

Abstract
In the pursuit of continuous improvement in the area of biomaterial design, blends of mixed-substituent polyphosphazenes and poly (lactic acid-glycolic acid) (PLGA) were prepared, and their morphology of phase distributions for the first time was studied. The degradation mechanism and osteocompatibility of the blends were also evaluated for their use as regenerative materials. Poly [(ethyl phenylalanato)25(glycine ethyl glycinato)75phosphazene](PNEPAGEG) and poly [(glycine ethyl glycinato)75(phenylphenoxy)25phosphazene](PNGEGPhPh) were blended with PLGA at various weight ratios to yield different blends, namely PNEPAGEG-PLGA 25:75, PNEPAGEG-PLGA 50:50, PNGEGPhPh-PLGA 25:75, and PNGEGPhPh-PLGA 50:50. The molecular interactions, domain sizes, and phase distributions of the blends were confirmed by atomic force microscopy (AFM) as the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends showed different domain sizes and phase distributions. Due to the extensive hydrogen bonding within the two polymer components, PNEPAGEG-PLGA exhibited small-sized domains and well-distributed morphology. While for the PNGEGPhPh-PLGA blends, the presence of phenylphenol (PhPh) caused the formation of PLGA large-sized domains as the PLGA formed a continuous phase and PNGEGPhPh constituted a dispersed phase. In addition to AFM results, scanning electron microscopy-energy dispersive X-ray spectrometry (SEM-EDS), differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA), and Fourier transform infrared spectroscopy (FTIR) results demonstrated the miscibility of the blends. The PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends presented in situ 3D interconnected porous structures upon degradation in phosphate-buffered saline (PBS) media at 37°C. However, the blends showed different mechanistic pathways to the formations of the pores. The difference in the erosion patterns could be attributed to the nature of molecular attractions that exist in the blends. Furthermore, the novel blends were able to support cell growth as compared to PLGA, and accommodate cell infiltrations, which ultimately augmented surface area for better cell-material interactions.
Keywords: regenerative biomaterials, biodegradable polymers, polyphosphazenes, polymer blends, poly (lactic-co-glycolic acid), biocompatible polymers
Graphic ABSTRACT:

INTRODUCTION
PLGA and its derivatives are FDA-approved materials used for many medically-related applications, and as such, they have been extensively investigated for regenerative engineering1–4. However, PLGA and formulations are not ideal biomaterial platforms and face limitations because of the lingering issues of acidic degradation products arising from bulk erosion4–7. The local accumulation of these acidic products (lactic and glycolic acids) often elicits a prolonged inflammatory response in the tissue microenvironment surrounding the implant, which may result in sudden structural failures5, 8. Also, the rise of regenerative engineering and its complexity and demands have caused a paradigm shift and stimulated innovation in the design of new biomaterials with diverse properties that can meet the ever-changing requirements of this approach9, 10. So an ideal polymeric biomaterial should possess excellent initial mechanical properties, appropriate degradability, neutral degradation products, and the ability to present interconnected porous structures for cell infiltration, tissue in-growth, and vascularization4, 11. However, no polymers have satisfied all the listed criteria. Since biomaterials are not a “one-size-fits-all” system, materials with a wide range of degradation rates and physicochemical properties are sought after. Hence, polyphosphazene-PLGA blends are gaining more attention from the materials science and engineering community as emerging biomaterials for tissue regeneration12–15. The blend of degradable polyphosphazene and PLGA would potentially combine the beneficial features of these two polymers16. The design flexibility, property tunability, and buffering degradation products of degradable polyphosphazene merged with the excellent properties of PLGA could lead to the development of a set of biomaterials with appropriate and desirable properties.
The objective of the present study was to develop polyphosphazene-PLGA blends using two different co-substituted polyphosphazenes (PNEPAGEG and PNGEGPhPh) and evaluate the molecular interactions within the phases, and access how the interactions would influence the physicochemical properties and degradation kinetics. To the best of our knowledge, this is the first time that a study on the molecular interactions and phase distributions morphology of a polyphosphazene-based blend is being carried out.
Also, in our previous study, the model for polyphosphazene-PLGA erosion was proposed to occur in three stages17. However, the present study reevaluated the erosion mechanism with two different kinds of polyphosphazene-PLGA blends (PNEPAGEG-PLGA and PNGEGPhPh-PLGA) and established the degradation mechanisms for each blend. The two blends were able to exhibit inherent pore-forming tendencies upon degradation, but each of them took a different erosion pathway to attain the desired porosity. As evidenced in SEM and FTIR results, the PNGEGPhPh-PLGA followed the three-step mode of degradation, whereas PNEPAGEG-PLGA showed a different degradation pattern that bypassed the stage of the formation of spheres through intramolecular hydrogen bonding between polyphosphazene chains.
Furthermore, the two blend systems demonstrated excellent osteocompatibility with degradation products whose pH values were higher than that of the pristine PLGA.
Generally, the results of this study revealed that the phase distribution morphology of polyphosphazene-PLGA blends and their degradation kinetics that led to the formation of 3D interconnected porous structures are dependent upon the side groups of the polyphosphazene components used and unique intermolecular and intramolecular hydrogen bonding interactions within the blends. Finally, with these evolving discoveries and widening landscape, polyphosphazene-based blends are expected to be the future of biomaterials and an advancing polymeric system that may eventually replace traditional ones such as PLGA for biomedical uses.
EXPERIMENTAL SECTION
Materials.
PNEPAGEG and PNGEGPhPh were synthesized as in our previous study18. PLGA (50:50, MW = 60800 g/mol) was purchased from Absorbables-Durect corporation and used as received. Chloroform and Tetrahydrofuran (THF) were obtained from Sigma-Aldrich and used without further purification.
Preparation of the blend.
Blends of PNEPAGEG-PLGA and PNGEGPhPh-PLGA were prepared using a mutual solvent, and test specimens were obtained from the fabricated blends (Table 1 & Figure S1). PLGA films were also produced with the same solvent and were used as controls. Briefly, PNEPAGEG/PNGEGPhPh and PLGA with the following weight ratios of PNEPAGEG/PNGEGPhPh to PLGA, 50:50, and 25:75 totaling 5g in weight were dissolved in 25ml of chloroform to obtain a homogeneous solution. The samples of the polymer solutions were subsequently poured into Teflon-lined Petri dishes, and then the solvent was allowed to evaporate slowly for 48 hrs. The blend films were further dried under vacuum for 48hrs, and film thickness around 0.5 mm was obtained for all films, including PLGA films, which were fabricated using the same procedures as those of the blends. Blends with more than 50% of PNEPAGEG or PNGEGPhPh could not mix properly and were exhibiting significant phase separations.
Table 1.
Blends fabricated using a mutual solvent approach
| PNEPAGEG : PLGA | PNGEGPhPh : PLGA |
|---|---|
| 25 : 75 | 25 : 75 |
| 50 : 50 | 50 : 50 |
Miscibility, surface morphology, and molecular interactions.
SEM-EDS –
Specimens for the blends were obtained from the various films, and their surfaces were sputter-coated with gold/palladium (Au/Pd) using a Hummer V sputtering system. The Au/Pd-coated samples were examined under an FEI Nova NanoSEM 450 to check their phase miscibility. Also, elemental mapping at the microstructural level was performed on the samples to access the phosphorus (P) and Nitrogen (N) distribution on the blends.
FTIR analysis –
FTIR spectrometer was used to analyze the blend films and to identify the formation and the nature of new physical interactions such as intermolecular and intramolecular hydrogen bonds. The analysis was operated in the wavelength range of 500–4000 cm−1 with a resolution of 5 wavenumbers and an average of 16 scans.
AFM analysis –
Atomic force microscopy was performed in tapping mode with an Asylum Research MFP-3D using Nanosensors PPP-FMAu-10 probes. The nominal scan parameters include a line rate from 0.75Hz to 1 Hz, set-point that is 60% - 80% of the free amplitude, and 256 × 256 pixel resolution. For each sample, phase, amplitude, and height images were acquired with specimen dimensions of 4μ × 4μ. The morphology, molecular interactions, and phase distributions of the blends were evaluated, and topographical pictures were taken. The phase-distance curves and histograms of the size distributions of the samples were also recorded.
DSC –
The glass transition temperatures (Tg) of the blends and polymer components were determined using a TA instruments DSC Q250 unit with Thermal Universal Analysis software. Briefly, samples of approximately 10mg were heated from −50°C to 130°C at heating and cooling rates of 10°c/min and 5°c/min respectively under 50ml/min of nitrogen gas and the resultant curves (heat flow versus temperature) were analyzed. The Tg was determined from the middle of the sloped region where there is a change in the heat capacity of the thermograms, and the third cycles for each specimen were utilized for the Tg measurement.
DMA –
Dynamic mechanical testing was conducted in tension on the blend samples with a DMA Q800 unit. The measurements were made using two different methods (temperature and force ramps) with specimen dimensions of L 12.81 mm, W 6.27 mm, T 0.13 mm. For the temperature ramp, the polymer films were subjected to a temperature range of from −30 to 80 °C at a constant strain amplitude of 0.1% with a heating rate of 3°C/min. Storage modulus-temperature curves were obtained and used to determine the brittle-ductile transitions and, ultimately, the miscibility of the blends. For the force ramp, stress-strain curves were generated at a constant temperature of 36 °C, and the ultimate tensile strength and elongation at break were calculated.
Contact angle measurement –
The contact angles of droplets of distilled deionized (DI) water on the surface of PLGA and blend films were determined using a Digital Contact Angle Measurement System equipped with a CCD camera (CAM 100 series, KSV instruments). The contact angle values were obtained from the temporal images of the water droplet on various sample surfaces.
Degradation and erosion mechanism.
Blend disks with dimensions of 0.5mm × 10mm (T × D) and approximate weight of 100 mg each were incubated in 10 ml of phosphate-buffered saline (PBS) at pH 7.4. The vials were maintained at 37°C in a water bath equipped with a mechanical shaker for 12 weeks at 250 rpm. At specific time points (2, 4, 7, 10, and 12 weeks), the samples were removed from PBS and were dried under vacuum for a week. The pH of the PBS media for each sample was recorded at various time points using a pH meter, and the percentage mass remaining with respect to time was calculated from the following equation.
Wt is the dry weight of the blend sample at each predetermined time point, and Wo is the initial dry weight of the sample before it was immersed in PBS media. IR spectra of the degrading materials were recorded using an FTIR spectrometer to monitor the formation and breakdown of bonds in the course of degradation of the materials. The morphological changes of the blend samples during degradation at various time points were visualized using SEM, and secondary electron images were obtained. The final compositions of the blends after the degradation study were ascertained using electrospray ionization mass spectrometry (ESI-MS). The ESI-MS data were acquired on a “Waters Q-ToF Premier” in positive ionization mode with external calibration using flow-injection analysis. The samples were dissolved in chloroform and infused at 20 μm per minute through the integrated syringe pump into the 0.1ml/min flow of methanol delivered by a Waters Alliance 2695 pump.
In Vitro Osteocompatibility.
Cell seeding –
The blend and PLGA films with dimensions of 0.5 mm × 10 mm (T × D) were subjected to pretreatment and sterilization processes by lyophilizing the samples in a lyophilizer for 48 hrs. They were then exposed to UV light for 15 minutes on each side and immersed in cell culture media (α-MEM) overnight. Osteoblast-like MC3T3-E1 cells (obtained from the Calvaria of a neonatal mouse) were employed in this study because MC3T3 cells are widely used in the evaluation of osteocompatibility. The cells were seeded onto the polymers and the PLGA control at a density of 3 × 104 cells per film. The cell-seeded materials were cultured in α-MEM supplemented with 10% FBS, 1% penicillin-streptomycin, and maintained in an incubator at 37°C with 5% CO2 and 95% humidified air for 1, 3, 7, 14, and 21 days.
Cell proliferation –
The quantitative analysis of the cell proliferation on the films was carried out with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium) (MTS, Promega) mitochondrial reduction. The basis for this assay is that it measures the ability of metabolically active cells to change tetrazolium-based compound, MTS, to a purple formazan product. The intensity of the resulting colored solution at an absorbance of 490 nm corresponds to the number of viable cells. Cells seeded onto the tissue-culture polystyrene (TCP) served as a positive control. Briefly, at each predetermined time point, the cell-seeded films were washed with PBS and transferred to a new 48-well tissue culture plate, and a mixture of the culture medium and MTS substrate (5:1) was added and incubated for 2hrs in a humidified atmosphere with 5% CO2 at 37°C. At the end of the 2hr-long incubation, the absorbance of the resulting solution was read at 490 nm using a Tecan spectraFluo Plus plate reader.
Cell morphology –
At days 1, 7, 14, and 21, cell-seeded films were taken out of the culture plate, transferred into a new 24-well plate, and gently washed with 1 mL of PBS to remove unattached cells. Cells on the films were fixed with 1% glutaraldehyde (1 mL) for 1hr at room temperature, and with 3% glutaraldehyde (1 mL) for 24hrs at 4°C. The films were then dehydrated sequentially using an increasing concentration of ethanol (30% 50%, 70%, 90%, and 100%) for 10 minutes each (1 mL quantity). The films were allowed to air-dry overnight at room temperature and stored in a desiccator for future use. For SEM visualization of cell morphology, the samples were sputter-coated with Au/Pd and examined under an FEI Nova NanoSEM 450.
Live/dead cell viability –
The viability of MC3T3 cells on the polymer films was examined using a Live/dead cell viability kit. In brief, the interaction of calcine AM and intracellular esterase of live cells results in bright green fluorescence. Whereas, ethidium homodimer-1 interacts with only dead cells with damaged membranes and binds up to their nucleic acids to produce a bright red fluorescence. The polymer samples were imaged at 2, 7, and 14 days using a Laser Scanning Confocal Microscope. (Zeiss LSCM 880).
Statistical Analysis.
All analyses were performed in triplicate or more per sample and quantitative data were presented as mean ± standard deviation (n ≥ 3). Statistical analysis was performed using Microsoft excel. The comparisons of the means were performed using a two sample t test with a significant difference of p < 0.05.
RESULTS AND DISCUSSION
Polyphosphazenes co-substituted with dipeptide side groups provide additional hydrogen bonding sites for intermolecular bonding with the carbonyl of the PLGA to ensure miscibility (Figure 1). The phenylalanine ethyl ester (EPA) and PhPh in PNEPAGEG and PNGEGPhPh respectively present hydrophobic features that can be utilized in fine-tuning the physicochemical and thermo-mechanical properties of the overall blend systems. This is due to the large aryloxy functional groups in phenylalanine and phenylphenol which are capable of π-π stacking19–21.
Figure 1.
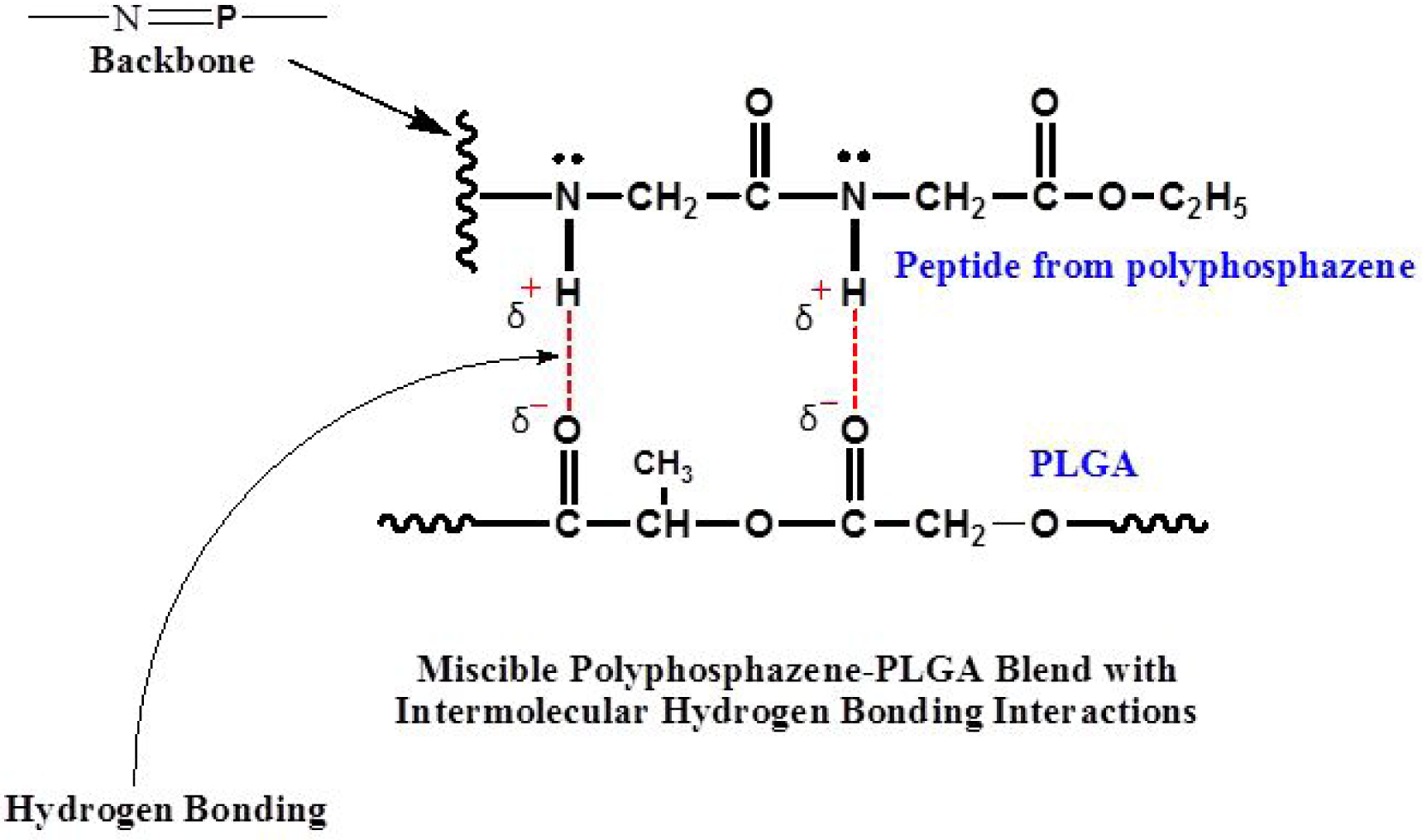
Intermolecular hydrogen bonding between the carbonyl of PLGA and glycylglycine dipeptide of the polyphosphazene enhances blend miscibility.
Miscibility, surface morphology, and molecular interactions.
Miscibility is essential in creating a polymer blend with uniform and expectable properties3. Analytical techniques such as AFM, SEM-EDS, FTIR, DSC, and DMA were employed to investigate the miscibility, surface morphology, and molecular interactions of the fabricated blends. Phase separation often occurs when a blend is immiscible or partially miscible22–24. As shown in Figure S1, the blends yielded uniform and smooth surfaces without any visual phase separation. The PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends exhibited light yellow and yellow coloration, respectively, as opposed to the colorless PLGA. The SEM images in Figure S2 confirmed the miscibility of the four blends as no phase separation was observed.
Even though the SEM images showed homogeneous material phases for all bend systems, but analysis by AFM showed that PNEPAGEG-PLGA and PNGEGPhPh-PLGA have different morphologies in the nanoscale. It was found that the AFM could visualize different phase distributions and domain sizes due to the molecular interactions between the PLGA molecules and side groups of the polyphosphazenes. As shown in Figure S3, AFM phase images are generated based on the variations of surface stiffness arising from the differences in viscoelastic properties between components of the blends. The distribution of phases in the blends is fully illustrated in the phase images. The results of the amplitude and height topographies are quite similar to that of the phase imaging as the deflections of the probes and penetrations of the cantilever tips due to the differences in local stiffness of the blends constitute the amplitude and height image contrasts respectively (Figure S4). PNEPAGEG-PLGA blends exhibited small-sized domains and well-distributed phase morphology due to their extensive hydrogen-bonding interactions. The extensive hydrogen bonding arises from the existence of more significant numbers of hydrogen bonding sites provided by glycylglycine ethyl ester and phenylalanine ethyl ester side groups of PNEPAGEG (Figure 2). For the PNGEGPhPh-PLGA blends, the presence of PhPh caused the formation of PLGA large-sized domains as PLGA formed a continuous phase, and PNGEGPhPh constituted the dispersed phase. The EPA has a higher number of hydrogen bonding sites than the PhPh, and thus PNEPAGEG polymers could form a better network of hydrogen-bonded molecules with PLGA. Besides the physical interactions provided by hydrogen bonds, there exists another interaction due to the similarity in functionalities. Ester-ester interactions could have some influence on the phase distributions in the blend. In other words, the refinement of the blends’ morphology is controlled by the compatibilizing effects of two interacting forces, such as intermolecular hydrogen bonding and ester-ester interactions. It is worth mentioning that EPA, unlike the PhPh, possesses ester functionalities for interactions with esters of PLGA and glycylglycine ethyl ester. These ester-ester interactions may have contributed to the well-distributed morphology of PNEPAGEG-PLGA blends. The graphical cross-section analyses represented in Figure S5 revealed topographic structures where, for PNEPAGEG-PLGA blends, the phases were evenly distributed, whereas, in the case of PNGEGPhPh-PLGA blends, homogeneity was less. Also, PNGEGPhPh-PLGA exhibited a narrow and sharp histogram, which signifies the definite difference between the distributions of sizes of the domains (Figure S6). The PNGEGPhPh-PLGA phases were preferably isolated from each other, while there was a continuous phase distribution for PNEPAGEG-PLGA whose histogram was broad. This means that there is a high distribution in the size of the domains/phases in PNEPAGEG-PLGA. These trends in the phase distributions of the blends conform to the results of the EDS elemental mapping, where the elements including the P and N were well distributed in the PNEPAGEG-PLGA blends while there is less distribution of P and N in PNGEGPhPh-PLGA blends (Figure S7).
Figure 2.
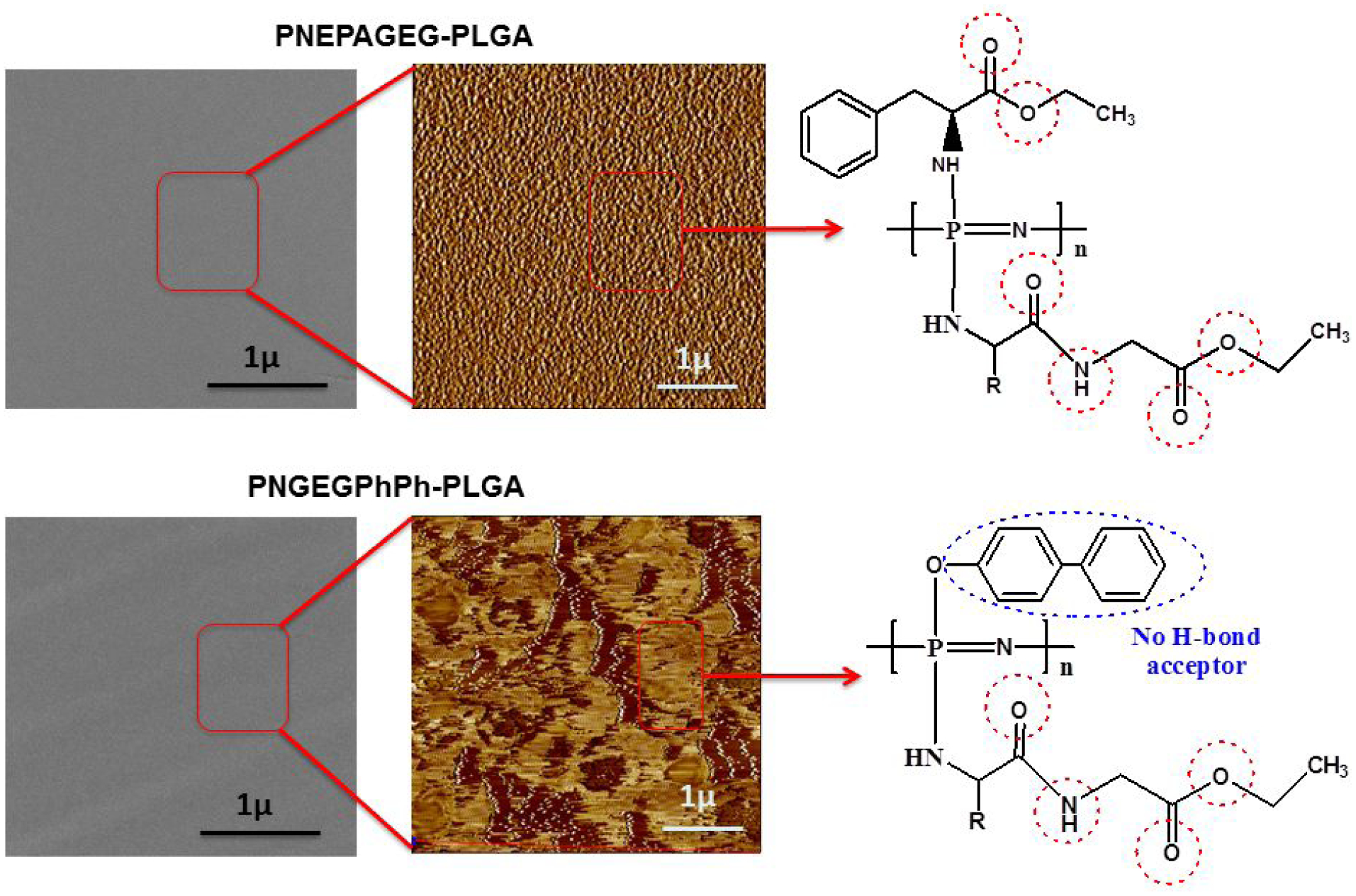
An image showing the molecular interactions of phases in the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends. There is no visual phase separation in the blends by SEM images and as such PNEPAGEG-PLGA and PNGEGPhPh-PLGA were miscible blends. AFM images indicate the phase distributions in the blends, and it appears that H-bonding and some degree of ester-ester interactions were the compatibilizing forces.
FTIR –
As shown in Figure S8, intermolecular hydrogen bonding was confirmed with a shift of C=O band from ~ 1750 to ~1739 cm−1 for PNEPAGEG-PLGA blends. The intensity of the C=O peak decreased as the amount of the PNEPAGEG increased (Figure S8a). The blends showed additional bands at ~1670 cm−1, which corresponds to the hydrogen-bonded carbonyl groups. Also, there was a disappearance of the intramolecular hydrogen-bonded secondary NH in PNEPAGEG polymers and the formation of intermolecular hydrogen-bonded secondary NH in the PNEPAGEG-PLGA blends (Figure S8b). The same band shift at 1750 cm−1 is also applicable to the PNGEGPhPh-PLGA blends, and there was a drastic drop in the intensity of the peaks as the PNGEGPhPh content was increased (Figure S9a). The hydrogen-bonded carbonyl groups at 1675 cm−1 of the PNGEGPhPh-PLGA blends were also observed for the both PNGEGPhPh-PLGA 25:75 and PNGEGPhPh-PLGA 50:50 blends. Furthermore, the PNGEGPhPh-PLGA blends showed the formation of strong (medium) and broad bands at 3300 cm−1, which corresponds to the intermolecular hydrogen-bonded secondary amine (Figure S9b). Before blending, weak and broad bands that correspond to intramolecular hydrogen-bonded secondary amines in the polyphosphazene (peptides) components were seen. For the fingerprint regions of the blends shown in Figure S10, sharp absorption bands at 697 cm−1 correspond to intramolecular H-bonded secondary amine within the side groups of polyphosphazene polymers. The incorporation of PLGA weakened the bands indicating the formation of intermolecular hydrogen bonds between the secondary amine of the polyphosphazenes’ peptide molecules and the carbonyl groups of PLGA. Therefore, the formation of intermolecular hydrogen bonds in the blends suggests that the blends were miscible.
DSC –
DSC thermograms for the parent polymers and the blends are shown in Figure 3. The results indicate that the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends exhibited single Tgs, which were intermediate between those of the two constituent polymers. The detection of single Tgs by the blends signifies miscibility, and the respective Tgs of the blends were above the physiological temperature (37°C). It was also found that the Tg increased with an increase in the content of the polyphosphazene components. This trend is expected and in agreement with our previous results.
Figure 3.

DSC thermograms showing that the blends exhibited single glass transition temperatures which are the average of that of the PLGA and polyphosphazene components, and increased with increasing polyphosphazene compositions. The generation of single glass transition temperatures by the blends signifies miscibility.
DMA –
The DMA technique usually supplements the information provided by the other techniques such as DSC. Figure S11a shows the storage modulus-temperature curves for the blends and PLGA generated from the DMA testing. The glass transition temperature of a material could be determined from the onset of the storage modulus drop. The blends presented a single Tg value indicating that the blends were totally miscible. Similar to the DSC analysis, there were linear relationships between the Tg of the blends and the polyphosphazene compositions. The Tg slightly increased as the amounts of polyphosphazene components were increased. Also, based on the stress-strain curves generated from the force ramp in Figure S11b, the blends showed enhanced mechanical properties (tensile strength and elongation) as compared to PLGA. An increase in the PNEPAGEG and PNGEGPhPh components increased the ultimate tensile strength. The extensive hydrogen bonding of the PNEPAGEG-PLGA as shown in AFM results in Figures 2 & S3 were instrumental to the high elongation at break of PNEPAGEG-PLGA blend.
Contact angle measurement –
The contact angle is a quantitative measure of the degree of hydrophilicity or hydrophobicity of a material surface, and it is inversely related to a material’s affinity for water and its capacity to undergo hydrolysis. The higher the contact angle, the less hydrophilic a material surface is, and the lower its tendency to degrade. In other words, the hydrolytic sensitivity of the side groups of the polyphosphazene polymers has significant effects on their surface contact angles and, ultimately, on their degradation rates. The results of the contact angle measurements indicated an increase in the contact angles as the PNEPAGEG and PNGEGPhPh polymers were added to the PLGA (Figure S12). This rise is presumably due to the presence of the PhPh and EPA in PNGEGPhPh and PNEPAGEG, respectively. Both side groups contain aromatic compounds that are known to be hydrophobic.
Degradation and erosion mechanism
Neutral bioactivity is highly desirable in biomaterials used for tissue regeneration, and indeed, the fabricated blends generated degradation products with pH values higher than that of the pristine PLGA. For instance, the pH of PLGA dropped drastically to 2.4 after 4 weeks, and this was attributed to the rapid PLGA hydrolysis to lactic acid and glycolic acid (Figure 4a). By contrast, the pH of the media in which the blends were placed was 62% higher than the pH of PLGA media after 4 weeks and 42% higher at the end of the12-week degradation study. These higher pH values of the blends’ degradation products suggest that the PLGA bulk degradation products were buffered to an extent by the PNEPAGEG and PNGEGPhPh hydrolysis products. This is consistent with our previous study, where it was found that ammonium phosphates generated from the hydrolytic breakdown of a polyphosphazene backbone constitute a natural buffer16, 18. The results of the percentage mass remaining shown in Figure 4b indicated that the blends retained ~12–24% of their original mass after 12 weeks of degradation, whereas PLGA encountered almost a total degradation after 7 weeks. During the first 7 weeks, the percentage mass losses for the blends were ~36–46% (Figure 4b), indicating slower degrading tendencies of the blends as compared to the PLGA. A decrease in the degradation of the blends was found as the content of the polyphosphazene component was increased. This decrease in the degradation of the blends was as a result of factors such as intermolecular and intramolecular hydrogen bonding, buffering effect of the polyphosphazenes’ degradation products, and the hydrophobic features of the side groups (EPA and PhPh) utilized in the design of PNEPAGEG and PNGEGPhPh polymers. Dismantling of the hydrogen bonds may take more time; the buffering effect or high pH will slow down hydrolysis, and the hydrophobic side groups will shield the backbone from being attacked by water and thereby retard the permeation of water molecules to the polymer backbone. An adjustment of the blends’ compositions provides efficient customization to attain suitable degradation rates. This is also consistent with the contact angle measurements in Figure S12, as changes in polyphosphazene compositions resulted in changes in hydrophobicity and contact angles, which consequently affected the overall degradation rates of the blend systems.
Figure 4.
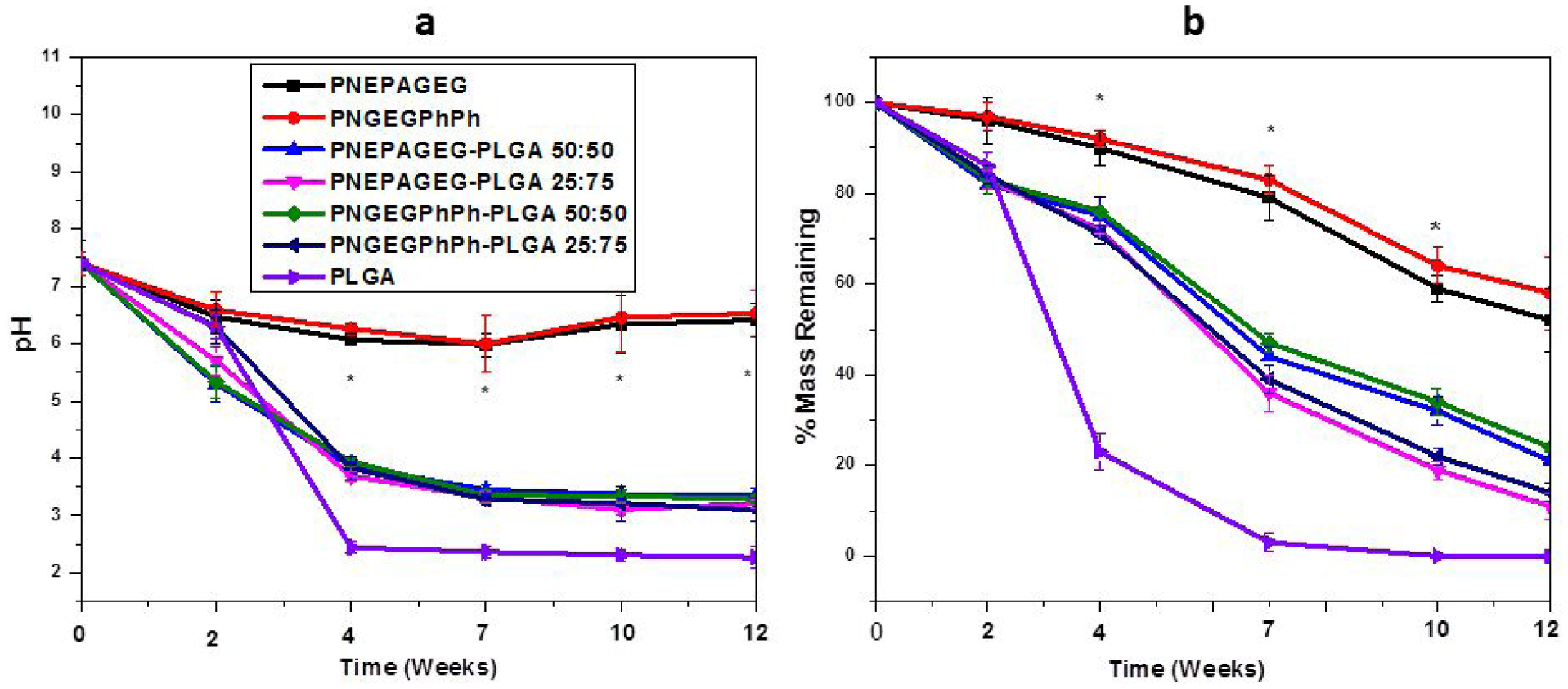
Hydrolytic degradation study of PNEPAGEG, PNGEGPhPh, PLGA and their blends in PBS medium at physiological conditions (pH 7.4, 37 °C) over 12 weeks. a) The pH of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA was higher than that of the PLGA, indicating the slight neutralization of the PLGA degradation products by the PNEPAGEG and PNGEGPhPh degradation products. b) Percentage of mass remaining of the blends and PLGA. More mass loss occurred for the PLGA than for the blends, suggesting that the presence of PNEPAGEG and PNGEGPhPh in the blends might have impeded degradation. (*) indicates a significant increase compared to PLGA. p ≥ 0.05.
Another significant thing about dipeptide-based polyphosphazene-PLGA blends lies in their inherent ability to present interconnected porosity through unique erosion. In this study, the two sets of blends were able to present the much-needed porosity through two different erosion modes. The SEM images of the blends during degradation in PBS in Figure 5 show the gradual formation of in situ 3D porous structures with respect to time. The initial morphology of the blends’ films was intact and smooth and then subsequently underwent morphological changes with time. For PNEPAGEG-PLGA, there was a manifestation of porosity after 4 weeks, and these interconnected porous structures extended to cover the whole area after 7 weeks (Figure 5a). The porosity observed in PNEPAGEG-PLGA appeared without the formation of polymer spheres, and the sizes of the pores range from macropores to micro and nanopores (<100 μm). In contrast, the PNGEGPhPh-PLGA generated its porosity based on the formation of an assemblage of microspheres that forms interconnectivity and with pore sizes less than a hundred microns (Figure 5b). Also, it was observed that resultant microspheres in PNGEGPhPh-PLGA were filled with micro and nanopores on the surface.
Figure 5.

Time-dependent morphological changes of PNEPAGEG-PLGA and PNGEGPhPh-PLGA films showing two mechanistic erosion pathways to the formation of dynamic pores when incubated in PBS at 37°C. The pore sizes were less than a hundred microns, which remained the same at all time points during degradation a) PNEPAGEG-PLGA yielded porosity upon degradation by avoiding the microsphere formation stage. b) The formation of polymer spheres is responsible for PNGEGPhPh-PLAG’s in situ porosity during degradation.
As proposed in our previous study, that this type of unique erosion process occurs in three stages, and we have further analyzed it with respect to an idealized three-step erosion process, which is as follow: 1) Breakdown of the intermolecular hydrogen bonds between the secondary amine of the peptide side group (of the polyphosphazene) and the carbonyl group of the PLGA. The permeation of water molecules and the fast degradation of PLGA trigger off the breakdown of the intermolecular hydrogen bonds between the two polymers. 2) The formation of intramolecular hydrogen bonding between the polyphosphazene molecules which may lead to the reorientation (self-assembly) of the system into polymer spheres. This reorientation is presumably due to the formation of hydrogen bonds within the polyphosphazene chains and, at the same time, the availability of the hydrophobic co-side group, which helps in temporarily keeping off water molecules. 3) The final breakdown of the intramolecular hydrogen bonds within the polyphosphazene molecules, which eventually results in complete degradation of the blend.
This unique erosion pattern is based on multi-phase degradation kinetics, where the PLGA component is expected to degrade faster than the polyphosphazene components, and hence, PLGA 50:50 was utilized. PLGA with a 50:50 monomer ratio undergoes rapid degradation because of its highly amorphous nature (low crystallinity)25–27. The amorphous nature arises from the fact that in balanced compositions of the monomers, there is a significant disruption of crystallinity as each monomer crystallizes differently with different crystal shape during production. Highly amorphous PLGA polymer, due to its high free volume, allows easy ingress of water molecules25.
The representative FTIR spectra confirm the erosion mechanism for PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends during a 12-week degradation study. For both blends at 0 weeks, there were initial intermolecular hydrogen-bonded carbonyl bands at 1670cm−1, indicating blend miscibility. After 2 weeks, the intermolecular hydrogen bonds were broken down as, evidenced by the disappearance of peaks at 1670 cm−1 for the PNEPAGEG-PLGA blends (Figure 6a). This disruption of intermolecular hydrogen bonds was also observed in PNGEGPhPh-PLGA blends (Figure 6b). A significant difference in the erosion mechanism between PNEPAGEG-PLGA and PNGEGPhPh-PLGA is witnessed after 4 weeks. The PNEPAGEG-PLGA blend did not show bands that correspond to the formation of intramolecular hydrogen bonds. In other words, there was no formation of polymer spheres because the reorientation of polyphosphazene components into microspheres in the degrading blend may require some attractive force such as intramolecular hydrogen interaction and water-repelling characteristics for it to occur. Conversely, FTIR spectra at 4 weeks and beyond for the PNGEGPhPh-PLGA blend illustrated the formation of intramolecular hydrogen bonds within PNGEGPhPh chains, leading to the self-assembly of the polymer molecules into microspheres. This is indicated in the bands around 1603 cm−1, and the intensity of the bands was found to increase until 12 weeks (Figure 6b).
Figure 6.
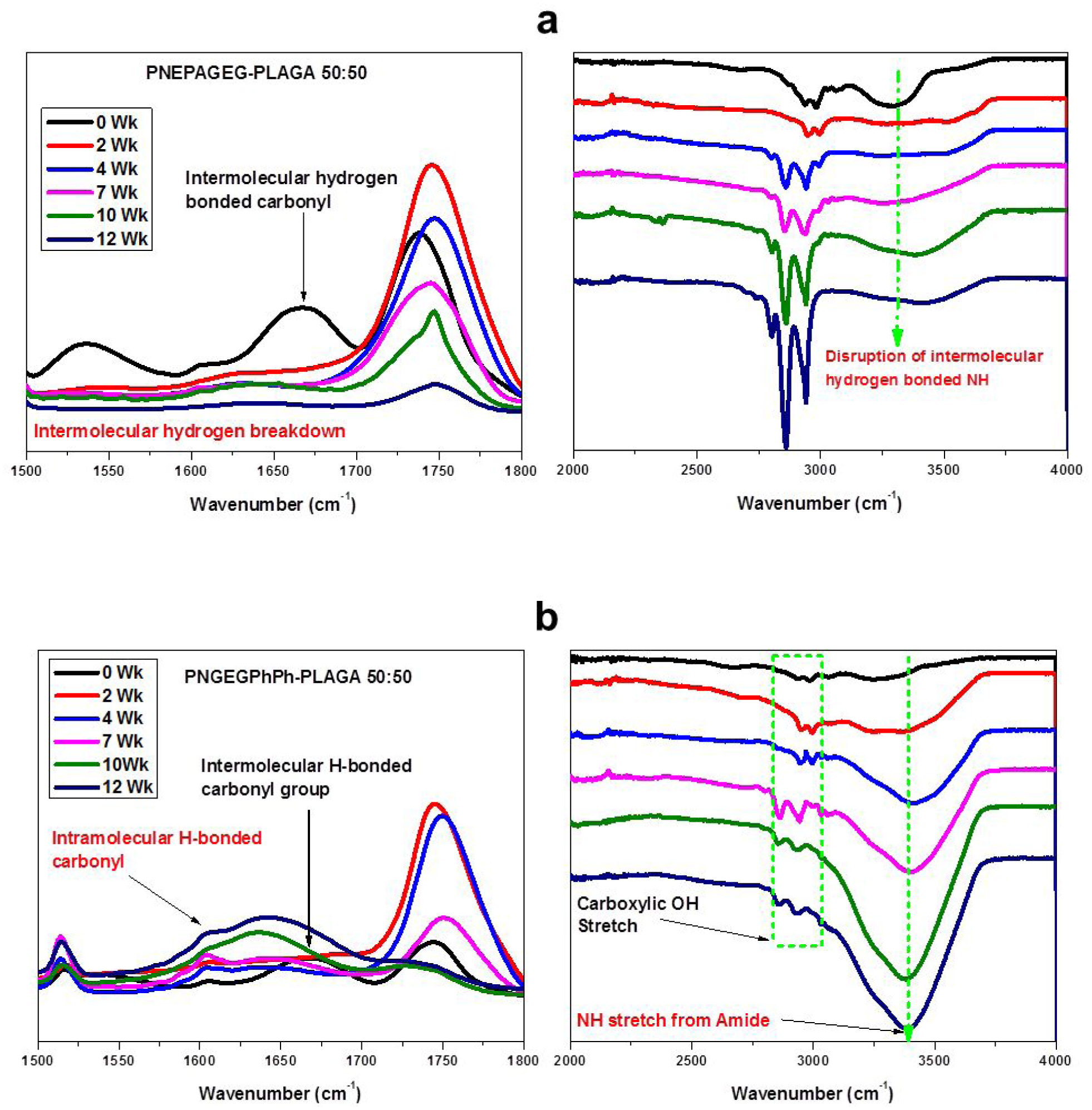
FTIR spectra dissecting the stages of the erosion mechanism. The blend erosion is controlled by intermolecular and intramolecular hydrogen bonds. a) PNEPAGEG-PLGA 50:50 b) PNGEGPhPh-PLGA 50:50.
The ESI-MS results in Figure 7 further confirmed the compositions of the microspheres formed after the 12-week degradation study. For PNGEGPhPh, several mass peaks were observed in the ESI-MS (Figure 7a), but the most intense of all was the peak at 523.2789 m/z. This particular peak at 523.2789 m/z corresponds to the PNGEGPhPh polymers since it is a common practice that the highest signal is taken as 100% abundance. Similarly, the mass spectrum for PLGA showed its major peak at 695.3990 m/z (Figure 7b), and this peak was prominent in spectra for the blend before degradation (Figure 7c) but later disappeared after the blend underwent 12-week degradation (Figure 7d). The disappearance of the PLGA peak at 695.3990 m/z and the presence of PNGEGPhPh peak at 523.2789 m/z suggest that the microspheres formed were mainly composed of PNGEGPhPh polymers. Similar results were obtained for the PNEPAGEG-PLGA blends where PLGA peak at 695.3990 m/z disappeared after 12 weeks.
Figure 7.
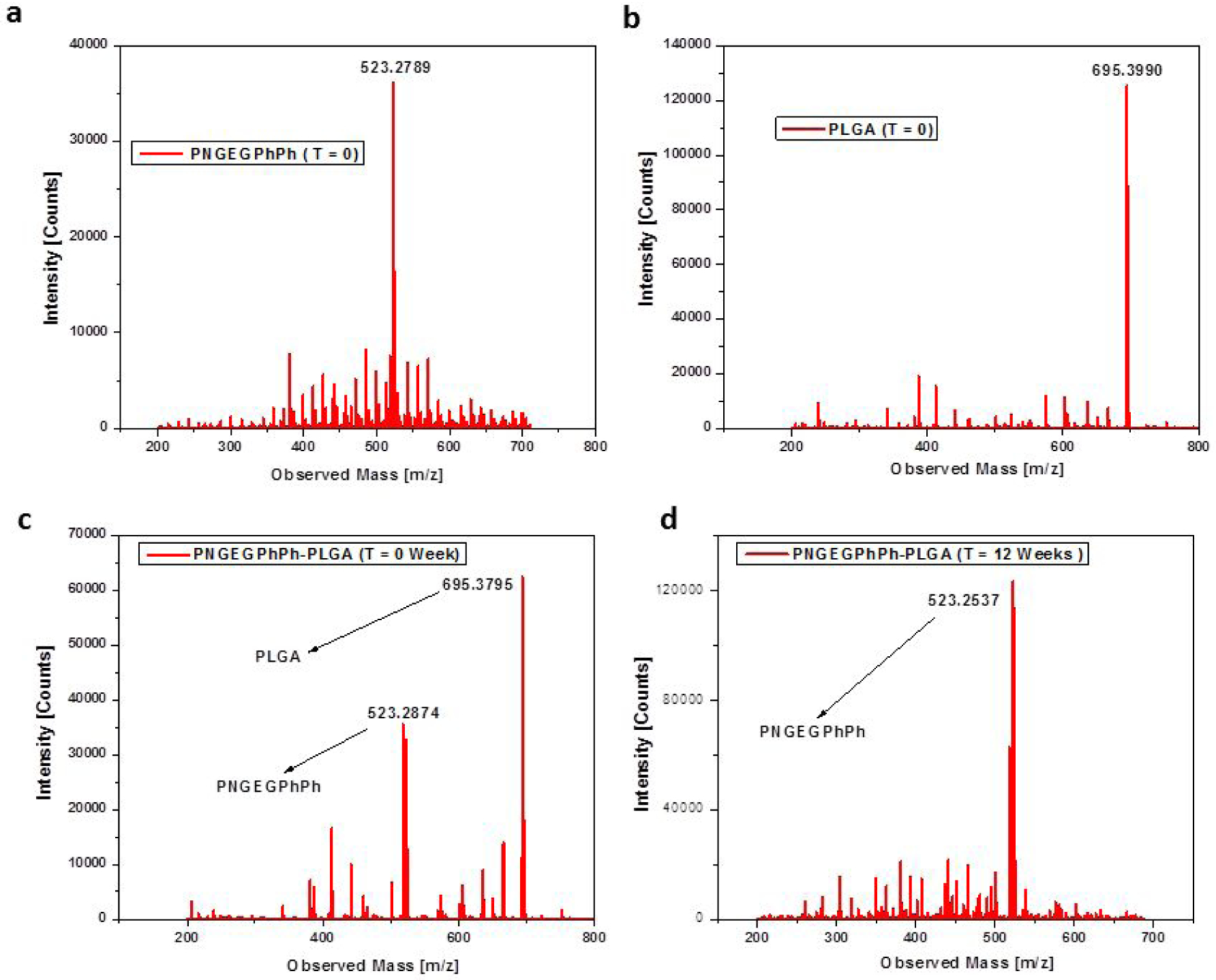
Electrospray ionization mass spectra of PNGEGPhPh, PLGA, and its blend, PNGEGPhPh-PLGA indicating that the polymer microspheres formed during degradation are mainly composed of polyphosphazene polymers. a) Mass spectrum with highest intensity at 523.2789 m/z corresponds to PNGEGPhPh b) Mass spectrum of PLGA with highest signal at 695.3990 m/z. c) Mass spectra of the PNGEGPhPh-PLGA blend before degradation showing the presence of the two polymer components. d) Mass spectrum of the blend after 12-week degradation showing that most PLGA molecules disintegrated leaving behind the PNGEGPhPh-based microspheres.
The degradation kinetics is highly dependent on the chemistry of the side groups of the polyphosphazenes and their interactions with PLGA. The use of phenylalanine amino acid ester or phenylphenol as co-substituents determines whether or not the erosion will ensue with the stage of microsphere formation. A slight adjustment in the hydrophobicity of the side groups could avert the formation of microspheres and alter the morphology of the matrix during degradation. For instance, PNGEGPhPh-PLGA with phenylphenol attached to the polyphosphazene backbone generated porosity during degradation via the formation of interconnected spheres because of the coalescence of hydrophobic phenylphenoxy groups. On the other hand, PNEPAGEG-PLGA with phenylalanine amino acid did not form spheres while degrading into porous structures because the phenylalanine amino acid is relatively less hydrophobic and has a single aryl ring. The compositions of the remaining materials in both PNEAPAGEG-PLGA and PNGEGPhPh-PLGA blends after the degradation period were mainly polyphosphazene polymers because the PLGA acted as a porogen that aided the formation of 3D voids. These 3D voids evolved into interconnected 3D pores, which promoted cell infiltration and augmented surface area for better cell-material interactions.
In Vitro Osteocompatibility.
Cell proliferation –
Based on the quantitative proliferation results from the MTS assay, the blends exhibited progressive cell growth as the cell numbers on the blends increased with culture time as compared to PLGA (Figure 8). However, the PLGA matrix showed lower osteoblast growth rates than the blends after 14 days. In other words, PLGA witnessed a small incremental change in cell number beyond 14 days due to the accumulation of its acidic degradation products. This is backed by our in vitro hydrolysis data. It was also found that an increase in the content of polyphosphazenes in the blends improved cell growth after 14 days. Moreover, TCPs presented outstanding cell growth, indicating a healthy cell population. The cell numbers on TCPS were significantly higher than that on all the polymeric matrices throughout the 21-day culture.
Figure 8.
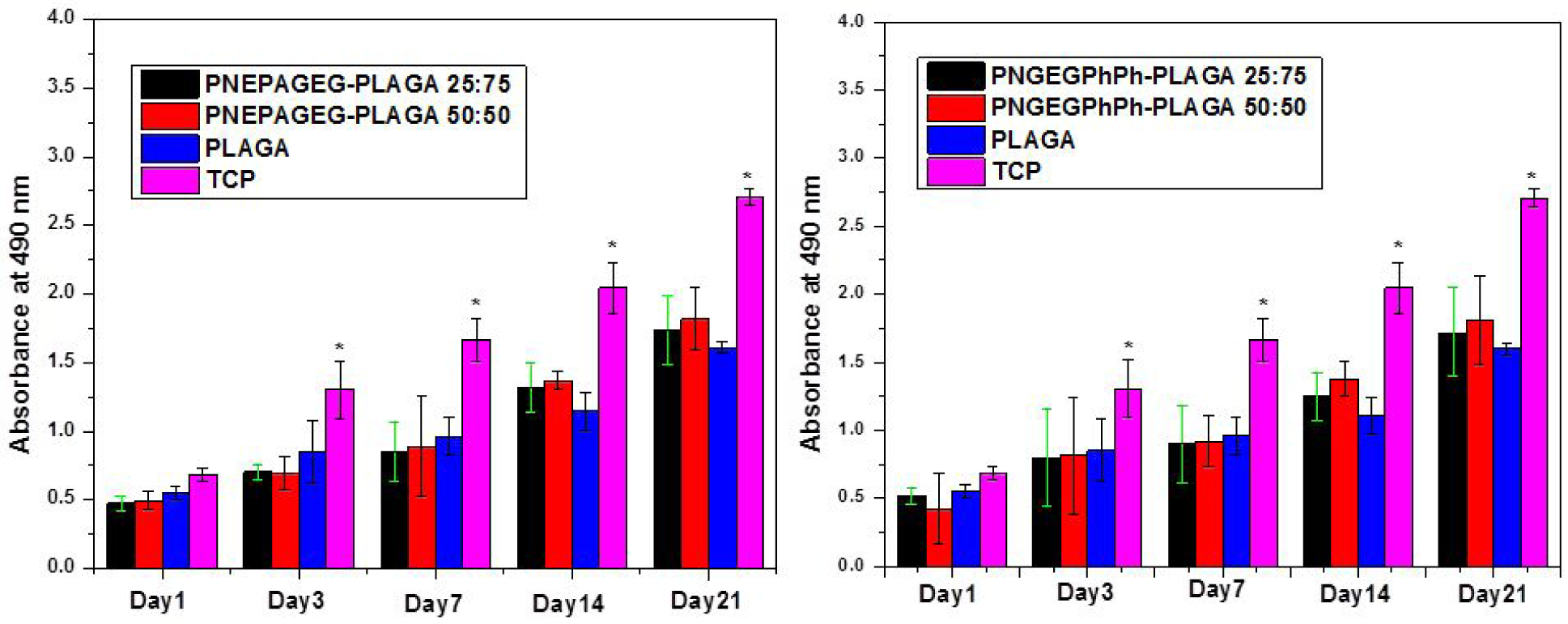
Cell proliferation measured by MTS assay. The intensity of the colored solution at 490 nm is directly related to the number of viable cells. The blends showed continuous cell growth in comparison to the pristine PLGA. (*) indicates significant increase compared to PLGA. p < 0.05.
Cell morphology –
The cell morphology by SEM illustrates a robust cell growth on the blends as the cells gradually spread out to cover the entire surface to form multilayers after 21 days of in vitro culture (Figure 9). Figure 10 shows the infiltration of cells within the micro/nanopores of the PNGEGPhPh-PLGA matrix in culture. This confirmed that the inherent porosity presented by the new blends could enhance cell infiltration, tissue in-growth, and augment the surface area for better cell-material interactions.
Figure 9.

SEM morphology of MC3T3 cells cultured on the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends after 21 days of in vitro studies. The blends supported cell attachment and growth throughout the culture period
Figure 10.

SEM micrographs demonstrating the infiltration of cells into the pores of the blend matrix during in vitro cell culture. The inherent porosity presented by the blends during degradation in the culture media could enhance cell infiltration, tissue in-growth, and surface area for better cell-material interactions.
Live/dead assay –
Live/Dead staining shows that the cells remained viable on the blends as compared to PLGA until 14 days in culture. There were sequential changes in the morphology of the cells on the matrices, as reported on the surfaces of other osteocompatible polymers28–30. The cells displayed a spindle-like morphology at day 2, whereas after 7 days, the cells appeared more elongated and attached to the matrices (Figure 11). Proliferation and SEM cell morphology results supported this observation.
Figure 11.
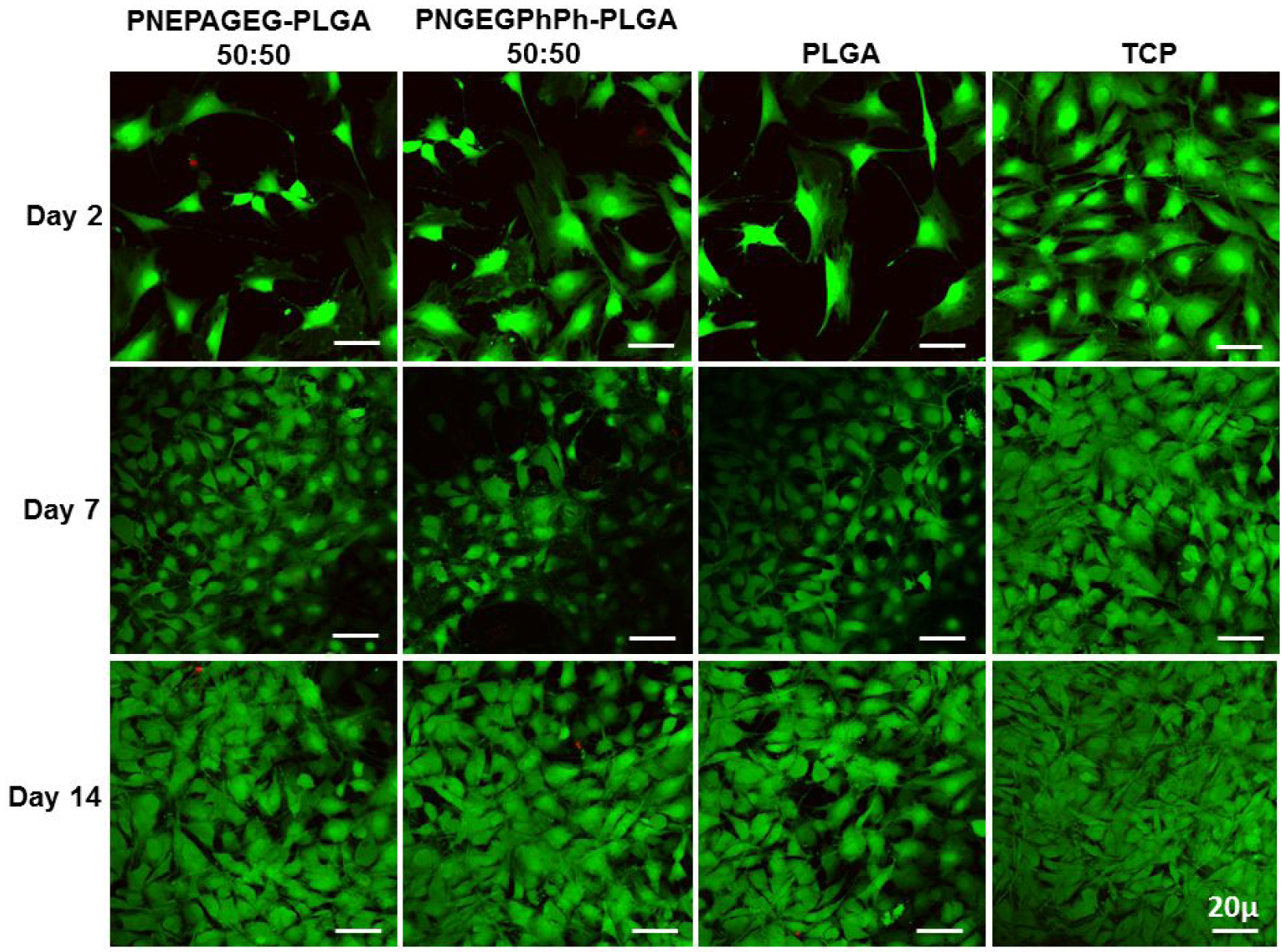
Fluorescence Live/dead staining of MC3T3 cells on PNEPAGEG-PLGA, PNGEGPhPh-PLGA, PLGA, and TCP. The results show that the cells remained viable on the blends until 14 days as compared to the PLGA. Green stains = live cells, and red stains = dead cells.
The results presented in this study have shed more light on the phase distribution morphology, molecular interactions within the blend phases, and the degradation mechanism that allows the formation of dynamic pores within the matrices. The unique intramolecular and intermolecular hydrogen bonding interactions presented by the peptide molecules of the polyphosphazene in the blends allowed the formation of miscible blends with distinct phase distribution morphologies. The chemistry of the side groups of the polyphosphazene and its interactions with PLGA at different compositions offer a platform for the design of a new class of polymeric materials that exhibit unique inherent pore-forming capacity and with a wide range of physicochemical properties.
Moreover, the blends were able to achieve near-neutral pH values during degradation as polyphosphazene degradation products stabilized the degradation products of PLGA. The self-neutralizing effects of the blends are essential for the development of tissues because neutral tissue microenvironment enhances the growth and well-being of cells and tissues. MTS, live/dead staining and cell morphology by SEM illustrated that cell adhesion, cell proliferation, and cell infiltration were well promoted on these promising blend matrices. For the first time, we successfully demonstrated the correlations between the polyphosphazene-based blends’ molecular interactions, phase distribution morphology, and degradation kinetics (which presented interconnected porosity). The critical properties of the new blends and constituent polymers are summarized in Table 2. We believe that this study will open up a wide range of possibilities in terms of manipulating biomaterials’ properties from a molecular level to achieve the desired performance.
Table 2.
Comparison of the properties of PNEPAGEG, PNGEGPhPh, pristine PLGA and their respective blends. The pH values represent the values recorded after 21 day degradation study.
| Polymer System | Tg(°C) | Tensile Strength (MPa) | Elongation at break (%) | Contact Angle (°) | Cell- Compatible | Degradation Rate | pH |
|---|---|---|---|---|---|---|---|
| PNEPAGEG | 108 | 11.2 | 1.02 | 83 | yes | Slowest | 6.4 |
| PNGEGPhPh | 95 | 6.4 | 2.5 | 89 | yes | Slowest | 6.5 |
| PNEPAGEG- PLGA 50:50 | 63 | 1.2 | 129.7 | 72 | yes | Moderate | 3.3 |
| PNGEGPhPh- PLGA 50:50 | 70 | 1.3 | 110 | 75 | yes | Moderate | 3.4 |
| PLGA | 38 | 0.5 | 87 | 52 | yes | Fastest | 2.1 |
CONCLUSION
In this study, blends of mixed-substituent polyphosphazenes (PNEPAGEG and PNGEGPhPh) and PLGA were fabricated at two weight ratios of 25:75 and 50:50 of PNEPAGEG/PNGEGPhPh to PLGA. The resulting blends, namely PNEPAGEG-PLGA 25:75, PNEPAGEG-PLGA 50:50, PNGEGPhPh-PLGA 25:75 and PNGEGPhPh-PLGA 50:50 were subjected to AFM, SEM-EDS, DSC, DMA, and FTIR analyses. Hydrogen bonding interactions were influential compatibilizing factors in these blends as they determined the miscibility, phase distribution morphology, and degradation mechanism. Extensive intermolecular hydrogen bonding through the dipeptide and amino acid esters contributed to the well-distributed morphology of PNEPAGEG-PLGA blends, and the presence of PhPh yielded different domain sizes for PNGEGPhPh-PLGA blends. Both blends exhibited inherent pore-forming abilities through two different modes of degradation with relatively higher pH values of the degradation products than that of pristine PLGA.
Furthermore, the in vitro studies indicated that the cell adhesion and proliferation on the blends were comparable to PLGA. It was discovered that the immanent 3D pore formation allowed the infiltration of the cells within the pores during culture, leading to increased material surface areas and enhanced material-cell interactions. Blends of glycylglycine ethyl ester-containing polyphosphazenes and PLGA are promising, and they represent a paradigm shift in biomaterial designs.
Supplementary Material
ACKNOWLEDGMENT
Support from NIH DP1 AR068147 and the Raymond and Beverly Sackler Center for Biomedical, Biological, Physical and Engineering Sciences is gratefully acknowledged. We thank Dr. Bryan D. Huey, Department Head, Materials Science and Engineering, and Luis Ortiz for their significant help in the AFM analysis. We also thank the following undergraduate students Riley Blumenfield and Ka-Jana Justin for their help.
Funding Sources
NIH DP1 AR068147.
ABBREVIATIONS
- PPHOS
Polyphosphazenes
- PNEPAGEG
Poly [(ethyl phenylalanato)25(glycine ethyl glycinato)75phosphazene]
- PNGEGPhPh
poly [(glycine ethyl glycinato)75(phenylphenoxy)25phosphazene](PNGEGPhPh)
- GEG
Glycylglycine ethyl ester
- EPA
Phenylalanine ethyl ester
- PhPh
Phenylphenol
- PLGA
poly (lactic-co-glycolic acid)
- AFM
Atomic Force Microscopy
- FTIR
Fourier-transform infrared spectroscopy
- DSC
Differential scanning calorimetry
- DMA
Dynamic mechanical analysis
- TGA
Thermal gravimetry analysis
- SEM
Scanning Electron Microscopy
- PBS
Phosphate-Buffered Saline
- Au/Pd
Gold/Palladium
- ESI-MS
Electrospray Ionization Mass Spectrometry
- TCP
Tissue-Culture Polystyrene
- FDA
The United States Food and Drug Administration
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting Information.
The Supporting Information is available free of charge on ACS publication website via http://pubs.acs.org
Photo images of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends and PLGA. SEM images of cell morphology on the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
AFM amplitude and height images of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
AFM histograms and graphs of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
EDS elemental mapping of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
FTIR spectra of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
DMA curves of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
Contact angle measurements of the PNEPAGEG-PLGA and PNGEGPhPh-PLGA blends.
The authors declare no competing financial interest.
REFERENCES
- 1.Allcock HR, Synthesis, Structures, and Emerging Uses for Poly(organophosphazenes). In Polyphosphazenes in Biomedicine, Engineering, and Pioneering Synthesis, American Chemical Society: 2018; Vol. 1298, pp 3–26. [Google Scholar]
- 2.Allcock HR; Morozowich NL Bioerodible Polyphosphazenes and their Medical Potential. Polym. Chem 2012, 3, 578–590. [Google Scholar]
- 3.Ogueri KS; Ivirico JLE; Nair LS; Allcock HR; Laurencin CT Biodegradable Polyphosphazene-Based Blends for Regenerative Engineering. Regener. Eng. Transl. Med 2017, 3, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogueri KS; Jafari T; Ivirico JLE; Laurencin CT Polymeric Biomaterials for Scaffold-Based Bone Regenerative Engineering. Regener. Eng. Transl. Med 2019, 5, 128–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sung H-J; Meredith C; Johnson C; Galis ZS The Effect of Scaffold Degradation Rate on Three-dimensional Cell Growth and Angiogenesis. Biomaterials 2004, 25, 5735–5742. [DOI] [PubMed] [Google Scholar]
- 6.Taylor M; Daniels A; Andriano K; Heller J Six Bioabsorbable Polymers: In Vitro Acute Toxicity of Accumulated Degradation Products. J. Appl. Biomater 1994, 5, 151–157. [DOI] [PubMed] [Google Scholar]
- 7.Fu K; Pack DW; Klibanov AM; Langer R Visual Evidence of Acidic Environment within Degrading Poly (lactic-co-glycolic acid)(PLGA) Microspheres. Pharm. Res 2000, 17, 100–106. [DOI] [PubMed] [Google Scholar]
- 8.Fabbri G; Brennan M; Manfredi M; Ban G Guided Bone Regeneration Technique in the Esthetic Zone: A Novel Approach using Resorbable PLLA-PGA Plates and Screw Fixation. A case report. Int J Periodontics Restorative Dent. 2009, 29, 543. [PubMed] [Google Scholar]
- 9.Laurencin CT; Khan Y Regenerative Engineering. Sci. Transl. Med 2012, 4, 160ed9–160ed9. [DOI] [PubMed] [Google Scholar]
- 10.Laurencin CT; Nair LS Regenerative Engineering: Approaches to Limb Regeneration and Other Grand Challenges. Regener. Eng. Transl. Med 2015, 1, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’brien FJ Biomaterials & Scaffolds for Tissue Engineering. Mater. Today 2011, 14, 88–95. [Google Scholar]
- 12.Ogueri KS; Laurencin CT Polyphosphazene-Based Biomaterials for Regenerative Engineering. In Polyphosphazenes in Biomedicine, Engineering, and Pioneering Synthesis, American Chemical Society: 2018; Vol. 1298, pp 53–75. [Google Scholar]
- 13.Allcock HR The Expanding Field of Polyphosphazene High Polymers. Dalton Trans. 2016, 45, 1856–1862. [DOI] [PubMed] [Google Scholar]
- 14.Deng M; Kumbar SG; Wan Y; Toti US; Allcock HR; Laurencin CT Polyphosphazene Polymers for Tissue Engineering: An Analysis of Material Synthesis, Characterization and Applications. Soft Matter 2010, 6, 3119–3132. [Google Scholar]
- 15.Polyphosphazene Polymer. In Encycl. Polym. Sci. Technol, pp 1–23. [Google Scholar]
- 16.Ogueri KS; Allcock HR; Laurencin CT Generational Biodegradable and Regenerative Polyphosphazene Polymers and their Blends with Poly (lactic-co-glycolic acid). Prog. Polym. Sci 2019, 101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng M; Nair LS; Nukavarapu SP; Kumbar SG; Jiang T; Weikel AL; Krogman NR; Allcock HR; Laurencin CT In situ Porous Structures: A Unique Polymer Erosion Mechanism in Biodegradable Dipeptide - based Polyphosphazene and Polyester Blends Producing Matrices for Regenerative Engineering. Adv funct mater. 2010, 20, 2794–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogueri KS; Ivirico Escobar JL; Li Z; Blumenfield R; Allcock HR; Laurencin CT Synthesis, Physicochemical Analysis and Side Group Optimization of Degradable Dipeptide-based Polyphosphazenes as Potential Regenerative Biomaterials. ACS Appl. Polym. Mater 2019, 1, 1568–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Headen TF; Howard CA; Skipper NT; Wilkinson MA; Bowron DT; Soper AK Structure of π− π Interactions in Aromatic Liquids. J. Am. Chem. Soc 2010, 132, 5735–5742. [DOI] [PubMed] [Google Scholar]
- 20.Molčanov K; Kojić-Prodić B Towards Understanding π-tacking Interactions Between Non-aromatic Rings. IUCrJ 2019, 6, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trujillo C; Sánchez - Sanz G A Study of π–π Stacking Interactions and Aromaticity in Polycyclic Aromatic Hydrocarbon/nucleobase Complexes. ChemPhysChem 2016, 17, 395–405. [DOI] [PubMed] [Google Scholar]
- 22.Markovic G; Visakh PM 1 - Polymer Blends: State of Art In Recent Developments in Polymer Macro, Micro and Nano Blends, Visakh PM; Markovic G; Pasquini D, Eds. Woodhead Publishing: 2017; pp 1–15. [Google Scholar]
- 23.Utracki LA, Polymer Blends: Fundamentals In Polypropylene: An A-Z reference, Karger-Kocsis J, Ed. Springer Netherlands: Dordrecht, 1999; pp 601–605. [Google Scholar]
- 24.Kim H; Kobayashi S; AbdurRahim MA; Zhang MJ; Khusainova A; Hillmyer MA; Abdala AA; Macosko CW Graphene/polyethylene Nanocomposites: Effect of Polyethylene Functionalization and Blending Methods. Polym. 2011, 52, 1837–1846. [Google Scholar]
- 25.Gentile P; Chiono V; Carmagnola I; Hatton PV An Overview of Poly (lactic-co-glycolic) acid (PLGA)-based Biomaterials for Bone Tissue Engineering. Int J Mol Sci. 2014, 15, 3640–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elmowafy EM; Tiboni M; Soliman ME Biocompatibility, Biodegradation and Biomedical Applications of Poly (lactic acid)/poly (lactic-co-glycolic acid) Micro and Nanoparticles. J. Pharm. Invest 2019, 1–34. [Google Scholar]
- 27.Makadia HK; Siegel SJ Poly lactic-co-glycolic acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polym. 2011, 3, 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen L; Yan C; Zheng Z Functional Polymer Surfaces for Controlling Cell Behaviors. Mater. Today 2018, 21, 38–59. [Google Scholar]
- 29.Lakard S; Herlem G; Propper A; Kastner A; Michel G; Valles-Villarreal N; Gharbi T; Fahys B Adhesion and Proliferation of Cells on New Polymers Modified Biomaterials. Bioelectrochemistry 2004, 62, 19–27. [DOI] [PubMed] [Google Scholar]
- 30.Pernagallo S; Unciti-Broceta A; Díaz-Mochón JJ; Bradley M Deciphering Cellular Morphology and Biocompatibility using Polymer Microarrays. Biomed. Mater 2008, 3, 034112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.