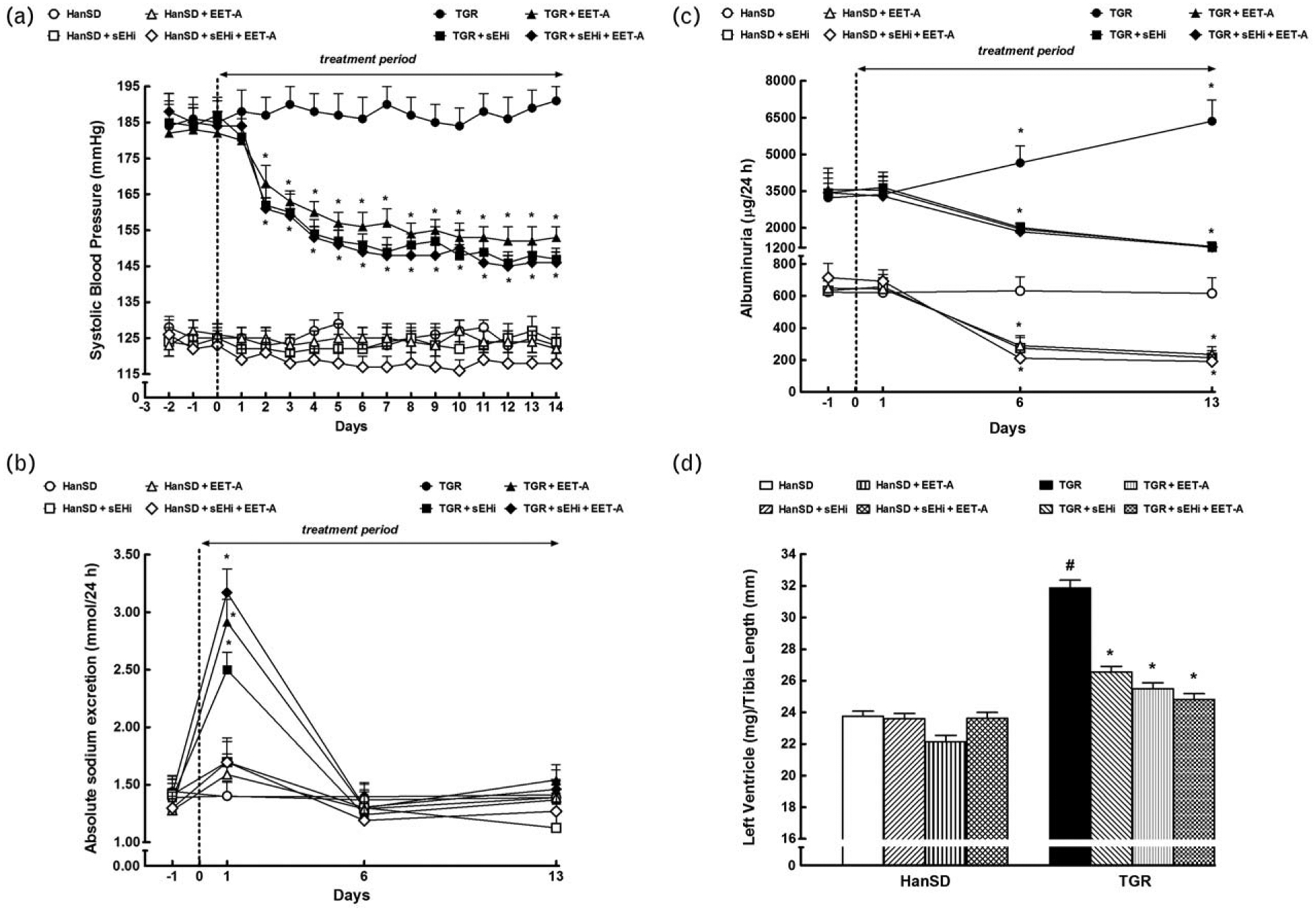
Time course of SBP (a), daily sodium excretion (b) and albuminuria (c), and the data on left ventricle weights normalized by tibia length (d), in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and effects of treatment with soluble epoxide hydrolase inhibitor (sEHi) or epoxyeicosatrienoic acid analogue (EET-A) given alone or combined. Values are shown as means±SEM. *P<0.05 versus basal values (or versus corresponding HanSD rats in the case of left ventricle weights). #P<0.05 versus corresponding treated TGR.
Figure 1b shows that in HanSD rats daily sodium excretion (UNaV) remained unchanged throughout the experiment and was not altered by any of the treatment regimes. In TGR, each of the three treatment variants elicited initial substantial increases in UNaV (day 1), which then returned to the values similar as observed in untreated TGR and HanSD rats (days 6 and 13).
Untreated TGR showed about five-fold higher albuminuria compared to HanSD rats (Fig. 1c). sEHi or EET-A given alone or combined decreased albuminuria in TGR and in HanSD rats to a similar degree, but on days 6 and 13 in treated TGR it was still significantly higher than in untreated HanSD rats.
As shown in Fig. 1d, untreated TGR exhibited severe LVH (evaluated as LVW/tibia length index) as compared with untreated HanSD rats (31.9±0.5 versus 23.4±0.3, P<0.05). sEHi or EET-A given alone or combined did not alter this index in HanSD rats, but elicited significant decreases in LVH in TGR; however, the posttreatment values were still significantly higher than those in untreated HanSD rats.
Effects of treatments on kidney tissue concentrations of cytochrome P-450-derived metabolites of arachidonic acid: epoxyeicosatrienoic acids, dihydroxyeicosatrienoic acid and hydroxyeicosatrienoic acid
Figure 2 summarizes the renal and myocardial availability of biologically active epoxygenase metabolites of arachidonic acid (expressed as the ratio of EETs to DHETEs) and of ω-hydroxylase metabolites of arachidonic acid (HETEs). The basal renal and myocardial ratios of the epoxygenase products (i.e. the ratios in untreated animals) were lower in TGR than in HanSD rats (0.69±0.09 versus 1.02±0.07 and 2.32±0.33 versus 3.79±0.41, respectively; P<0.05 in both cases) (Fig. 2a and d). sEHi given alone and combined increased these ratios in TGR as well as in HanSD rats (P<0.05 for both strains); however, as shown more clearly in Fig. 2b and e, the increases in renal and myocardial EETs/DHETEs ratios were markedly greater in TGR than in HanSD rats. In contrast, EET-A alone did not alter renal and myocardial EETs/DHETEs ratios, similarly in TGR or HanSD rats (Fig. 2a and d). There were no significant differences in the renal and LV myocardial tissue concentration of biologically active ω-hydroxylase metabolites of arachidonic acid (HETEs) between TGR and HanSD rats and the values were not altered by any of the treatment regimes (Fig. 2c and f).
FIGURE 2.
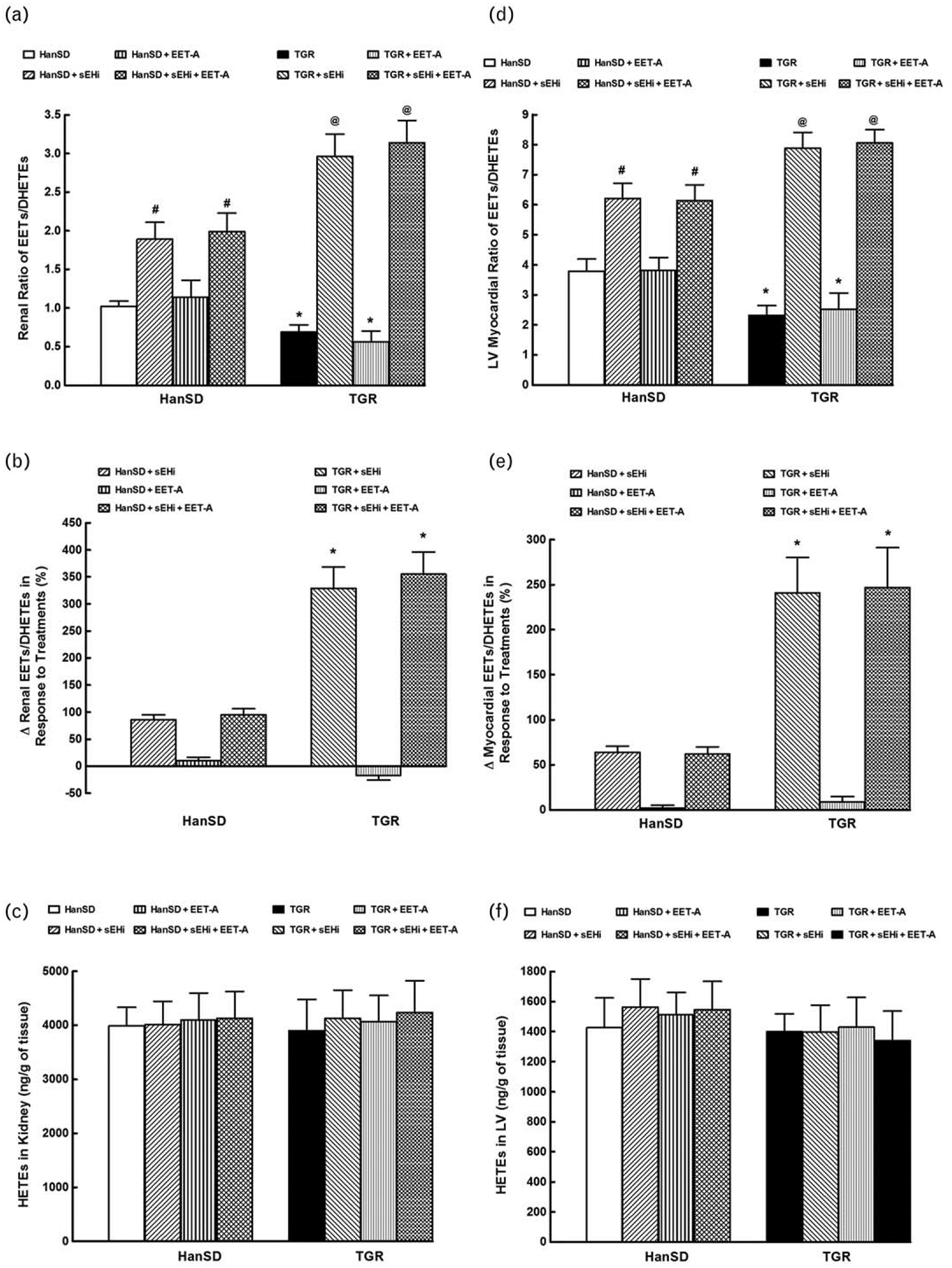
Kidney (a) and left ventricle (LV) myocardial (d) epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETEs) ratios, changes in renal (b) and myocardial EETs/DHETEs (e), kidney (c) and LV (f) hydroxyeicosatrienoic acids (HETEs) concentrations in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats, (TGR) and effects of soluble epoxide hydrolase inhibitor (sEHi) and epoxyeicosatrienoic acid analogue (EET-A) given alone or combined.*P<0.05 versus unmarked values. #P<0.05 versus untreated HanSD rats. @P<0.05 versus all the other groups.
Effects of treatments on kidney and myocardial protein expression of CYP2C23, CYP4A1 and soluble epoxide hydrolase inhibitor
Densitometric analysis showed that, when normalized for β-actin, there were no significant differences in CYP2C23, sEH and CYP4A protein expression in the renal cortex between TGR and HanSD rats, and the values were not significantly altered by any treatment regime (Fig. 3a–c). Likewise, there were no significant differences in CYP2C23 and sEH protein expression in the LV between TGR and HanSD rats (Fig. 3d and e). On the other hand, CYP4A protein expression in the LV was significantly higher in TGR than in HanSD; neither value was significantly altered by any of the treatment regimes (Fig. 3f).
FIGURE 3.

Kidney (a) and left ventricle (LV) myocardial (d) CYP2C23, kidney soluble epoxide hydrolase (sEH) and LV myocardial (b and e) and kidney and LV myocardial (c and f) CYP4A protein expression in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and effects of soluble epoxide hydrolase inhibitor (sEHi) or epoxyeicosatrienoic acid analogue (EET-A) given alone or combined sEHi and EET-A treatment.*P<0.05 versus unmarked values.
Effects of treatments on plasma and kidney tissue components of the renin–angiotensin system
As shown in Fig. 4a, plasma ANG II levels were significantly higher in untreated TGR than in HanSD rats (21±2 versus 10±2 fmol/ml, P<0.05). sEHi or EET-A given alone or combined significantly increased plasma ANG II levels in HanSD rats but did not alter them in TGR.
FIGURE 4.
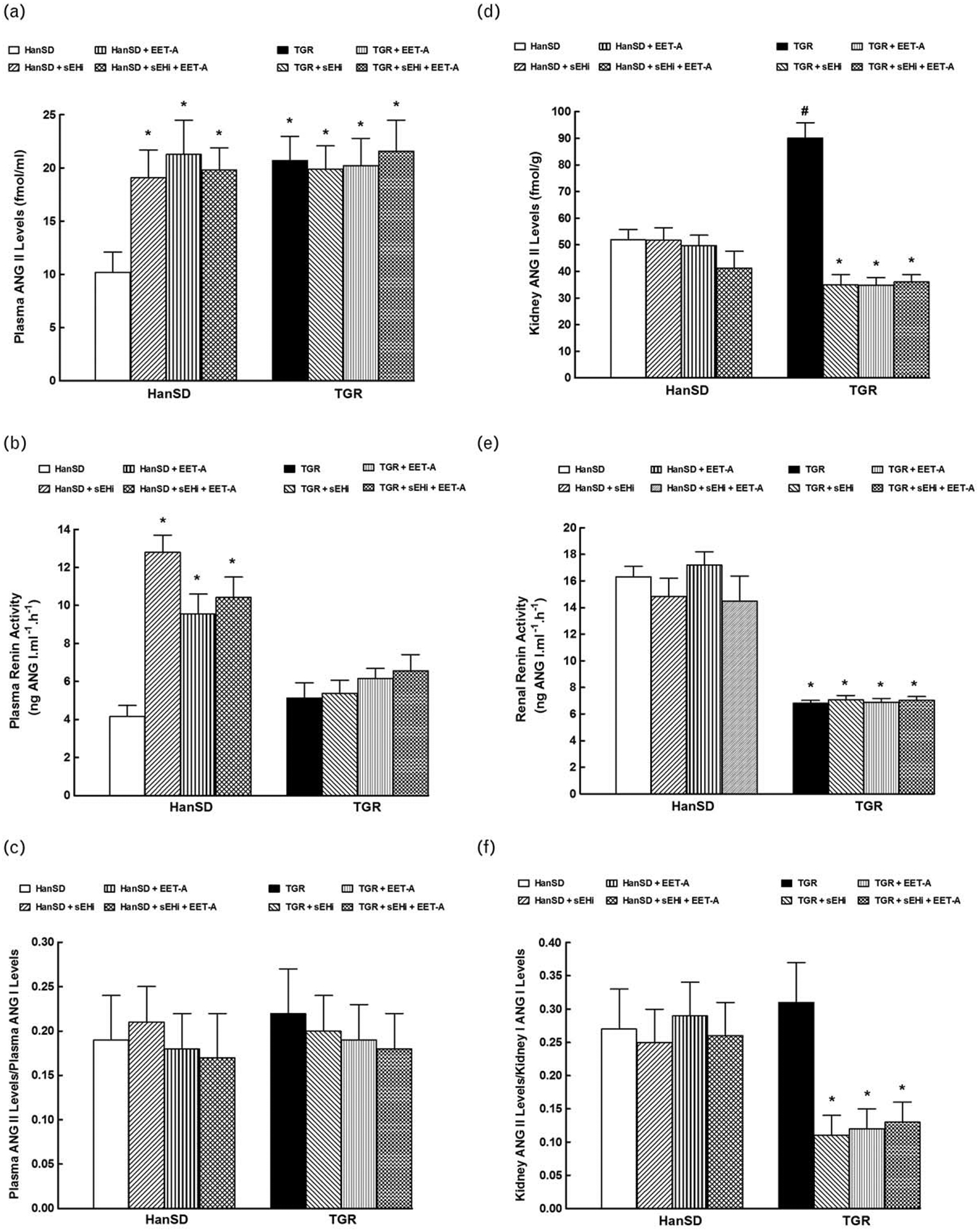
Plasma angiotensin II (ANG II) levels (a), plasma renin activity (b), the ratio of plasma ANG II to angiotensin I (ANG I) levels (c), kidney ANG II levels (d), renal renin activity (e), and the ratio of kidney ANG II to ANG I levels (f), in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and effects of treatment with soluble epoxide inhibitor (sEHi) and epoxyeicosatrienoic acid analogue (EET-A) alone and combined sEHi and EET-A treatment.*P<0.05 versus untreated HanSD rats. #P<0.05 versus all the other groups.
Figure 4b shows no significant baseline differences in plasma PRA between TGR and untreated HanSD rats. As in the case of plasma ANG II, sEHi or EET-A given alone or combined increased PRA in HanSD rats but did not significantly change it in TGR.
There was no significant difference in plasma ANG II to angiotensin I (ANG I) ratio, an index of plasma angiotensin-converting enzyme (ACE) activity [28,58], between TGR and HanSD rats (Fig. 4c). sEHi or EET-A given alone or combined did not alter this ratio in TGR or HanSD rats.
As shown in Fig. 4d, kidney ANG II concentrations were significantly higher in untreated TGR than in untreated HanSD rats (90±6 versus 52±4 fmol/g, P<0.05). sEHi or EET-A given alone or combined did not change kidney ANG II in HanSD rats but in each of TGR treatment groups significant decreases were seen, even below the values observed in untreated HanSD rats (35±4, 35±3 and 36±3 versus 52±4, respectively, P<0.05 in each case).
Figure 4e shows that RRA was significantly lower in untreated TGR than in untreated HanSD rats. sEHi or EET-A applied alone or combined did not alter RRA in TGR or HanSD rats.
There was no significant difference in baseline kidney ANG II to ANG I ratio, an index in intrarenal ACE activity [61], between TGR and HanSD rats (Fig. 4f). sEHi or EET-A applied alone or combined did not alter this ratio in HanSD rats but elicited profound decreases in TGR.
Effects of treatments on the indices of the severity of myocardial ischemia/reperfusion injury
As shown in Fig. 5a, the normalized area at risk (area at risk, expressed as the percentage of the LV) was between 41 and 50% and did not significantly differ between experimental groups. Similarly, myocardial infarct size normalized to area at risk did not significantly differ between TGR and HanSD rats (73±2 versus 78±2%); sEHi or EET-A given alone or combined did not alter it in TGR or HanSD rats (Fig. 5b). Representative images of myocardial infarction induced by 20-min coronary artery occlusion and 3-h reperfusion in individual experimental groups are shown in online Supplemental Figure 1, http://links.lww.com/HJH/A909. The supplemental Figure 2, http://links.lww.com/HJH/A909 shows examples of ECG tracings.
FIGURE 5.
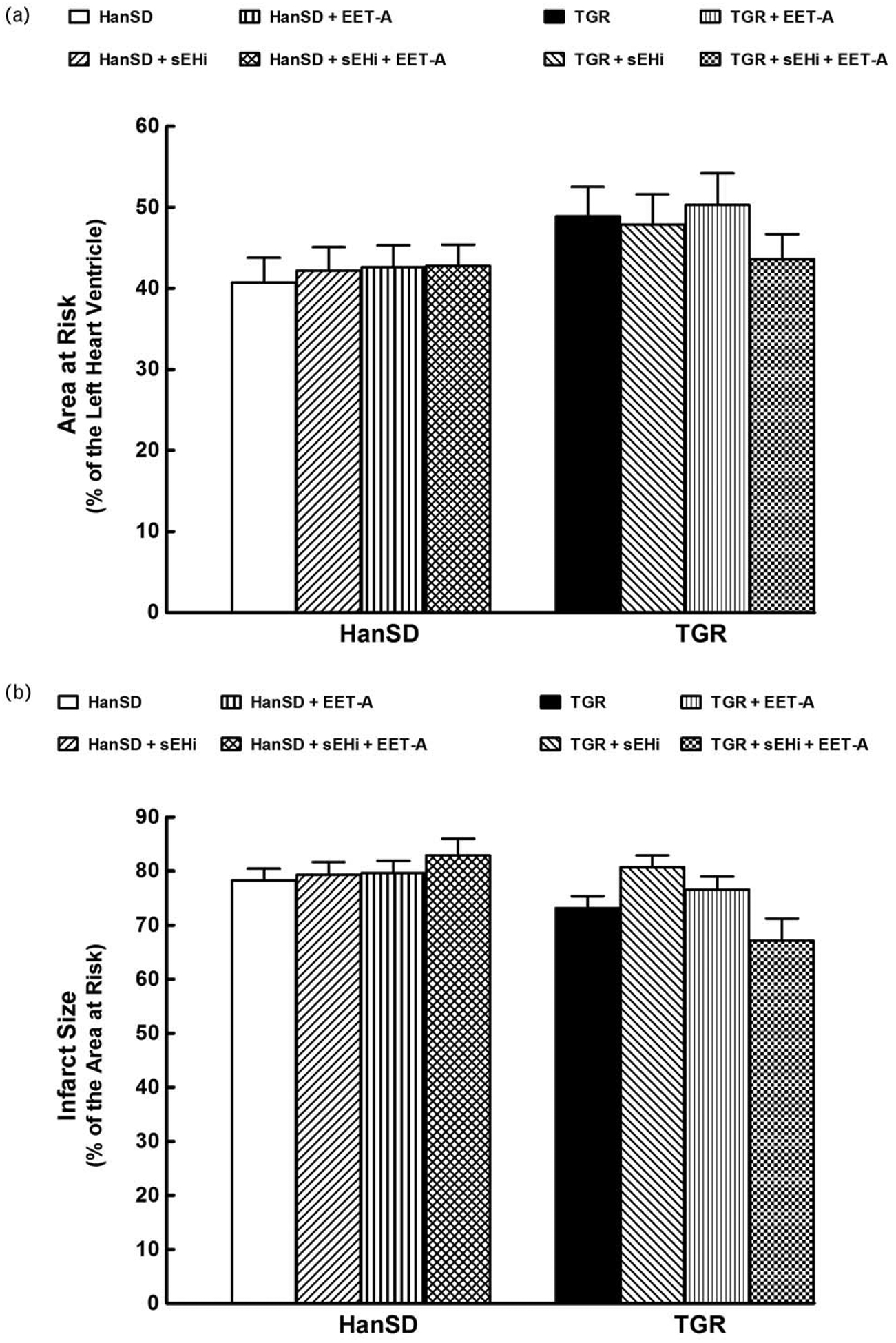
Myocardial area at risk normalized to the size of the left heart ventricle (a), infarct size expressed as a percentage of the area at risk (b) in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and effects of treatment with soluble epoxide hydrolase inhibitor (sEHi) or epoxyeicosatrienoic acid analogue (EET-A) given alone or combined. Values are shown as means±SEM.
Untreated TGR showed a trend towards elevation of the overall incidence of ischemia-induced ventricular fibrillation and towards an increase of the score of ischemic arrhythmias compared to untreated HanSD rats (Fig. 6a and b). The treatment with sEHi or EET-A alone or combined did not significantly influence the overall incidence of ventricular fibrillation in HanSD rats; however, the frequency of sustained ventricular fibrillation decreased markedly after each of the three treatment variants (Fig. 6a). In TGR, EETs-based therapies significantly decreased the overall incidence of ventricular fibrillation due to a lower incidence of both reversible and sustained ventricular fibrillation (Fig. 6a).
FIGURE 6.
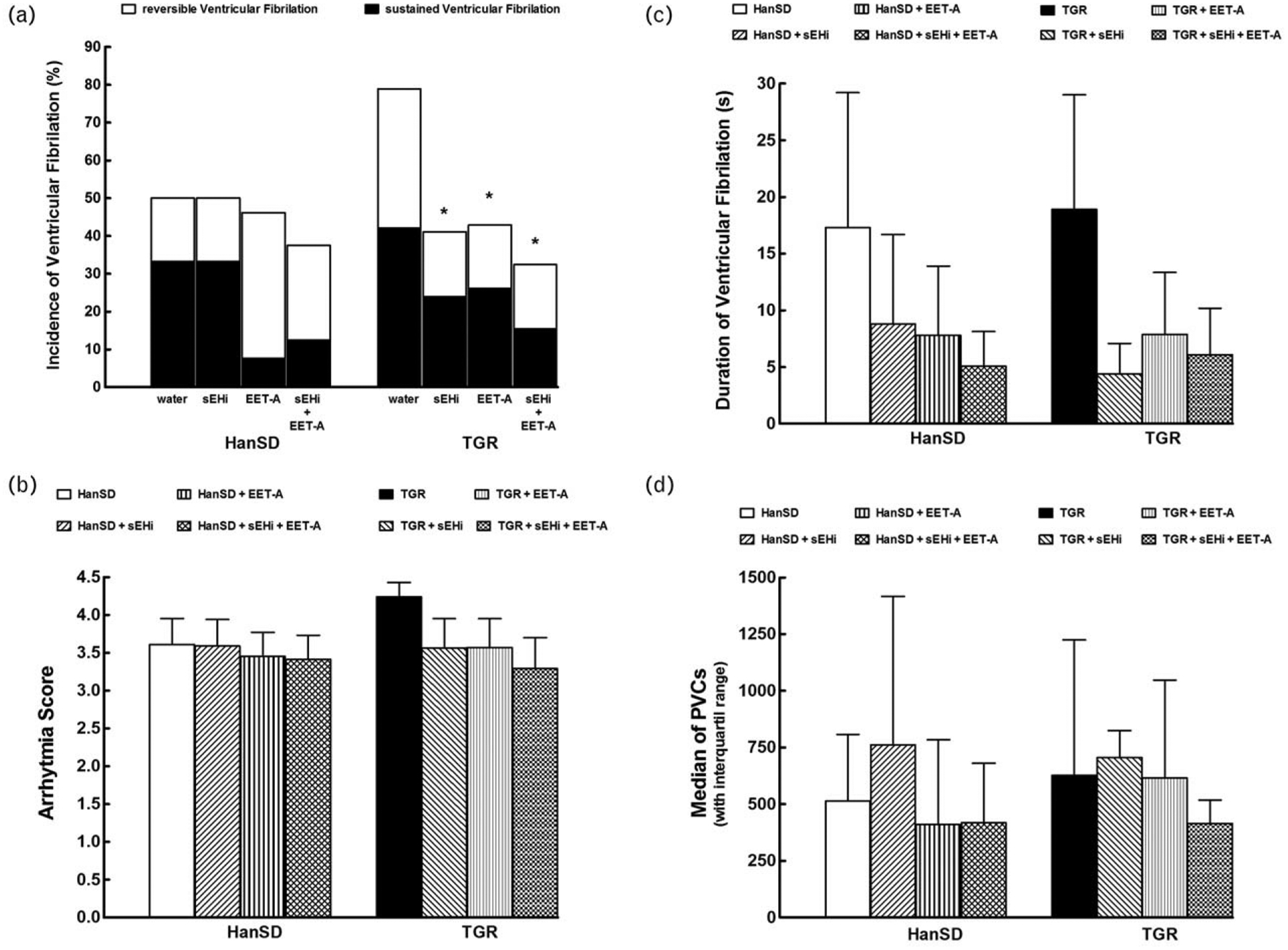
Incidence of ventricular fibrillation, both reversible and sustained (a), score of ischemic arrhythmias (b), duration of reversible ventricular fibrillation during 20-min coronary artery occlusion (c), and total number of premature ventricular complexes (PVCs) (d) in Hannover Sprague–Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and effects of treatment with soluble epoxide inhibitor (sEHi) or epoxyeicosatrienoic acid analogue (EET-A) given alone or combined. Values are shown as means±SEM (graphs b and c) or as median with interquartile range (graph d) *P<0.05 versus untreated TGR.
As shown in Fig. 6b, in TGR the treatment with sEHi or EET-A alone or combined tended to lower the score of ischemic arrhythmias, which suggests a beneficial effect of EETs-based therapies on the incidence of life-threatening ischemic arrhythmias in this strain.
EETs-based therapies induced a trend towards a reduction of the duration of reversible ventricular fibrillation in HanSD rats as well as in TGR, but these effects were not statistically significant due to high variability of the values in most of the groups (Fig. 6c).
Figure 6d shows that there were no significant differences in the total number of ischemic PVCs between untreated HanSD rats and TGR; the EETs-based therapy did not alter it, similarly in HanSD rats or TGR.
DISCUSSION
The first important finding of the present study is that chronic treatment with either sEHi or EET-A elicited antihypertensive and organ-protective actions in TGR, but the combined treatment with sEHi and EET-A brought no additional beneficial effects. Explicitly, sEHi as well as EET-A decreased BP to similar and almost normotensive levels, and attenuated albuminuria and cardiac hypertrophy in TGR: no such effects were seen in HanSD rats. In addition, we confirmed here our previous findings that TGR in the phase of established hypertension, when compared to normotensive HanSD rats, exhibit reduced tissue availability of biologically active epoxygenase products – EETs. While we have not established that the observed deficiency in EETs and activation of the RAS are causally related, our findings are consistent with the notion that intrarenal deficiency of biologically active EETs is a permissive factor in the development and maintenance of ANG II-dependent hypertension [25,26,36,37,40,49]. Our demonstration that chronic treatment with sEHi significantly increased tissue availability of EETs both in TGR and in HanSD rats suggests very strongly that sEH-mediated conversion of EETs to DHETEs is the main factor determining the availability of EETs. The increases in EETs content were substantially greater in TGR than in HanSD rats, indicating that in this ANG II-dependent form of hypertension sEH activity is indeed increased, even though tissue sEH protein expression was not found elevated [25,26,36,37,40,49].
What are the mechanism(s) responsible for blood pressure-lowering and organ protective effects of chronic treatment with sEHi and EET-A in Ren-2 renin transgenic hypertensive rats?
One should be aware that EETs are known to be an important factor in the control of the systemic vascular tone and also of renal tubular transport of sodium. EETs-induced vasodilation is mediated by stimulating large-conductance calcium-activated potassium channels and this was also clearly shown in studies employing EET-A [55,56,67]. Moreover, it was demonstrated that EETs as well as EET-A oppose the vasoconstrictor actions of ANG II [56,68]. Therefore, it is conceivable that one potential reason for the BP-lowering effects of chronic sEHi and EET-A treatment was EETs-mediated or EET-A-mediated attenuation of previously documented exaggerated peripheral and renal vascular responsiveness to ANG II [69].
However, there is also vast evidence that BP-lowering properties of EETs are related to their action on tubular sodium reabsorption [36,37,48,55,57,61]: they were shown to inhibit the proximal transport by blocking the sodium–hydrogen exchanger and the cortical collecting duct transport by blocking epithelial sodium channel [70,71]. Likewise, EET-A was shown to inhibit epithelial sodium channel and increase renal sodium excretion in two different models of ANG II-dependent hypertension [55–57]. This evidence and our present data showing, admittedly transient, increases in UNaV after sEHi or EET-A treatment suggest another possible mechanism of BP-lowering (active at least at the initial phase), one related to direct inhibitory influence of increased intrarenal EETs or EET-A concentrations on renal tubular sodium transport.
Of critical importance is our demonstration that in TGR chronic sEHi or EET-A treatment distinctly suppressed intrarenal ANG II without altering its plasma levels: the intrarenal augmentation is claimed to be the main mechanism responsible for the development and especially maintenance of ANG II-dependent hypertension [72]. In this context, it is important to briefly introduce the concept about the critical role of intrarenal RAS in the pathophysiology of hypertension. Guyton and coworkers almost 50 years ago introduced a hypothesis that was based on findings that increases in renal perfusion pressure are immediately accompanied by increased sodium and water excretion, which Guyton named pressure-natriuresis mechanism [73–76]. Based on the tremendous capacity of the kidney to excrete sodium and water, all BP-increasing incentives ranging from increased heart rate to elevated peripheral vascular resistance should be offset via this mechanism, which should have sufficient gain to limit intravascular volume and thereby maintain BP in normotensive range [73–76]. Further detailed studies have shown that the RAS regulates a diversity of renal hemodynamic and transport processes, which helps protect the organism against life-threatening loss of salt and fluid volume and arterial hypotension [72,75,77]. ANG II exerts pleiotropic actions, such as glomerular arteriolar constriction with reduction of glomerular filtration, enhancement of tubular sodium reabsorption and modulation of tubuloglomerular feedback mechanism [72,75,77]. While this makes the RAS very efficient in its physiological control functions, its powerful actions can disturb the pressure-natriuresis mechanism and contribute to the pathophysiology of hypertension [72,75,77,78]. In addition, apart from the circulating (endocrine) RAS, the intrarenal RAS, a local autocrine/paracrine system can change its activity independent of the circulating form [72,78]. There is evidence that inappropriately activated intrarenal RAS represents a final event leading to the impairment of pressure-natriuresis mechanism and BP responses to various hypertensiogenic stimuli [72–78]. Nevertheless, it must be admitted that while this ‘Guyton concept’ combined with notion of the critical mediatory role of intrarenal RAS in BP control has been widely accepted [73–75], it is contradicted by some scientific groups which provide research indicating that BP control and pathophysiology of hypertension depend on neural and vascular regulatory pathways [79,80]. Nevertheless, despite the above controversies, our data suggest that the BP-lowering effect of sEHi or EET-A treatment were secondary to suppression of intrarenal ANG II rather than a direct effect of an enhancement of intrarenal EETs. In addition, our present findings indicate that chronic sEHi or EET-A treatment suppressed ANG II production at the ACE level, which fits the recent hypothesis that ACE-mediated ANG II formation is an important factor in the development of various forms of experimental hypertension [81]. The findings are also in agreement with our recent studies showing that in inbred transgenic rats with inducible malignant ANG II-dependent hypertension (strain Cyp1a1-Ren-2) the sEHi or EET-A treatment also blocked RAS activity at the ACE level [57,61]. In those studies we found that the treatment with either sEHi or EET-A decreased kidney ACE activity that was measured either directly by a commercially available kit for radioimmunoassay or indirectly, as a ratio of ANG II to ANG I. The latter is now recognized as a very reliable index of tissue ACE activity [61,82]. Also in our present study we found that that the treatment with either sEHi or EET-A decreased kidney ANG II to ANG I ratio (see Fig. 4f). Therefore, it can be hypothesized that the two drugs exhibit ACE inhibitory properties, apparent at least in models of hypertension induced by increasing RAS activity. The limitation of our present as well as earlier studies is that the mechanism of the purported ACE inhibition was not explored: further studies are required to address this issue. Nevertheless, our present results strengthen recent suggestions that interaction at various levels of CYP eicosanoids and RAS is critically important for the final consequences of RAS actions [83].
Taken together, we propose that the mechanism underlying BP-lowering effect of chronic sEHi and EET-A treatment was primarily a suppression of inappropriately high intrarenal ANG II concentration dependent on inhibition of ACE activity. In the initial phase of treatment natriuretic action of EETs could contribute to the overall hypotensive effect.
Aside from the influence on BP, the mechanism of the organ protective effects of chronic sEHi and EET-A treatment requires consideration. It has to be admitted that both drugs were reported to exert anti-inflammatory and anti-apoptotic actions and thus were potentially renoprotective [36–39]. On the other hand, it was recently shown that in the ANG II-dependent model used here and likely in other similar hypertension models the cardioprotective and renoprotective effects are dominantly BP-dependent [29,84]. In our opinion the organ protective effects of chronic sEHi and EET-A treatment in TGR are indeed secondary to BP lowering.
Why combination of chronic sEHi and EET-A treatment was not more effective than any of single treatments?
As discussed earlier, when baseline EETs generation is impaired, as observed in many renal and cardiovascular diseases [50–52], the efficiency of sEHi treatment would be limited. Hence the rationale for application EETs-agonists (such as EET-A), whose effectiveness would not depend on the initial rate of endogenous EETs formation [52–55]. However, in our TGR model endogenous EETs formation was not compromised: low tissue concentrations of EETs was probably due to increased sEH-mediated conversion of EETs to DHETEs. The failure of sEHi to show antihypertensive effects additional to those induced by EET-A supports the contention that the effects of sEHi on BP are mediated by the induced changes in EETs levels. Nevertheless, this conclusion is valid only for our transgenic model of ANG II-dependent hypertension. Additional benefits of combined treatment might still be observed in salt-sensitive hypertension or in renal injury induced by radiation or cis-platinum treatment, the states where reduced tissue EETs is the result of both decreased formation and increased degradation by sEH [50–52,85]. In this context, it is important to emphasize that our concept of possible additional benefit of combined sEHi and EET-A treatment was based on the expectation that pharmacologically-induced enhancement of EETs by sEHi would raise endogenous EETs component, and EET-A treatment (exogenous component) should further increase EETs actions. However, our present findings disprove this paradigm and strongly suggest that at the dose applied the biological effects of EET-A were already maximal and addition of endogenous EETs did not enhance cardioprotection or BP-lowering). Notably, this conclusion is valid only for our present TGR model.
The second important set of findings in the present study relates to cardioprotective effects of the treatments. First, we found that the infarct size after acute ischemia/reperfusion insult did not significantly differ between TGR and HanSD rats. Furthermore, chronic treatment with sEHi or EET-A, applied alone or combined, did not reduce infarct size in either strain. These data could on the first sight suggest that TGR were not more sensitive to ischemia/reperfusion injury compared to HanSD rats.
It will here be noticed that the need for evaluation of potential novel cardioprotective treatment strategies in experimental animal models was not fully realized until some 10 years ago. Obviously, attention was focused on the models that simulate well known cardiovascular risks and modifying risk factors for human ischemic heart disease, such as hypertension, hyperlipidemia, established atherosclerosis, heart failure and age factor. It was proposed that preclinical studies should be conducted in models that exhibit some associated comorbidities [9,27]. Actually, the preclinical studies performed so far employed usually the models with one comorbidity only. This limitation pertains also to the present study: it is clear that future research should employ models combining more risks factors, for example hypertension and diet-induced hyperlipidemia [9,27]. Furthermore, it has been demonstrated that EETs have anti-inflammatory effects and the treatment with EET-A proved organ-protective by reducing oxidative stress and inflammation [85]. Therefore, it is likely that also in our study the anti-inflammatory actions of sEHi or EET-A treatment could play an important role, however, this issue was not addressed here. While this is an obvious limitation, yet since arterial hypertension affects more than 1.4 billion people worldwide [86] and is considered the major independent risk factor for myocardial ischemia/reperfusion injury, we decided to focus on the role of hypertension and of RAS hyperactivity as plausible risk factors for myocardial ischemia/reperfusion injury. In this context, it should be remembered that analysis of the data from Framingham cohort studies indicates a clear relationship between LVH and cardiovascular morbidity and mortality [11], in agreement with the early report on the ‘stone heart’ phenomenon detected during cardiothoracic surgery [87]. This forms a basis for the long-standing belief that the hypertrophic myocardium exhibits decreased tolerance to ischemia/reperfusion injury. Indeed, many experimental studies showed that animals with LVH are more vulnerable to myocardial ischemia/reperfusion injury compared with control animals [15–20]. However, experimental studies did not disclose increased post-ischemia/reperfusion infarct size in the hypertrophied heart [21–27]. Moreover, we showed recently that, in the early stage of hypertension, in two ANG II-dependent models the myocardial resistance to ischemia/reperfusion injury was even higher, and that was seen in the hearts with marked LVH [25,26]. Apparently, the role of hypertension and LVH as determinants of myocardial infarct size is complex, perhaps one of a ‘double-edged sword’. Depending on the phase of hypertension and the degree of LVH some cardioprotective mechanism(s) could first be activated but later the myocardial sensitivity to ischemia/reperfusion injury would increase. This could be particularly the case in models of hypertension dependent on activation of endogenous RAS [25,26].
Nevertheless, our data show that the incidence and severity of ischemia/reperfusion arrhythmias were higher in TGR, in agreement with earlier reports on similar augmentation in hearts with LVH [27]. Apparently, when infarct size and arrhythmias are both taken into consideration, the TGR with established LVH (even without signs of heart failure) exhibit increased myocardial sensitivity to life-threatening ventricular arrhythmias. Moreover, sEHi or EET-A given alone or combined did not alter the incidence of ventricular fibrillation in HanSD rats but significantly reduced them in TGR. It has been reported previously that elevation of myocardial EETs levels by transgenic over-expression of CYP epoxygenase or pharmacological inhibition of sEH protect hearts against a harmful electric remodeling, ventricular tachyarrhytmias and atrial fibrillation in mouse hearts after myocardial infarction. This was also the case during maladaptive cardiac hypertrophy induced by chronic pressure overload elicited by transverse aortic constriction [88–90]. Therefore, the antiarrhythmic action of EET-based therapies observed in the present study could provide a new pharmacological strategy in ischemia/reperfusion treatment. This is a key finding because ischemia/reperfusion-induced ventricular arrhythmias remain the major cause of sudden cardiac death after acute myocardial infarction [91].
The explanation why combination of chronic sEHi and EET-A treatment was not more effective in lowering BP than single treatments (see above) seems equally valid for the cardioprotective effects. Also in the myocardium the crucial change after either treatment was increasing tissue concentrations of EETs, as shown in Fig. 2d and e. Assuming that each of the two treatments exerts a maximal possible effect, combining the two treatments would not further enhance cardioprotection.
It is still conceivable that in some disorders and experimental models combined chronic sEHi and EET-A treatment would exhibit additive antihypertensive, renoprotective and cardioprotective actions. Indeed, previous studies have shown that tissue EETs deficiency results either from decreased formation or increased degradation of EETs [36–41,50–52]. Therefore, whenever both causes for the deficiency are present, the combined treatment could be more effective. It is clear that increased ANG II concentrations upregulate sEH activity [40,92]. Because ANG II-dependent hypertension is also associated with increased oxidative stress and tissue inflammation [93], which are considered an important factor diminishing endogenous EETs production [37,39], it was reasonable to suspect that decreased tissue concentration of EETs in the sustained phase of hypertension in TGR is the consequence of both reduced generation and increased degradation of these agents. However, our present data clearly show that tissue EETs deficiency at this stage results largely from enhanced sEH-mediated conversion of EETs do DHETEs.
Study limitations
Apart from the above discussed drawbacks, our present study has two major limitations.
The first one is the lack of molecular (cellular) assessment of the cardioprotective effects of treatment; this could help clarify the reason(s) why chronic treatment with sEHi or EET-A, applied alone or combined, did not reduce infarct size in either strain. It is known that mitochondria provide the primary source of energy in the myocardium and ischemia/reperfusion injury can cause irreversible mitochondrial damage [30–32]. It is now recognized that EETs have a role in minimizing the loss of mitochondrial membrane potential and limit MPTP opening during reperfusion, thus contributing to overall cardiac protection under various stress situations [31,35]. However, it remains unknown how EETs-induced actions maintain mitochondrial membrane potential and/or whether the preservation is through direct or indirect effects on mitochondria. It has been shown that EETs-induced effects are mediated via numerous pathways: for example by activation of the sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels, the calcium-activated potassium channel, the phosphatidylinositol 3-kinase/protein kinase B, and many others [34,35]. Therefore, it is obvious that more studies are needed, with a focus on the molecular (cellular) mechanism(s) responsible for ischemia/reperfusion injury. The present work provides a necessary background for such studies.
The second limitation relates to the absence of comprehensive evaluation of pharmacological properties of the chronic sEHi and EET-A treatment, to clarify if the doses of sEHi and EET-A which result only in half maximal concentrations of the drugs would actually cause additive BP-lowering effects? A positive answer can be assumed but appropriate studies (NB: time consuming and expensive) should address this issue.
In summary, we found that chronic treatment with either sEHi or EET-A exhibits antihypertensive actions in TGR, most probably dependent on the suppression by augmented EETs (endogenous or exogenous) of inappropriately high intrarenal ANG II content; the mechanism consists of ACE-inhibitory effect of EETs. In the initial phase of sEHi or EET-A treatment their direct renal transport-inhibitory and natriuretic action could contribute to BP lowering. Second, our data show that in TGR both sEHi and EET-A reduced the incidence of life-threatening ventricular fibrillations induced by myocardial ischemia. Third, somewhat surprisingly, combined sEHi and EET-A treatment did not provide additive antihypertensive, renoprotective or antiarrhythmic effects.
In conclusion, we believe that the treatment targeting CYP-dependent epoxygenase pathway of arachidonic acid should be considered in attempts to develop new pharmacological strategies to achieve concomitant antihypertensive and cardioprotective effects in patients with ischemic heart disease. However, combined treatment with sEHi and EET-A should probably be confined to situations when tissue deficiency of EETs is the consequence of both decreased formation and increased degradation of these active metabolites.
Supplementary Material
ACKNOWLEDGEMENTS
EET-A has been jointly patented by the Medical College of Wisconsin and UT Southwestern.
This study was primarily supported by grant no. 15-07544S awarded to Z.H. by the Czech Science Foundation (GAČR). J.R.F. was supported in part by the Robert A. Welch Foundation (I-001). J.D.I. and this work was partially supported by a National Institute of Health (NIH) grant (DK103616) and a Dr Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee grant.
Abbreviations:
- ACE
angiotensin-converting enzyme
- ANG I
angiotensin I
- ANG II
angiotensin II
- BP
blood pressure
- CYP
cytochrome P-450
- DHETE
dihydroxyeicosatrienoic acid
- EET-A
epoxyeicosatrienoic acid analogue
- EETs
epoxyeicosatrienoic acids
- HanSD
transgene-negative, normotensive Hannover Sprague–Dawley rats
- HETE
hydroxyeicosatrienoic acid
- LV
left ventricle
- LVH
left ventricle hypertrophy
- LVW
left ventricle weight
- MPTP
mitochondrial permeability transition pore
- PRA
plasma renin activity
- PVC
premature ventricular complexe
- RAS
renin–angiotensin system
- RRA
renal renin activity
- sEH
soluble epoxide hydrolase
- sEHi
soluble epoxide hydrolase inhibitor
- TGR
Ren-2 renin transgenic hypertensive rats
- UNaV
daily sodium excretion
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
REFERENCES
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357:121–135. [DOI] [PubMed] [Google Scholar]
- 2.Altamirano F, Wang ZV, Hill JA. Cardioprotection in ischaemia-reperfusion injury: novel mechanisms and clinical translation. J Physiol 2015; 17:3773–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulluck H, Yellon DM, Hausenloy DJ. Reducing myocardial infarct size: challenges and future opportunities. Heart 2016; 102:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heusch G Critical issues for the translation of cardioprotection. Circ Res 2017; 120:1477–1486. [DOI] [PubMed] [Google Scholar]
- 5.Kloner RA, Hale SL, Dai W, Shi J. Cardioprotection: where to from here? Cardiovasc Drugs Ther 2017; 31:53–61. [DOI] [PubMed] [Google Scholar]
- 6.Cabrera-Fuentes HA, Aragones J, Bernhagen J, Boening A, Boisvert WA, Botker HE, et al. From basic mechanisms to clinical applications in heart protection, new players in cardiovascular diseases and cardiac theranostics: meeting report from the third international symposium on ‘New frontiers in cardiovascular research’. Basic Res Cardiol 2016; 111:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heusch G, Libby P, Gersch B, Yellon D, Bohm M, Lopaschuk G, Opie L. Cardiovascular remodeling in coronary artery disease and heart failure. Lancet 2014; 383:1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Pharmacol Rev 2014; 66:1142–1174. [DOI] [PubMed] [Google Scholar]
- 10.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol 2011; 301: H1723–H1741. [DOI] [PubMed] [Google Scholar]
- 11.Prisant LM. Hypertensive heart disease. J Clin Hypertens (Greenwich) 2005; 7:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alderman MH, Ooi WL, Cohen H, Madhavan S, Sealey JE, Laragh JH. Plasma renin activity: a risk factor for myocardial infarction in hypertensive patients. Am J Hypertens 1997; 10:1–8. [DOI] [PubMed] [Google Scholar]
- 13.Mozaffari MS, Liu JY, Abebe W, Baban B. Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am J Cardiovasc Dis 2013; 3:180–196. [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal V, Gupta JK, Qureshi SS, Vishwakarma VK. Role of cardiac renin angiotensin system in ischemia reperfusion injury and preconditioning of heart. Indian Heart J 2016; 68:856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molgaard S, Faricelli B, Salomonsson M, Engstrom T, Treiman M. Increased myocardial vulnerability to ischemia-reperfusion injury in the presence of left ventricular hypertrophy. J Hypertens 2016; 34: 513–523. [DOI] [PubMed] [Google Scholar]
- 16.Anderson PG, Bishop SP, Dignerness SB. Transmural progression of morphological changes during ischemia and reperfusion in the normal and hypertrophied heart. Am J Pathol 1987; 129:152–167. [PMC free article] [PubMed] [Google Scholar]
- 17.Minor T, Isselhard W, Sturz J. Recovery of healthy and hypertrophied hearts after global ischemia and gradual reperfusion. Ann Cardiol Angeol 1994; 43:395–399. [PubMed] [Google Scholar]
- 18.Snoeckx LH, van der Vusse GJ, Coumans WA, Willemsen PH, van der Nagel T, Reneman RS. Myocardial function in normal and spontaneously hypertensive rats during reperfusion after a period of global ischaemia. Cardiovasc Res 1986; 20:67–75. [DOI] [PubMed] [Google Scholar]
- 19.Hearse DJ, Stewart DA, Green DG. Myocardial susceptibility to ischemic damage: a comparative study of disease models in the rat. Eur J Cardiol 1978; 7:437–450. [PubMed] [Google Scholar]
- 20.Ledvényiová-Farkašová V, Bernátová I, Balis P, Puzserova A, Bartekova M, Gablovsky I, Ravingerova T. Effects of crowding stress on tolerance to ischemia-reperfusion injury in young male and female hypertensive rats: molecular mechanisms. Can J Physiol Pharmacol 2015; 93:793–802. [DOI] [PubMed] [Google Scholar]
- 21.Mozaffari MS, Schaffer SW. Effect of hypertension and hypertension-glucose intolerance on myocardial ischemic injury. Hypertension 2003; 42:1042–1049. [DOI] [PubMed] [Google Scholar]
- 22.Saupe KW, Lim CC, Ingwall JS, Apstein CS, Eberli FR. Comparison of hearts with 2 types of pressure-overload left ventricular hypertrophy. Hypertension 2000; 35:1167–1172. [DOI] [PubMed] [Google Scholar]
- 23.Wagner C, Ebner B, Tillack D, Strasser RH, Weinbrenner C. Cardioprotection by ischemic postconditioning is abrogated in hypertrophied myocardium of spontaneously hypertensive rats. J Cardiovasc Pharmacol 2013; 61:35–41. [DOI] [PubMed] [Google Scholar]
- 24.Matsuhisa S, Otani H, Okazaki T, Yamashita K, Akita Y, Sato D, et al. Angiotensin II type 1 receptor blocker preserves tolerance to ischemia-reperfusion injury in Dahl salt-sensitive rat heart. Am J Physiol 2008; 294:H2473–H2479. [DOI] [PubMed] [Google Scholar]
- 25.Neckář J, Kopkan L, Husková Z, Kolář F, Papoušek F, Kramer HJ, et al. Inhibition of soluble epoxide hydrolase by cis-4-[4–(3–adamantan–I–ylureido)cyclohexyl–oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci 2012; 122:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alánová P, Husková Z, Kopkan L, Sporková A, Jíchová Š, Neckář J, et al. Orally active epoxyeicosatrienoic acid analog does not exhibit antihypertensive and reno- or cardioprotective actions in two-kidney, one-clip Goldblatt hypertensive rats. Vascul Pharmacol 2015; 73:45–56. [DOI] [PubMed] [Google Scholar]
- 27.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 2007; 59:418–458. [DOI] [PubMed] [Google Scholar]
- 28.Husková Z, Kramer HJ, Vaňourková Z, Červenka L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J Hypertens 2006; 24:517–527. [DOI] [PubMed] [Google Scholar]
- 29.Kujal P, Čertíková-Chábová V, Vernerová Z, Walkowska A, Kompanowska-Jezierska E, Sadowski J, et al. Similar renoprotection after renin-angiotensin-dependent and -independent antihypertensive therapy in 5/6-nephrectomized Ren-2 transgenic rats: are there blood pressure-independent effects? Clin Exp Pharmacol Physiol 2010; 37: 1159–1169. [DOI] [PubMed] [Google Scholar]
- 30.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 2015; 78:129–141. [DOI] [PubMed] [Google Scholar]
- 31.Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ. Role of the MPTP in conditioning the heart translability and mechanism. Br J Pharmacol 2015; 172:2074–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Javadov S, Jang S, Parodi-Rullán R, Khuchua Z, Kuznetsov AV. Mitochondrial permeability transition in cardiac ischemia-reperfusion: whether cyclophilin D is a viable target for cardioprotection? Cell Mol Life Sci 2017; 74:2795–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross GJ, Hsu A, Pfeiffer A, Nithipatikom K. Roles of endothelial nitric oxide synthase (eNOS) and mitochondrial permeability transition pore (MPTP) in epoxyeicosatrienoic acid (EET)-induced cardioprotection against infarction in intact rat hearts. J Mol Cell Cardiol 2013; 59:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol 2014; 74:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamieson KL, Endo T, Darwesh AM, Samokhvalov V, Seubert JM. Cytochrome P450-derived eicosanoids and heart function. Pharmacol Ter 2017; 179:47–83. [DOI] [PubMed] [Google Scholar]
- 36.Imig JD. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 2015; 65:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elmarakby AA. Reno-protective mechanisms of epoxyeicosatrienoic acids in cardiovascular disease. Am J Physiol 2012; 302:R321–R330. [DOI] [PubMed] [Google Scholar]
- 38.Fan F, Muoya Y, Roman RJ. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens 2015; 24:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fleming I The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol Rev 2014; 66:1106–1140. [DOI] [PubMed] [Google Scholar]
- 40.Ai D, Shyy JYJ, Zhu Y. Linking an insect enzyme to hypertension: angiotensin II-epoxide hydrolase interactions. Kidney Int 2010; 77:88–92. [DOI] [PubMed] [Google Scholar]
- 41.He J, Wang C, Zhu Y, Ai D. Soluble epoxide hydrolase: a potential target for metabolic diseases. J Diabetes 2016; 8:305–313. [DOI] [PubMed] [Google Scholar]
- 42.Jamieson KL, Samokhvalov V, Akhnokh MK, Lee K, Cho WJ, Takawale A, et al. Genetic deletion of soluble epoxide hydrolase provides cardioprotective responses following myocardial infarction in aged mice. Prostaglandins Other Lipid Mediat 2017; 132:47–58. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Morisseau C, Wang JF, Yang T, Falck JR, Hammock BD, Wang MH. Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am J Physiol 2007; 293:F342–F349. [DOI] [PubMed] [Google Scholar]
- 44.Honetschlägerová Z, Sporková A, Kopkan L, Husková Z, Hwang SH, Hammock BD, et al. Inhibition of soluble epoxide hydrolase improves the impaired pressure-natriuresis relationship and attenuates the development of hypertension and hypertension-associated end-organ damage in Cyp1a1-Ren-2 transgenic rats. J Hypertens 2011; 29:1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Carroll MA, Chander PN, Falck JR, Sangras B, Stier CT. Soluble epoxide hydrolase inhibitor, AUDA, prevents early salt-sensitive hypertension. Front Biosci 2008; 13:3480–3487. [DOI] [PubMed] [Google Scholar]
- 46.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J 2010; 24:3770–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sporková A, Kopkan L, Varcabová A, Husková Z, Hwang SH, Hammock BD, et al. Role of cytochrome P450 metabolites in the regulation of renal function and blood pressure in 2-kidney, 1-clip hypertensive rats. Am J Physiol 2011; 300:R1468–R1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honetschlägerová Z, Husková Z, Vaňourková Z, Sporková A, Kramer HJ, Hwang SH, et al. Renal mechanisms contributing to the antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats with inducible hypertension. J Physiol 2011; 589:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varcabová Š, Husková Z, Kramer HJ, Hwang SH, Hammock BD, Imig JD, et al. Antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats is mediated by suppression of the intrarenal renin-angiotensin system. Clin Exp Pharmacol Physiol 2013; 40:273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma YH, Schwartzman ML, Roman RJ. Altered renal P-450 metabolism of arachidonic acid in Dahl salt-sensitive rats. Am J Physiol 1994; 267 (2 Pt 2):R579–R589. [DOI] [PubMed] [Google Scholar]
- 51.Kaergel E, Muller DN, Honeck H, Theuer J, Shagdarsuren E, Mullaly A, et al. P450-dependent arachidonic acid metabolism and angiotensin II-induced renal damage. Hypertension 2002; 40:273–279. [DOI] [PubMed] [Google Scholar]
- 52.Imig JD. Targeting epoxides for organ damage in hypertension. J Cardiovasc Pharmacol 2010; 56:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falck JR, Kodela R, Manne R, Atcha R, Puli N, Dubasi N, et al. 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem 2009; 52:5069–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki RV, et al. Development of epoxyeiocastrienoic acids analogs with in vivo antihypertensive actions. Front Physiol 2010; 1:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hye Khan MA, Pavlov TS, Christain SV, Neckář J, Staruschenko A, Gauthier KM, et al. Epoxyeicosatrienoic acid analogue lowers blood pressure through vasodilatation and sodium channel inhibition. Clin Sci 2014; 127:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hye Khan MA, Falck JR, Manthati VL, Campbell WB, Imig JD. Epoxyeicosatrienoic acid analog attenuates angiotensin II hypertension and kidney injury. Front Pharmacol 2014; 5:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jíchová Š, Kopkan L, Husková Z, Doleželová Š, Neckář J, Kujal P, et al. Epoxyeicosatrienoic acid analog attenuates the development of malignant hypertension, but does not reverse it once established: a study in Cyp1a1-Ren-2 transgenic rats. J Hypertens 2016; 34:2008–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mullins JJ, Peter J, Ganten D. Fulminant hypertension in transgenic rats harboring the mouse Ren-2 gene. Nature 1990; 344:541–544. [DOI] [PubMed] [Google Scholar]
- 59.Lee MA, Bohm M, Paul M, Bader M, Ganten U, Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol 1996; 270 (6 Pt 1):E919–E929. [DOI] [PubMed] [Google Scholar]
- 60.Dvořák P, Kramer HJ, Backer A, Malý J, Kopkan L, Vaněčková I, et al. Blockade of endothelin receptors attenuates end-organ damage in homozygous hypertensive Ren-2 transgenic rats. Kidney Blood Press Res 2004; 27:248–258. [DOI] [PubMed] [Google Scholar]
- 61.Sporková A, Jíchová Š, Husková Z, Kopkan L, Nishiyama A, Hwang SH, et al. Different mechanisms of acute versus long-term antihypertensive effects of soluble epoxide hydrolase inhibition: Studies in Cyp1a1-Ren-2 transgenic rats. Clin Exp Pharmacol Physiol 2014; 41:1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurements in humans and experimental animals. Part 2: Blood pressure measurements in experimental animals. Hypertension 2005; 45:299–310. [DOI] [PubMed] [Google Scholar]
- 63.Jíchová Š, Doleželová Š, Kopkan L, Kompanowska-Jezierska E, Sadowski J, Červenka L. Fenofibrate attenuates malignant hypertension by suppression of the renin-angiotensin system: a study in Cyp1a1-Ren-2 transgenic rats. Am J Med Sci 2016; 352: 618–630. [DOI] [PubMed] [Google Scholar]
- 64.Neckář J, Svatoňová A, Weissová R, Drahota Z, Zajíčková P, Brabcová I, et al. Selective replacement of mitochondrial DNA increases the cardioprotective effects of chronic continuous hypoxia in spontaneously hypertensive rats. Clin Sci 2017; 131:865–881. [DOI] [PubMed] [Google Scholar]
- 65.Alánová P, Chytilová A, Neckář J, Hrdlička J, Míčová P, Holcerová K, et al. Myocardial ischemic tolerance in rats subjected to endurance exercise training during adaptation to chronic hypoxia. J Appl Physiol 2017; 122:1452–1461. [DOI] [PubMed] [Google Scholar]
- 66.Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia, infarction, and reperfusion. Cardiovasc Res 1988; 22:447–455. [DOI] [PubMed] [Google Scholar]
- 67.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent response. Pfluegers Arch 2010; 459:881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal microvascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonamide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens 2001; 19:983–992. [DOI] [PubMed] [Google Scholar]
- 69.Jacinto S, Mullins JJ, Mitchell KD. Enhanced renal vascular responsiveness to angiotensin II in hypertensive Ren-2 transgenic rats. Am J Physiol 1999; 276 (2 Pt 2):F315–F322. [DOI] [PubMed] [Google Scholar]
- 70.Madhun ZT, Goldthwait DA, McKay D, Hopfer U, Douglas JG. An epoxygenase metabolite of arachidonic acid mediates angiotensin II-induced rises in cytosolic calcium in rabbit proximal tubule epithelial cells. J Clin Invest 1991; 88:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakairi Y, Jacobson HR, Noland DT, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol 1995; 268 (5 Pt 2): F931–F939. [DOI] [PubMed] [Google Scholar]
- 72.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59:251–287. [DOI] [PubMed] [Google Scholar]
- 73.Guyton AC, Hall JE, Coleman TG, Manning RD Jr, Norman RA Jr. The dominant role of the kidneys in long-term arterial pressure regulation in normal and hypertensive states In: Laragh JH, Brenner BM, editors. Hypertension: pathophysiology, diagnosis and management. New York, NY: Raven Press; 1995. pp. 1311–1328. [Google Scholar]
- 74.Cowley AW, Roman RJ. The role of the kidney in hypertension. JAMA 1996; 275:1581–1589. [PubMed] [Google Scholar]
- 75.Hall JE, Granger JP, Hall ME. Physiology and pathophysiology of hypertension In: Albeprn RJ, Caplan MJ, Moe OW, editors. Seldin and Giebisch’s the kidney physiology and pathophysiology, 5th ed. Academic Press; 2013. pp. 1319–1352. [Google Scholar]
- 76.Crowley SD, Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest 2014; 124:2341–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mitchell KD, Navar LG. Intrarenal actions of angiotensin II in the pathogenesis of experimental hypertension In: Laragh JH, Brenner BM, editors. Hypertension: pathophysiology, diagnosis and management. New York, NY: Raven Press; 1995. pp. 1437–1449. [Google Scholar]
- 78.Yang T, Xu Ch. Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J Am Soc Nephrol 2017; 28:1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Averina VA, Othmer HG, Fink GD, Osborn JW. A mathematical model of salt-sensitive hypertension: the neurogenic hypothesis. J Physiol 2015; 14:3065–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feng W, Dell’Italia LJ, Sanders PW. Novel paradigms of salt and hypertension. J Am Soc Nephrol 2017; 28:1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernstein KE, Giani JF, Shen XZ, Gonzalez-Villalobos RA. Renal angiotensin-converting enzyme and blood pressure control. Curr Opin Nephrol Hypertens 2014; 23:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Husková Z, Kopkan L, Červenková L, Doleželová Š, Vaňourková Z, Škaroupková P, et al. Intrarenal alterations of the angiotensin-converting enzyme type 2/angiotensin 1–7 complex of the renin-angiotensin system do not alter the course of malignant hypertension in Cyp1a1-Ren-2 transgenic rats. Clin Exp Pharmacol Physiol 2016; 43:438–449. [DOI] [PubMed] [Google Scholar]
- 83.Garcia V, Schwartzman ML. Recent developments on the vascular effects of 20-hydroxyeicosatrienoic acid. Curr Opin Nephrol Hypertens 2017; 26:74–82. [DOI] [PubMed] [Google Scholar]
- 84.Carlstrom M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev 2015; 95:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hye Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J 2013; 27:2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.ESH/ESC Task Force for the Management of Arterial Hypertension. 2013 Practice guidelines for the management of arterial hypertension of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC): ESH/ESC Task Force for the Management of Arterial Hypertension. J Hypertens 2013; 31:1925–1938. [DOI] [PubMed] [Google Scholar]
- 87.Cooley DA, Reul GJ, Wukasch DC. Ischemic contracture of the heart: ‘stone heart’. Am J Cardiol 1972; 29:575–577. [DOI] [PubMed] [Google Scholar]
- 88.Li N, Liu JY, Timofeyev V, Qiu H, Hwang SH, Tuteja D, et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: insight gained using metabolomics approaches. J Moll Cell Cardiol 2009; 47:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wesphal C, Spallek B, Konkel A, Marko L, Qadri F, DeGraff LM, et al. CYP2J2 overexpresion protects against arrhythmia susceptibility in cardiac hypertrophy. PLosONE 2013; 8: e73490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sirish P, Li N, Timofeyev V, Zhang XD, Wang L, Yang J, et al. Molecular mechanisms and new treatment paradigm for atrial fibrillation. Circ Arrhythm Electrophysiol 2016; 9:e003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Glinge C, Sattler S, Jabbari R, Tfelt-Hansen J. Epidemiology and genetics of ventricular fibrillation during acute myocardial infarction. J Geriatr Cardiol 2016; 13:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, et al. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A 2007; 104: 9018–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol 2003; 284:R893–R912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.